 Jessica Y. Morales1
Jessica Y. Morales1 Cassandra M. Young-Stubbs1
Cassandra M. Young-Stubbs1 Caroline G. Shimoura1
Caroline G. Shimoura1 William R. Kem2
William R. Kem2 Victor V. Uteshev3
Victor V. Uteshev3 Keisa W. Mathis1*
Keisa W. Mathis1*- 1Department of Physiology and Anatomy, University of North Texas (UNT) Health Science Center, Fort Worth, TX, United States
- 2Department of Pharmacology and Therapeutics, University of Florida, Gainesville, FL, United States
- 3Department of Pharmacology and Neuroscience, University of North Texas (UNT) Health Science Center, Fort Worth, TX, United States
There is a critical need for safe treatment options to control inflammation in patients with systemic lupus erythematosus (SLE) since the inflammation contributes to morbidity and mortality in advanced disease. Endogenous neuroimmune mechanisms like the cholinergic anti-inflammatory pathway can be targeted to modulate inflammation, but the ability to manipulate such pathways and reduce inflammation and end organ damage has not been fully explored in SLE. Positive allosteric modulators (PAM) are pharmacological agents that inhibit desensitization of the nicotinic acetylcholine receptor (α7-nAChR), the main anti-inflammatory feature within the cholinergic anti-inflammatory pathway, and may augment α7-dependent cholinergic tone to generate therapeutic benefits in SLE. In the current study, we hypothesize that activating the cholinergic anti-inflammatory pathway at the level of the α7-nAChR with systemic administration of a partial agonist, GTS-21, and a PAM, PNU-120596, would reduce inflammation, eliminating the associated end organ damage in a mouse model of SLE with advanced disease. Further, we hypothesize that systemic α7 ligands will have central effects and improve behavioral deficits in SLE mice. Female control (NZW) and SLE mice (NZBWF1) were administered GTS-21 or PNU-120596 subcutaneously via minipumps for 2 weeks. We found that the increased plasma dsDNA autoantibodies, splenic and renal inflammation, renal injury and hypertension usually observed in SLE mice with advanced disease at 35 weeks of age were not altered by GTS-21 or PNU-120596. The anxiety-like behavior presented in SLE mice was also not improved by GTS-21 or PNU-120596. Although no significant beneficial effects of α7 ligands were observed in SLE mice at this advanced stage, we predict that targeting this receptor earlier in the pathogenesis of the disease may prove to be efficacious and should be addressed in future studies.
Introduction
Chronic inflammation is inevitable in systemic lupus erythematosus (SLE) and it contributes to the morbidity and mortality of the disease (1–3). Nearly all organ systems are affected by inflammation during SLE. For example, there is renal injury due to nephritis that can result in hypertension as well as central nervous system inflammation that can result in unfavorable behavioral phenotypes, among others (4, 5). Because chronic inflammation is an underlying mechanism in SLE and its complications, it is a justifiable target to improve outcomes. The current standard of care in SLE patients includes glucocorticoid therapy that provides overall immunosuppression but comes with adverse side effects like weight gain, mood swings, upset stomach, headache, dizziness, insomnia, depression. The use of biologics, an important class of drugs that usually target one cytokine, has not been effective in SLE clinical trials possibly because multiple immune cells and cytokines are involved in the disease process. Therefore, there is a critical need for safe alternative treatment options to broadly control inflammation in patients with SLE.
The cholinergic anti-inflammatory pathway is a neuroimmune mechanism by which the autonomic nervous system controls inflammatory processes (6–9). The cholinergic anti-inflammatory pathway has been shown to be relevant in many disease models, including autoimmune diseases (10–17). In this pathway, stimulation of the vagus nerve increases splenic nerve activity and ultimately inhibits the release of pro-inflammatory cytokines from splenic macrophages and other immune cells. The latter requires activation of the α7 subunit-containing nicotinic acetylcholine receptor (α7-nAChR) expressed in these splenic immune cells. It is well-accepted that the α7-nAChR is a critical component of this nerve-to-spleen pathway (15); the lack of α7-nAChRs exacerbates inflammatory diseases (15, 18–21). There are no studies that dispute the protective role of α7-nAChR stimulation in reducing inflammation: in fact, the benefits of α7-nAChR stimulation are explained in studies of the classic pathway described by Kevin Tracey as well as the alternative tissue-specific pathways described by others (22–25). For these reasons, drugs that can augment activation of α7-nAChRs may prove to be a viable therapeutic option for many diseases associated with chronic inflammation, and especially SLE. Indeed, our preliminary and published studies support this hypothesis by demonstrating that sub-chronic administration (7 days) of nicotine, a non-selective α7-nAChR agonist, protected kidneys by significantly reducing renal inflammation and renal injury in a mouse model of SLE (26). However, nicotine cannot be used as a therapy in humans because of its addictive and toxic nature. Thus, a solid proof-of-principle study is required to demonstrate that non-addictive, non-toxic cholinergic agents can be as effective as nicotine in producing significant anti-inflammatory efficacy in SLE but without severe side effects. Furthermore, as these recent results are promising so it would benefit the field to determine if targeting the α7-nAChR by global means can also be beneficial in other organs like the central nervous system and alter behavior in SLE.
Positive allosteric modulators (PAM) and selective partial agonists of α7-nAChRs can be used to augment endogenous α7-mediated cholinergic tone and thus, augment α7-dependent anti-inflammatory activity. In this study, we used GTS-21, a partial agonist, and PNU-120596 (i.e., PNU), a prototypical type-II α7 PAM, to test their effects on chronic inflammation in a mouse model of SLE. The protective anti-inflammatory efficacy of these classes of α7 ligands has been established in various animal models of stroke and traumatic brain injury (27–37). Since α7-nAChRs are essential in the anti-inflammatory response within the cholinergic anti-inflammatory pathway, we hypothesize that systemic administration of α7 ligands will be a successful therapeutic strategy in SLE by reducing peripheral inflammation and improving behavior in a mouse model of SLE. Because of the ubiquitous expression of α7-nAChRs in neuronal, glial and immune tissues, selective α7 agents are uniquely positioned as promising neuroprotective, anti-inflammatory and pro-cognitive combinational treatments.
