 Meropi Karakioulaki
Meropi Karakioulaki Eleni Papakonstantinou1,2
Eleni Papakonstantinou1,2 Daiana Stolz
Daiana Stolz- 1Clinic of Respiratory Medicine and Pulmonary Cell Research, University Hospital of Basel, Basel, Switzerland
- 2First Laboratory of Pharmacology, Department of Medicine, School of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, Greece
Common to several allergic diseases is the generation of immunoglobulin E (IgE) by plasma cells, when exposed to an innocuous antigen. Asthma and chronic obstructive pulmonary disease (COPD) are two prevalent chronic airway inflammatory diseases. Asthma is mediated in some patients through eosinophilic inflammatory mechanisms that include allergic sensitization and Th2-mediated immune airway response. COPD, on the other hand is mainly considered a Th1-mediated inflammatory process with neutrophilic predominance or a non-Th2 inflammation, occasionally associated with the presence of airway bacteria or viruses. IgE production appears to play an important role in the development of both COPD and asthma, as it has been associated to respiratory symptoms, lung function, bacterial and viral infections, airway remodeling and bronchial hyperreactivity in both diseases. The aim of this review is to summarize all current data concerning the role of specific and total IgE in COPD and asthma and to highlight similarities and differences in view of possible therapeutic interventions.
Immunology of Asthma and COPD
T lymphocytes are distinguished by the presence of cell membrane molecules known as cluster of differentiation 4 (CD4) and CD8. The CD4 T-lymphocytes are also known as helper T cells and can be further subdivided into Th1 and Th2, producing, respectively, Th1- and Th2-type cytokines (1). Th1-type cytokines induce the proinflammatory response that leads to the elimination of intracellular parasites and to the propagation of autoimmune responses (1). The Th2-type cytokines, such as interleukins (IL)- 4, 5, and 13 are associated with the production of IgE and with the promotion of eosinophilic responses in atopy (1).
Common to several allergic diseases is the generation of immunoglobulin E (IgE) by plasma cells, when exposed to an innocuous antigen (2). Upon initial allergen exposure, antigen presenting cells capture, process and present allergen peptides to T-cells. In presence of IL-4 or IL-13, T-cells acquire the Th2-phenotype, proliferate and engage B-cells to differentiate into plasma cells, which produce IgEs (3) (Figure 1). IgEs bind almost irreversibly to the high affinity IgE receptor (FcεRI) on the surface of mast cells or basophils to create “allergen receptors” (2, 3). A subsequent exposure to the same antigen leads to cross-linking of the IgE:FcεRI complex, degranulation of the cells and release of inflammatory mediators, leading to the early phase response (EPR) (2, 3). EPR occurs immediately after exposure and is accompanied by symptoms such as rhinorrhea, sneezing, nasal congestion and itching (4). The ensuing effector cell infiltration into the tissue encompasses the late-phase response (LPR), along with the continuing production of IgE (4) (Figure 2).
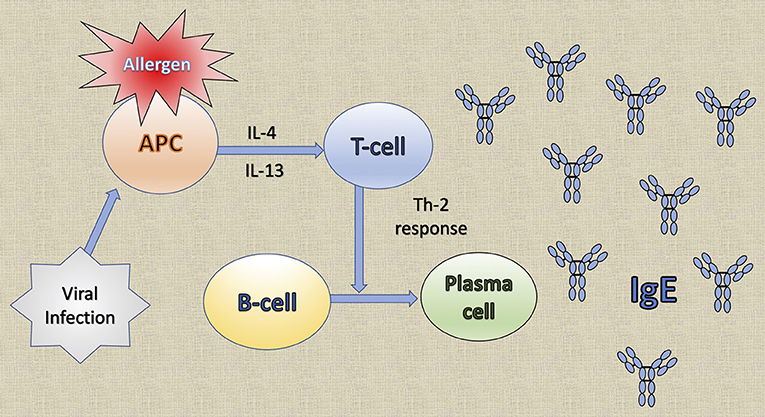
Figure 1. The generation of immunoglobulin E (IgE) by plasma cells. Upon initial allergen exposure, antigen presenting cells (APC) capture, process and present allergen peptides to T-cells. In the presence of interleukin (IL)-4 or IL-13, T-cells acquire the Th2 phenotype, proliferate and stimulate B-cells to differentiate into plasma cells that produce IgE. Viral infections induce the production of the T cell chemoattractant chemokine ligand 28 (CCL28) by dendritic cells and consequently recruit Th2 cells that lead to IgE production.
