 Mirjam Kiener1,2,3
Mirjam Kiener1,2,3 Nuria Roldan3
Nuria Roldan3 Carlos Machahua1,4
Carlos Machahua1,4 Arunima Sengupta5
Arunima Sengupta5 Thomas Geiser1,4
Thomas Geiser1,4 Olivier Thierry Guenat1,5,6
Olivier Thierry Guenat1,5,6 Manuela Funke-Chambour1,4Nina Hobi3*†
Manuela Funke-Chambour1,4Nina Hobi3*† Marianna Kruithof-de Julio2,3,7†
Marianna Kruithof-de Julio2,3,7†- 1Department of Pulmonary Medicine, Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland
- 2Department for BioMedical Research DBMR, Urology Research Laboratory, University of Bern, Bern, Switzerland
- 3Alveolix AG, Swiss Organs-on-Chip Innovation, Bern, Switzerland
- 4Department for BioMedical Research DBMR, Department of Pulmonary Medicine, Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland
- 5Organs-on-Chip Technologies, ARTORG Center for Biomedical Engineering, University of Bern, Bern, Switzerland
- 6Department of General Thoracic Surgery, Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland
- 7Organoid Core, Department for BioMedical Research, University of Bern, Bern, Switzerland
The coronavirus disease 2019 (COVID-19) pandemic has caused considerable socio-economic burden, which fueled the development of treatment strategies and vaccines at an unprecedented speed. However, our knowledge on disease recovery is sparse and concerns about long-term pulmonary impairments are increasing. Causing a broad spectrum of symptoms, COVID-19 can manifest as acute respiratory distress syndrome (ARDS) in the most severely affected patients. Notably, pulmonary infection with Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2), the causing agent of COVID-19, induces diffuse alveolar damage (DAD) followed by fibrotic remodeling and persistent reduced oxygenation in some patients. It is currently not known whether tissue scaring fully resolves or progresses to interstitial pulmonary fibrosis. The most aggressive form of pulmonary fibrosis is idiopathic pulmonary fibrosis (IPF). IPF is a fatal disease that progressively destroys alveolar architecture by uncontrolled fibroblast proliferation and the deposition of collagen and extracellular matrix (ECM) proteins. It is assumed that micro-injuries to the alveolar epithelium may be induced by inhalation of micro-particles, pathophysiological mechanical stress or viral infections, which can result in abnormal wound healing response. However, the exact underlying causes and molecular mechanisms of lung fibrosis are poorly understood due to the limited availability of clinically relevant models. Recently, the emergence of SARS-CoV-2 with the urgent need to investigate its pathogenesis and address drug options, has led to the broad application of in vivo and in vitro models to study lung diseases. In particular, advanced in vitro models including precision-cut lung slices (PCLS), lung organoids, 3D in vitro tissues and lung-on-chip (LOC) models have been successfully employed for drug screens. In order to gain a deeper understanding of SARS-CoV-2 infection and ultimately alveolar tissue regeneration, it will be crucial to optimize the available models for SARS-CoV-2 infection in multicellular systems that recapitulate tissue regeneration and fibrotic remodeling. Current evidence for SARS-CoV-2 mediated pulmonary fibrosis and a selection of classical and novel lung models will be discussed in this review.
Introduction
Coronavirus disease 2019 (COVID-19) is a zoonotic disease caused by the novel Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 is the seventh coronavirus known to infect humans. Human coronavirus strains HKU1, OC43, NL63 and 229E cause mild symptoms similar to the common cold, while SARS-CoV and Middle East Respiratory Syndrome coronavirus (MERS-CoV) can result in severe viral pneumonia with a high mortality and have been responsible for two epidemic outbreaks in the twenty-first century (1). Compared to SARS-CoV and MERS-CoV, SARS-CoV-2 is more easily transmitted from human to human which has allowed it to evolve into a worldwide pandemic (2). SARS-CoV-2 enters the human body via the respiratory tract and reaches its initial main target organ, the lung. About one-fourth to one-third of hospitalized patients develop severe complications and require treatment in the intensive care unit for ~10 days or longer (3, 4), which risks a global collapse of the health care system. Countermeasures including curfews to limit the spread of SARS-CoV-2 have caused dramatic economic losses (5). Despite improved management of critically ill patients (6) this situation can only be resolved by effective treatment strategies and COVID-19 vaccines. Four COVID-19 vaccines have already been approved in Europe (ema.europa.eu) and various other vaccines are currently being developed or have entered late-phase clinical trials (7). In parallel, inhibitory compounds are tested for re-purposing (8, 9). In vitro models of the respiratory tract have significantly contributed to screening for promising drug candidates such as remdesivir, camostat, imatinib, and Retro-2.1 and have helped elucidating the molecular mechanisms of host-pathogen interactions in more detail (10–15). Increasing knowledge about the course of COVID-19 raised concerns regarding its long-term consequences. Experts warn that SARS-CoV-2 might cause long-lasting or persisting interstitial pulmonary fibrosis, an incurable clinical condition marked by abnormal fibrogenesis in the alveolar wall resulting in a progressive reduction of pulmonary function and gas exchange in the lung (16). Recent studies show that severe or critically ill COVID-19 survivors have reduced diffusion capacity and oxygenation levels compared to mildly or moderately sick patients 4 months after infection (17). Whether these impairments resolve, remain or evolve into persisting pulmonary fibrosis is currently unknown.
This review focuses on the clinical course of COVID-19 in the lung and relates the pathology to the underlying molecular biology. Furthermore, we will discuss interstitial pulmonary fibrosis, with idiopathic pulmonary fibrosis (IPF) as the worst example, and how COVID-19 may lead to pulmonary fibrosis. Finally, we will review available in vivo and in vitro models of lung fibrosis and SARS-CoV-2 infection to propose the most suited advanced in vitro models for studying COVID-19-associated pulmonary fibrosis.
Pathogenesis of COVID-19 in the Lung
Fundamental Processes of Breathing: The Biology and Regeneration of the Lung Epithelium
The respiratory tract is continuously exposed to inhaled particles and pathogens. Therefore, it is lined by a highly specialized epithelium, which can be divided into conducting airways and alveoli based on their location and primary function. The pseudostratified epithelium in the proximal airways harbors secretory club and goblet cells, which produce a protective layer of mucus toward the lumen. The terminally differentiated ciliated cells convey the mucus layer upwards to clear trapped particles. Basal cells are able to differentiate into secretory or ciliated cells and are therefore considered to represent the progenitor cells of the airway epithelium, though most cell types of the airway epithelium are highly plastic [(18); Figure 1A]. On the distal end, the conducting airways branch into bronchioles and ultimately in the alveoli. These sac-shaped units represent one of the largest body surfaces in constant contact with the environment essential for efficient gas exchange. About 95% of the alveolar surface is covered by highly specialized flattened type I alveolar epithelial (ATI) cells (19). They form an ultra-thin epithelial-blood barrier with the pulmonary microvasculature endothelial cells, supporting efficient oxygen and CO2 passive diffusion (20). Together with ATI cells, type II alveolar epithelial (ATII) cells are the main constituents of the highly differentiated alveolar epithelium, which closely interacts with surrounding cells in the niche including alveolar macrophages (AMϕ), microvascular cells, and fibroblasts (Figure 1B).
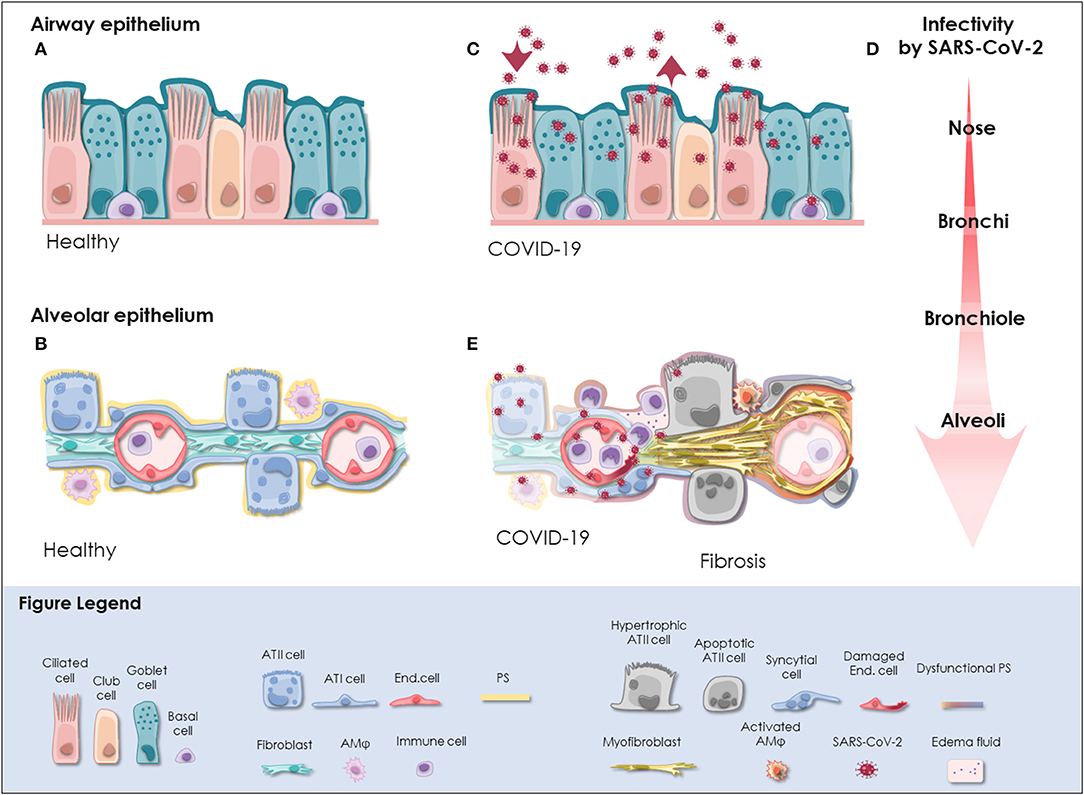
Figure 1. SARS-CoV-2 infection in the respiratory tract. (A) In the pseudostratified epithelium of the airways, secretory goblet, and club cells produce mucus, which is transported by ciliated cells to clear trapped particles and protect the lung from micro-injuries and infection. Basal cells reside at the lamina propria and comprise progenitor cells. The composition and frequency of the individual cell types is variable among the distinct anatomical sites in the nose, trachea, bronchi, and bronchioles. (B) The alveolar epithelium is specialized for gas exchange with flattened ATI cells forming an ultra-thin (~2 μm) epithelial-endothelial barrier allowing oxygen and CO2 diffusion. Cuboidal ATII cells are considered as progenitor cells of ATI cells and fulfill vital functions by the production of pulmonary surfactant (PS), which lowers surface tension and prevents alveolar collapse. Lung fibroblasts are essential to maintain the ATII stem cell niche. Resident alveolar macrophages (AMφ) and immune cells defend the epithelium from infection. (C) SARS-CoV-2 initially infects the airway epithelium. The virus can efficiently replicate in ciliated and secretory cells resulting in the shedding of high viral titers and mild to moderate COVID-19 symptoms. (D) The respiratory epithelium exhibits differential susceptibility to SARS-CoV-2 infection. In correlation with ACE2 expression, SARS-CoV-2 infection is most efficient in the upper airways, particularly in the nasal epithelium. Infectivity gradually decreases toward the alveoli. However, when SARS-CoV-2 reaches the alveoli it can result in severe manifestation of COVID-19. (E) Upon reaching the alveoli, SARS-CoV-2 infects alveolar epithelial cells and endothelial cells and causes viral pneumonia. Cytopathic effects of SARS-CoV-2 are evident as syncytial and apoptotic alveolar epithelial cells resulting in the breakdown of pulmonary surfactant and barrier integrity. In some patients, alveolar damage culminates in life-threatening microvascular activation and an imbalanced immune response. Tissue regeneration takes place already during acute COVID-19 as indicated by fibrin deposition, ATII cell hyperplasia and alveolar wall thickening. Moreover, severely ill COVID-19 patients exhibit radiological signs of fibrosis even months after recovery indicative for the induction of COVID-19-associated fibrosis. ACE2, angiotensin-converting enzyme 2; ATI cell, type I alveolar epithelial cell; ATII cell, type II alveolar epithelial cell; COVID-19, coronavirus disease 2019; End. cell, endothelial cell; AMφ, alveolar macrophage; PS, pulmonary surfactant; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
ATII cells are cuboidal cells often located at the edges of the alveolar sacs and, as opposed to the flat and large ATI cells, account for a small fraction of the alveolar surface. ATII cells produce pulmonary surfactant, a lipid-protein complex with exceptional surface tension lowering properties (21). By doing so, they sustain the breathing function and protect the delicate alveolar structure from collapsing upon exhalation (21). ATII cells also have a role in innate immunity and take part in surfactant recycling. But most importantly, ATII cells are capable of self-renewal and differentiation into ATI cells, which allows re-epithelization upon injury [reviewed in (22)].
Already in 1977, Mason and Williams termed ATII cells as “the defender of the alveolus” for their central role in lung homeostasis (23). However, what exactly defines an ATII cell has been a matter of discussion for years (24). In vitro, isolated human ATII cells behave as facultative stem cells giving rise to alveolar organoids containing multiple cell types (25, 26). Recent studies have suggested that different ATII subtypes may coexist within what has been classically considered as ATII cells, including the proposed alveolar epithelial progenitors comprising TM4SF1+ cells which are highly responsive to Wnt signaling (26).
Rising evidence supports the role of other cell types in alveolar tissue repair together with ATII cells. A subset of Hopx+ ATI cells has been suggested as a source of ATII cells via transdifferentiation upon injury (27). Other studies have proven that a rare basal-like p63+ Krt5+ epithelial cell population migrates to sites of injury in the distal lung to re-create the damaged barrier in the mouse [reviewed in (28)]. In humans, such a population has not been found to date, but bronchiolization is a common histologic finding after injury. In addition, the contribution of basaloid cells in the repair process is supported by the finding of basaloid cells in the damaged areas of patients suffering from IPF, an aggressive form of progressive interstitial pulmonary fibrosis with unknown cause, although several risk factors have been identified [reviewed in (28)]. Recently, EpCAM+ CD73+ epithelial cells, which localize at the basal membrane of the respiratory and alveolar epithelium, have also been suggested as progenitors for both, pseudostratified mucociliary and mature alveolar epithelium in the postnatal and adult human lung (29).
Further, the contribution of stromal cells to ATII cell stemness maintenance and tissue repair cannot be neglected. Lung fibroblasts have been shown to support progenitor ATII cell characteristics in vitro and in vivo in mice (25, 30, 31) and human (25), underscoring the relevance of Wnt signals as determinants for ATII cell fate. On the other hand, fibroblasts, and myofibroblasts are also responsible for extracellular matrix (ECM) deposition and wound closure upon alveolar injury (32).
In summary, repair in the alveolar epithelium is characterized by an acute inflammatory phase, progenitor differentiation and migration, wound closure and finally, resolution (33). Upon injury, ATII cells behave as facultative stem cells and activate their regenerative response becoming hyperplastic. These ATII cells will either self-renew, migrate to the site of injury and differentiate into ATI cells, or undergo apoptosis. These processes depend on the balance of different mediators and a complex cell-cell crosstalk in which stromal cells and AMφ are crucial players (34). Some studies point at the pro-inflammatory and oxidative environment as a driving force for differentiation and repair in the mouse [reviewed in (22)], with Wnt signaling as a key regulator for ATII cell differentiation (35). Further, the relevance of ATII cells in the repair process is highlighted by studies in which ATII-targeted damage or cellular intrinsic alterations, rather genetic or due to aging, lead to aberrant tissue remodeling (36, 37).
It is also important to consider that the lung is subject to mechanical stress and deformation which is essential for several key biological events such as lung development (38) and pulmonary surfactant secretion (39–41). The correlation between alveolar inflation to the corresponding increase in alveolar surface area is still debatable. Nevertheless, during restful breathing, also termed as tidal breathing (defined as 40–80% of TLC, total lung capacity), alveolar linear strain has been suggested to go from 4 to 10% (42–45), up to even higher than 20% during exercise or deep sighs (42, 43, 45, 46). Hence, local mechanical tension and stiffness changes which occur along the repair process converge with the forces supporting breathing (38). In fact, breathing-like cyclic strain has been proven to influence the regenerative epithelial response as shown by wound closure experiments in vitro (47–51). Mechanical ventilation with high tidal volumes, on the other hand, has been observed to amplify lung damage in animal models and in ventilated patients suffering from different respiratory pathologies (52, 53). In fact, mechanical stress has been suggested as an important factor for fibrogenesis (54). Considering this, protective ventilation protocols have been adopted to prevent ventilation-associated lung injury in COVID-19 patients (55).