Materials and Methods
Experimental Design
Female and SLE (NZBWF1) and control (NZW/LacJ) mice were obtained from Jackson Laboratories (Bar Harbor, ME) at 4–6 weeks of age and aged in a temperature-controlled facility with free access to food and water. Control and SLE mice were randomly assigned to 3 groups each: control/saline, control/GTS, control/PNU, SLE/saline, SLE/ GTS, and SLE/PNU. At 33 weeks of age, drugs were administered subcutaneously via Alzet mini-pumps for 2 weeks. Blood samples were taken (retroorbitally, ~100 μL) and the beginning and end of the study in order to measure anti-dsDNA autoantibodies associated with SLE via ELISA and albuminuria was obtained weekly following 24 h metabolic cage collection of urine. Both anti-dsDNA autoantibodies and albuminuria are important determinants of disease severity in disease. Body weight was monitored throughout the study and at the end of experiments, animals were euthanized and tissues harvested and stored for later biochemical analysis. All animal studies utilized ARRIVE guidelines (38) and were approved by the University of North Texas Health Science Center's Institutional Animal Care and Use Committee (IACUC) in accordance with National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Drugs
GTS-21 was made in house at the University of Florida as described previously (39). PNU-120596 was purchased from Selleckchem (Houston, TX). Drugs were dissolved in dimethyl sulfoxide (DMSO; vehicle) to create an 80 mM stock solution that was kept frozen at −30°C until the day of experiments. The amount of DMSO injected in each animal did not exceed 0.5 ml/kg. Analytical data for GTS-21 including melting point have been reported previously (39). Purity was ≥98% as determined by proton NMR analysis. In the current study, GTS-21 was administered subcutaneously at a dose of 15 mg/kg/day and PNU-120596 was administered subcutaneously at a dose of 10 mg/kg/day. Drugs administered at 4–20 mg/kg/day were found effective in other studies (37, 40–42).
dsDNA Autoantibody Detection
Anti-double-stranded DNA (dsDNA) autoantibodies were measured in plasma samples acquired at the end of the study via ELISA (Alpha Diagnostic International, San Antonio, TX, USA) per the manufacturer's instructions as previously described (4, 43–46).
Assessment of Peripheral Inflammation
Splenic as well as renal cortical and medullary TNF-α expression were assessed via Western blot and normalized to total protein as previously described (46).
Indices of Renal Injury and Blood Pressure Measurements
Urine was collected throughout the study, which allowed assessment of urinary albumin by ELISA and calculation of albumin excretion rate as previously reported (26, 46, 47).
At 35 weeks of age, a subset of anesthetized mice was implanted with carotid artery catheters in order to measure mean arterial pressure before euthanization as previously described (43–47). Blood pressure was collected in conscious mice for 1.5 h between the hours of 8 am and 12 noon for 2 consecutive days.
Behavioral Assessment
Standard assays for sensorimotor and emotional deficits were conducted in a subset of mice pre- (day 0) and post- (day 15) treatment to assess behavioral effects of GTS-21 and PNU in SLE mice. Each mouse was tested at both time points to minimize the number of animals used and correlate performance in various assays. The open field (OF) test was used to evaluate sensorimotor deficits and anxiety. Anxiety was additionally evaluated in the light-dark box (LDB) test. Depression was evaluated in the tail suspension (TS) test. The following order of tests was followed in each experimental day: ≥0.5 h accommodation→OF→≥1 h rest→LDB→ ≥1 h rest→TS. In OF experiments, each mouse was placed for 11 min in an opaque chamber (40.5 × 40.5 × 30.5 cm) made of polyvinyl chloride. The spontaneous locomotion within the chamber was recorded by a video camera mounted above the chamber and analyzed by All-Maze Video Tracking Software (Orchid Scientific Ltd, Maharashtra, India). Forward locomotion in centimeters, sleep time and time spent in the chamber center were determined. After OF tests, the animals were given at least 1 h rest before subjecting them to LDB tests. In LDB experiments, each mouse was placed for 10 min in an opaque acrylic chamber (43 × 29 × 20 cm). One half of the chamber was exposed to direct light from above, while the other half was protected from light by an opaque carton lid. The amount of time the mouse spent in each chamber was recorded by a video camera mounted above the chamber. In TS experiments, each mouse was suspended in the air for 6 min by its tail taped to a plastic cord attached to a horizontal bar mounted to a stand to ensure that 10–15 cm distance separates the mouse from the surface underneath. The animal's behavior was recorded on a video camera and analyzed offline. The total immobility time was measured. Behavioral tests were initiated between the hours of 7 and 8 am, but could last throughout the daylight hours. All behavioral protocols have been approved by the IACUC at UNTHSC.
Statistical Analysis
Statistical analysis was achieved using SigmaPlot 11.0 (Systat, Richmond, CA, USA). Data are calculated as mean ± standard error of the mean. T-test or two-way ANOVA, with or without repeated measures followed by the appropriate post-hoc test, was used to determine differences between groups, with a p < 0.05 indicating a significant difference. The statistical test used for each data set is included within each figure legend.
Results
Body weight at the conclusion of the study was higher in SLE mice compared to controls, and SLE mice also exhibited splenomegaly (Table 1). GTS-21 and PNU had no effect on body weight or spleen weight. There were no differences between any of the groups in kidney weight or heart weight (Table 1; both normalized to body weight).
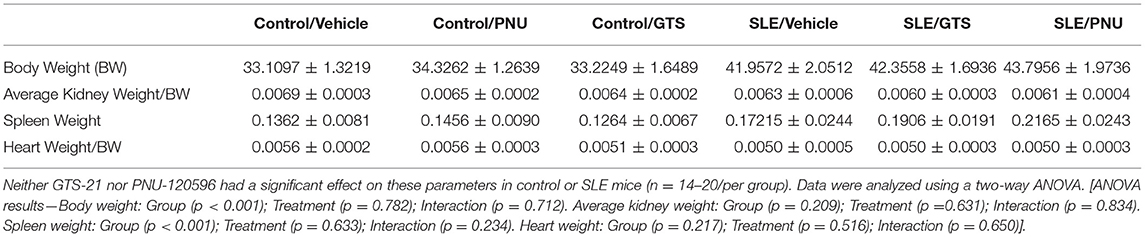
Table 1. Body and tissue weights (in grams) for control and SLE mice treated with either vehicle, GTS-21, or PNU-120596.
Disease severity, as indicated by plasma dsDNA autoantibodies (activity units), was higher in SLE mice compared to controls (Figure 1; p < 0.001). There was no significant effect of treatment (p = 0.429) and there was no interaction (p = 0.993) to allow the determination of differences within groups.
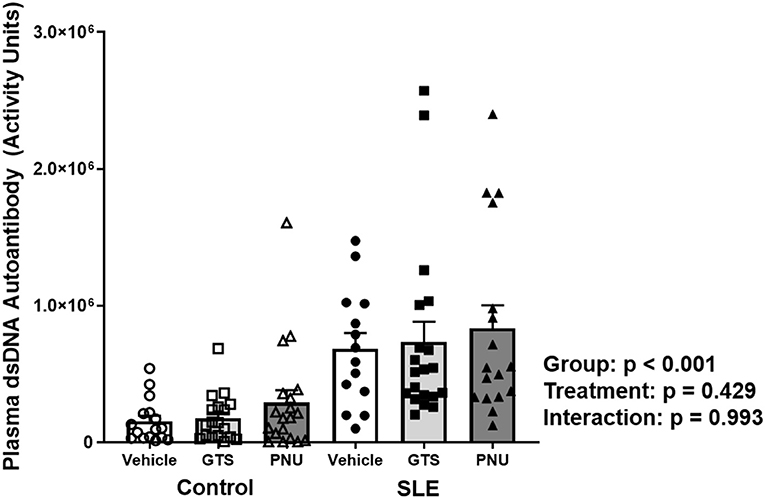
Figure 1. Plasma levels of dsDNA autoantibodies (activity units) in control and SLE mice treated with either vehicle, GTS-21, or PNU-120596. Neither GTS-21 nor PNU-120596 had a significant effect on albumin excretion rate in control or SLE mice (n = 14–20 per group). Data were analyzed using a two-way ANOVA and the results of this analysis are listed on the graph.