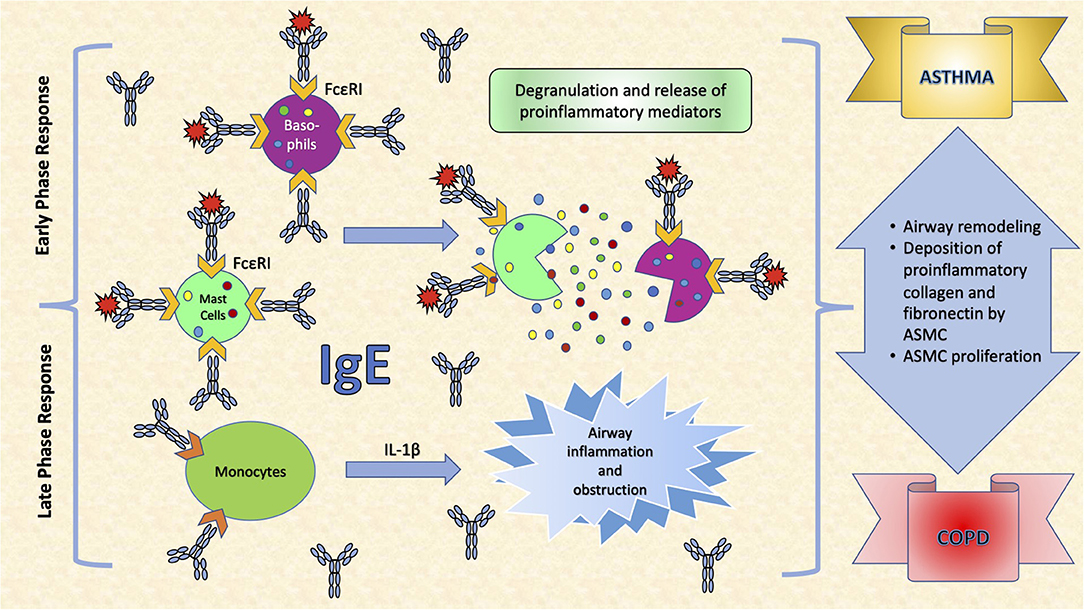
Figure 2. The effects of IgE in the respiratory system. IgE binds almost irreversibly to the high affinity IgE receptor (FcεRI) on the surface of mast cells or basophils to create allergen receptors. A subsequent exposure to the antigen leads to cross-linking of the IgE:FcεRI complex, degranulation of the cells and release of inflammatory mediators, leading to symptoms such as rhinorrhea, sneezing, nasal congestion and itching. Interleukin (IL)-1β is a very important pro-inflammatory cytokine that mediates the inflammatory response, and its expression may be induced in monocytes, as a response to increased IgE levels. This leads to airway inflammation and obstruction, an important determinant in COPD. Moreover, IgEs increase airway remodeling by increasing deposition of pro-inflammatory collagens and fibronectin by airway smooth muscle cells (ASMC) and stimulating their proliferation.
Asthma and chronic obstructive pulmonary disease (COPD) are two common chronic airway inflammatory diseases. Asthma is often mediated through an eosinophilic
inflammatory mechanism that includes allergic sensitization and Th2-mediated immune airway responses (5). COPD, on the other hand is mainly considered a Th1-mediated inflammatory process with neutrophilic predominance or a non-Th2 inflammation that is occasionally associated with the presence of airway bacteria or viruses (6) (Table 1).

Table 1. Comparison of IgE production and the allergic profile in asthma and COPD patients.
Atopy in Asthma and COPD
IgE-mediated sensitivity of inhaled allergens is strongly associated with asthma (9), but this is not true for all asthma cases (8). Allergic asthma is the most common asthma phenotype, which is defined by the presence of allergic sensitization (25), or by a correlation between allergen exposure and asthmatic symptoms (26) but may overlap with other phenotypes (27). Patients with allergic asthma are more likely to report seasonal variations of their symptoms (25). Although it might present at any age, patients with allergic asthma are usually younger than those with non-allergic asthma (25, 26, 28, 29). In school-age children, aeroallergen sensitization is more frequent in children with severe asthma, rather than in children with mild-to-moderate asthma (30). In adults, allergic asthma has been associated with a greater FEV1 reversibility, a higher sputum eosinophil count, higher FENO levels and more exacerbations in the past year (31). Moreover, exercise-induced bronchospasm is more frequent and severe in allergic rather than in non-allergic asthma (32). Conversely, some studies suggest that allergic asthma is less severe than non-allergic (25, 26, 33–37), or cast doubt on the association between asthma severity and atopic status (38–41). Indeed, the different phenotypes of asthma are an important determinant in that matter, as the role of allergy differs in early onset asthma when compared to late onset asthma (42, 43). Accordingly, young asthmatics mostly present with allergic asthma, while late onset asthma is usually severe, steroid resistant and not related to allergy (42–44). Total serum IgE, peripheral eosinophilia levels, as well as Th2-type responses are usually higher in allergic vs. non-allergic asthma (27, 32, 45).