Besides stretch, the alveolar niche sustains other mechanical forces such as shear stress and surface tension. At the alveolar epithelium, surface tension, and the so-called interfacial stress dominate particularly at low volumes (45). These forces stem from the continuous change in area exposed to the air, its associated fluid oscillation and cell-induced deformation (45). Interfacial stress alone has been observed to be deleterious for ATII cells in vitro, however, it has been also proven to constitute a powerful signal for pulmonary surfactant release in addition to cyclic stretch (56, 57). Pulmonary surfactant efficiency in lowering surface tension is tightly associated to its lipid and protein composition, which adapts very quickly to meet different respiratory demands (58, 59), and has been suggested to be refined along breathing cycles in a mechanism assisted by surfactant proteins (60–64). In the context of surfactant exhaustion, higher surface tension may then act as a trigger for further surfactant release to restore alveolar homeostasis. This system fails in pathological conditions in which aberrant surfactant composition (65–67) contributes to associated higher surface tension and repetitive tissue damage (54), thus stressing the relevance of surfactant and ATII as a secreting cell in addition to its role in repair.
Altogether, this evidence highlights the complexity of alveolar epithelial repair and the central role played by ATII cells. Hence, we speculate that targeted ATII cell injury such as that caused by SARS-CoV-2 infection may increase alveolar susceptibility to injury and aberrant tissue repair, with severe long-term consequences even after disease resolution.
Molecular Mechanisms of SARS-CoV-2 Infection in the Lung
The initial step of coronavirus infection involves binding of the viral spike (S) protein to the compatible receptor on the surface of the target cell (68). Like the closely related SARS-CoV, SARS-CoV-2 uses angiotensin-converting enzyme 2 (ACE2) as an essential entry receptor into human cells. In contrast to SARS-CoV, the receptor binding domain (RBD) of SARS-CoV-2 S protein even exhibits higher binding affinity for human ACE2 (69–72) but seems to be less exposed possibly enabling immune evasion (70). As a consequence, ACE2 affinity of SARS-CoV and SARS-CoV-2 full-length S protein is comparable enabling both viruses to attach to human ACE2 but not to other coronavirus entry receptors such as aminopeptidase N (APN) and dipeptidyl peptidase 4 (DPP4) (11, 73, 74).
Subsequently, proteolytic cleavage of the S protein exposes the fusion domain and enables virus entry into the host cell (75). Multiple proteases can fulfill this function such as transmembrane protease serine subtype 2 (TMPRSS2) and cathepsin B/L in case of SARS-CoV (76–79) or TMPRSS2, cathepsin L, and Furin for MERS-CoV (80, 81). Strong evidence for SARS-CoV-2 S protein priming by TMPRSS2 and cathepsin L has been gathered in vitro (11, 69, 82).
Therefore, cells co-expressing ACE2 and TMPRSS2 can potentially be infected by SARS-CoV-2. Single cell RNA sequencing data analysis has revealed ACE2 expressing cells in multiple organs, though it is generally expressed at low levels. This suggests that ACE2 expression is the limiting factor for SARS-CoV-2 infection (83, 84). However, enriched expression of ACE2 protein and co-expression with TMPRSS2 potentially renders alveolar epithelial cells and enterocytes particularly vulnerable to SARS-CoV-2 (83–85). Accordingly, SARS-CoV-2 RNA is detected prominently in the respiratory tract but occasionally also in the feces and blood of COVID-19 patients (86–88). In the respiratory tract, SARS-CoV-2 is detected in diagnostic samples and tissue specimens from different anatomical sites implying that it can replicate throughout the airway and lung epithelium (74, 89–91). Despite overall low ACE2 expression levels in the respiratory tract, about 20% of lung cells have been found to express ACE2 mRNA (82). The highest levels of ACE2 are reached in the nasal epithelium and gradually decrease from the proximal airways toward the distal lung (82, 83, 92). Accordingly, viral yields are higher in nasal swabs than throat swabs indicating that the nasal epithelium is the initial site of SARS-CoV-2 infection, replication, and shedding (74). The infection can propagate further as ACE2 and TMPRSS2 expression is found throughout the airway epithelium, particularly in ciliated and secretory cells (83, 92). Correspondingly, ciliated cells and goblet cells in the trachea and bronchi are efficiently infected by SARS-CoV-2, whereas basal cells are permissive for SARS-CoV-2 to a lower extent [(13, 14, 82, 93, 94); Figure 1C]. The finding that SARS-CoV-2 does not infect ciliated cells of distal lung organoids but exhibits a strong tropism for club cells seems contradictory (95). However, the cell tropism of SARS-CoV-2 might shift among different anatomical sites given the highly variable infection efficiencies reported for in vitro cultured ciliated, goblet and club cells (14, 82, 95). Moreover, ACE2 is upregulated upon interferon (INF) stimulation to protect the tissue during acute lung injury (96). Despite inducing an imbalanced immune response and delayed IFN signaling (97), we cannot rule out that SARS-CoV-2 infection itself might trigger INF-mediated upregulation of ACE2 promoting infection. Taken together, it is likely that SARS-CoV-2 initially infects and replicates in the nasal epithelium, particularly in ciliated cells, achieves high titers in the proximal airways and reaches the alveoli by aspiration through the airways [(74, 82, 90); Figure 1D].
In the alveoli, SARS-CoV-2 can be detected in ATI and ATII cells, endothelial cells and immune cells of deceased COVID-19 patients, which is in line with experimental findings from 3D in vitro models (82, 95, 98–101). Infection of ATI cells, endothelial cells and alveolar immune cells presumably results in a disturbed immune response and persistent inflammation (98–100). However, based on the analysis of single cell RNA sequencing datasets and in vitro infectivity experiments it has been suggested that ATII cells represent the primary target of SARS-CoV-2 in the alveoli [(82, 83, 92, 95, 100–102); Figure 1E]. Notably, increased susceptibility of an ATII cell subpopulation has been consistently reported by in vitro studies (99, 101). Gene expression profiling revealed an apoptotic signature and a strong downregulation of ATII-specific genes including surfactant proteins in heavily infected ATII cell models (101–103). In line with in vitro data, the induction of apoptotic pathways paralleled by a significant downregulation of surfactant protein transcripts is also apparent in ATII cells of COVID-19 patients (103) suggesting that SARS-CoV-2 infection results in the loss of ATII cell identity and function. Ultimately, this potentially leads to reduced surfactant production and consequently, alveolar collapse, massive tissue damage, and scaring (54). Therefore, further investigations on this fatal course of the disease are critical. To date, it is unclear whether these highly infected cells secrete viral particles, what are the immunological and clinical consequences and why a subpopulation of ATII cells seems to be more vulnerable than others. Possibly, SARS-CoV-2 relies on different entry mechanisms among different cell types and subsets. For example, it has been shown that TMPRSS2 is critical for SARS-CoV-2 entry in ATII cells but cathepsin B/L seems to be dispensable (102). Furthermore, as opposed to SARS-CoV, SARS-CoV-2 can exploit a wider range of host factors for cell entry, which can act synergistically with initial ACE2 attachment and TMPRSS2 cleavage. Detailed resolution of the sequence and structure of SARS-CoV-2 S protein has revealed only 73% similarity to SARS-CoV S protein RBD (104) and the presence of a multibasic site at the S1/S2 subunit boundary of SARS-CoV-2 S protein, which creates a novel furin cleavage site (70, 71, 105). Accordingly, furin overexpression enhances SARS-CoV-2 uptake (82) and has a cumulative effect with TMPRSS2 and cathepsin L on virus entry (70). Processing of the SARS-CoV-2 S protein by furin or other members of the proprotein convertase subtilisin kexin (PCSK) family might be highly relevant during SARS-CoV-2 infection of ATII cells as a recent meta-analysis of human lung single-cell RNA sequencing datasets has demonstrated significant co-expression of ACE2 and PCSK proteases in lung cells (85). Importantly, S protein processing by furin generates a RRAR motif at the S1 C-terminus which is able to bind to Nuropilin-1 (NRP1) and Nuropilin-2 (NRP2) (106). While ACE2 is still required for initial attachment of the virus to the cell surface, NRP1 depletion significantly reduces SARS-CoV-2 uptake (106). Notably, deletion of the multibasic S1/S2 site in SARS-CoV-2 S protein decreases the infection efficiency in human lung cells (105) and attenuates pathogenicity in animal models (107). Whether this is due to the loss of interaction with NRP1 and NRP2 remains to be demonstrated. However, NRP1 and NRP2 are upregulated in the lung tissue of COVID-19 patients (108), which might promote disease progression.
These data indicate that ACE2 expression is critical for SARS-CoV-2 infection and mediates initial attachment (71, 105). At the same time, activation of SARS-CoV-2 S protein by TMPRSS2, cathepsin L, and furin allows it to interact with surface molecules other than ACE2 (70, 106). This is likely to confer wider tissue tropism and promotes transmissibility of SARS-CoV-2. Once SARS-CoV-2 has entered the host cell and released its positive-sense single-stranded RNA genome into the cytoplasm, viral non-structural proteins are translated to generate the viral replication and transcription complex (RTC). Furthermore, coronavirus proteins hijack the translation machinery of the host cell and favor translation of viral mRNA over cellular mRNA, inhibit the innate antiviral IFN response and interfere with normal cell function (109). Infection of alveolar cells potentially results in the most critical disease manifestation due to abrogation of ATII cell function and stimulation of an inflammatory response.
Acute Pathologic Manifestation of COVID-19 in the Lungs
SARS-CoV-2 infection results in a complex symptomatology associated with mild, moderate and severe illness or might even take an asymptomatic course (110–113). In non-hospitalized patients testing positive for SARS-CoV-2 infection, the most prevalent symptoms include cough, dyspnea, loss of smell or taste, fever and chills, myalgia, headache, body aches, sinus congestion, sore throat, nausea, diarrhea and dizziness (110, 111). Surprisingly, subclinical lung opacities and diffuse consolidation have been detected on computed tomography (CT) scans in more than half of asymptomatic COVID-19 patients (112, 114). Moreover, histologic alterations in the alveoli including edema, protein and fibrin exudate, ATII cell hyperplasia and fibroblast proliferation, inflammatory clusters and multinucleated giant cells have been reported in two pre-symptomatic cases of COVID-19 (115). Radiologic lung abnormalities seem to resolve in mildly to moderately symptomatic COVID-19 patients but the regeneration process in these patients is scarcely studied (116, 117).
In contrast, about one-third of patients—mainly elderly men with underlying comorbidities—have a severe course of the disease with a high case fatality rate (3, 4, 118–121). Host factors rather than viral factors seem to be the significant determinants for disease severity. Pre-existing comorbidities, old age, male sex, and blood group other than O have been associated with a higher susceptibility to SARS-CoV-2 and risk for a severe disease course (118, 119, 122, 123). Furthermore, clinical parameters at hospitalization are critical predictors of severe illness. These include elevated levels of coagulation markers (e.g., D-dimers) in the blood (124) and lymphocytopenia, which correlates with increased interleukin (IL)-6 and IL-8 levels and a higher risk of cytokine storm (121). Autopsies have revealed that SARS-CoV-2 infects multiple organs including upper airways, lung, heart, kidney, the vasculature and the brain (125) and as a consequence can manifest extra-pulmonary [reviewed in (126)].
However, most commonly severe COVID-19 patients develop viral pneumonia and suffer from fever, fatigue, dry cough, myalgia and dyspnea (3, 4). In these patients, SARS-CoV-2 replicates in the upper airways and the distal lung, where it causes life-threatening damage to the alveoli (90, 98, 125, 127, 128). Nearly all hospitalized COVID-19 patients present with ground-glass opacities with or without consolidation on chest CT scans that gradually worsen before death (3, 4, 119, 120, 129). Critically ill patients usually develop acute respiratory distress syndrome (ARDS) (3, 4). ARDS can be provoked by various direct or indirect pulmonary insults. Infection, including viral infection, is a major cause for ARDS, being pneumonia the most common underlying pathology (130). ARDS is defined as the clinical manifestation of diffuse alveolar damage (DAD) (131). Correspondingly, typical histologic patterns of DAD including hyaline membrane formation, fibrin exudates, syncytial alveolar epithelial cells, diffuse ATII cell hyperplasia and the replacement of ATI cells by cuboidal ATII-like cells are apparent in the lungs of deceased COVID-19 patients [(100, 120, 125, 129); Figure 1E]. The recent description of two differential pathologic patterns in the lungs of deceased COVID-19 patients suggests that both, direct cytopathic effect of SARS-CoV-2 and a deleterious inflammatory immune response, can cause fatal alveolar damage (132). As a consequence, marked hypoxia develops and results in the enlargement of the pulmonary vasculature, blood vessel activation and coagulopathies with formation of micro-thrombi in multiple organs (6, 98, 108, 125). About 2% of hospitalized COVID-19 patients ultimately succumb to the disease with respiratory or multi-organ failure as a major cause of death (127, 128, 133). However, it is currently not known whether severely affected COVID-19 survivors will fully recover or may suffer from complications in the resolution phase of ARDS. First results after 4 months indicate that the diffusion capacity is reduced in COVID-19 patients after severe or critical disease (17).
Pulmonary Fibrosis: A Long-Term Complication of COVID-19?
Alveolar Damage as a Cause of Interstitial Pulmonary Fibrosis
It has been reported that ARDS can lead to lasting physical impairment after 5 years of follow up (134), including fibrotic pulmonary changes as a consequence of abnormal wound healing (135). Acute alveolar damage (e.g., from viral infection) is followed by the activation of inflammatory and apoptotic responses (136, 137). The alveolar epithelial cell damage triggers a cascade of reactions, including the release of pro-inflammatory cytokines, to activate local immune responses and controlled fibroblast proliferation as well as interstitial fibrogenesis, to initiate primary wound healing mechanisms (138, 139). These effects will normally be reconstituted by recovery of the basal lamina, re-epithelialization of the alveolar epithelium (140), and the degradation as well as clearance of ECM proteins (141). A precise and controlled repair mechanism following alveolar damage is crucial to terminate progression of the lung remodeling toward pulmonary fibrosis.
However, sustained alveolar injuries together with possible intrinsic factors, such as genetic mutations [e.g., MUC5B, SFTPC, TERT/TERC or TERF-1; (142–145)] or an accelerated aging phenotype (146), can impair the capability of alveolar epithelial cells to proliferate and orderly cover the defect. This provokes chronic alveolar damage that can eventually trigger an uncontrolled fibrotic response (147). This impaired wound healing can generate a disequilibrium in favor of the pro-fibrotic factors such as tumor necrosis factor alpha (TNF-α), platelet-derived growth factor (PDGF) or transforming growth factor beta (TGF-β), which will mediate the development and further progression of lung fibrosis (148). Particularly, TGF-β has an essential role in activating fibrotic mechanisms, inducing the perpetuation of exaggerated wound repair (149).
The aberrant wound healing response can lead to additional loss of alveolar epithelial cells by apoptosis (150), induce lung fibrosis by activation of a pro-fibrotic profile in macrophages (151) and maintain the unruly activation and regulation of fibrotic lung fibroblasts mediated by TGF-β (152). This dysfunctional alveolar re-epithelialization favors the uncontrolled proliferation of lung fibroblasts and secretion of ECM proteins that consolidate the fibrotic change (153). Indeed, viral lung infections can trigger DAD on top of interstitial lung diseases (ILD) which is a common histological feature in some stages of ILD progression (154, 155). Given the development of DAD that manifests as ARDS in severely sick COVID-19 patients, it remains to be investigated whether the alveolar wound healing response will eventually result in pulmonary fibrosis and in its worst form IPF.