To test whether PNU and GTS-21 treatments had any effect on inflammation via the α7-nAChR component of the cholinergic anti-inflammatory pathway, we measured TNF-α in the spleen and kidneys. Splenic, renal cortical, and renal medullary TNF-α were higher in SLE mice compared to controls (Figures 2A–C; all p < 0.001). There was no significant effect of treatment (cortical, p = 0.877; medullary, p = 0.172; splenic, p = 0.191) for either set of data and there were no interactions to allow determination of differences within groups.
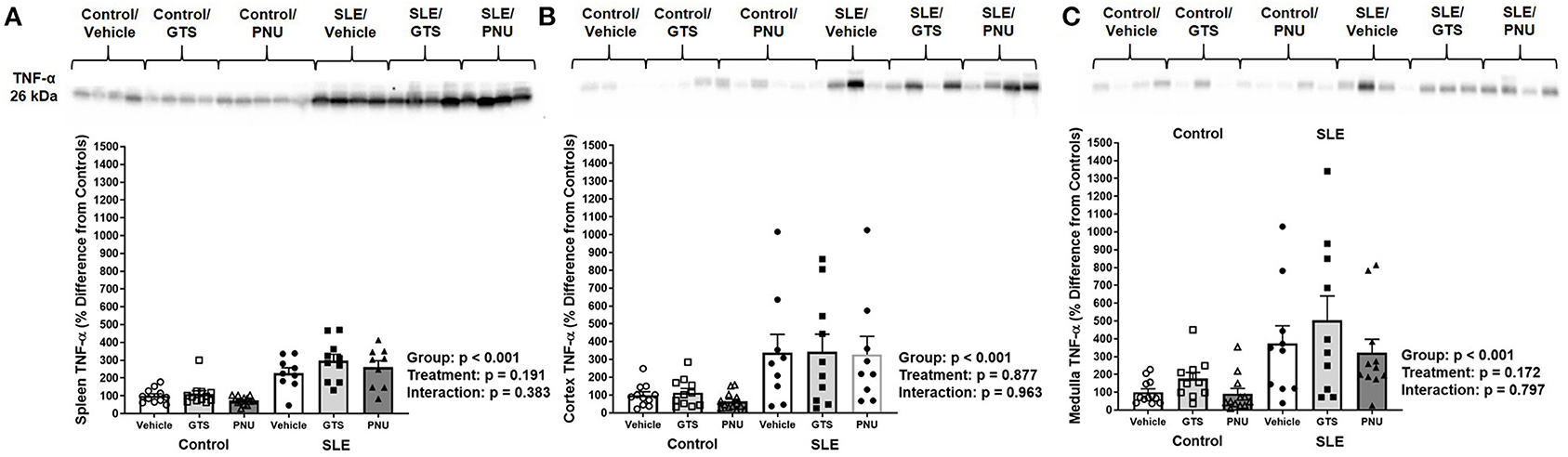
Figure 2. Splenic (A), renal cortical (B), and medullary (C) TNF-α (presented as percent difference from controls) in control and SLE mice treated with either vehicle, GTS-21, or PNU-120596. Neither GTS-21 nor PNU-120596 had a significant effect on renal or splenic inflammation in control or SLE mice (n = 9–13 per group). Data were analyzed using a two-way ANOVA and the results of this analysis are listed on the graph. Representative blots are included above each figure. The molecular weight of TNF-α is 26 kDa.
Chronic inflammation contributes to end organ damage during SLE, so we determined the effect of PNU and GTS-21 on the renal injury and hypertension that develops in SLE. Albumin excretion rate, an index of renal injury, was higher in SLE mice compared to controls (Figure 3A; p = 0.044). There was no significant effect of treatment (p = 0.439) and there was no interaction (p = 0.440) to allow the determination of differences within groups. SLE mice were hypertensive with a higher mean arterial pressure than controls (Figure 3B; p < 0.001). There was no significant effect of treatment (p = 0.775) and there was no interaction (p = 0.370) to allow the determination of differences within groups.
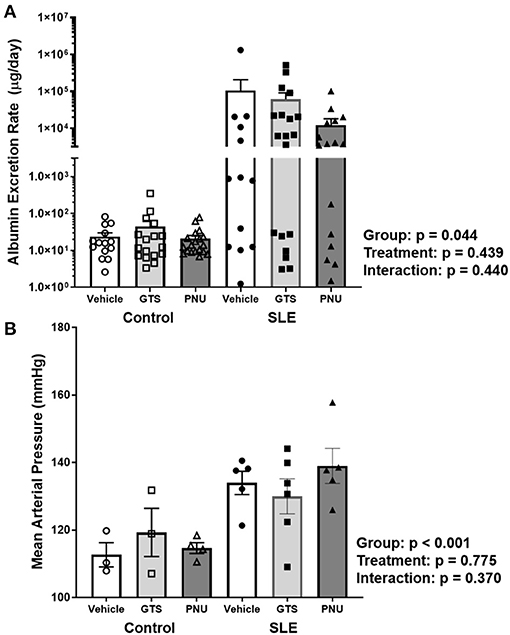
Figure 3. (A) Albumin excretion rate (mg/day) in control and SLE mice treated with either vehicle, GTS-21, or PNU-120596. Neither GTS-21 nor PNU-120596 had a significant effect on albumin excretion rate in control or SLE mice (n = 13–19 per/group) Data were analyzed using a two-way ANOVA and the results of this analysis are listed on the graph. Note: the y-axis is presented as a log scale. (B) Mean arterial pressure (mmHg) in control and SLE mice treated with either vehicle, GTS-21, or PNU-120596. Neither GTS-21 nor PNU-120596 had a significant effect on mean arterial pressure in conscious and freely moving control or SLE mice (n = 3–6 per group). Data were analyzed using a two-way ANOVA and the results of this analysis are listed on the graph.
To test anxiety like behavior in SLE and possible anxiolytic effects of PAM, animals were submitted to the open field test. SLE mice had less locomotor activity than controls pre-treatment suggesting a baseline anxiety-like behavior in SLE mice (Figure 4A; p < 0.001). While SLE mice had even less locomotor activity post-treatment (Figure 4B), PNU and GTS-21 had no significant effect in SLE mice. Since locomotion was decreased further in all SLE mice post-treatment, this suggests that age or disease progression can be responsible for the decrease in locomotor activity in these animals.