In a study of Bafadhel et al., atopy was defined as a positive skin prick test and/or elevated allergen specific IgEs and it showed a prevalence of 34% among COPD patients (46). Jin et al. showed that even among 273 COPD patients without obvious atopy, the prevalence of elevated total IgE was 47.3% (47). A meta-analysis by Putcha et al. indicated that 35% of the COPD patients (N = 403) from the SPIROMICS cohort and 36% of the COPD patients (N = 696) from the COPDGene cohort presented with atopy, defined as positive sensitization to any of the 10 indoor and outdoor allergens measured in the study (48). There was an almost 50% overlap between atopic status with COPD with asthmatic characteristics (defined as self-report of doctor diagnosed asthma in patients with COPD) in both cohorts (48). Moreover, COPD individuals with non-atopic asthmatic characteristics had the most impaired symptom scores (SGRQ = 4.2, 95% CI: 0.4–7.9; CAT score = 2.8, 95% CI: 0.089–5.4) and highest risk of exacerbations (incidence rate ratio = 1.41, 95% CI: 1.05–1.88), compared to the group without atopy or asthma, while COPD individuals with atopy and atopic asthma were not at increased risk of adverse outcomes (48).
A recent study in an Asian cohort recruited across three countries demonstrated that specific IgEs produced against a broad range of allergens (pollens, house dust mite, cockroach, and fungi) were increased in COPD (n = 466) when compared to controls (n = 51) (49). House dust mite (B. tropicalis, D. pteronyssinus, D. farinae) and grass pollens (pooids and panicoids) presented the highest specific IgE-binding intensities in COPD (49). A significant number of COPD patients demonstrated sensitization to fungi (n = 249, 55.8%) and house dust mites (n = 229, 51.3%) (49). Frequent exacerbators showed significantly increased specific IgE-binding to crude fungal allergens (Curvularia, Penicillium, A. fumigatus) and the cockroach allergen Bl. germanica, but no significant specific IgE-binding to pollens, house dust mites or the cockroach allergen Pr. americana (49). On the other hand, no association between sensitization status and the COPD GOLD stage (lung function) or GOLD group (ABCD) was detected, but a highly sensitized, fungal predominant subgroup of COPD patients demonstrated the worse clinical outcome, with greatest symptoms (median CAT score = 16, IQR = 10–22, P < 0.01), poorest lung function (FEV1 = 41.1% predicted, IQR = 32.5–57.0%, p < 0.01) and increased exacerbation rate (IRR 2.01, 95%CI = 1.44–2.81, p < 0.001) (49). Additionally, there was a significantly increased systemic IgE response to a number of outdoor air fungi (Schizophyllum, p < 0.01; Aspergillus, p < 0.001; Penicillium, p < 0.001; Byssochlamys, p < 0.001; and Cladosporium, p < 0.01) in COPD patients when compared to controls and an increased number of exacerbations was detected in the COPD patients that were sensitized against air-fungi (IRR = 2.29, 95%CI = 1.12–4.68, p < 0.01) (49). The study indicated that the abundance of indoor air and surface allergens positively correlates with COPD symptoms (r = 0.75, p < 0.01) and negatively with lung function (r = −0.61, p < 0.01), suggesting that indoor air and surfaces represent a potential source of fungal allergen exposure (49). However, blood eosinophil counts were not different among the high, moderate and low-sensitized patients (49). Notably, in this study patients were enrolled from Singapore, Hong Kong, and Malaysia (49). All these countries are located in South Asia where the temperature and humidity differ dramatically from other Asian and Western countries. It is important to note here that the prevalence and role of fungal sensitization in tropical climate countries may not be the same in temperate climate ones and further studies elucidating this difference should be conducted.
Total IgE serum levels have been previously associated with a longitudinal decline of lung function (FEV1/FVC), independent of smoking and asthma status (50) and therefore atopy is potentially associated with the risk for COPD (50). Several studies suggest that in COPD patients, IgE-mediated sensitization to environmental allergens plays an important role to the pathogenesis of the disease, as it has been associated with severe symptoms or deteriorating lung function (10, 11, 47, 51, 52). Fattahi et al. demonstrated that atopy in COPD was associated with a higher prevalence of cough and phlegm, but not with FEV1 decline (10). When compared to non-allergic controls, patients with increased serum total IgE have a more severe or longer history of respiratory symptoms, such as dyspnoea and a greater impairment of lung function (47, 51). Additionally, COPD individuals with allergic sensitization have been shown to have increased respiratory symptoms and exacerbation rates (52).
Another study indicated that in COPD, IL-1β and IgE serum levels correlate with clinical aspects of disease severity and suggested that the production of both IgE and IL-1β may be related to smoking, which affects airway obstruction (53). IL-1β is a very important pro-inflammatory cytokine that mediates the inflammatory response and its expression may be induced in monocytes, as a result of increased IgE levels (53) (Figure 3). However, in that study, no correlation between IL-1β and IgE was observed, probably due to the small sample size (30 COPD patients, 30 healthy controls) (53).
Figure 3. Graphic abstract of the review.