Emerging Evidence of COVID-19-Associated Lung Fibrosis
Long-term follow-up data on recovered COVID-19 patients is currently emerging and insights gained from earlier coronavirus epidemics can allow to predict likely scenarios. The first coronavirus epidemic of the twenty-first century has been caused by SARS-CoV, the causative agent of severe acute respiratory syndrome (SARS). SARS is an illness that shows typical infection-related symptoms, including fever and pneumonitis, with a recovery time in most patients after 1–2 weeks following the infection. Up to one third of SARS patients can develop severe pulmonary complications, requiring oxygen therapy (156). The acute phase of SARS starts with acute lung damage and edema, bronchiolar sloughing of ciliated epithelial cells and the deposition of hyaline-rich alveolar membranes, which clinically manifests with impaired oxygen exchange. A progressive phase during the following 2–5 weeks is characterized by fibrin deposition and infiltration of inflammatory cells and fibroblasts. In the last stage, after 1–2 months, pulmonary fibrosis consolidates with collagen deposition and fibroblast proliferation in the interstitial spaces (157–159).
The extent of fibrosis can be a sign of SARS severity and illness duration, as demonstrated in post-mortem studies (160, 161). Radiological features of fibrosis after SARS have been observed at 3 and 6 months after infection in around 30% of the cases, findings that have been confirmed by another study in survivors (162). Ground glass opacities were found 1 month after diagnosis in 45% of SARS patients, underlining the possibility to find early signs of fibrosis in those patients (163). Moreover, a patient's age can also be a critical risk factor in the fibrotic manifestation and long-term damage as older SARS patients have an increased risk for lung fibrosis (164).
Another coronavirus infection—the Middle East respiratory syndrome (MERS), shows a similar clinical outcome as SARS. However, radiological abnormalities are more common in MERS (90–100%) than SARS (60–100%), and MERS patients have a higher incidence of ARDS with a higher case fatality rate (~36%). For both diseases, risk factors like age and male sex are associated with poorer disease outcomes (165).
Early evidence implies that, similarly to SARS and MERS, fibrotic remodeling and scaring occurs in the lungs of severely ill COVID-19 patients. An alarmingly large number of COVID-19 patients reported persistent symptoms, mainly fatigue and dyspnea, even months after first diagnosis in multiple independent surveys (166–168). In line, radiological signs of fibrosis become apparent as early as 3 weeks after diagnosis (169) and persist over months (170, 171). After 3 months, impaired diffusion capacity and persisting radiological abnormalities are observed in many survivors, while others recover completely (172–176). Further studies are ongoing whether radiological and functional impairments are chronic and even progressing. Worrisomely, lung autopsies of deceased COVID-19 patients have revealed the aberrant localization of mucus to the alveolar parenchyma, pathologic signs of proliferative DAD and thickening of the alveolar wall, particularly after a long severe phase (82, 120, 125). These findings suggest that COVID-19 induces lung abnormalities including cases with pulmonary fibrosis. Notably, virus-induced cell fusion has been shown to induce cellular senescence (177). Giant cells are a pronounced feature in COVID-19 lungs which might be due to furin-mediated cleavage of the SARS-CoV-2 S protein at the plasma membrane of ACE2 expressing cells resulting in syncytial alveolar epithelial cells (98). Potentially, this results in the acquisition of a senescent alveolar epithelial cell phenotype that can provoke inflammation and fibrosis (178–180). Moreover, intussusceptive angiogenesis occurs to a greater extent in pulmonary COVID-19 as compared to influenza A pneumonia, suggesting activation of tissue regeneration that follows similar patterns as in pulmonary fibrosis (108).
The possibility to use early anti-fibrotic strategies is currently being investigated (16). The principal feature of anti-fibrotic treatment is preventing the worsening of the disease by slowing down the fibrotic progression in established lung fibrosis, and potentially influencing the cytokine storm by anti-inflammatory effects of these drugs (181). Currently, some clinical studies are investigating both available anti-fibrotic treatments in patients with COVID-19 (recruiting phase): pirfenidone (NCT04282902, NCT04607928) and nintedanib (NCT04541680, NCT04619680). These results will provide us with new insights into the relevance of the fibrotic changes in COVID-19 and the effectiveness of anti-fibrotic treatment to improve the management of those patients in the future. In parallel, novel treatment strategies might be discovered in vitro, particularly in light of the recent advances in the field of ex vivo tissue cultures, lung organoids, and bioengineered microfluidic devices to study lung fibrosis.
How Can We Study COVID-19-Associated Pulmonary Fibrosis?
In vivo and in vitro Models of Pulmonary Fibrosis
Despite tremendous research efforts for pharmacological interventions over the past decade, pulmonary fibrosis remains one of the most challenging diseases to manage clinically. Although a single model is unable to mirror the progressive and irreversible nature of lung fibrosis, they provide valuable mechanistic insights into fibrogenesis. Animal experimental models have been widely used to understand the complex fibrotic responses and perform early pre-clinical testing for anti-fibrotic drugs. Among them, the bleomycin-induced pulmonary fibrosis model has been most widely used since the 1970's as the classical standard and best characterized in vivo fibrosis model (182). Contrary to human pulmonary fibrosis, bleomycin-induced fibrosis is temporary, and its inflammatory aspect justified criticism to accurately represent the pathophysiological process in IPF. Aside from bleomycin, fluorescein isothiocyanate (FITC) has also been widely used to induce experimental lung fibrosis which results in alveolar injury and acute fibrotic reaction that persist up to 24 weeks. Occupational exposure to environmental risk factors has been extensively associated with pulmonary fibrosis (183). Reports suggested that inhalation of silica and asbestos particles in rats results in fibrotic nodule formation which closely mimics prominent features of silicosis and asbestosis in humans with long-term occupational exposure (184, 185). Additionally, whole thorax irradiation in mice has been invaluable to study early inflammatory responses in radiation-induced lung fibrosis (186). It is well-established that IPF includes genetic predisposition affecting genes encoding e.g., surfactant protein-C (SP-C) (187), SP-A (188), Mucin-5B (MUC5B) (189), telomerase reverse transcriptase (TERT), and telomerase RNA component (TERC) (145). These known mutations have paved the way for genetically modified animal models of pulmonary fibrosis. Furthermore, intratracheal delivery of pro-fibrotic cytokines like TGF-β1 (190), TNF-α (191), and IL-1β (192) by adenovirus and lentivirus vectors have been extensively used to recreate mild early inflammation and rapid onset of lung fibrosis in mouse models. Despite the fact that animal models cannot fully recapitulate the complex, progressive and irreversible nature of lung fibrosis in humans, they remain the first line for preclinical testing in lack of appropriate alternatives. Nevertheless, animal models have been proven valuable for gaining a better mechanistic understanding of fibrogenesis, assessing lung function in the course of pulmonary fibrosis and performing pharmacokinetic studies (Figure 2).
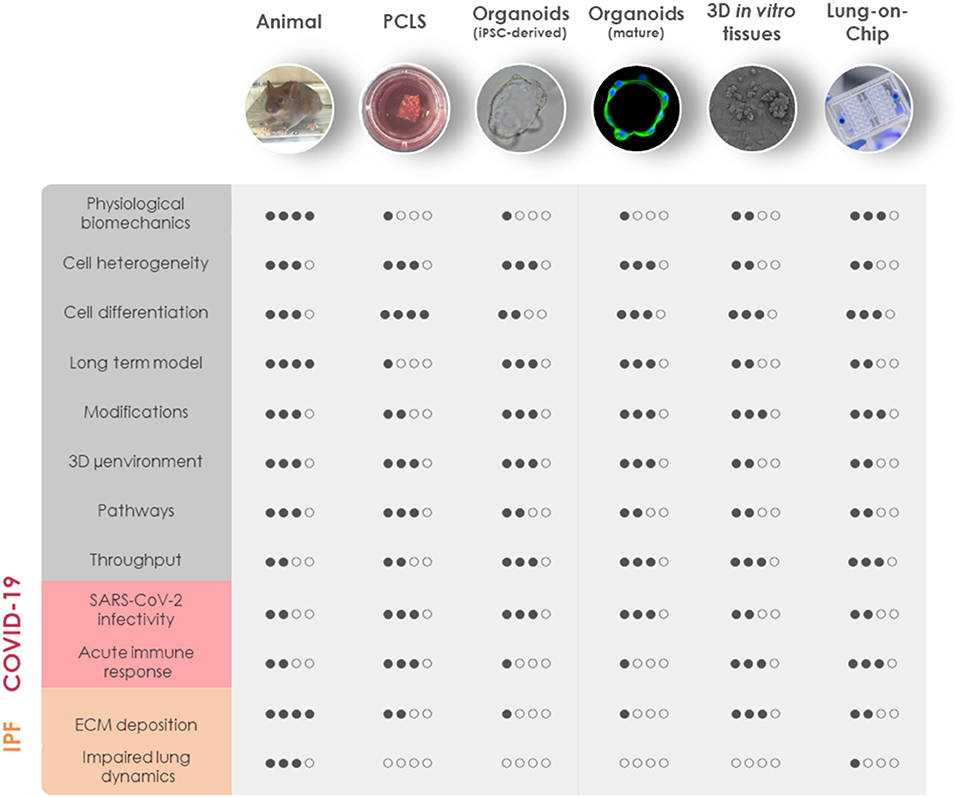
Figure 2. Comparison of in vivo and 3D in vitro lung models for COVID-19 and fibrosis research. General aspects of experimental animal models and advanced in vitro models including PCLS, iPSC-derived organoids, mature organoids, 3D in vitro tissues, and LOC are rated based on similarities to human physiology (physiological biomechanics, cell heterogeneity, cell differentiation, long-term model, and 3D microenvironment), genetic manipulation (modifications), the possibility for mechanistic investigations (pathways), and throughput capabilities (throughput). Their applicability to model the diseased state of the lung has been evaluated separately for COVID-19 and IPF. COVID-19, coronavirus disease 2019; IPF, idiopathic pulmonary fibrosis; iPSC, induced pluripotent stem cells; LOC, lung-on-chip; μenvironment, microenvironment; PCLS, precision-cut lung slices; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
However, most of our understanding of lung fibrosis stems solely from in vitro studies, typically relying on the activation of fibroblasts with pro-fibrotic cytokines in cellular models. Although in vitro fibrosis models represent a robust platform to study cell-specific responses to soluble cues in a controlled setting, cells in vivo are embedded in a complex 3D microenvironment with varied mechanical cues, cell-ECM interactions, differential polarity, and growth factor gradients. Given the strong involvement of fibroblasts and ECM in the pathology of fibrotic diseases, it is particularly important to maintain tissue architecture in human-derived models of fibrosis. Fibrotic tissue explants from patients suffering from a fibroproliferative skin disease, have been shown to retain viability for several days in ex vivo tissue culture, allowing to study molecular mechanisms of fibrosis and test novel therapeutic strategies (193). Recently, precision-cut lung slices (PCLS) have garnered increasing attention as a novel lung ex vivo fibrosis model. Overcoming the classical limitation for the study of human lung cells in 2D cell culture models, PCLS are able to spatially retain the native lung architecture along with fundamental ECM composition, stiffness and responsiveness together with viable lung resident cell populations [(194); Figure 2]. PCLS derived from healthy lung tissue resections closely mimic fibrotic-like changes including increased ECM deposition and alveolar remodeling when induced with a pro-fibrotic cocktail (195). A study in 2018 has reported that induction with TGF-β1 resulted in increased deposition of collagen and ECM proteins in 2 mm3 sections of human lung parenchymal tissues within 1 week in culture (196). The close recapitulation of pathologic processes and the possibility to culture tissue from IPF patients allows to study drug responses ex vivo. Interestingly, nintedanib and pirfenidone exhibit distinct anti-fibrotic potential in mouse and human PCLS underscoring the need for human-derived models of IPF (197). Notch1 inhibition in PCLS derived from IPF patients has shown significant improvement in surfactant protein processing along with decreased ECM deposition and an overall reversal of fibrosis (198). In addition, a study for inhalation-based anti-fibrotic therapies has utilized advanced 3D printing technologies to develop a replica for Ear-Nose-Throat which has been connected to an ex vivo porcine respiratory tract within a sealed chamber. To mimic fibrosis-related alterations, mechanical properties of the lung parenchyma have been modified by reduction of lung compliance and passive ventilation which allowed them to analyze in vivo aerosol regional deposition in a fibrosis-mimicking environment (199). Although a key advantage in using human tissues is the exclusion of cross-species heterogeneity, the significant limiting factor of ex vivo tissue culture is the constant need for fresh tissues. Generally, they are not readily available as the tissues are mostly obtained from “end-stage” pulmonary fibrosis patients after lung transplantation or healthy surrounding tissue from tumor resections used for artificially induced early fibrotic changes ex vivo. Moreover, the complexities associated with long-term cultivation of the lung explants makes it difficult to standardize PCLS technique for high-throughput testing. Nevertheless, PCLS can be useful to investigate specific aspects of pulmonary fibrosis and viral infection directly in human lung tissue (Figure 2).
Efforts have been undertaken to generate easily accessible, controlled model systems that provide structural and cellular complexity but hold the possibility to increase the throughput. Different cell types in the lung contribute to the pathology of fibrosis and hence the choice of the cell system is an important consideration for in vitro studies (200). Moreover, recent studies have focused more on using mechanically tunable substrates over standard extremely stiff (106 kPa) cell culture plastic dishes. Several studies have demonstrated that increased substrate stiffness directly influences (myo)fibroblast activation, differentiation and ECM deposition (201, 202). Instead, seeding fetal-derived fibroblasts on hydrogel beads to mimic the structure of alveolar sacs recreates the patchy areas of myofibroblast proliferation, contraction, and interstitial thickening upon TGF-β1 stimulation as it is observed in IPF patients (203). Tests for novel IPF medication and mechanistic studies on fibroblast invasion of IPF patients have also been undertaken in self-assembled pneumospheres comprising heterogeneous cell populations (204). Additionally, biocompatible and biodegradable cross-linked polymer like Matrigel is a widely used substrate for 3D lung cell culture and organoid modeling for fibrosis. A recent study has analyzed transcriptional signatures of fibrotic lung organoids in order to identify aberrantly expressed genes (205). While multicellular organoids closely capture the minute details of cell-cell and cell-ECM interactions and physiological cellular organization they lack vasculature and air-liquid interface (ALI) [(206); Figure 2]. Recently, it has been shown that ALI promotes differentiation of human pluripotent stem cell (hPSC)-derived alveolar epithelial progenitor cells into ATII-like cells and reduces their transdifferentiation into ATI cells, which occurs in submerged cultures (207). Stimulation with a pro-fibrotic cocktail results in the loss of SPC+ ATII cells paralleled by an increase in MUC5B+ goblet-like cells mimicking the bronchialization process occurring in the alveoli of IPF patients (207). However, these models still lack biomechanical stimulation (Figure 2).
Advanced microfluidic technologies have been able to overcome these limitations with the development of lung-on-chip (LOC) devices (208). Organ-on-Chip technology is a new field emerging only recently as a system to model human tissues for the application in research and pharmacology. Despite the development of multiorgan systems, it remains challenging to apply the technique for gaining insights into systemic effects and standardize the models for pre-clinical testing. Moreover, microfluidic systems require the optimization of many factors such as the ECM, medium and scaffold material to support optimal cell growth. However, the complexity of Organ-on-Chip technology is also a chance allowing the modulation of a variety of biological, physical, and chemical factors in a controlled and closed system (209). A microfluidic device recreating the alveolar epithelium in ALI and in close contact with a microchannel, that is lined by endothelial cells and perfused with human full blood, has been employed to study pulmonary vascular inflammation and microthrombus formation (210). Furthermore, tiny wounds can be induced to the alveolar epithelium on chip either by trypsin or gastric-like content to mimic alveolar damage taking place in IPF (211) and wound-healing (212). Moreover, micro-tissues generated from human lung fibroblasts have been shown to exhibit enhanced contractility, stiffness and expression of alpha smooth muscle actin (α-SMA), pro-collagen, and EDA fibronectin in response to TGF-β, effects that have been reversed by treatment with pirfenidone (213).
Due to the importance of cyclic stretch for tissue regeneration after lung injury, Stucki et al. developed a breathing LOC model with primary human alveolar epithelial cells and lung endothelial cells. This system incorporates key mechanical forces of the alveoli including 3D cyclical stretch (corresponding to 8% linear strain) and surface tension (through the exposure to ALI) to recreate the complex alveolar microenvironment of the air-blood barrier [(214, 215); Figure 2]. Further advances in these models aiming at integrating pathophysiological stretch and introducing the often-neglected pulmonary surfactant warrant a bright future for accurate in vitro models of the alveolus. However, the availability of optimal biological material (e.g., high-quality tissue specimens from the relevant anatomical site, high cell viability, and physiological ECM composition) is often challenging and therefore requires further methodological advances in cell culture and tissue processing.