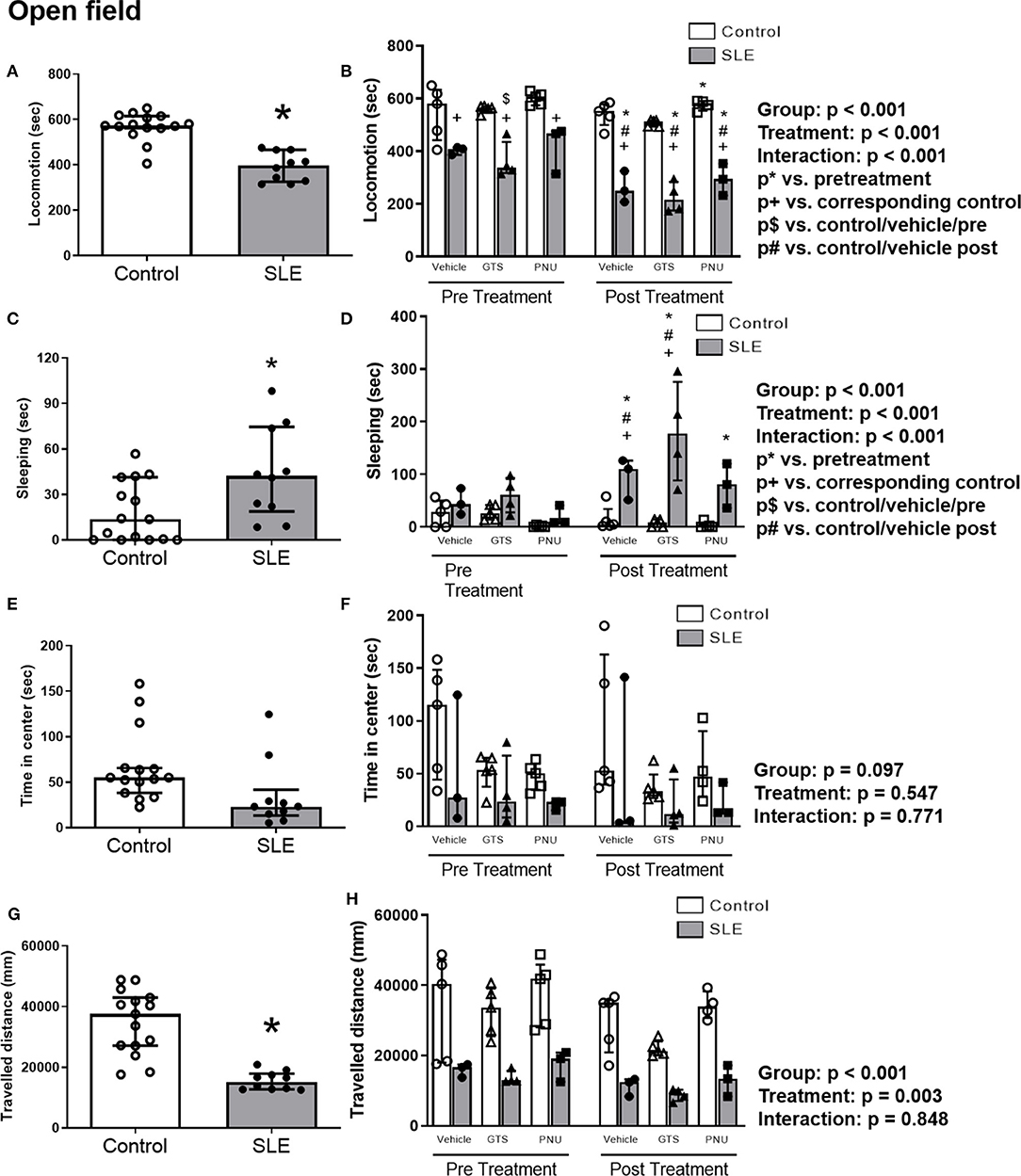
Figure 4. Open field test in control and SLE mice pre-treatment (A,C,E,G) and post-treatment with GTS-21 and PNU-120596 (B,D,F,H). The pre-treatment data is a comparison between all control and all SLE mice at baseline. A t-test was used to determine significant differences between SLE and control mice pretreatment. SLE mice had lower locomotion (A), spent a larger amount of time sleeping (C), and traveled less during the test than controls (G) (all p < 0.05). A two-way ANOVA with repeated measures was used to determine differences between all groups post-treatment (B,D,F,H; n = 3–5 per group). The results of each ANOVA appears on the graph.
SLE mice had more sleeping time during the open field test compared to controls pre-treatment (Figure 4C, p = 0.02). Sleeping time increased in SLE mice post-treatment in all treatment groups (vehicle, GTS-21 and PNU) compared to pre-treatment (Figure 4D, p < 0.05), but only vehicle- and GTS-21-treated SLE mice had higher sleeping time than corresponding controls post-treatment.
There were no differences observed across groups for time spent in center during the open field test (Figures 4E,F), but distance traveled was decreased in SLE mice compared to controls pre-treatment (Figure 4G, p < 0.001). Although there was a significant effect of treatment (p = 0.003), there was no interaction (p = 0.848) to allow determination of differences within groups in the traveled distance measurements (Figure 4H).
In order to test unconditioned anxiety response through spontaneous exploratory behaviors, animals underwent to the light-dark box test. There were no significant differences between SLE and controls pre-treatment (Figure 5A). There was also no effect of treatment (p = 0.300) and no interaction (p = 0.836; Figure 5B) to allow determination of differences within groups in % out quantification post-treatment. Although significant treatment effect (p = 0.045) there was no interaction (p = 0.777) to allow determination of differences within groups in T-out entrance.
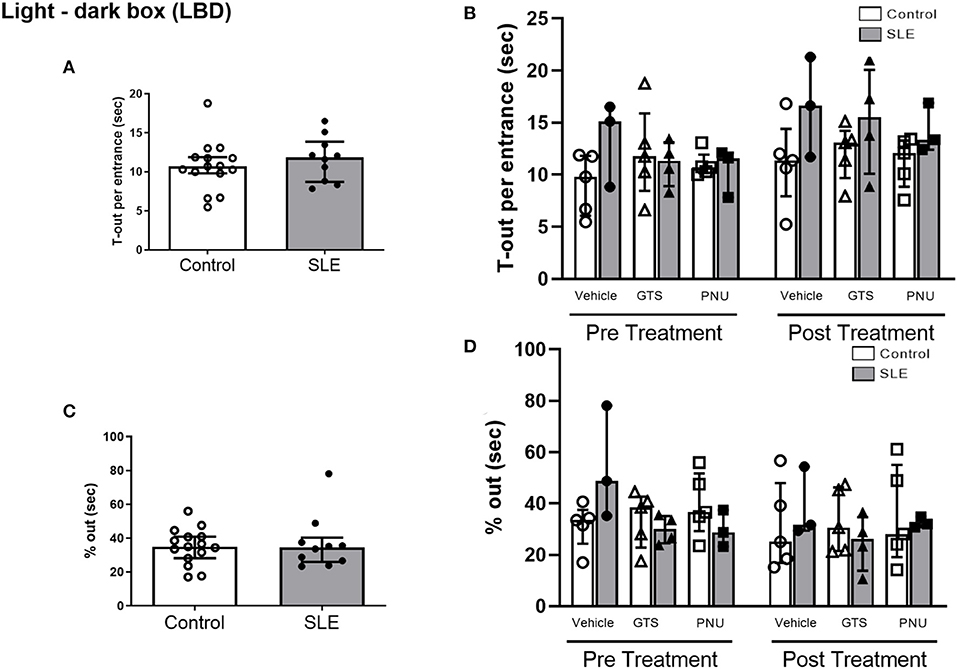
Figure 5. Light-dark box test in control and SLE mice pre-treatment (A,C) and post-treatment with GTS-21 and PNU-120596 (B,D). The pre-treatment data is a comparison between all control and all SLE mice at baseline. A t-test was used to determine significant differences between SLE and control mice pretreatment and there were no differences (A,C). A two-way ANOVA with repeated measures was used to determine differences between all groups post treatment (B,D; n = 3–5 per group). The results of each ANOVA appears on the graph.