The Dutch hypothesis proposed in 1,961 states that there are common risk host factors for asthma and COPD, including airway hyperresponsiveness and atopy and that disease manifestations also depend on external factors, such as exposures (54). Indeed, the finding of a bronchial epithelial Th2- inflammatory gene expression signature in some COPD individuals suggests shared mechanisms with asthma (12). This implies that Th2-mediated airway responses may be important in COPD patients without clinical history of asthma (12). Bozek et al. demonstrated that neither the prevalence nor the profile of IgE-dependent sensitization to inhaled allergens differed between asthma and COPD (8). They pointed out that there are similar cytokine profiles in asthma and COPD and this could indicate that both Th2 and Th1 cells are involved in the immunopathology of these diseases (8).
Additionally, severe COPD and allergic asthma were characterized by a similar overexpression of FcεRI on plasmacytoid dendritic cells (DC) and this was in turn related to the reduction of asthma and COPD with asthmatic characteristics exacerbations following omalizumab treatment, as omalizumab suppresses this receptor (13–15). In view of the effect of anti-IgE therapies on asthma exacerbations, trials investigating the effect of anti-IgE on exacerbations of COPD without asthmatic characteristics are warranted. It appears that COPD patients display an overexpression of the FcεRI receptor on DC and this highlights the important role of IgE in the development and progression of COPD (55). Current smokers display an increased expression of the FcεRI receptor on myeloid and plasmacytoid DC, when compared to never smokers (55). Moreover, the overexpression of the FcεRI receptor on plasmacytoid DC is similar between patients with severe COPD and patients with allergic asthma, while in COPD patients it is associated with increased serum levels of total IgE, worse GOLD stage and worse lung function (55).
Hersh et al. reported that total IgE levels were higher in 541 patients with self-report of doctor's diagnosis of asthma before the age of 40, compared to 598 controls without any airflow obstruction (7). Moreover, when the asthma patients of that study were tested for six specific IgEs against indoor aeroallergens [cat and dog dander, dust mite (D. farinae and D. pteronyssinus), German cockroach and mold mix] they were found to be more frequently positive at least to one specific IgE when compared to controls (49.9% of asthma patients vs. 30.3% of controls, p < 0.05) (7). However, total IgE levels among 899 patients with COPD were not significantly different from total IgE levels of 598 controls without any airflow obstruction (7). Moreover, when COPD patients were tested for the same six specific IgEs against indoor aeroallergens, they were not found more frequently positive with regard to any IgE when compared to controls (24.5% of COPD patients vs. 30.3% of controls, p > 0.05). A higher proportion of current smokers had elevated total IgE levels and at least one positive specific IgE, when compared to former smokers (7). This is in agreement with the study of Omenaas et al., who reported potential effects of smoking on IgE levels (56).
Interplay Between Atopy and Viral Infections in Asthma and COPD
Several observational studies have associated allergic sensitization by 1 year of age with respiratory viral infections in infancy (57–59), and severe respiratory infections in early age have been associated with a higher prevalence of asthma in later childhood (60, 61). The association between viral infections and allergic disease is not unreasonable, because the immune response against many viral infections includes the production of specific IgEs against viral pathogens (62–65). Therefore, anti-IgE therapy was shown to prevent viral exacerbations of asthma (66) and the exacerbation number was reduced even when omalizumab was given 4–6 weeks before children return to school in fall, especially in those who had recently experienced an asthma attack (67).
A viral infection can attract several cell types, such as Th1 cells, CD8 cells, neutrophils and DC, to the site of inflammation, and they can all participate in the allergic response of the patient (68) (Figure 1). Moreover, viral infections affect the expression of receptors involved in the allergic response (68) and can induce several inflammatory mediators, including TGF-β, neutrophilic elastase and several cytokines, which have an effect in the remodeling process of the lung (68). Thereby, they might produce long term effects on the structure of the developing lung, leading to suboptimal lung growth and function and increasing the risk of airway narrowing and the development of clinical asthma (68). That is why respiratory viral infections have similar symptoms with allergic diseases, like allergic rhinitis, and they clearly exacerbate asthma (69).
The etiology of IgE production during viral infections is not yet clearly understood, as IgE production is normally a result of Th2-biased responses (70). In severe viral infections, however, an antiviral Th1-biased response may lead to a proatopic Th2 response and the link between virus and atopy appears to be through the production of IgEs (71, 72). It has been demonstrated that IgEs are produced against a large number of human viral pathogens (73–80). Results from mouse model studies indicate that severe viral infections induce the production of antiviral IgEs that bind to the FcεRI receptor of DCs (71, 81–84). The receptor is then crosslinked by an antigen, the DCs produce the T cell chemoattractant chemokine ligand 28 (CCL28) and recruit IL-13-producing Th2 cells that can contribute to asthma (71, 81–84). CCL28 was found elevated in the lungs of patients with asthma (71, 85) (Figure 1).
Specifically, the Respiratory Syncytial Virus (RSV) was suggested to enhance Th2 sensitization to aeroallergens (86). Some studies have demonstrated that there is a correlation between the titer of antiviral IgEs and the severity of RSV symptoms, such as wheezing in infants and recurrent wheezing or development of asthma in older children (62, 64, 87). Moreover, infants requiring hospitalization for severe RSV infection in the first 6 months of life, have a nearly 20-fold increased risk of developing asthma (88).