In summary, recent advancements in bio-engineered tissue and cell culture highlights promising platforms for lung fibrosis modeling and drug testing in a clinically-relevant setup (Figure 2). Importantly, lung fibrosis models that are compatible with SARS-CoV-2 infection models will enable investigations on the regenerative phase of COVID-19.
Modeling SARS-CoV-2 Infection and Pathogenesis in the Respiratory Tract
In vivo models of viral infection integrate the full complexity of virulence factors, local and systemic immune responses and recovery. Therefore, animal models are particularly useful to test anti-inflammatory compounds and vaccines to combat infection (216). However, mice, the most widely available laboratory animals, are naturally resistant to SARS-CoV-2 infection (217, 218). The inability of SARS-CoV-2 to bind to murine ACE2 (74) poses the need to study COVID-19 in humanized mouse models expressing human ACE2 (217–220). SARS-CoV-2 infection in these mouse models results in weight loss, pneumonia and pathologic alterations in the lung tissue. However, the organ tropism and severity of symptoms varies among the models depending on the promoter to control human ACE2 expression. Mouse models expressing human ACE2 under the control of murine ACE2 develop rather mild symptoms and all animals spontaneously recover (217, 218). In contrast, severe pneumonia develops in mice expressing human ACE2 under the control of HFH4 or KRT18 promoter (219, 220). However, it remains arguable if these models correctly recapitulate SARS-CoV-2 tissue tropism given non-endogenous ACE2 expression patterns. Alternatively, mutation of the SARS-CoV-2 S protein or serial passaging in mice generates adapted virus to bind to murine ACE2 and infect the murine host (221, 222). These models might better resemble natural host-pathogen interactions in immunocompetent mice and result in mild pneumonia, however, it is unclear whether the mechanisms of mouse-adapted SARS-CoV-2 pathology can be translated to human.
Other animal species are naturally susceptible to SARS-CoV-2 (223). SARS-CoV-2 infects and replicates in ferrets but it is restricted to the upper respiratory tract allowing transmission studies but causing only mild symptoms (224, 225). Natural SARS-CoV-2 infection in golden hamsters and non-human primates involves the distal lung, however, results only in mild to modest pneumonitis and all infected animals spontaneously recover (226–231). Altogether, animal models recapitulate aspects of human COVID-19 such as an age-related risk to develop more severe disease as it has been demonstrated in mice and non-human primates (221, 222, 228, 230, 231). However, there are important differences between laboratory animal models and human COVID-19 pathogenesis (Figure 2). None of the available in vivo models captures the drastic hypoxia and associated coagulopathy, vascular inflammation and multi-organ failure as seen in severely ill COVID-19 patients. Mostly, SARS-CoV-2 infection takes a milder course in experimental animals or results in death by different pathologic mechanisms than in humans. This is likely due to a distinct distribution and affinity of ACE2 and TMPRSS2 for the SARS-CoV-2 S protein and fundamental differences in the immune system (232). Therefore, it is mandatory to complement in vivo data with findings garnered in vitro from human-derived models.
Essential knowledge about SARS-CoV-2 entry receptors, replication kinetics and cell-intrinsic immune response has been gained from in vitro studies using cell lines such as ACE2 overexpressing HeLa cells, the intrinsically IFN-deficient Vero E6 green monkey kidney cells or cancer cell lines (11, 74, 233). However, the relevance of SARS-CoV-2-induced gene expression changes in lung cancer cell lines such as A549 and Calu-3 remains arguable since these cells have lost the expression of Nkx-2.1, which is a master transcription factor for lung epithelial differentiation (234). Moreover, the lack of native ACE2 expression in some widely studied cell lines (e.g., HeLa, A549) and the absence of an intrinsic innate IFN response in Vero E6 cells hinders the assessment of normal physiological anti-viral responses (74, 235, 236). For this reason, the pathologic consequences of SARS-CoV-2 infection should ideally be studied in clinically relevant human cell systems.
Organoids generated from intestine, liver, microvasculature, kidney and airways are susceptible to SARS-CoV-2 infection. These studies have provided more comprehensive information on the SARS-CoV-2 target cell types and innate immune responses that are elicited by the virus (93, 237–240). Moreover, they provided functional evidence for the broad tissue tropism of SARS-CoV-2 as concluded earlier from the in silico analysis of ACE2 expression patterns among different organs and cell types (83). Due to the extraordinary difficulty to model the alveoli in vitro most studies aiming to elucidate the mechanisms of SARS-CoV-2 infection in its primary replication site have focused on the nasal, tracheal or bronchial airway epithelium. Bronchial organoids have been employed to identify target cell types in the upper airways and develop drug screening protocols. However, they do not support efficient SARS-CoV-2 infection (15, 101). This seems to be in disagreement with the higher ACE2 expression and susceptibility to SARS-CoV-2 infection in the upper airway epithelium as compared to alveoli (82). A possible explanation for this discrepancy might be the enrichment of basal progenitor cells in bronchial organoid cultures, which are not the primary target of SARS-CoV-2. Therefore, ALI cultures are more suitable to study SARS-CoV-2 in the upper airways. Human bronchial epithelial cells differentiate into functional ciliated and secretory cells in ALI cultures to form a pseudostratified epithelium capable of mucus production and cilia movement (241). They are efficiently infected by SARS-CoV-2 and produce high viral titers enabling functional studies and drug screening [(14, 82, 242); Figure 2]. In addition, a LOC model of the human bronchial epithelium under constant flow in the blood vessel chamber has recently been developed to study influenza A virus and SARS-CoV-2 infection and has led to the identification of candidate antiviral compounds (243). Hence, drug testing in a LOC device might further refine the number of candidate drugs (Figure 2).
In contrast, the alveoli are more challenging to reconstruct. Freshly isolated ATII cells rapidly transdifferentiate into ATI-like cells and are gradually lost in 2D in vitro cultures (244). A more stable ATII cell phenotype can be achieved by deriving ATII cells from induced hPSCs (245). These ATII cells can be maintained as organoids for prolonged cell culture but their main limitation is the fetal gene expression signature (10, 246). Seeding hPSC-derived ATII cells in ALI monolayers increases the degree of maturation, facilitates infection from the apical side and maintains the ATII cells, most likely due to the addition of the CHIR99021 Wnt agonist to the medium (102). SARS-CoV-2 infection elicits a rapid NF-κB, TNF, IL-6, and IL-2 signaling driven inflammatory response in infected ATII cells but induces only a modest and delayed IFN response (10, 102). This indicates that hPSC-derived ATII cell models capture the intrinsic antiviral response of ATII cells but not the full spectrum of COVID-19. Nevertheless, hPSC-derived ATII-like ALI cultures are a useful tool to study early events of SARS-CoV-2 infection and discover compounds with anti-viral activity in the alveolar setting (247). Co-culture models of hPSC-derived lung organoids and hPSC-derived macrophages indicate that in this setting macrophages are essential producers of IFN-γ and drive protective or damaging immune responses (248). A major limitation of hPSC-derived alveolar models for high-throughput drug testing is their time- and cost-intensive derivation that involves a multi-step differentiation protocol [(249); Figure 2]. As a consequence, hPSC lines are usually generated from a few donors and maintained for the derivation of ATII cells, which results in a rather homogeneous genetic background. Therefore, they neglect individual genetic predispositions such as polymorphisms in IFN pathway genes or in mucus production and regeneration which might have an impact on disease severity and the fibrotic response after acute phase (250–253). A more heterogeneous cellular composition has been achieved by differentiating fetal lung-derived SOX2+SOX9+ bud tip progenitor cell organoids in 2D ALI cultures. They comprise alveolar-like and bronchial-like cell types and are readily infected by SARS-CoV-2 (254). However, this model meets similar limitations as hPSC-derived alveolar models due to the limited access to donor material and the derivation of differentiated bronchioalveolar ALI cultures from few organoid lines.
The patient-to-patient variability can be captured by adult stem cell derived alveolar organoid models. Alveolar organoids have been generated from HTII-280+-enriched ATII cells (101, 103) or mixed alveolar epithelial cells (13, 100). They maintain an ATII cell subpopulation during prolonged culture. In the intact organoids, ACE2 entry receptor faces the lumen while the basolateral side is exposed to the external milieu. In order to infect the organoids with SARS-CoV-2, the apical side has to be exposed either by apical-out polarization in suspension (95) or mechanical and chemical dissociation for the infection as single cells (13, 101) or as 2D monolayers (100). In contrast to hPSC-derived ATII cell monolayers, adult stem cell derived alveolar organoids nearly entirely lose the ATII cell population upon culture in 2D monolayers (100). Interestingly, it has been shown that ATII cells from dissociated organoids preferentially transdifferentiate into ATI cells in short-term submerged culture resulting in an alveolar-like epithelium. In contrast, maintaining the same cells in long-term ALI culture results in the replacement of alveolar epithelial cells by ciliated and goblet cells to form a pseudostratified airway-like epithelium (100). Notably, the authors describe sustained SARS-CoV-2 infection in submerged alveolar-like monolayers while ALI airway-like monolayers show a slow initial infection followed by exponential viral replication starting on day 2 post-infection. Furthermore, an innate immune response signature that resembles the gene expression signature in the lungs of deceased COVID-19 patients is induced in alveolar-like epithelium cultures (100). The authors conclude that the proximal airway components of the model are important for SARS-CoV-2 infectivity, while transdifferentiating alveolar epithelial cells subsequently recapitulate the host response (100). Multiple studies have reported the induction of an IFN response and upregulation of inflammatory (NF-κB) pathways upon SARS-CoV-2 infection in alveolar organoids (13, 100, 101, 103). This characteristic allows to study IFN treatment, which has been administered to COVID-19 patients (255), in a physiologically relevant site. Interestingly, IFN treatment on alveosphere cultures induces apoptotic markers, upregulates ACE2 and TMPRSS2 expression and reduces the production of surfactant protein in ATII cells suggesting a potential positive effect on SARS-CoV-2 propagation. Nevertheless, IFN pre-treatment of alveospheres at lower doses impairs SARS-CoV-2 infection implying a preventive effect of IFN treatment in COVID-19 patients (103). Despite their utility for screening anti-viral and anti-inflammatory compounds, a major limitation of organoids is the lack of immune cells, a vascular-epithelial compartment and the ability to monitor epithelial barrier integrity (Figure 2).
LOC models have been applied in order to capture the complex physical and cellular microenvironment of the alveoli. They allow co-culture systems, temporal monitoring of trans-epithelial electrical resistance (TEER; a measure of barrier function) and integration of mechanical stretch or shear stress (256). The first study on SARS-CoV-2 infection in a bioengineered alveolus has employed an epithelial-endothelial co-culture approach under shear stress, which resulted in the differential expression of SARS-CoV-2 entry receptors as compared to 2D monolayers (99). Interestingly, infection with low SARS-CoV-2 titers induces a downregulation of NRP1 and ACE2 but an upregulation of TMPRSS2 expression in alveolar epithelial cells illustrating how multiple factors affect SARS-CoV-2 susceptibility and contribute to SARS-CoV-2 spread in the distal lung (99). In agreement with inefficient SARS-CoV-2 infection kinetics in alveolar cultures (13, 82), unproductive SARS-CoV-2 infection has also been observed in the alveolar layer in the LOC (99). However, SARS-CoV-2 infection elicits strikingly different responses among the cell types in co-culture. The alveolar cell layer remains largely intact, in line with previous findings in organoid models (101). In contrast, vascular injury is evident by 3 days post-infection resulting in damaged barrier function (99). This study provides evidence that the lung microvasculature is an essential contributor to model COVID-19 pathology in vitro, particularly in the maintenance of a prolonged pro-inflammatory response and IL-6 secretion (99). Disruption of the air-blood barrier in an alveolus-on-chip SARS-CoV-2 infection model and translocation of the virus to the vascular compartment has recently been demonstrated by another group, though the mechanism of vascular damage seems to be different (257). In the future, the application of LOC co-culture models will provide more mechanistic insights into host-pathogen interaction and inflammation-mediated damage. Moreover, LOC devices are superior to pure epithelial mono-cultures in predicting the protective effect of drugs on barrier integrity and inflammation [(99); Figure 2].
Modeling Alveolar Epithelium Regeneration and COVID-19-Associated Fibrotic Tissue Remodeling
Most in vitro studies on COVID-19 have focused on the acute phase of the disease. However, uncontrolled inflammatory and early fibrotic signatures are typically found in post-mortem lung autopsies of COVID-19 patients after a long disease course (132, 258). Radiologic abnormalities, including small airway abnormalities, are also found in the lungs of recovering COVID-19 patients but their consequences on the patient's quality of life are of yet unknown (170). Therefore, it will be important to dissect the sequela of virus clearance, resolution of the immune response and tissue regeneration in more detail. Unfortunately, it may take years until large patient cohorts have been followed-up for a sufficient amount of time to conclude on the clinical course of fibrotic remodeling. Therefore, it is mandatory to experimentally study lung fibrosis in the aftermath of COVID-19 in dedicated SARS-CoV-2 infection models. For this purpose, a pre-requisite is a model system of the alveoli that supports multicellular composition including epithelial, mesenchymal and immune cells and remains stable over an extended period of time to monitor the acute phase of COVID-19 and subsequent progression to fibrosis.
Animal models provide the complete tissue microenvironment and systemic context for temporally controlled infection. Humanized ACE2 mice develop COVID-19 and pathologic tissue remodeling has been confirmed in their lungs ~3 days post-infection (217, 219, 220). These studies have been conducted in healthy young or old animals, however, other predispositions than age have not yet been investigated in vivo in the context of COVID-19. Multiple groups have demonstrated that viral infection exacerbates IPF in the bleomycin mouse model (259–261). It will be interesting to investigate COVID-19-associated fibrosis in bleomycin-induced or genetically predisposed (e.g., MUC5B, SP-C, and TERT) animal models. Treatment with anti-fibrotic therapy and prolonged monitoring will eventually identify effective candidate compounds and unravel the mechanisms of tissue regeneration after acute lung injury. Nevertheless, it is questionable that these findings can directly translate to human COVID-19 due to the fundamental differences in lung architecture, regeneration, and immune response [(262); Figure 2]. Pre-clinical drug testing is expected to be more accurate in macaques due to the phylogenic proximity to humans (228, 263). However, few animal experiments are performed in non-human primates due to logistic, financial, and ethical concerns posing the need for reliable in vitro infection models.
With intact alveolar structures and preserved local immune responses, PCLS from animals have proven their utility in the study of viral infection such as influenza A viruses (264). Although pulmonary explants of human proximal and distal airways are susceptible to SARS-CoV-2 infection (265), PCLS technique has not yet been used to investigate COVID-19. Hence, PCLS can be potentially employed to study the action of anti-fibrotic agents within SARS-CoV-2-infected alveolar epithelium. For instance, senolytic combination treatment (dasatinib and quercetin) possesses anti-fibrotic activity in mouse PCLS (266) and might be an interesting treatment strategy in the context of COVID-19 given the acquisition of a senescence-like phenotype in SARS-CoV-2 infected syncytial alveolar epithelial cells. Despite the applicability of PCLS for the screening of anti-fibrotic agents, the utility of this model is limited for this particular disease (267). PCLS only permits to study the local immune response, ignoring the important recruitment of circulating immune cells. In addition, short term of culture limits investigations over time (Figure 2).
Alternatively, pulmonary fibrosis has been studied in organoid models, which can be maintained in culture for several weeks and passages. hPSC-derived lung organoids that contain a mix of epithelial and mesenchymal cells have been genetically engineered to develop Hermansky-Pudlak syndrome, a clinical condition with similarities to IPF, and have been sequenced to discover potential new drug targets (205). Despite accurate recapitulation of the disease phenotype and a gene expression signature of early fibrosis, the fibrotic organoids lack some major hallmarks of IPF such as activation of TGF-β signaling. Transcriptomic and histologic alterations of IPF have recently been modeled in hPSC-derived ATII-like cell cultures maintained in ALI (207). Given that alveolar epithelial progenitors, an essential contributor of aberrant wound healing, are enriched in hPSC-derived models they have been suggested as suitable model to study pulmonary fibrosis (207). However, since fibrosis predominantly occurs in senescent lungs, it is suboptimal to study fibrosis in an embryonic stem cell derived model system. Therefore, it will be interesting to study fibrotic changes in mixed mature long-term organoid culture after SARS-CoV-2 infection.