To verify the depressive like behavior in SLE mice and the possible effects of PAM, the tail suspension test was performed. There were no differences in the duration of immobility during the tail suspension test between control and SLE animals in pre-treatment (Figure 6A) and although there was an effect of treatment within the groups (p < 0.001) there was no interaction (p = 0.948) (Figure 6B).
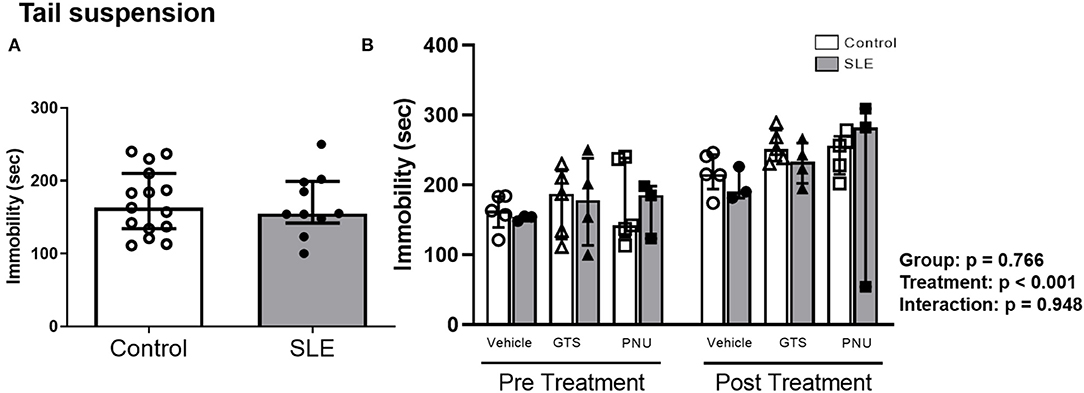
Figure 6. Tail suspension test in control and SLE mice pre-treatment (A) and post-treatment with GTS-21 and PNU-120596 (B). The pre-treatment data is a comparison between all control and all SLE mice at baseline. A t-test was used to determine significant differences between SLE and control mice pretreatment. There was not a difference in immobility during the suspension test between SLE mice and controls. A two-way ANOVA with repeated measures was used to determine differences between all groups post treatment (B; n = 3-5 per group). The results of each ANOVA appears on the graph.
Discussion
The current study revealed that the use of α7-nAChR modulators in mice with advanced SLE has minimal beneficial effects. The severity of SLE was not altered by either GTS-21 or PNU. In addition, renal inflammaton and renal injury seen in untreated SLE mice with advanced disease were not improved following GTS-21 or PNU administration. The deficits in behavior noted in untreated SLE mice were minimally altered by administration of α7 ligands at this late stage.
The current study was planned as a post-treatment; theoretically this study mimics a late-stage SLE patient with active disease searching for therapy to reverse the vicious sequelae of events resulting from the chronic inflammation. The NZBWF1 mice used in this study closely mirrors human SLE (48–50). These mice typically start to develop nuclear autantibodies like dsDNA autoantibodies at ~20 weeks of age and these autoantibodies then deposit into tissues like the kidney and initiate inflammatory responses. We have shown in our published data that these inflammatory responses lead to chronic inflammation in the kidney in the form of lupus nephritis and that this inflammation contributes to the hypertension and renal injury frequently seen in these mice by 35 weeks of age (51). Prior studies indicate that systemic administration of nicotine at this late stage was beneficial in reducing renal inflammation and injury (26), but because of the toxic and addictive nature of nicotine, we aimed to determine the efficacy of systemic administration of a less toxic, yet potent alternative—a partial agonists and a selective α7 PAM. We demonstrated in the current study that global GTS-21 and PNU did not have significant effects on splenic and renal inflammation in SLE mice with advance disease, and therefore did not protect from the associated renal injury and hypertension (Figures 3A,B).
In our previously published study we hypothesized that chronic infusion of nicotine would activate the cholinergic anti-inflammatory pathway at the level of splenic α7-nAChRs (26). We know now that the cholinergic anti-inflammatory pathway is indeed impaired in female SLE mice (46, 51). As a PAM, PNU-120596 is inactive in the absence of α7 agonists meaning that a basal cholinergic tone is needed in order to PAM have an effect (29). We have demonstrated that SLE model has a dampened vagal tone (46), so perhaps PNU-120596 was not effective in decreasing inflammation and hypertension due to the lack of cholinergic tone and α7 agonists present in this advanced stage in female SLE mice. Further, there are 9-α (α1-7, α 9-10), 4-β (β1–4), and γ, δ, and ε subunits of the nAChR on the plasma membranes of skeletal muscle cells, neurons, and other non-neuronal cells (52). Therefore, receptors other than α7-nAChR could be involved in the beneficial effects of chronic nicotine and these other receptors may not be altered by GTS-21. Nevertheless, the enthusiam for studying augmented activation of the α7 receptor remains since we only tested the ability of the drugs to reverse advanced SLE. Studies to test the efficacy of α7 ligands at different stages of SLE are still warranted. In addition, a new prototypical metagonist has become available and we hypothesize that this α7 ligand will be more efficacious than previously used α7 ligands since it is highly effective in reducing inflammation and inflammatory pain in multiple rodent models (53). This agent activates primarily non-ionotropic α7-dependent pathways by inducing extremely prolonged states of nearly complete desensitization (i.e., non-conductive states) of α7 nAChRs (54). Thus, our future studies will test a novel concept and hypothesis that robust α7 desensitization and activation of α7-nAChR non-conductive states will effectively inhibit chronic inflammation associated with physiological and behavioral deficits in a mouse model of SLE.