Human rhinovirus (HRV) is also known to be a major cause of asthma exacerbations in infants and infection with HRV is associated with a higher incidence of asthma onset (61, 89–91). The prevalence of virus detection in adult asthma exacerbations was demonstrated in the range of 41–78% (92), and HRV appears to be a frequent cause of those exacerbations (93). Accordingly, in suburban children, from birth up to 3 years of age, who were at risk of asthma (one parent with asthma or allergy) and showed that the most important risk factor for development of wheeze by the third year of life was a symptomatic rhinovirus infection (94).
Children with parainfluenza infection in the first year of life had higher odds of developing asthma in their second year of life (95). In another study, infants younger than 3 months, infected with RSV, influenza or parainfluenza had comparable increases in their Th2 cytokine profiles, implicating that there are similarities between RNA respiratory viruses in their ability to push forward an atopic predisposition (96). A cohort of 90,000 children was examined and an increased risk of asthma was found in children with episodes of bronchiolitis during non-winter months (97). It was also found that patients born 4 months before the winter virus peak, had an increased risk of developing asthma, suggesting that the timing of the viral infection plays an important role in the development of early onset asthma (98).
A growing body of evidence implicates viral respiratory tract infections as the predominant risk factor associated with exacerbations of COPD and the development of chronic airway disease (99, 100). Most exacerbations of COPD are triggered by either bacterial or viral infections or a combination of both (101), since 40–80% of acute exacerbations of COPD (AECOPD) that frequently require hospitalization are attributed to viral respiratory tract infections (99). AECOPD associated with symptoms of a common cold have been shown to have a more sudden onset and longer recovery times than AECOPD without cold symptoms (102). Additionally, COPD patients having more frequent exacerbations experience nearly double the number of colds compared to patients experiencing fewer exacerbations (103) and the presence of cold symptoms is associated with a 15% risk of AECOPD (104). On the other hand, in a recent study, Stolz et al. have shown that the prevalence of viral infections during a stable period of COPD is low and that the risk of exacerbations following the onset of common cold symptoms depends on the particular virus associated with the event and is significant only for parainfluenza 3 (105).
Interplay Between Atopy and Bacterial Infection in Asthma and COPD
There are a few reports regarding the relationship between asthma attacks and bacterial infections (106–109). Viral and bacterial infections were observed in 70% of inpatients with an asthma exacerbation in clinical practice, and infection with Streptococcus pneumoniae has been related to adult asthma exacerbation (110).
Staphylococcous aureus is a Gram+ coccus that colonizes humans, as well as domestic animals and is a common opportunistic pathogen (111). At least 20 serologically distinct staphylococcal superantigens have been described that include staphylococcal enterotoxins (SEs) A through V and toxic shock syndrome toxin-1 (TSST-1) (112, 113). Of the more than 20 staphylococcal enterotoxins, SEA and SEB are the best characterized and are also regarded as superantigens, because they are capable of binding to multiple types of the variable region on the beta chain of the T-cell receptor and stimulate large populations of T cells (polyclonal activation of T cells) (112, 114). The result of this massive T cell activation is a cytokine bolus leading to an acute toxic shock (115). Moreover, SEA and SEB induce polyclonal IgE formation (SEA-IgE and SEB-IgE), associated with allergic multi-morbidity in adolescents (116) and they may also activate B-cells, eosinophils, epithelial cells and others, resulting in a cytokine storm locally in the tissue and the generation of a strong inflammatory response (117).
Staphylococcous aureus is frequently found colonizing patients with Th2-biased diseases, such as atopic dermatitis and chronic rhinosinusitis with nasal polyps (118–122). It releases proteins that facilitate bacterial invasion and colonization and that exert immunosuppressive action on the mucosal environment (123–126). Asthmatic patients have increased IgE reactivity specific to various secreted S. aureus proteins (117). There is evidence that S. aureus is persistently colonizing the nasal mucosa of patients, protected by a biofilm or hiding inside immune cells (121) and is constantly producing a panel of factors that could initiate and aggravate a Th2-biased immune response (127). Chronic exposure to S. aureus secreted proteins could also hinder the resolution of inflammation, fostering chronification (127).
A number of studies have demonstrated in the past that sensitization to staphylococcal enterotoxins is associated with asthma severity (16–19), asthma exacerbations (17), asthma control and age of asthma onset (20, 21). There is only one study that associates increased SE-IgE to COPD exacerbations and COPD control but without defining the allergic profile of those COPD patients (22) (Table 1).
Haemophilus influenzae (NTHi) triggers histamine release through both IgE- and non-IgE-dependent mechanisms (128) from cells of the respiratory mucosa sensitized to the bacterium (129, 130). Specific anti-NTHi IgEs occur at a low level in healthy subjects, and patients with chronic bronchitis and moderate-severe COPD have elevated specific anti-NTHi IgEs compared to healthy controls, with higher anti-NTHi IgE levels in those with most severe disease. Additionally, specific anti-NTHi IgE levels are greater in those with moderate-severe COPD than in those with chronic bronchitis (131).