Potentially more physiologically relevant are ALI models, that can mimic SARS-CoV-2 infection by inhalation and provide a scaffold for epithelial/mesenchymal co-cultures. Yet it is not easy to avoid transdifferentiation of ATII cells, the main alveolar target cells of SARS-CoV-2 and drivers of fibrosis. Recently, the EpiAlveolar 3D tissue culture system has been developed to model micro-particle inhalation and the resulting pro-fibrotic events in the alveolar epithelium [(268); Figure 2]. Repeated exposure over an extended time period is possible, which implies that these models could also be applied to study the progression of acute COVID-19 to pulmonary fibrosis. However, it remains to be tested how efficiently SARS-CoV-2 infects these 3D alveolar tissue culture models and whether immune cells are required to induce fibrosis.
A higher degree of complexity can be achieved in LOC models. In the future, they might enable the investigation of cellular, mechanical, and chemical processes resulting in complex multi-factorial diseases such as pulmonary fibrosis. As compared to conventional cell culture, LOC models support stable cell culture systems over an extended time period, which is particularly relevant for studying pulmonary fibrosis, a disease of older age. However, as compared to the in vivo situation, long-term culture of cells in microfluidic devices remains a limiting factor to date. Nevertheless, aspects of lung inflammatory and fibrotic processes can be studied under nearly physiological conditions in such microfluidic devices. Importantly, it has been shown that substrate stiffness, porosity, and physiologic stretch has to be taken into account when studying tissue regeneration and assessing efficacy of therapeutic compounds (50). In addition, LOC models are well-suited to establish co-cultures enabling to study the sequence of molecular events in COVID-19 at the delicate air-blood barrier in the presence of alveolar resident cell types and peripheral immune cells (Figure 2). Taken together, advanced in vitro models have the potential to provide highly relevant data to discover novel effective treatment strategies for COVID-19, identify predictive biomarkers for a severe course of the disease and elucidate the mechanisms of lung repair in recovering patients.
Conclusion
Refined treatment strategies for recently emerged SARS-CoV-2 are becoming available and have improved the management of COVID-19 patients. Evidence has been gathered that particularly critically ill COVID-19 patients suffer from pulmonary dysfunction even months after diagnosis and may possibly never fully recover. SARS-CoV-2 infection of alveolar epithelial cells and an imbalanced inflammatory response result in DAD and trigger a fibrotic response to regenerate the epithelial barrier and lung function. To date, it is not clear whether fibrosis will develop and consequently resolve or progress. IPF, the most severe form of interstitial pulmonary fibrosis, is a fatal disease and few treatment options exist to slow down the progression of chronic fibrosis. It is therefore crucial, to delineate the early mechanisms that drive fibrotic progression after virus clearance. In vivo models capture aspects of human COVID-19 and have been classically used to study progression of IPF. However, due to inter-species anatomical and immunological differences, the translation of basic and pre-clinical animal research to patient-relevant insights often fails (182). The systemic manifestation of COVID-19 and the complex host-pathogen interactions highlight the importance of human-derived models to study the underlying mechanisms of the disease. Lung organoids promote cell-cell and cell-matrix interactions and provide a robust SARS-CoV-2 infection model of the alveoli. Due to their capacity to propagate and the relatively cost-efficient culture methods, patient-derived organoids enable medium-throughput drug screening for precision medicine in cancer and other diseases (269, 270). We expect that personalized therapy will also improve the management of severely ill COVID-19 patients. However, organoids are not ideally suited to construct complex co-cultures or study biomechanical forces, which might be a pre-requisite to gain more insights into COVID-19-associated pulmonary fibrosis. In this regard, PCLS, 3D in vitro tissue culture, and LOC are complementary models to lung organoids providing a higher degree of microenvironmental cues. These models have been used successfully to study IPF and are expectedly permissive for SARS-CoV-2 infection. Moreover, LOC holds the unique opportunity to study SARS-CoV-2 mediated alveolar injury at ALI and under stretch. It is well-known that stretch can significantly alter epithelial barrier permeability (215), ATII cell function and tissue regeneration (41, 271, 272). However, studies about the impact of stretch on the acute and recovery phase of COVID-19 are still lacking. The combinational application of advanced in vitro models is expected to generate meaningful data on the molecular processes taking place in the lungs of COVID-19 patients and providing insights into disease course before patient data from large cohorts become available. Hopefully, this knowledge will help to improve patient care and prevent fibrosis at an early stage of progression.
Author Contributions
MK, NR, CM, AS, TG, OG, MF-C, NH, and MK-dJ contributed to the review, writing, and revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research received financial support from the Bern Center for Precision Medicine (BCPM), the Horizon 2020 (Eurostars) program (project AIM4Doc) and the Lungenliga Bern.
Conflict of Interest
NR and NH are employed by AlveoliX AG. OG and TG are shareholder and in the scientific board of AlveoliX AG. MK and MK-dJ are collaborators of AlveoliX AG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
α-SMA, α-smooth muscle actin; ACE2, angiotensin-converting enzyme 2; ALI, air-liquid interface; AMϕ, alveolar macrophage; APN, aminopeptidase N; ARDS, Acute Respiratory Distress Syndrome; ATI, type I alveolar epithelial cell; ATII, type II alveolar epithelial cell; COVID-19, coronavirus disease 2019; CT scan, computed tomography scan; DAD, diffuse alveolar damage; DPP4, dipeptidyl peptidase 4; ECM, extracellular matrix; FITC, fluorescein isothiocyanate; hPSC, human pluripotent stem cell; IFN, interferon; IL, interleukin; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; LOC, lung-on-chip; MERS, Middle East Respiratory Syndrome; MERS-CoV, Middle East Respiratory Syndrome coronavirus; MUC5B, mucin-5B; NRP1, neuropilin-1; NRP2, neuropilin-2; PCLS, precision-cut lung slices; PCSK, proprotein convertase subtilisin kexin; PDGF, platelet-derived growth factor; RBD, receptor-binding domain; RTC, replication and transcription complex; S protein, spike protein; SARS, Severe Acute Respiratory Syndrome; SARS-CoV, Severe Acute Respiratory Syndrome coronavirus; SARS-CoV-2, Severe Acute Respiratory Syndrome coronavirus 2; SP-A, surfactant protein A; SP-C, surfactant protein C; TEER, transepithelial electrical resistance; TERC, telomerase RNA component; TERF-1, telomeric repeat-binding factor 1; TERT, telomerase reverse transcriptase; TGF-β, transforming growth factor β; TLC, total lung capacity; TMPRSS2, transmembrane protease serine subtype 2; TNF-α, tumor necrosis factor α.
References
1. Fung TS, Liu DX. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol. (2019) 73:529–57. doi: 10.1146/annurev-micro-020518-115759
2. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med. (2020) 382:1199–207. doi: 10.1056/nejmoa2001316
3. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
4. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA J Am Med Assoc. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
5. World Bank. Global Economic Prospects, June 2020. Washington, DC: World Bank (2020). doi: 10.1596/978-1-4648-1553-9
6. Gibson PG, Qin L, Puah SH. COVID-19 acute respiratory distress syndrome (ARDS): clinical features and differences from typical pre-COVID-19 ARDS. Med J Aust. (2020) 213:54–6.e1. doi: 10.5694/mja2.50674
7. Krammer F. SARS-CoV-2 vaccines in development. Nature. (2020) 586:516–27. doi: 10.1038/s41586-020-2798-3
8. Kandimalla R, John A, Abburi C, Vallamkondu J, Reddy PH. Current status of multiple drug molecules, and vaccines: an update in SARS-CoV-2 therapeutics. Mol Neurobiol. (2020) 57:4106–16. doi: 10.1007/s12035-020-02022-0
9. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA J Am Med Assoc. (2020) 323:1824–36. doi: 10.1001/jama.2020.6019
10. Han Y, Duan X, Yang L, Nilsson-Payant BE, Wang P, Duan F, et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature. (2021) 589:270–5. doi: 10.1038/s41586-020-2901-9
11. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
12. Holwerda M, V'kovski P, Wider M, Thiel V, Dijkman R. Identification of an antiviral compound from the pandemic response box that efficiently inhibits sars-cov-2 infection in vitro. Microorganisms. (2020) 8:1–19. doi: 10.3390/microorganisms8121872
13. Mulay A, Konda B, Garcia G, Yao C, Beil S, Villalba JM, et al. SARS-CoV-2 infection of primary human lung epithelium for COVID-19 modeling and drug discovery. Cell Rep. (2021) 109055. doi: 10.1016/j.celrep.2021.109055
14. Pizzorno A, Padey B, Julien T, Trouillet-Assant S, Traversier A, Errazuriz-Cerda E, et al. Characterization and treatment of SARS-CoV-2 in nasal and bronchial human airway epithelia. Cell Rep Med. (2020) 1:100059. doi: 10.1016/j.xcrm.2020.100059
15. Suzuki T, Itoh Y, Sakai Y, Saito A, Okuzaki D, Motooka D, et al. Generation of human bronchial organoids for SARS-CoV-2 research. bioRxiv [Preprint]. (2020) doi: 10.1101/2020.05.25.115600
16. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir. Med. (2020) 8:807–15. doi: 10.1016/S2213-2600(20)30225-3
17. Guler SA, Ebner L, Beigelman C, Bridevaux P-O, Brutsche M, et al. Pulmonary function and radiological features four months after COVID-19: first results from the national prospective observational Swiss COVID-19 lung study. Eur Respir J. (2021) 2003690. doi: 10.1183/13993003.03690-2020
18. Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med. (2014) 20:822–32. doi: 10.1038/nm.3642
19. Schneider JP, Wrede C, Hegermann J, Weibel ER, Mühlfeld C, Ochs M. On the topological complexity of human alveolar epithelial type 1 cells. Am J Respir Crit Care Med. (2019) 199:1153–6. doi: 10.1164/rccm.201810-1866LE
20. Weibel ER. On the tricks alveolar epithelial cells play to make a good lung. Am J Respir Crit Care Med. (2015) 191:504–13. doi: 10.1164/rccm.201409-1663OE
21. Cañadas O, Olmeda B, Alonso A, Pérez-Gil J. Lipid-protein protein-protein interactions in the pulmonary surfactant system their role in lung homeostasis. Int J Mol Sci. (2020) 21:3708. doi: 10.3390/ijms21103708
22. Olajuyin AM, Zhang X, Ji H.-L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. (2019) 5:63. doi: 10.1038/s41420-019-0147-9
23. Mason RJ, Williams MC. Type II alveolar cell. Defender of the alveolus. Am Rev Respir Dis. (1977) 115:81–91. doi: 10.1164/arrd.1977.115.S.81
24. Beers MF, Moodley Y. When is an alveolar type 2 cell an alveolar type 2 cell? A conundrum for lung stem cell biology and regenerative medicine. Am J Respir Cell Mol Biol. (2017) 57:18–27. doi: 10.1165/rcmb.2016-0426PS
25. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. (2013) 123:3025–36. doi: 10.1172/JCI68782
26. Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. (2018) 555:251–5. doi: 10.1038/nature25786
27. Jain R, Barkauskas CE, Takeda N, Bowie EJ, Aghajanian H, Wang Q, et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat Commun. (2015) 6:6727. doi: 10.1038/ncomms7727
28. de Mello Costa MF, Weiner AI, Vaughan AE. Basal-like progenitor cells: a review of dysplastic alveolar regeneration and remodeling in lung repair. Stem Cell Reports. (2020) 15:1015–25. doi: 10.1016/j.stemcr.2020.09.006
29. Wang L, Dorn P, Simillion C, Froment L, Berezowska S, Tschanz SA, et al. EpCAM+CD73+ mark epithelial progenitor cells in postnatal human lung and are associated with pathogenesis of pulmonary disease including lung adenocarcinoma. Am J Physiol Lung Cell Mol Physiol. (2020) 319:L794–809. doi: 10.1152/AJPLUNG.00279.2019
30. Hegab AE, Arai D, Gao J, Kuroda A, Yasuda H, Ishii M, et al. Mimicking the niche of lung epithelial stem cells and characterization of several effectors of their in vitro behavior. Stem Cell Res. (2015) 15:109–21. doi: 10.1016/j.scr.2015.05.005
31. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. (2018) 359:1118–23. doi: 10.1126/science.aam6603
32. Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. (2013) 229:298–309. doi: 10.1002/path.4104
33. Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. (2010) 298:L715–31. doi: 10.1152/ajplung.00361.2009
34. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
35. Raslan AA, Yoon JK. WNT signaling in lung repair and regeneration. Mol Cells. (2020) 43:774–83. doi: 10.14348/molcells.2020.0059
36. Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. (2010) 181:254–63. doi: 10.1164/rccm.200810-1615OC
37. Piñeiro-Hermida S, Autilio C, Martínez P, Bosch F, Pérez-Gil J, Blasco MA. Telomerase treatment prevents lung profibrotic pathologies associated with physiological aging. J Cell Biol. (2020) 219:1–21. doi: 10.1083/jcb.202002120
38. Waters CM, Roan E, Navajas D. Mechanobiology in lung epithelial cells: measurements, perturbations, and responses. Compr Physiol. (2012) 2:1–29. doi: 10.1002/cphy.c100090
39. Ashino Y, Ying X, Dobbs LG, Bhattacharya J. [Ca(2+)](i) oscillations regulate type II cell exocytosis in the pulmonary alveolus. Am J Physiol Lung Cell Mol Physiol. (2000) 279:L5–13. doi: 10.1152/ajplung.2000.279.1.L5
40. Dietl P, Haller T, Mair N, Frick M. Mechanisms of surfactant exocytosis in alveolar type II cells in vitro and in vivo. News Physiol Sci. (2001) 16:239–43. doi: 10.1152/physiologyonline.2001.16.5.239
41. Edwards YS. Stretch stimulation: its effects on alveolar type II cell function in the lung. Comp Biochem Physiol A Mol Integr Physiol. (2001) 129:245–60. doi: 10.1016/s1095-6433(01)00321-x
42. Gil J, Bachofen H, Gehr P, Weibel ER. Alveolar volume-surface area relation in air- and saline-filled lungs fixed by vascular perfusion. J Appl Physiol. (1979) 47:990–1001. doi: 10.1152/jappl.1979.47.5.990
43. Mercer RR, Laco JM, Crapo JD. Three-dimensional reconstruction of alveoli in the rat lung for pressure-volume relationships. J Appl Physiol. (1987) 62:1480–7. doi: 10.1152/jappl.1987.62.4.1480
44. Tschumperlin DJ, Margulies SS. Alveolar epithelial surface area-volume relationship in isolated rat lungs. J Appl Physiol. (1999) 86:2026–33. doi: 10.1152/jappl.1999.86.6.2026
45. Knudsen L, Ochs M. The micromechanics of lung alveoli: structure and function of surfactant and tissue components. Histochem Cell Biol. (2018) 150:661–76. doi: 10.1007/s00418-018-1747-9
46. Fredberg JJ, Kamm RD. Stress transmission in the lung: pathways from organ to molecule. Annu Rev Physiol. (2006) 68:507–41. doi: 10.1146/annurev.physiol.68.072304.114110
47. Desai LP, Chapman KE, Waters CM. Mechanical stretch decreases migration of alveolar epithelial cells through mechanisms involving Rac1 and Tiam1. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L958–65. doi: 10.1152/ajplung.90218.2008