In a set of behavioral assays, we explored the therapeutic utility of sub-chronic GTS-21 and PNU exposure. The drug-like properties of GTS-21 (EC50~2.9 μM) (55) and PNU (EC50~1.5 μM) (56) are advantageous for central nervous system therapeutics, including rapid brain penetration and efficacy. PNU-120596 (<30 mg/kg) was found to be safe in rodents (57, 58), but GTS-21 was found to be safe in humans as major side effects have not been detected after oral administration of large doses of GTS-21 (e.g., <450 mg/day) (59, 60). GTS-21 is rapidly metabolized to its 4-hydroxy metabolite, 4OH-GTS-21. In a direct comparison study of 4OH-GTS-21 vs. GTS-21 (55): 4OH-GTS-21 was found to be 30–50% more efficacious and potent than GTS-21 for both rat [EC50(4OH-GTS-21)~1.6 μM vs. EC50(GTS-21)~2.9 μM] and human [EC50(4OH-GTS-21)~4.0 μM vs. EC50(GTS-21)~6.0 μM] α7-nAChRs. 4OH-GTS-21 was found to be less efficacious to cause residual inhibition than GTS-21 for both rat and human α7-nAChRs with IC50 = 356 μM (rat) and >1 mM (human) as compared to IC50 = 25 μM (rat) and 82 μM (human) for GTS-21 (55). These electrophysiological assays revealed 4OH-GTS-21 as a superior α7 agonist as compared to GTS-21 and suggested that therapeutic effects of GTS-21 may largely reflect its pro-drug activity rather than direct agonism.
While exogenous orthostatic α7-nAChR agonists like GTS-21 present valuable pharmacological tools, therapeutic approaches that could amplify the brain's innate anti-inflammatory efficacy present potentially powerful therapeutic alternatives and have not been fully explored (29). α7 PAM only amplify α7-dependent endogenous cholinergic tone in a spatially and temporally restricted manner (29), creating a potential for differential efficacy and an improved safety profile as compared to α7 agonists that activate α7-nAChRs indiscriminately. Thus, α7 PAM-based treatments in chronic inflammatory disorders like SLE may enhance endogenous auto-therapy by converting endogenous acetylcholine into potent therapeutic agent. Because α7 PAM are inactive in the absence of α7 agonists, α7 PAM can be accurately delivered to the site of inflammation by a routine systemic administration: α7 PAM will be homogeneously distributed throughout the body by circulation, but their therapeutic action will occur only near the site and time of elevated levels of endogenous acetylcholine (e.g., vagus nerve stimulation) (29). Therefore, α7-PAM introduce a novel therapeutic concept that is based on a substantively different mechanism: inflammation-induced recruitment and potentiation of endogenous α7-dependent cholinergic anti-inflammatory pathway (29, 61).
Evidence in the literature shows a high prevalence of behavioral disorders in SLE patients as well as SLE mice (62–64). NZBWF1 mice manifest anxiety-like phenotypes and decreased locomotor activity. This phenotype is exacerbated by interferon-α suggesting that inflammation has an important role on deficits in behavior in this model (65). In the present study, behavioral assays detected significant differences between control and SLE groups (Figures 4–6), but both GTS-21 and PNU failed to generate therapeutic benefits in either group. This suggests that at the time of treatment (weeks 33–35) sensorimotor and emotional deficits may have matured to the point of no return. In the context of obtained data it would be informative to determine the effects of chronic exposure to GTS-21 and PNU at earlier time points (e.g., weeks 10–30) to test the hypothesis that α7 therapies may impede the disease development.
Although we found the GTS-21 and PNU did not contribute significantly in reducing inflammation and improving behavior, there were some limitations to our current study and notable findings that suggest that treating SLE mice at an earlier stage with potent α7 ligands could be beneficial. First, the end of the study coincides with the average lifespan of female SLE mice (i.e, NZBWF1 mice) used in this study −35 weeks—and it is known that SLE pathology and morbidity worsens with age. It is likely that SLE mice at this advanced stage have reached a point of irreversibility in renal inflammation and renal injury and this explains why we saw no effect of α7 ligands. Future studies that use an earlier timeline for administration of α7 ligands, e.g., at 20 weeks of age when plasma dsDNA autoantibodies start to develop, could inform us if this novel therapy is efficacious in preventing the development and maintenance of chronic inflammation and therefore protecting from end stage renal disease and hypertension among other morbidities in SLE. Lastly, we measured renal and splenic TNF in this study, a common inflammatory mediator involved in renal disease and hypertension. A more involved temporal analysis of renal immune cells and inflammatory mediators would aid in determining if modulating the α7-nAChR is indeed a viable therapeutic option in SLE.
The studies discussed here within pertaining to manipulation of the cholinergic anti-inflammatory pathway are highly clinically relevant. Manipulation of the components of this pathway via pharmacological means should prove to be efficacious in human SLE as in mouse models. The current study focused on pharmacological activation of the downstream α7nAChR, but stimulation of the efferent vagus nerve is the main trigger that turns on this anti-inflammatory pathway and can be targeted pharmacologically. We have shown that pharmacological potentiation of the efferent vagus nerve attenuates blood pressure in female SLE mice (46), but other studies involving vagus nerve stimulation have already been explored in humans. A recently completed clinical trial exploring transcutaneous auricular vagus nerve stimulation demonstrated its effectiveness in reducing pain and fatigue in patients with SLE (66). Together, these findings are confirmation of the importance of the vagally-mediated cholinergic anti-inflammatory pathway in SLE and they highlight the need for understanding the mechanisms involved.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by University of North Texas Health Science Center's Institutional Animal Care and Use Committee (IACUC).