Atopy and Airway Remodeling, Inflammation and Bronchial Hyperreactivity in Asthma and COPD
In allergic asthma, IgE increases airway remodeling by increasing deposition of pro-inflammatory collagens and fibronectin by airway smooth muscle cells (ASMC), as well as by stimulating their proliferation (132) (Figure 2). When ASMC are exposed in vitro to serum from allergic patients, their proliferation, as well as the deposition of collagen type-I (48 h) and of fibronectin (24 h) are stimulated (133). All these can be prevented by exposure of ASMC to allergic serum that is pre-incubated for 1 h with anti-IgE antibodies (omalizumab) (133).
Fang et al. have investigated the effect of IgE on human primary ASMC remodeling in the absence of allergens (non-immune IgE) in vitro (134). They reported that non-immune IgE (produced in vitro from a monoclonal hybridoma cell line and not induced by allergens) was sufficient to stimulate ASMC remodeling by upregulating microRNA-21-5p, which in turn downregulated phosphatase and tensin homolog (PTEN) expression and supported mammalian target of rapamycin (mTOR) signaling (134). Interestingly, they pointed out that the inhibition of microRNA-21-5p increased PTEN and reduced ASMC remodeling (134). Therefore, the suppression of microRNA-21-5p may present a therapeutic target to reduce airway wall remodeling (134).
Airway hyperreactivity is a general term to describe the increased response of the airways to bronchoconstricting agonists, such as methacholine or histamine, a hallmark of asthma (135). In COPD, the response to bronchoconstrictors is related to the degree of baseline airflow obstruction (136), in contrast to patients with asthma, without fixed airflow obstruction (23, 137, 138). There is an evident heterogeneity of remodeling in COPD patients in that the pathology may involve large or small airways (inflammation and increased wall-thickness), loss of small airways and emphysema (135). It is also recognized that COPD patients who have increased airway responsiveness are most commonly atopic and present with a higher degree of reversibility, as well as with eosinophilic airway inflammation, higher response to corticosteroids and faster decline in FEV1 (135). Moreover, airway hyperreactivity, can be an independent predictor of COPD development in the general population (139) and also a risk factor for rapid progression of airway obstruction in patients with mild COPD (24). However, it decreases after smoking cessation (140) (Table 1).
Remodeling of the airways is a well-recognized feature of COPD, depended on COPD severity based on post-bronchodilator FEV1 (141). There are several pathological changes in the lungs of COPD patients including: thickness of the airway wall and the airway smooth muscle layer (although not to the extent seen in asthma), increased blood vessel density, hypersecretion of mucus, metaplasia of the epithelial cells, enlargement of the submucosal glands, loss of terminal and respiratory bronchioles and enlargement and destruction of the alveoli, as well as neutrophilic inflammation and infiltration of CD8 T-lymphocytes (142–144). However, the effect of atopy on airway remodeling in COPD patients has not been studied yet. Understanding better this issue is of clinical importance, as it may assist us to apply the proper medical intervention for atopic and non-atopic COPD patients.
Interplay Between IGG and IGE-Mediated Inflammatory Responses
A recent study demonstrated that IgG antibodies play a key role in controlling IgE-mediated inflammatory responses in patients with nasal polyps, by interfering with allergens potentially binding to cell-bound IgEs (145). Depletion of IgG from nasal polyp tissue homogenates resulted in an increase in IgE-facilitated allergen binding to B cells but also enhanced FcεRI-mediated allergen-driven basophil activation and histamine release (145). A similar response was observed in relation to SE-IgEs (145). In fact, IgG repertoires share extensively the same antigen targets with IgE repertoires in both allergic and non-allergic subjects and nasal polyps are characterized by abundant clonally related IgG- and IgE- secreting plasma cells (145).
Allergen-specific IgGs are competing with IgEs for allergen binding, thereby decreasing the allergen-induced effector cell activation (3). This suggests that augmenting the allergen specific IgG/IgE ratio could be effective in preventing immediate hypersensitivity responses and reducing allergic symptoms (3, 146).
Conclusions and Future Perspectives
In this review we summarized current data concerning the role of IgE in COPD and asthma (Figure 3). It appears that IgE production plays an important role in the development of both diseases. Interestingly, allergic sensitization and viral infections in infancy have been associated with a higher prevalence of asthma later in life, as well as with the most important determinant for COPD, namely the level of airflow limitation in late adulthood. Anti-IgE therapy (omalizumab) prevents viral exacerbations of asthma and COPD with asthmatic characteristics. Moreover, allergen-specific IgGs are competing with IgEs for allergen binding and augmenting the allergen specific IgG/IgE ratio could be effective in preventing immediate hypersensitivity responses and reducing allergic symptoms. Additionally, the suppression of microRNA-21-5p may present a therapeutic target to reduce airway wall remodeling induced by IgE. Further studies that will analyze the allergic profile of COPD patients in association with lung function, disease severity, outcome, exacerbations and airway remodeling are warranted, in order to examine a potential use of anti-IgE treatment for COPD patients.