48. Crosby LM, Luellen C, Zhang Z, Tague LL, Sinclair SE, Waters CM. Balance of life and death in alveolar epithelial type II cells: proliferation, apoptosis, and the effects of cyclic stretch on wound healing. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L536–46. doi: 10.1152/ajplung.00371.2010
49. Ito Y, Correll K, Schiel JA, Finigan JH, Prekeris R, Mason RJ. Lung fibroblasts accelerate wound closure in human alveolar epithelial cells through hepatocyte growth factor/c-Met signaling. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L94–105. doi: 10.1152/ajplung.00233.2013
50. Felder M, Trueeb B, Stucki AO, Borcard S, Stucki JD, Schnyder B, et al. Impaired wound healing of alveolar lung epithelial cells in a breathing lung-on-a-chip. Front Bioeng Biotechnol. (2019) 7:3. doi: 10.3389/fbioe.2019.00003
51. van Riet S, Ninaber DK, Mikkers HMM, Tetley TD, Jost CR, Mulder AA, et al. In vitro modelling of alveolar repair at the air-liquid interface using alveolar epithelial cells derived from human induced pluripotent stem cells. Sci Rep. (2020) 10:5499. doi: 10.1038/s41598-020-62226-1
52. Oeckler RA, Hubmayr RD. Ventilator-associated lung injury: a search for better therapeutic targets. Eur Respir J. (2007) 30:1216–26. doi: 10.1183/09031936.00104907
53. Rocco PRM, Marini JJ. What have we learned from animal models of ventilator-induced lung injury? Intensive Care Med. (2020) 46:2377–80. doi: 10.1007/s00134-020-06143-x
54. Sehlmeyer K, Ruwisch J, Roldan N, Lopez-Rodriguez E. Alveolar dynamics and beyond - the importance of surfactant protein c and cholesterol in lung homeostasis and fibrosis. Front Physiol. (2020) 11:386. doi: 10.3389/fphys.2020.00386
55. Lentz S, Roginski MA, Montrief T, Ramzy M, Gottlieb M, Long B. Initial emergency department mechanical ventilation strategies for COVID-19 hypoxemic respiratory failure and ARDS. Am J Emerg Med. (2020) 38:2194–202. doi: 10.1016/j.ajem.2020.06.082
56. Ravasio A, Hobi N, Bertocchi C, Jesacher A, Dietl P, Haller T. Interfacial sensing by alveolar type II cells: a new concept in lung physiology? Am J Physiol Physiol. (2011) 300:C1456–65. doi: 10.1152/ajpcell.00427.2010
57. Hobi N, Ravasio A, Haller T. Interfacial stress affects rat alveolar type II cell signaling and gene expression. Am J Physiol Cell Mol Physiol. (2012) 303:L117–29. doi: 10.1152/ajplung.00340.2011
58. Doyle IR, Morton S, Crockett AJ, Barr HA, Davidson KG, Jones MJ, et al. Composition of alveolar surfactant changes with training in humans. Respirology. (2000) 5:211–20. doi: 10.1046/j.1440-1843.2000.00251.x
59. Orgeig S, Morrison JL, Daniels CB. Evolution, development, and function of the pulmonary surfactant system in normal and perturbed environments. Compr Physiol. (2015) 6:363–422. doi: 10.1002/cphy.c150003
60. Pastrana-Rios B, Flach CR, Brauner JW, Mautone AJ, Mendelsohn R. A direct test of the “squeeze-out” hypothesis of lung surfactant function. External reflection FT-IR at the air/wave interface. Biochemistry. (1994) 33:5121–7. doi: 10.1021/bi00183a016
61. Nag K, Munro JG, Inchley K, Schürch S, Petersen NO, Possmayer F. SP-B refining of pulmonary surfactant phospholipid films. Am J Physiol Cell Mol Physiol. (1999) 277:L1179–89. doi: 10.1152/ajplung.1999.277.6.L1179
62. Hobi N, Giolai M, Olmeda B, Miklavc P, Felder E, Walther P, et al. A small key unlocks a heavy door: the essential function of the small hydrophobic proteins SP-B and SP-C to trigger adsorption of pulmonary surfactant lamellar bodies. Biochim Biophys Acta Mol Cell Res. (2016) 1863:2124–34. doi: 10.1016/j.bbamcr.2016.04.028
63. Roldan N, Nyholm TKM, Slotte JP, Pérez-Gil J, García-Álvarez B. Effect of lung surfactant protein SP-C and SP-C-promoted membrane fragmentation on cholesterol dynamics. Biophys J. (2016) 111:1703–13. doi: 10.1016/j.bpj.2016.09.016
64. Roldan N, Pérez-Gil J, Morrow MR, García-Álvarez B. Divide andamp; conquer: surfactant protein SP-C and cholesterol modulate phase segregation in lung surfactant. Biophys J. (2017) 113:847–59. doi: 10.1016/j.bpj.2017.06.059
65. Günther A, Schmidt R, Nix F, Yabut-Perez M, Guth C, Rosseau S, et al. Surfactant abnormalities in idiopathic pulmonary fibrosis, hypersensitivity pneumonitis and sarcoidosis. Eur Respir J. (1999) 14:565–73. doi: 10.1034/j.1399-3003.1999.14c14.x
66. Schmidt R, Meier U, Yabut-Perez M, Walmrath D, Grimminger F, Seeger W, et al. Alteration of fatty acid profiles in different pulmonary surfactant phospholipids in acute respiratory distress syndrome and severe pneumonia. Am J Respir Crit Care Med. (2001) 163:95–100. doi: 10.1164/ajrccm.163.1.9903029
67. Hobi N, Siber G, Bouzas V, Ravasio A, Pérez-Gil J, Haller T. Physiological variables affecting surface film formation by native lamellar body-like pulmonary surfactant particles. Biochim Biophys Acta Biomembr. (2014) 1838:1842–50. doi: 10.1016/j.bbamem.2014.02.015
68. Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. (2016) 3:237–61. doi: 10.1146/annurev-virology-110615-042301
69. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. (2020) 11:1620. doi: 10.1038/s41467-020-15562-9
70. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. (2020) 117:11727–34. doi: 10.1073/pnas.2003138117
71. Walls AC, Park Y, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–92.e6. doi: 10.1016/j.cell.2020.02.058
72. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C.-L., et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 367:1260–63. doi: 10.1126/science.abb2507
73. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. (2020) 5:562–9. doi: 10.1038/s41564-020-0688-y
74. Zhou P, Yang X.-L., Wang X.-G., Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
75. Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. (2015) 202:120–34. doi: 10.1016/j.virusres.2014.11.021
76. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA. (2005) 102:11876–81. doi: 10.1073/pnas.0505577102
77. Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. (2010) 84:12658–64. doi: 10.1128/JVI.01542-10
78. Glowacka I, Bertram S, Müller MA, Allen P, Soilleux E, Pfefferle S, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. (2011) 85:4122–34. doi: 10.1128/JVI.02232-10
79. Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. (2011) 85:873–82. doi: 10.1128/JVI.02062-10
80. Shirato K, Kawase M, Matsuyama S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol. (2013) 87:12552–61. doi: 10.1128/JVI.01890-13
81. Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA. (2014) 111:15214–9. doi: 10.1073/pnas.1407087111
82. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. (2020) 182:429–46.e14. doi: 10.1016/j.cell.2020.05.042
83. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
84. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. (2020) 14:185–92. doi: 10.1007/s11684-020-0754-0
85. Muus C, Luecken MD, Eraslan G, Sikkema L, Waghray A, Heimberg G, et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med. (2021) 27:546–59. doi: 10.1038/s41591-020-01227-z
86. Pan Y, Zhang D, Yang P, Poon LLM, Wang Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect Dis. (2020) 20:411–2. doi: 10.1016/S1473-3099(20)30113-4
87. Wang W, Xu Y, Gao R, Lu R, Han K, Wu G, et al. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. (2020) 323:1843–4. doi: 10.1001/jama.2020.3786
88. Zhang W, Du RH, Li B, Zheng XS, Yang XL, Hu B, et al. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect. (2020) 9:386–9. doi: 10.1080/22221751.2020.1729071
89. Martines RB, Ritter JM, Matkovic E, Gary J, Bollweg BC, Bullock H, et al. Pathology and pathogenesis of SARS-CoV-2 associated with fatal coronavirus disease, United States. Emerg Infect Dis. (2020) 26:2005–15. doi: 10.3201/eid2609.202095
90. Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. (2020) 581:465–9. doi: 10.1038/s41586-020-2196-x
91. Zeng Z, Xu L, Xie XY, Yan H, Xie BJ, Xu WZ, et al. Pulmonary pathology of early-phase COVID-19 pneumonia in a patient with a benign lung lesion. Histopathology. (2020) 77:823–31. doi: 10.1111/his.14138
92. Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T, et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. (2020) 39:e105114. doi: 10.15252/embj.20105114
93. Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, et al. SARS-CoV-2 productively infects human gut enterocytes. Science. (2020) 369:50–4. doi: 10.1126/science.abc1669
94. Ravindra NG, Alfajaro MM, Gasque V, Habet V, Wei J, Filler RB, et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium. bioRxiv. (2020) 1–39. doi: 10.1101/2020.05.06.081695
95. Salahudeen AA, Choi SS, Rustagi A, Zhu J, de la O. SM, et al. Progenitor identification and SARS-CoV-2 infection in long-term human distal lung organoid cultures. bioRxiv. (2020). doi: 10.1101/2020.07.27.212076
96. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. (2020) 181:1016–35.e19. doi: 10.1016/j.cell.2020.04.035
97. Blanco-Melo D, Nilsson-Payant BE, Liu W.-C., Uhl S, Hoagland D, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. (2020) 181:1036–45.e9. doi: 10.1016/j.cell.2020.04.026
98. Bussani R, Schneider E, Zentilin L, Collesi C, Ali H, Braga L, et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine. (2020) 61:103104. doi: 10.1016/j.ebiom.2020.103104
99. Thacker VV, Sharma K, Dhar N, Mancini G.-F., Sordet-Dessimoz J, et al. Rapid endothelialitis and vascular inflammation characterise SARS-CoV-2 infection in a human lung-on-chip model. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.08.10.243220
100. Tindle C, Fuller M, Fonseca A, Taheri S, Ibeawuchi S.-R., et al. Adult Stem Cell-derived Complete Lung Organoid Models Emulate Lung Disease in COVID-19. bioRxiv Prepr. Serv. Biol. (2020). doi: 10.1101/2020.10.17.344002
101. Youk J, Kim T, Evans KV, Jeong YI, Hur Y, Hong SP, et al. Three-dimensional human alveolar stem cell culture models reveal infection response to SARS-CoV-2. Cell Stem Cell. (2020) 27:905–919.e10. doi: 10.1016/j.stem.2020.10.004
102. Huang J, Hume AJ, Abo KM, Werder RB, Villacorta-Martin C, Alysandratos KD, et al. SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell. (2020) 27:962–73.e7. doi: 10.1016/j.stem.2020.09.013
103. Katsura H, Sontake V, Tata A, Kobayashi Y, Edwards CE, Heaton BE, et al. Human lung stem cell-based alveolospheres provide insights into SARS-CoV-2-mediated interferon responses and pneumocyte dysfunction. Cell Stem Cell. (2020) 27:890–904.e8. doi: 10.1016/j.stem.2020.10.005
104. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat. Med. (2020) 26:450–2. doi: 10.1038/s41591-020-0820-9
105. Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. (2020) 78:779–84.e5. doi: 10.1016/j.molcel.2020.04.022
106. Daly JL, Simonetti B, Klein K, Chen K.-E., Williamson MK, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. (2020) 370:861–5. doi: 10.1126/science.abd3072
107. Lau SY, Wang P, Mok BWY, Zhang AJ, Chu H, Lee ACY, et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg Microbes Infect. (2020) 9:837–42. doi: 10.1080/22221751.2020.1756700
108. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
109. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. (2020) 19:155–70. doi: 10.1038/s41579-020-00468-6
110. Adorni F, Prinelli F, Bianchi F, Giacomelli A, Pagani G, Bernacchia D, et al. Self-Reported symptoms of SARS-CoV-2 infection in a nonhospitalized population in italy: cross-sectional study of the EPICOVID19 web-based survey. JMIR Public Heal Surveill. (2020) 6:e21866. doi: 10.2196/21866
111. Bergquist SH, Partin C, Roberts DL, O'Keefe JB, Tong EJ, Zreloff J, et al. Non-hospitalized adults with COVID-19 differ noticeably from hospitalized adults in their demographic, clinical, social characteristics. SN Compr Clin Med. (2020) 2:1–9. doi: 10.1007/s42399-020-00453-3
112. Long QX, Tang XJ, Shi QL, Li Q, Deng HJ, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med. (2020) 26:1200–04. doi: 10.1038/s41591-020-0965-6
113. Oran DP, Topol EJ. Prevalence of asymptomatic SARS-CoV-2 infection : a narrative review. Ann Intern Med. (2020) 173:362–7. doi: 10.7326/M20-3012
114. Inui S, Fujikawa A, Jitsu M, Kunishima N, Watanabe S, Suzuki Y, et al. Chest CT findings in cases from the cruise ship diamond princess with coronavirus disease (COVID-19). Radiol Cardiothorac Imaging. (2020) 2:e200110. doi: 10.1148/ryct.2020200110
115. Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) Pneumonia in Two Patients With Lung Cancer. J Thorac Oncol. (2020) 15:700–4. doi: 10.1016/j.jtho.2020.02.010
116. Lan L, Xu D, Ye G, Xia C, Wang S, Li Y, et al. Positive RT-PCR test results in patients recovered from COVID-19. JAMA. (2020) 323:1502–3. doi: 10.1001/jama.2020.2783
117. Rogliani P, Calzetta L, Coppola A, Puxeddu E, Sergiacomi G, D'Amato D, et al. Are there pulmonary sequelae in patients recovering from COVID-19? Respir Res. (2020) 21:286. doi: 10.1186/s12931-020-01550-6
118. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. (2020) 130:2620–9. doi: 10.1172/JCI137244
119. Rodriguez-Morales AJ, Cardona-Ospina JA, Gutiérrez-Ocampo E, Villamizar-Peña R, Holguin-Rivera Y, Escalera-Antezana JP, et al. Clinical, laboratory and imaging features of COVID-19: a systematic review and meta-analysis. Travel Med Infect Dis. (2020) 34:101623. doi: 10.1016/j.tmaid.2020.101623
120. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
121. Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature. (2020) 583:437–40. doi: 10.1038/s41586-020-2355-0
122. Severe Covid-19 GWAS Group, Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, et al. Genomewide association study of severe covid-19 with respiratory failure. N Engl J Med. (2020) 383:1522–34. doi: 10.1056/NEJMoa2020283
123. Zietz M, Zucker J, Tatonetti NP. Associations between blood type and COVID-19 infection, intubation, and death. Nat Commun. (2020) 11:5761. doi: 10.1038/s41467-020-19623-x
124. Yao Y, Cao J, Wang Q, Shi Q, Liu K, Luo Z, et al. D-dimer as a biomarker for disease severity and mortality in COVID-19 patients: a case control study. J Intensive Care. (2020) 8:49. doi: 10.1186/s40560-020-00466-z
125. Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. (2020) 77:198–209. doi: 10.1111/his.14134
126. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med. (2020) 26:1017–32. doi: 10.1038/s41591-020-0968-3
127. Sorci G, Faivre B, Morand S. Explaining among-country variation in COVID-19 case fatality rate. Sci Rep. (2020) 10:18909. doi: 10.1038/s41598-020-75848-2
128. Wang K, Qiu Z, Liu J, Fan T, Liu C, Tian P, et al. Analysis of the clinical characteristics of 77 COVID-19 deaths. Sci Rep. (2020) 10:16384. doi: 10.1038/s41598-020-73136-7
129. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
130. Bauer TT, Ewig S, Rodloff AC, Müller EE. Acute respiratory distress syndrome and pneumonia: a comprehensive review of clinical data. Clin Infect Dis. (2006) 43:748–56. doi: 10.1086/506430
131. Cardinal-Fernández P, Lorente JA, Ballén-Barragán A, Matute-Bello G. Acute respiratory distress syndrome and diffuse alveolar damage. New insights on a complex relationship. Ann Am Thorac Soc. (2017) 14:844–50. doi: 10.1513/AnnalsATS.201609-728PS
132. Nienhold R, Ciani Y, Koelzer VH, Tzankov A, Haslbauer JD, Menter T, et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat Commun. (2020) 11:5086. doi: 10.1038/s41467-020-18854-2