Author Contributions
VU and KM conceived and designed study. JM, CY-S, VU, and KM performed experiments. JM, CY-S, CS, VU, and KM analyzed data. JM, CY-S, CS, VU, and KM interpreted results. JM, CY-S, CS, VU, and KM prepared figures. VU and KM drafted manuscript. JM, CY-S, CS, WK, VU, and KM edited and revised manuscript. JM, CY-S, CS, WK, VU, and KM approved final version of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was partially funded by the Lupus Research Alliance (550778 to KM) and the National Institutes of Health (1K01HL139859 to KM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. (2008) 358:929–39. doi: 10.1056/NEJMra071297
2. Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Rheumatol. (2010) 6:317–25. doi: 10.1038/nrrheum.2010.60
4. Mathis KW, Wallace K, Flynn ER, Maric-Bilkan C, LaMarca B, Ryan MJ. Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension. (2014) 64:792–800. doi: 10.1161/HYPERTENSIONAHA.114.04006
5. Bialas A, Presumey J, Das A, van der Poel C, Lapchak P, Mesin L, et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature. (2017) 546:539–43. doi: 10.1038/nature22821
7. Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Investig. (2007) 117:289–96. doi: 10.1172/JCI30555
8. Rosas-Ballina M, Olofsson PS, Ochani M, Ochani M, Valdes-Ferrer S, Levine YA, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. (2011) 334:98–101. doi: 10.1126/science.1209985
9. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, et al. Splenic nerve is required for cholienrgic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci USA. (2008) 105:11008–13. doi: 10.1073/pnas.0803237105
10. Sadis C, Teske G, Stokman G, Kubjak C, Claessen N, Moore F, et al. Nicotine protects kidney from renal ischemia/reperfusion injury through the cholinergic anti-inflammatory pathway. PLoS ONE. (2007) 2:e469. doi: 10.1371/journal.pone.0000469
11. Leib C, Goser S, Luthje D, Ottl R, Tretter T, Lasitschka F, et al. Role of the cholinergic anti-inflammatory pathway in murine autoimmune myocarditis. Circul Res. (2011) 109:130–40. doi: 10.1161/CIRCRESAHA.111.245563
12. Li D-J, Evans RG, Yang Z-W, Song S-W, Wang P, Ma X-J, et al. Dysfunction of the cholinergic anti-inflammatory pathway mediated organ damage in hypertension. Hypertension. (2011) 57:298–307. doi: 10.1161/HYPERTENSIONAHA.110.160077
13. Levine Y, Koopman F, Faltys M, Caravaca A, Bendele A, Zitnik R, et al. Neurostimulation of the cholinergic anti-inflammatory pathway ameliorates disease in rat collagen-induced arthritis. PLoS ONE. (2014) 9:e104530. doi: 10.1371/journal.pone.0104530
14. Borovikova L, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. (2000) 405:458–62. doi: 10.1038/35013070
15. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor a7 subunit is an essential regulator of inflammation. Nature. (2003) 421:384–7. doi: 10.1038/nature01339
16. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. (2004) 10:1216–21. doi: 10.1038/nm1124
17. Martelli D, Yao S, McKinley M, McAllen R. Reflex control of inflammation by sympathetic nerves, not vagus. J Physiol. (2014) 592:1677–86. doi: 10.1113/jphysiol.2013.268573
18. Chen J-K, Zhao T, Ni M, Li D-J, Tao X, Shen F-M. Downregulation of α7 nicotinic acetylcholine receptor in two-kidney one-clip hypertensive rats. Biomed Central Cardiovasc Disord. (2012) 12:38. doi: 10.1186/1471-2261-12-38
19. Inoue T, Abe C, Sung S, Moscalu S, Jankowski J, Huang L, et al. Vagus nerve stimulation mediates protection from kidney ischemia-reperfusion injury through a7nAChR+ splenocytes. J Clin Investig. (2016) 126:1939–52. doi: 10.1172/JCI83658
20. Truong L, Trostel J, Garcia G. Absence of nicotinic acetycholine receptor a7 subunit amplifies inflammation and accelerates onset of fibrosis: an inflammatory kidney model. FASEB J. (2015) 29:3558–70. doi: 10.1096/fj.14-262493
21. Koopman F, Chavan S, Miljko S, Grazio S, Sokolovic S, Schuurman P, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. PNAS. (2016) 113:1–6. doi: 10.1073/pnas.1605635113
23. Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov. (2005) 4:673–84. doi: 10.1038/nrd1797
24. Goverse G, Stakenborg M, Matteoli G. The intestinal cholinergic anti-inflammatory pathway. J Physiol. (2016) 594:5771–80. doi: 10.1113/JP271537
25. Matteoli G, Gomez-Pinilla P, Nemethova A, Giovangiulio M, Cailotto C, van Bree S, et al. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut. (2014) 63:938–48. doi: 10.1136/gutjnl-2013-304676
26. Fairley A, Mathis K. Cholinergic agonists reduce blood pressure in a mouse model of systemic lupus erythematosus. Physiol Rep. (2017) 5:e13213. doi: 10.14814/phy2.13213
27. Sun F, Johnson S, Jin K, Uteshev V. Boosting endogenous resistance of brain to ischemia. Mol Neurobiol. (2016) 54:2045–59. doi: 10.1007/s12035-016-9796-3
28. Kalappa B, Sun F, Johnson S, Jin K, Uteshev V. A positive allosteric modulator of a7 nAChRs augments neuroprotective effects of endogenous nicotinic agonists in cerebral ischaemia. Br J Pharmacol. (2013) 169:1862–78. doi: 10.1111/bph.12247
29. Uteshev V. The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur J Pharmacol. (2014) 727:181–5. doi: 10.1016/j.ejphar.2014.01.072
30. Gatson J, Simpkins J, Uteshev V. High therapeutic potential of positive allosteric modulation of a7 nAChRs in a rat model of traumatic brain injury: proof-of-concept. Brain Res Bull. (2015) 112:35–41. doi: 10.1016/j.brainresbull.2015.01.008
31. Uteshev V. Allosteric modulation of nicotinic acetylcholine receptors: the concept and therapeutic trends. Curr Pharm Des. (2016) 22:1986–97. doi: 10.2174/1381612822666160201115341
32. Uteshev V, Tenovuo O, Gaidhani N. The cholinergic potential, the vagus nerve and challenges in treatment of traumatic brain injury. Curn Pharm Des. (2016) 22:2083–92. doi: 10.2174/1381612822666160127111630
33. Han Z, Shen F, He Y, Degos V, Camus M, Maze M, et al. Activation of a-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro-inflammatory macrophages and oxidative stress. PLoS ONE. (2014) 9:e105711. doi: 10.1371/journal.pone.0105711
34. Han Z, Li L, Wang L, Degos V, Maze M, Su H. Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J Neurochem. (2014) 131:498–508. doi: 10.1111/jnc.12817
35. Dash P, Zhao J, Kobori N, Redell J, Hylin M, Moore A. Activation of alpha 7 cholinergic nicotinic receptors reduce blood-brain barrier permeability following experimental traumatic brain injury. J Neurosci. (2016) 36:2809–18. doi: 10.1523/JNEUROSCI.3197-15.2016
36. Sun F, Jin K, Uteshev V. A type-II positive allosteric modulator of a7 nAChRs reduces brain injury and improves neurological function after focal cerebral ischemia in rats. PLoS ONE. (2013) 8:e73581. doi: 10.1371/journal.pone.0073581
37. Gaidhani N, Kem WR, Uteshev VV. Spleen is not required for therapeutic effects of 4OH-GTS-21, a selective α7 nAChR agonist, in the sub-acute phase of ischemic stroke in rats. Brain Res. (2020) 4:147196. doi: 10.1016/j.brainres.2020.147196
38. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. BMC Vet Res. (2020) 16:242. doi: 10.1186/s12917-020-02451-y
39. Kem WR, Mahnir VM, Prokai L, Papke RL, Cao X, LeFrancois S, et al. Hydroxy metabolites of the Alzheimer's drug candidate 3-[(2,4-dimethoxy)benzylidene]-anabaseine dihydrochloride (GTS-21): their molecular properties, interactions with brain nicotinic receptors, and brain penetration. Mol Pharmacol. (2004) 65:56–67. doi: 10.1124/mol.65.1.56
40. Ren K, King MA, Liu J, Siemann J, Altman M, Meyers C, et al. The α7 nicotinic receptor agonist 4OH-GTS-21 protects axotomized septohippocampal cholinergic neurons in wild type but not amyloid-overexpressing transgenic mice. Neuroscience. (2007) 148:230–7. doi: 10.1016/j.neuroscience.2007.05.013
41. Meyer EM, Tay ET, Papke RL, Meyers C, Huang GL, de Fiebre CM. 3-[2,4-Dimethoxybenzylidene]anabaseine (DMXB) selectively activates rat α7 receptors and improves memory-related behaviors in a mecamylamine-sensitive manner. Brain Res. (1997) 768:49–56. doi: 10.1016/S0006-8993(97)00536-2
42. Nanri M, Yamamoto J, Miyake H, Watanabe H. Protective effect of GTS-21, a novel nicotinic receptor agonist, on delayed neuronal death induced by ischemia in gerbils. Jpn J Pharmacol. (1998) 76:23–9. doi: 10.1254/jjp.76.23
43. Mathis KW, Venegas-Pont M, Masterson C, Wasson K, Ryan MJ. Blood pressure in a hypertensive mouse model of SLE is not salt-sensitive. Am J Physiol Regul Integr Comp Physiol. (2011) 301:1281–5. doi: 10.1152/ajpregu.00386.2011
44. Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension. (2012) 59:673–9. doi: 10.1161/HYPERTENSIONAHA.111.190009
45. Mathis KW, Venegas-Pont M, Flynn E, Williams J, Maric-Bilkan C, Dwyer T, et al. Hypertension in an experimental model of systemic lupus erythematosus occures independently of the renal nerves. Am J Physiol Regul Integr Comp Physiol. (2013) 301:R711–19. doi: 10.1152/ajpregu.00602.2012
46. Pham G, Wang L, Mathis K. Pharmacological potentiation of the efferent vagus nerve attenuates blood pressure and renal inury in a murine mouse model of systemic lupus erythematosus. Am J Physiol. (2018) 315:1261–71. doi: 10.1152/ajpregu.00362.2017
47. Pham G, Shimoura C, Chaudhari S, Lima C, Kulp D, Mathis K. Chronic unilateral cervical vagotomy reduces renal inflammation, blood pressure, and renal injury in a mouse model of lupus. Am J Physiol Renal. (2020) 319:F155–61. doi: 10.1152/ajprenal.00201.2020
48. Perry D, Sang A, Yin Y, Zheng Y-Y, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol. (2011) 2011:1–9. doi: 10.1155/2011/271694
49. Richard ML, Gilkeson G. Mouse models of lupus: what they tell us and what they don't. Lupus Sci Med. (2018) 5:e000199. doi: 10.1136/lupus-2016-000199
50. Dubois E, Horowitz R, Dmeopoulos H, Teplitz R. NZB/NZW mice as a model of systemic lupus erythematosus. JAMA. (1966) 195:145–9. doi: 10.1001/jama.1966.03100040091025
51. Mathis K. An impaired neuroimmune pathway promotes the development of hypertension in systemic lupus erythematosus. AJP Regul. (2015) 309:1074–7. doi: 10.1152/ajpregu.00143.2015
52. Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, et al. Expression and function of the cholinergic system in immune cells. Front Immunol. (2017) 8:1085. doi: 10.3389/fimmu.2017.01085
53. Papke RL, Bagdas D, Kulkarni AR, Gould T, AlSharari SD, Thakur GA, et al. The analgesic-like properties of the α7 nAChR silent agonist NS6740 is associated with non-conducting conformations of the receptor. Neuropharmacology. (2015) 91:34–42. doi: 10.1016/j.neuropharm.2014.12.002
54. Gaidhani N, Tucci FC, Kem WR, Beaton G, Uteshev VV. Therapeutic efficacy of α7 ligands after acute ischemic stroke is linked to conductive states of α7 nAChRs. Br J Pharmacol. (2021) 178:1684–704. doi: 10.1111/bph.15392
55. Papke RL, Porter Papke JK. Comparative pharmacology of rat and human α7 nAChR conducted with net charge analysis. Br J Pharmacol. (2002) 137:49–61. doi: 10.1038/sj.bjp.0704833
56. Grønlien JH, Håkerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, et al. Distinct profiles of α7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. (2007) 72:715–24. doi: 10.1124/mol.107.035410
57. Uteshev V. Are positive allosteric modulators of α7 nAChRs clinically safe? J Neurochem. (2016) 136:217–9. doi: 10.1111/jnc.13236
58. Gaidhani N, Uteshev VV. Treatment duration affects cytoprotective efficacy of positive allosteric modulation of α7 nAChRs after focal ischemia in rats. Pharmacol Res. (2018) 136:121–32. doi: 10.1016/j.phrs.2018.09.001
59. Kem WR. The brain α7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer's disease: studies with DMXBA (GTS-21). Behav Brain Res. (2000) 113:169–81. doi: 10.1016/S0166-4328(00)00211-4
60. Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, et al. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology. (2003) 28:542–1. doi: 10.1038/sj.npp.1300028
61. Pavlov VA, Tracey KJ. Neural circuitry and immunity. Immunol Res. (2015) 63:38–57. doi: 10.1007/s12026-015-8718-1
62. Kong X, Zhang Z, Fu T, Ji J, Yang J, Gu Z. TNF-α regulates microglial activation via the NF-κB signaling pathway in systemic lupus erythematosus with depression. Int J Biol Macromol. (2019) 125:892–900. doi: 10.1016/j.ijbiomac.2018.12.146
63. Lapter S, Marom A, Meshorer A, Elmann A, Sharabi A, Vadai E, et al. Amelioration of brain pathology and behavioral dysfunction in mice with lupus following treatment with a tolerogenic peptide. Arthritis Rheum. (2009) 60:3744–54. doi: 10.1002/art.25013
64. McHugh J. Systemic lupus erythematosus: IFN drives synapse loss via microglia. Nat Rev Rheumatol. (2017) 13:449. doi: 10.1038/nrrheum.2017.109
65. Zeng J, Meng X, Zhou P, Yin Z, Xie Q, Zou H, et al. Interferon-α exacerbates neuropsychiatric phenotypes in lupus-prone mice. Arthritis Res Ther. (2019) 21:205. doi: 10.1186/s13075-019-1985-9
66. Aranow C, Atish-Fregoso Y, Lesser M, Mackay M, Anderson E, Chavan S, et al. Transcutaneous auricular vagus nerve stimulation reduces pain and fatigue in patients with systemic lupus erythematosus: a randomised, double-blind, sham-controlled pilot trial. Ann Rheum Dis. (2021) 80:203–8. doi: 10.1136/annrheumdis-2020-217872
Keywords: SLE, cholinergic anti-inflammatory pathway, positive allosteric modulators, renal injury, renal inflammation, behavior
Citation: Morales JY, Young-Stubbs CM, Shimoura CG, Kem WR, Uteshev VV and Mathis KW (2021) Systemic Administration of α7-Nicotinic Acetylcholine Receptor Ligands Does Not Improve Renal Injury or Behavior in Mice With Advanced Systemic Lupus Erythematosus. Front. Med. 8:642960. doi: 10.3389/fmed.2021.642960
Received: 17 December 2020; Accepted: 18 March 2021;
Published: 13 April 2021.
Edited by:
Alain Le Moine, Université Libre de Bruxelles, BelgiumReviewed by:
Claude Sadis, Centre Hospitalier Universitaire de Tivoli, BelgiumErin Taylor, University of Mississippi Medical Center, United States
Copyright © 2021 Morales, Young-Stubbs, Shimoura, Kem, Uteshev and Mathis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keisa W. Mathis, a2Vpc2EubWF0aGlzQHVudGhzYy5lZHU=