Author Contributions
MK and DS: conception of the review topic. MK, EP, AG, and DS: literature review, writing of the manuscript, finalization of the manuscript, and approval of the submitted article. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Clinic of Respiratory Medicine and Pulmonary Cell Research, University Hospital of Basel, Basel, Switzerland.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Berger A. Th1 and Th2 responses: what are they? BMJ. (2000) 321:424. doi: 10.1136/bmj.321.7258.424
2. Martorano LM, Grayson MH. Respiratory viral infections and atopic development: from possible mechanisms to advances in treatment. Eur J Immunol. (2018) 48:407–14. doi: 10.1002/eji.201747052
3. Orengo JM, Radin AR, Kamat V, Badithe A, Ben LH, Bennett BL, et al. Treating cat allergy with monoclonal IgG antibodies that bind allergen and prevent IgE engagement. Nat Commun. (2018) 9:1421. doi: 10.1038/s41467-018-03636-8
4. Hansen I, Klimek L, Mosges R, Hormann K. Mediators of inflammation in the early and the late phase of allergic rhinitis. Curr Opin Allergy Clin Immunol. (2004) 4:159–63. doi: 10.1097/00130832-200406000-00004
5. George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis. (2016) 7:34–51. doi: 10.1177/2040622315609251
6. Albertson TE, Chenoweth JA, Pearson SJ, Murin S. The pharmacological management of asthma-chronic obstructive pulmonary disease overlap syndrome (ACOS). Expert Opin Pharmacother. (2020) 21:213–31. doi: 10.1080/14656566.2019.1701656
7. Hersh CP, Zacharia S, Prakash Arivu Chelvan R, Hayden LP, Mirtar A, Zarei S, et al. Immunoglobulin E as a biomarker for the overlap of atopic asthma and chronic obstructive pulmonary disease. Chronic Obstr Pulm Dis. (2020) 7:1–12. doi: 10.15326/jcopdf.7.1.2019.0138
8. Bozek A, Rogala B. IgE-dependent sensitization in patients with COPD. Ann Agric Environ Med. (2018) 25:417–20. doi: 10.26444/aaem/83413
9. Mathur SK, Viswanathan RK. Relevance of allergy in adult asthma. Curr Allergy Asthma Rep. (2014) 14:437. doi: 10.1007/s11882-014-0437-5
10. Fattahi F, Ten Hacken NH, Lofdahl CG, Hylkema MN, Timens W, Postma DS, et al. Atopy is a risk factor for respiratory symptoms in COPD patients: results from the EUROSCOP study. Respir Res. (2013) 14:10. doi: 10.1186/1465-9921-14-10
11. Neves MC, Neves YC, Mendes CM, Bastos MN, Camelier AA, Queiroz CF, et al. Evaluation of atopy in patients with COPD. J Bras Pneumol. (2013) 39:296–305. doi: 10.1590/S1806-37132013000300006
12. Christenson SA, Steiling K, van den Berge M, Hijazi K, Hiemstra PS, Postma DS, et al. Asthma-COPD overlap. Clinical relevance of genomic signatures of type 2 inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 191:758–66. doi: 10.1164/rccm.201408-1458OC
13. Stoll P, Ulrich M, Bratke K, Garbe K, Virchow JC, Lommatzsch M. Imbalance of dendritic cell co-stimulation in COPD. Respir Res. (2015) 16:19. doi: 10.1186/s12931-015-0174-x
14. Yalcin AD, Celik B, Yalcin AN. Omalizumab (anti-IgE) therapy in the asthma-COPD overlap syndrome (ACOS) and its effects on circulating cytokine levels. Immunopharmacol Immunotoxicol. (2016) 38:253–6. doi: 10.3109/08923973.2016.1173057
15. Maltby S, Gibson PG, Powell H, McDonald VM. Omalizumab treatment response in a population with severe allergic asthma and overlapping COPD. Chest. (2017) 151:78–89. doi: 10.1016/j.chest.2016.09.035
16. Kim SH, Yang SY, You J, Lee SB, You J, Chang YS, et al. Association of specific immunoglobulin E to staphylococcal enterotoxin with airway hyperresponsiveness in asthma patients. Tuberc Respir Dis. (2016) 79:295–301. doi: 10.4046/trd.2016.79.4.295
17. Sintobin I, Siroux V, Holtappels G, Pison C, Nadif R, Bousquet J, et al. Sensitisation to staphylococcal enterotoxins and asthma severity: a longitudinal study in the EGEA cohort. Eur Respir J. (2019) 54:1900198. doi: 10.1183/13993003.00198-2019
18. Bachert C, van Steen K, Zhang N, Holtappels G, Cattaert T, Maus B, et al. Specific IgE against Staphylococcus aureus enterotoxins: an independent risk factor for asthma. J Allergy Clin Immunol. (2012) 130:376–81.e8. doi: 10.1016/j.jaci.2012.05.012