133. Horwitz LI, Jones SA, Cerfolio RJ, Francois F, Greco J, Rudy B, et al. Trends in COVID-19 risk-adjusted mortality rates. J Hosp Med. (2020) 16:90–2. doi: 10.12788/jhm.3552
134. Herridge MS, Tansey CM, Matté A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. (2011) 364:1293–304. doi: 10.1056/NEJMoa1011802
135. Nöbauer-Huhmann IM, Eibenberger K, Schaefer-Prokop C, Steltzer H, Schlick W, Strasser K, et al. Changes in lung parenchyma after acute respiratory distress syndrome (ARDS): assessment with high-resolution computed tomography. Eur Radiol. (2001) 11:2436–43. doi: 10.1007/s003300101103
136. Yang J, Hooper WC, Phillips DJ, Tondella ML, Talkington DF. Induction of proinflammatory cytokines in human lung epithelial cells during Chlamydia pneumoniae infection. Infect. Immun. (2003) 71:614–20. doi: 10.1128/iai.71.2.614-620.2003
137. Drakopanagiotakis F, Xifteri A, Polychronopoulos V, Bouros D. Apoptosis in lung injury and fibrosis. Eur Respir J. (2008) 32:1631–8. doi: 10.1183/09031936.00176807
138. Kligerman SJ, Franks TJ, Galvin JR. From the radiologic pathology archives: organization and fibrosis as a response to lung injury in diffuse alveolar damage, organizing pneumonia, and acute fibrinous and organizing pneumonia. Radiographics. (2013) 33:1951–75. doi: 10.1148/rg.337130057
139. Venosa A, Tomer Y, Jamil S, Beers MF. Modeling immune effector responses in alveolar epithelial cell driven lung injury and fibrosis. J Immunol. (2020) 204(1 Supplement):74.1
140. Yanagi S, Tsubouchi H, Miura A, Matsumoto N, Nakazato M. Breakdown of epithelial barrier integrity and overdrive activation of alveolar epithelial cells in the pathogenesis of acute respiratory distress syndrome and lung fibrosis. Biomed Res Int. (2015) 2015:573210. doi: 10.1155/2015/573210
141. Glasser SW, Hagood JS, Wong S, Taype CA, Madala SK, Hardie WD. Mechanisms of lung fibrosis resolution. Am J Pathol. (2016) 186:1066–77. doi: 10.1016/j.ajpath.2016.01.018
142. Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. (2016) 1:e86704. doi: 10.1172/jci.insight.86704
143. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. (2018) 9:5363. doi: 10.1038/s41467-018-07768-9
144. Venosa A, Katzen J, Tomer Y, Kopp M, Jamil S, Russo SJ, et al. Epithelial expression of an interstitial lung disease-associated mutation in surfactant protein-c modulates recruitment and activation of key myeloid cell populations in mice. J Immunol. (2019) 202:2760–71. doi: 10.4049/jimmunol.1900039
145. Bilgili H, Białas AJ, Górski P, Piotrowski WJ. Telomere abnormalities in the pathobiology of idiopathic pulmonary fibrosis. J Clin Med. (2019) 8:1232. doi: 10.3390/jcm8081232
146. Yin L, Zheng D, Limmon GV, Leung NHN, Xu S, Rajapakse JC, et al. Aging exacerbates damage and delays repair of alveolar epithelia following influenza viral pneumonia. Respir Res. (2014) 15:116. doi: 10.1186/s12931-014-0116-z
147. Kasper M, Barth K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci Rep. (2017) 37:1–18. doi: 10.1042/BSR20171301
148. Agostini C, Gurrieri C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. (2006) 3:357–63. doi: 10.1513/pats.200601-010TK
149. Chambers RC, Mercer PF. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann Thorac Soc. (2015) 12(Suppl.) 1:S16–20. doi: 10.1513/AnnalsATS.201410-448MG
150. Fujiwara K, Kobayashi T, Fujimoto H, Nakahara H, D'Alessandro-Gabazza CN, Hinneh JA, et al. Inhibition of cell apoptosis and amelioration of pulmonary fibrosis by thrombomodulin. Am J Pathol. (2017) 187:2312–22. doi: 10.1016/j.ajpath.2017.06.013
151. Kim KK, Dotson MR, Agarwal M, Yang J, Bradley PB, Subbotina N, et al. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. (2018) 9:1056. doi: 10.1038/s41419-018-1074-z
152. Epstein Shochet G, Brook E, Bardenstein-Wald B, Shitrit D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir Res. (2020) 21:56. doi: 10.1186/s12931-020-1319-0
153. Chilosi M, Poletti V, Murer B, Lestani M, Cancellieri A, Montagna L, et al. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of deltaN-p63. Lab Invest. (2002) 82:1335–45. doi: 10.1097/01.lab.0000032380.82232.67
154. Kaarteenaho R, Kinnula VL. Diffuse alveolar damage: a common phenomenon in progressive interstitial lung disorders. Pulm Med. (2011) 2011:1–10. doi: 10.1155/2011/531302
155. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med. (2016) 194:265–75. doi: 10.1164/rccm.201604-0801CI
156. Tsui PT, Kwok ML, Yuen H, Lai ST. Severe acute respiratory syndrome: clinical outcome and prognostic correlates. Emerg Infect Dis. (2003) 9:1064–9. doi: 10.3201/eid0909.030362
157. Cheung OY, Chan JWM, Ng CK, Koo CK. The spectrum of pathological changes in severe acute respiratory syndrome (SARS). Histopathology. (2004) 45:119–24. doi: 10.1111/j.1365-2559.2004.01926.x
158. Ketai L, Paul NS, Wong KTT. Radiology of Severe Acute Respiratory Syndrome (SARS): the emerging pathologic-radiologic correlates of an emerging disease. J Thorac Imaging. (2006) 21:276–83. doi: 10.1097/01.rti.0000213581.14225.f1
159. Gu J, Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol. (2007) 170:1136–47. doi: 10.2353/ajpath.2007.061088
160. Tse GMK, To KF, Chan PKS., Lo AWI, Ng KC, Wu A, Lee N, et al. Pulmonary pathological features in coronavirus associated severe acute respiratory syndrome (SARS). J Clin Pathol. (2004) 57:260–5. doi: 10.1136/jcp.2003.013276
161. Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol. (2005) 18:1–10. doi: 10.1038/modpathol.3800247
162. Hui DS, Joynt GM, Wong KT, Gomersall CD, Li TS, Antonio G, et al. Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax. (2005) 60:401–9. doi: 10.1136/thx.2004.030205
163. Ngai JC, Ko FW, Ng SS, To K.-W., Tong M, et al. The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status. Respirology. (2010) 15:543–50. doi: 10.1111/j.1440-1843.2010.01720.x
164. Antonio GE, Wong KT, Hui DSC, Wu A, Lee N, Yuen EHY, et al. Thin-section CT in patients with severe acute respiratory syndrome following hospital discharge: preliminary experience. Radiology. (2003) 228:810–5. doi: 10.1148/radiol.2283030726
165. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. (2016) 14:523–34. doi: 10.1038/nrmicro.2016.81
166. Carfì A, Bernabei R, Landi F, Gemelli, Against COVID-19 Post-Acute Care Study Group. Persistent symptoms in patients after acute COVID-19. JAMA. (2020) 324:603–5. doi: 10.1001/jama.2020.12603
167. Goërtz YMJ, Van Herck M, Delbressine JM, Vaes AW, Meys R, Machado FVC, et al. Persistent symptoms 3 months after a SARS-CoV-2 infection: the post-COVID-19 syndrome? ERJ Open Res. (2020) 6:00542-2020. doi: 10.1183/23120541.00542-2020
168. Weerahandi H, Hochman KA, Simon E, Blaum C, Chodosh J, Duan E, et al. Post-discharge health status and symptoms in patients with severe COVID-19. medRxiv. (2020) 2:37–41. doi: 10.1101/2020.08.11.20172742
169. Shi H, Han X, Jiang N, Cao Y, Alwalid O, Gu J, et al. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. (2020) 20:425–34. doi: 10.1016/S1473-3099(20)30086-4
170. Ebner L, Funke-Chambour M, von Garnier C, Ferretti G, Ghaye B, Beigelman-Aubry C. Imaging in the aftermath of COVID-19: what to expect. Eur Radiol. (2020). doi: 10.1007/s00330-020-07465-6. [Epub Ahead of Print].
171. Zhao YM, Shang YM, Song WB, Li QQm, Xie H, Xu QF, et al. Follow-up study of the pulmonary function and related physiological characteristics of COVID-19 survivors three months after recovery. EClinicalMedicine. (2020) 25:100463. doi: 10.1016/j.eclinm.2020.100463
172. Lerum TV, Aaløkken TM, Brønstad E, Aarli B, Ikdahl E, Lund KMA, et al. Dyspnoea, lung function and CT findings three months after hospital admission for COVID-19. Eur Respir J. (2020). doi: 10.1183/13993003.03448-2020. [Epub Ahead of Print].
173. Liang L, Yang B, Jiang N, Fu W, He X, Zhou Y, et al. Three-month follow-up study of survivors of coronavirus disease 2019 after discharge. J Korean Med Sci. (2020) 35:e418. doi: 10.3346/jkms.2020.35.e418
174. Shah AS, Wong AW, Hague CJ, Murphy DT, Johnston JC, Ryerson CJ, et al. A prospective study of 12-week respiratory outcomes in COVID-19-related hospitalisations. Thorax. (2021) 76:402–4. doi: 10.1136/thoraxjnl-2020-216308
175. Sonnweber T, Sahanic S, Pizzini A, Luger A, Schwabl C, Sonnweber B, et al. Cardiopulmonary recovery after COVID-19 - an observational prospective multi-center trial. Eur Respir J. (2020) 10:2003481. doi: 10.1183/13993003.03481-2020
176. van der Sar-van der Brugge S, Talman S, Boonman-de Winter L, de Mol M, Hoefman E, van Etten RW, et al. Pulmonary function and health-related quality of life after COVID-19 pneumonia. Respir Med. (2020) 176:106272. doi: 10.1016/j.rmed.2020.106272
177. Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, et al. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. (2013) 27:2356–66. doi: 10.1101/gad.227512.113
178. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. doi: 10.1038/nrm3823
179. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. (2015) 21:1424–35. doi: 10.1038/nm.4000
180. Hansel C, Jendrossek V, Klein D. Cellular senescence in the lung: the central role of senescent epithelial cells. Int J Mol Sci. (2020) 21:1–21. doi: 10.3390/ijms21093279
181. Vitiello A, Pelliccia C, Ferrara F. COVID-19 patients with pulmonary fibrotic tissue: clinical pharmacological rational of antifibrotic therapy. SN Compr Clin Med. (2020) 2:1–4. doi: 10.1007/s42399-020-00487-7
182. Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Königshoff M, Kolb M, et al. An official american thoracic society workshop report: use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol. (2017) 56:667–79. doi: 10.1165/rcmb.2017-0096ST
183. Trethewey SP, Walters GI. The role of occupational and environmental exposures in the pathogenesis of idiopathic pulmonary fibrosis: a narrative literature review. Medicina. (2018) 54:108. doi: 10.3390/medicina54060108
184. Padilla-Carlin DJ, Schladweiler MCJ, Shannahan JH, Kodavanti UP, Nyska A, Burgoon LD, et al. Pulmonary inflammatory and fibrotic responses in Fischer 344 rats after intratracheal instillation exposure to Libby amphibole. J Toxicol Environ Health A. (2011) 74:1111–32. doi: 10.1080/15287394.2011.586940
185. Shoeb M, Mustafa GM, Joseph P, Umbright C, Kodali V, Roach KA, et al. Initiation of pulmonary fibrosis after silica inhalation in rats is linked with dysfunctional shelterin complex and DNA damage response. Sci Rep. (2019) 9:471. doi: 10.1038/s41598-018-36712-6
186. Jin H, Yoo Y, Kim Y, Kim Y, Cho J, Lee YS. Radiation-induced lung fibrosis: preclinical animal models and therapeutic strategies. Cancers. (2020) 12:1–24. doi: 10.3390/cancers12061561
187. Nureki S.-I., Tomer Y, Venosa A, Katzen J, Russo SJ, et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest. (2018) 128:4008–24. doi: 10.1172/JCI99287
188. Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet. (2016) 25:1457–67. doi: 10.1093/hmg/ddw014
189. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. (2011) 364:1503–12. doi: 10.1056/NEJMoa1013660
190. Boutanquoi PM, Burgy O, Beltramo G, Bellaye PS, et al. TRIM33 prevents pulmonary fibrosis by impairing TGF-β1 signalling. Eur Respir J. (2020) 55:1901346. doi: 10.1183/13993003.01346-2019
191. Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA, et al. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest. (1995) 96:250–9. doi: 10.1172/JCI118029
192. Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. (2001) 107:1529–36. doi: 10.1172/JCI12568
193. Karkampouna S, Kruithof BPT, Kloen P, Obdeijn MC, van der Laan AM, Tanke HJ, et al. Novel ex vivo culture method for the study of dupuytren's disease: effects of TGFβ type 1 receptor modulation by antisense oligonucleotides. Mol Ther Nucleic Acids. (2014) 3:e142. doi: 10.1038/mtna.2013.69
194. Gerckens M, Alsafadi HN, Wagner DE, Lindner M, Burgstaller G, Königshoff M. Generation of human 3D Lung Tissue Cultures (3D-LTCs) for disease modeling. J Vis Exp. (2019). doi: 10.3791/58437
195. Alsafadi HN, Staab-Weijnitz CA, Lehmann M, Lindner M, Peschel B, Königshoff M, et al. An ex vivo model to induce early fibrosis-like changes in human precision-cut lung slices. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L896–902. doi: 10.1152/ajplung.00084.2017
196. Roach KM, Sutcliffe A, Matthews L, Elliott G, Newby C, Amrani Y, et al. A model of human lung fibrogenesis for the assessment of anti-fibrotic strategies in idiopathic pulmonary fibrosis. Sci Rep. (2018) 8:342. doi: 10.1038/s41598-017-18555-9
197. Lehmann M, Buhl L, Alsafadi HN, Klee S, Hermann S, Mutze K, et al. Differential effects of nintedanib and pirfenidone on lung alveolar epithelial cell function in ex vivo murine and human lung tissue cultures of pulmonary fibrosis. Respir Res. (2018) 19:175. doi: 10.1186/s12931-018-0876-y
198. Wasnick R, Korfei M, Piskulak K, Henneke I, Wilhelm J, Mahavadi P, et al. Restored alveolar epithelial differentiation and reversed human lung fibrosis upon Notch inhibition. bioRxiv [Preprint]. (2019). doi: 10.1101/580498
199. Montigaud Y, Périnel S, Dubus JC, Leclerc L, Suau M, Goy C, et al. Development of an ex vivo respiratory pediatric model of bronchopulmonary dysplasia for aerosol deposition studies. Sci Rep. (2019) 9:5720. doi: 10.1038/s41598-019-42103-2
200. Sundarakrishnan A, Chen Y, Black LD, Aldridge BB, Kaplan DL. Engineered cell and tissue models of pulmonary fibrosis. Adv Drug Deliv Rev. (2018) 129:78–94. doi: 10.1016/j.addr.2017.12.013
201. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L344–57. doi: 10.1152/ajplung.00300.2014
202. Asano S, Ito S, Takahashi K, Furuya K, Kondo M, Sokabe M, et al. Matrix stiffness regulates migration of human lung fibroblasts. Physiol Rep. (2017) 5:1–11. doi: 10.14814/phy2.13281
203. Wilkinson DC, Alva-Ornelas JA, Sucre JMS, Vijayaraj P, Durra A, Richardson W, et al. Development of a three-dimensional bioengineering technology to generate lung tissue for personalized disease modeling. Stem Cells Transl. Med. (2017) 6:622–33. doi: 10.5966/sctm.2016-0192
204. Surolia R, Li FJ, Wang Z, Li H, Dsouza K, Thomas V, et al. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI insight. (2019) 4:e123253. doi: 10.1172/jci.insight.123253
205. Strikoudis A, Cieślak A, Loffredo L, Chen Y.-W., Patel N, et al. Modeling of fibrotic lung disease using 3d organoids derived from human pluripotent stem cells. Cell Rep. (2019) 27:3709–23.e5. doi: 10.1016/j.celrep.2019.05.077
206. Yanagihara T, Chong SG, Vierhout M, Hirota JA, Ask K, Kolb M. Current models of pulmonary fibrosis for future drug discovery efforts. Expert Opin Drug Discov. (2020) 15:931–41. doi: 10.1080/17460441.2020.1755252
207. Schruf E, Schroeder V, Le HQ, Schönberger T, Raedel D, Stewart EL, et al. Recapitulating idiopathic pulmonary fibrosis related alveolar epithelial dysfunction in a human iPSC-derived air-liquid interface model. FASEB J. (2020) 34:7825–46. doi: 10.1096/fj.201902926R
208. Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. (2010) 328:1662–8. doi: 10.1126/science.1188302
209. Low LA, Mummery C, Berridge BR, Austin CP, Tagle DA. Organs-on-chips: into the next decade. Nat Rev Drug Discov. (2020). doi: 10.1038/s41573-020-0079-3. [Epub Ahead of Print].
210. Jain A, Barrile R, van der Meer AD, Mammoto A, Mammoto T, De Ceunynck K, et al. Primary human lung alveolus-on-a-chip model of intravascular thrombosis for assessment of therapeutics. Clin Pharmacol Ther. (2018) 103:332–40. doi: 10.1002/cpt.742
211. Felder M, Stucki AO, Stucki JD, Geiser T, Guenat OT. The potential of microfluidic lung epithelial wounding: towards in vivo-like alveolar microinjuries. Integr Biol. (2014) 6:1132–40. doi: 10.1039/c4ib00149d
212. Felder M, Sallin P, Barbe L, Haenni B, Gazdhar A, Geiser T, et al. Microfluidic wound-healing assay to assess the regenerative effect of HGF on wounded alveolar epithelium. Lab Chip. (2012) 12:640–6. doi: 10.1039/c1lc20879a
213. Asmani M, Velumani S, Li Y, Wawrzyniak N, Hsia I, Chen Z, et al. Fibrotic microtissue array to predict anti-fibrosis drug efficacy. Nat Commun. (2018) 9:2066. doi: 10.1038/s41467-018-04336-z
214. Stucki AO, Stucki JD, Hall SRR, Felder M, Mermoud Y, Schmid RA, et al. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip. (2015) 15:1302–1310. doi: 10.1039/c4lc01252f
215. Stucki JD, Hobi N, Galimov A, Stucki AO, Schneider-Daum N, Lehr CM, et al. Medium throughput breathing human primary cell alveolus-on-chip model. Sci Rep. (2018) 8:14359. doi: 10.1038/s41598-018-32523-x
216. Leist SR, Schäfer A, Martinez DR. Cell and animal models of SARS-CoV-2 pathogenesis and immunity. Dis Model Mech. (2020) 13:dmm046581. doi: 10.1242/dmm.046581
217. Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. (2020) 583:830–3. doi: 10.1038/s41586-020-2312-y
218. Sun SH, Chen Q, Gu HJ, Yang G, Wang YX, Huang XY, et al. A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe. (2020) 28:124–33.e4. doi: 10.1016/j.chom.2020.05.020
219. Jiang RD, Liu MQ, Chen Y, Shan C, Zhou YW, Shen XR, et al. Pathogenesis of SARS-CoV-2 in transgenic mice expressing human angiotensin-converting enzyme 2. Cell. (2020) 182:50–8.e8. doi: 10.1016/j.cell.2020.05.027
220. Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat Immunol. (2020) 21:1327–35. doi: 10.1038/s41590-020-0778-2
221. Dinnon KH, Leist SR, Schäfer A, Edwards CE, Martinez DR, Montgomery SA, et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature. (2020) 586:560–6. doi: 10.1038/s41586-020-2708-8
222. Gu H, Chen Q, Yang G, He L, Fan H, Deng YQ, et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science. (2020) 369:1603–7. doi: 10.1126/science.abc4730
223. Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B, et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science. (2020) 368:1016–20. doi: 10.1126/science.abb7015
224. Kim Y.-I., Kim S.-G., Kim S.-M. -H., Park S.-J., Yu K.-M., et al. Infection and Rapid Transmission of SARS-CoV-2 in Ferrets. Cell Host Microbe. (2020) 27:704–709.e2. doi: 10.1016/j.chom.2020.03.023
225. Park SJ, Yu KM, Kim YI, Kim SM, Kim EH, Kim SG, et al. Antiviral efficacies of FDA-approved drugs against SARS-CoV-2 infection in ferrets. MBio. (2020) 11:1–10. doi: 10.1128/mBio.01114-20
226. Chan JFW, Zhang AJ, Yuan S, Poon V, Chan CCS, Lee ACY, et al. Simulation of the clinical and Pathological Manifestations of Coronavirus Disease 2019 (COVID-19) in a Golden Syrian Hamster Model: implications for disease pathogenesis and transmissibility. Clin Infect Dis. (2020) 71:2428–46. doi: 10.1093/cid/ciaa325
227. Chandrashekar A, Liu J, Martinot AJ, McMahan K, Mercado NB, Peter L, et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science. (2020) 369:812–7. doi: 10.1126/science.abc4776
228. Lu S, Zhao Y, Yu W, Yang Y, Gao J, Wang J, et al. Comparison of nonhuman primates identified the suitable model for COVID-19. Signal Transduct Target Ther. (2020) 5:157. doi: 10.1038/s41392-020-00269-6
229. Munster VJ, Feldmann F, Williamson BN, van Doremalen N, Pérez-Pérez L, Schulz J, et al. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature. (2020) 585:268–72. doi: 10.1038/s41586-020-2324-7
230. Rockx B, Kuiken T, Herfst S, Bestebroer T, Lamers MM, Oude Munnink BB, et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science. (2020) 368:1012–5. doi: 10.1126/science.abb7314
231. Yu P, Qi F, Xu Y, Li F, Liu P, Liu J, et al. Age-related rhesus macaque models of COVID-19. Anim Model Exp Med. (2020) 3:93–7. doi: 10.1002/ame2.12108
232. Ehaideb SN, Abdullah ML, Abuyassin B, Bouchama A. Evidence of a wide gap between COVID-19 in humans and animal models: a systematic review. Crit Care. (2020) 24:594. doi: 10.1186/s13054-020-03304-8
233. Chu H, Chan JFW, Yuen TTT, Shuai H, Yuan S, Wang Y, et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: an observational study. Lancet Microbe. (2020) 1:e14–23. doi: 10.1016/S2666-5247(20)30004-5
234. Abo KM, Ma L, Matte T, Huang J, Alysandratos KD, Werder RB, et al. Human iPSC-derived alveolar and airway epithelial cells can be cultured at air-liquid interface and express SARS-CoV-2 host factors. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.06.03.132639
235. Emeny JM, Morgan MJ. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol. (1979) 43:247–52. doi: 10.1099/0022-1317-43-1-247
236. Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. (2005) 79:14614–21. doi: 10.1128/JVI.79.23.14614-14621.2005
237. Krüger J, Groß R, Conzelmann C, Müller JA, Koepke L, Sparrer KMJ, et al. Drug inhibition of SARS-CoV-2 replication in human pluripotent stem cell-derived intestinal organoids. Cell Mol Gastroenterol Hepatol. (2020) 11:935–48. doi: 10.1016/j.jcmgh.2020.11.003
238. Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. (2020) 181:905–13.e7. doi: 10.1016/j.cell.2020.04.004
239. Stanifer ML, Kee C, Cortese M, Zumaran CM, Triana S, Mukenhirn M, et al. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep. (2020) 32:107863. doi: 10.1016/j.celrep.2020.107863
240. Zhao B, Ni C, Gao R, Wang Y, Yang L, Wei J, et al. Recapitulation of SARS-CoV-2 infection and cholangiocyte damage with human liver ductal organoids. Protein Cell. (2020) 11:771–5. doi: 10.1007/s13238-020-00718-6
241. Jonsdottir HR, Dijkman R. Coronaviruses and the human airway: a universal system for virus-host interaction studies. Virol J. (2016) 13:24. doi: 10.1186/s12985-016-0479-5
242. V'kovski P, Gultom M, Kelly JN, Steiner S, Russeil J, Mangeat B, et al. Disparate temperature-dependent virus-host dynamics for SARS-CoV-2 and SARS-CoV in the human respiratory epithelium. PLoS Biol. (2021) 19:e3001158. doi: 10.1371/journal.pbio.3001158
243. Si L, Bai H, Rodas M, Cao W, Oh CY, Jiang A, et al. Human organ chip-enabled pipeline to rapidly repurpose therapeutics during viral pandemics. bioRxiv. (2020) 02115:212–41. doi: 10.1101/2020.04.13.039917
244. Fuchs S, Hollins AJ, Laue M, Schaefer UF, Roemer K, Gumbleton M, et al. Differentiation of human alveolar epithelial cells in primary culture: morphological characterization and synthesis of caveolin-1 and surfactant protein-C. Cell Tissue Res. (2003) 311:31–45. doi: 10.1007/s00441-002-0653-5
245. Tamò L, Hibaoui Y, Kallol S, Alves MP, Albrecht C, Hostettler KE, et al. Generation of an alveolar epithelial type II cell line from induced pluripotent stem cells. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L921–32. doi: 10.1152/ajplung.00357.2017
246. Chen YW, Huang SX, de Carvalho ALRT, Ho SH, Islam MN, Volpi S, et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat Cell Biol. (2017) 19:542–9. doi: 10.1038/ncb3510
247. Hekman RM, Hume AJ, Goel RK, Abo KM, Huang J, Blum BC, et al. Actionable cytopathogenic host responses of human alveolar type 2 cells to SARS-CoV-2. Mol Cell. (2020) 80:1104–22.e9. doi: 10.1016/j.molcel.2020.11.028
248. Duan F, Guo L, Yang L, Han Y, Thakur A, Nilsson-Payant BE, et al. Modeling COVID-19 with human pluripotent stem cell-derived cells reveals synergistic effects of anti-inflammatory macrophages with ACE2 inhibition against SARS-CoV-2. Res Square [Preprint]. (2020). doi: 10.21203/rs.3.rs-62758/v1
249. Jacob A, Vedaie M, Roberts DA, Thomas DC, Villacorta-Martin C, Alysandratos KD, et al. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat. Protoc. (2019) 14:3303–32. doi: 10.1038/s41596-019-0220-0
250. Chan K, Zheng J, Mok Y, Li Y, Liu YN, Chu CM, et al. SARS: prognosis, outcome and sequelae. Respirology. (2003) 8:S36–40. doi: 10.1046/j.1440-1843.2003.00522.x
251. Rogers AJ, Solus JF, Hunninghake GM, Baron RM, Meyer NJ, Janz DR, et al. MUC5B promoter polymorphism and development of acute respiratory distress syndrome. Am J Respir Crit Care Med. (2018) 198:1342–5. doi: 10.1164/rccm.201801-0140LE
252. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. (2020) 370:eabd4585. doi: 10.1126/science.abd4585
253. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. (2020) 370: eabd4570. doi: 10.1126/science.abd4570
254. Lamers MM, van der Vaart J, Knoops K, Riesebosch S, Breugem TI, Mykytyn AZ, et al. An organoid-derived bronchioalveolar model for SARS-CoV-2 infection of human alveolar type II-like cells. EMBO J. (2021) 40:e105912. doi: 10.15252/embj.2020105912
255. Haji Abdolvahab M, Moradi-Kalbolandi S, Zarei M, Bose D, Majidzadeh-A K, Farahmand L, et al. Potential role of interferons in treating COVID-19 patients. Int Immunopharmacol. (2021) 90:107171. doi: 10.1016/j.intimp.2020.107171
256. Guenat OT, Geiser T, Berthiaume F. Clinically relevant tissue scale responses as new readouts from organs-on-a-chip for precision medicine. Annu Rev Anal Chem. (2020) 13:111–33. doi: 10.1146/annurev-anchem-061318-114919
257. Deinhardt-Emmer S, Böttcher S, Häring C, Giebeler L, Henke A, Zell R, et al. SARS-CoV-2 causes severe epithelial inflammation and barrier dysfunction. J Virol. (2021). doi: 10.1128/JVI.00110-21. [Epub Ahead of Print]
258. Wu M, Chen Y, Xia H, Wang C, Tan CY, Cai X, et al. Transcriptional and proteomic insights into the host response in fatal COVID-19 cases. Proc Natl Acad Sci USA. (2020) 117:28336–43. doi: 10.1073/pnas.2018030117
259. Lok SS, Haider Y, Howell D, Stewart JP, Hasleton PS, Egan JJ. Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur Respir J. (2002) 20:1228–32. doi: 10.1183/09031936.02.00272902
260. Wang L, Cheng W, Zhang Z. Respiratory syncytial virus infection accelerates lung fibrosis through the unfolded protein response in a bleomycin-induced pulmonary fibrosis animal model. Mol Med Rep. (2017) 16:310–6. doi: 10.3892/mmr.2017.6558
261. Huang S, Goplen NP, Zhu B, Cheon IS, Son Y, Wang Z, et al. Macrophage PPAR-γ suppresses long-term lung fibrotic sequelae following acute influenza infection. PLoS ONE. (2019) 14:e0223430. doi: 10.1371/journal.pone.0223430
262. Miller AJ, Spence JR. In vitro models to study human lung development, disease and homeostasis. Physiology. (2017) 32:246–60. doi: 10.1152/physiol.00041.2016
263. Johansen MD, Irving A, Montagutelli X, Tate MD, Rudloff I, Nold MF, et al. Animal and translational models of SARS-CoV-2 infection and COVID-19. Mucosal Immunol. (2020) 13:877–91. doi: 10.1038/s41385-020-00340-z
264. Krimmling T, Schwegmann-Weßels C. Comparison of mono- and co-infection by swine influenza A viruses and porcine respiratory coronavirus in porcine precision-cut lung slices. Res Vet Sci. (2017) 115:470–7. doi: 10.1016/j.rvsc.2017.07.016
265. Hui KPY, Cheung MC, Perera RAPM, Ng KC, Bui CHT, Ho JCW, et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir Med. (2020) 8:687–95. doi: 10.1016/S2213-2600(20)30193-4
266. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J. (2017) 50:1602367. doi: 10.1183/13993003.02367-2016
267. Liu G, Betts C, Cunoosamy DM, Åberg PM, Hornberg JJ, Sivars KB, et al. Use of precision cut lung slices as a translational model for the study of lung biology. Respir Res. (2019) 20:162. doi: 10.1186/s12931-019-1131-x
268. Barosova H, Maione AG, Septiadi D, Sharma M, Haeni L, Balog S, et al. Use of epialveolar lung model to predict fibrotic potential of multiwalled carbon nanotubes. ACS Nano. (2020) 14:3941–56. doi: 10.1021/acsnano.9b06860
269. Berkers G, van Mourik P, Vonk AM, Kruisselbrink E, Dekkers JF, de Winter-de Groot KM, et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. (2019) 26:1701–8.e3. doi: 10.1016/j.celrep.2019.01.068
270. Karkampouna S, La Manna F, Benjak A, Kiener M, De Menna M, Zoni E, et al. Patient-derived xenografts and organoids model therapy response in prostate cancer. Nat. Commun. (2021) 12:1117. doi: 10.1038/s41467-021-21300-6
271. Sanchez-Esteban J, Cicchiello LA, Wang Y, Tsai SW, Williams LK, Torday JS, et al. Mechanical stretch promotes alveolar epithelial type II cell differentiation. J. Appl. Physiol. (2001) 91:589–95. doi: 10.1152/jappl.2001.91.2.589
Keywords: COVID-19, interstitial pulmonary fibrosis, SARS-CoV-2, alveolar regeneration, organoids, lung-on-chip, precision-cut lung slices, in vitro lung models
Citation: Kiener M, Roldan N, Machahua C, Sengupta A, Geiser T, Guenat OT, Funke-Chambour M, Hobi N and Kruithof-de Julio M (2021) Human-Based Advanced in vitro Approaches to Investigate Lung Fibrosis and Pulmonary Effects of COVID-19. Front. Med. 8:644678. doi: 10.3389/fmed.2021.644678
Received: 21 December 2020; Accepted: 01 April 2021;
Published: 07 May 2021.
Edited by:
Elena Lopez-Rodriguez, Charité – Universitätsmedizin Berlin, GermanyReviewed by:
Ilias Papanikolaou, General Hospital of Corfu, GreeceSonia Giambelluca, Charité – Universitätsmedizin Berlin, Germany
Copyright © 2021 Kiener, Roldan, Machahua, Sengupta, Geiser, Guenat, Funke-Chambour, Hobi and Kruithof-de Julio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Hobi, bmluYS5ob2JpQGFsdmVvbGl4LmNvbQ==
†These authors have contributed equally to this work