19. Elabras JF, Mello FC, Lupi O, Bica BE, Papi JA, Franca AT. Staphylococcal superantigen-specific IgE antibodies: degree of sensitization and association with severity of asthma. J Bras Pneumol. (2016) 42:356–61. doi: 10.1590/S1806-37562016000000010
20. Tanaka A, Suzuki S, Ohta S, Manabe R, Furukawa H, Kuwahara N, et al. Association between specific IgE to Staphylococcus aureus enterotoxins A and B and asthma control. Ann Allergy Asthma Immunol. (2015) 115:191–7.e2. doi: 10.1016/j.anai.2015.06.017
21. Tomassen P, Jarvis D, Newson R, Van Ree R, Forsberg B, Howarth P, et al. Staphylococcus aureus enterotoxin-specific IgE is associated with asthma in the general population: a GA(2)LEN study. Allergy. (2013) 68:1289–97. doi: 10.1111/all.12230
22. Rohde G, Gevaert P, Holtappels G, Borg I, Wiethege A, Arinir U, et al. Increased IgE-antibodies to Staphylococcus aureus enterotoxins in patients with COPD. Respir Med. (2004) 98:858–64. doi: 10.1016/j.rmed.2004.02.012
23. Karakioulaki M, Koletsa T, Papakonstantinou E, Jahn K, Tamm M, Stolz D. Histopathological comparison of endobronchial biopsies from different pulmonary lobes of severe asthmatic patients. Chest. (2020) 158:923–8. doi: 10.1016/j.chest.2020.04.029
24. Tashkin DP, Altose MD, Connett JE, Kanner RE, Lee WW, Wise RA. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The lung health study research group. Am J Respir Crit Care Med. (1996) 153:1802–11. doi: 10.1164/ajrccm.153.6.8665038
25. Romanet-Manent S, Charpin D, Magnan A, Lanteaume A, Vervloet D. Allergic vs. nonallergic asthma: what makes the difference. Allergy. (2002) 57:607–13. doi: 10.1034/j.1398-9995.2002.23504.x
26. Kanani AS, Broder I, Greene JM, Tarlo SM. Correlation between nasal symptoms and asthma severity in patients with atopic and nonatopic asthma. Ann Allergy Asthma Immunol. (2005) 94:341–62. doi: 10.1016/S1081-1206(10)60985-4
27. Schatz M, Rosenwasser L. The allergic asthma phenotype. J Allergy Clin Immunol Pract. (2014) 2:645–8. doi: 10.1016/j.jaip.2014.09.004
28. Novak N, Bieber T. Allergic and non-allergic forms of atopic diseases. J Allergy Clin Immunol. (2003) 112:252–62. doi: 10.1067/mai.2003.1595
29. Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. (2006) 368:804–13. doi: 10.1016/S0140-6736(06)69290-8
30. Fitzpatrick AM, Gaston BM, Erzurum SC, Teague WG. Severe Asthma Research Program. Features of severe asthma in school-age children: atopy and increased exhaled nitric oxide. J Allergy Clin Immunol. (2006) 118:1218–25. doi: 10.1016/j.jaci.2006.08.019
31. Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. (2008) 178:218–24. doi: 10.1164/rccm.200711-1754OC
32. Virdkow J-C, Kroegel C, Walker C, Matthys H. Cellular and immunological markers of allergic and intrinsic bronchial asthma. Lung. (1994) 172:313–4. doi: 10.1007/BF00172846
33. Kitch BT, Levy BD, Fanta CH. Late onset asthma: epidemiology diagnosis and treatment. Drugs Aging. (2000) 17:385097. doi: 10.2165/00002512-200017050-00005
34. The ENFUMOSA. Study Group. The ENFUMOSA cross-sectional European multicenter study of the clinical phenotype of chronic severe asthma. Eur Respir J. (2003) 22:470–7. doi: 10.1183/09031936.03.00261903
35. Gaga M, Papageorgiou N, Yiourgioti G, Karydi P, Liapikou A, Bitsakou H, et al. Risk factors and characteristics associated with severe and difficult to treat asthma phenotype: an analysis of the ENFUMOSA group of patients based on the SCRHS questionnaire. Clin Exp Allergy. (2005) 35:954–9. doi: 10.1111/j.1365-2222.2005.02281.x
36. Moore WC, Bleecker ER, Curran-Everett D, Erzurum SC, Ameredes BT, Bacharier L, et al. Characterization of the severe asthma phenotype by the national heart, lung, and blood institute's severe asthma research program. J Allergy Clin Immunol. (2007) 119:405–13. doi: 10.1016/j.jaci.2006.11.639
37. Amelink M, de Groot JC, de Nijs SB, Lutter R, Zwinderman AH, Sterk PJ, et al. Severe adult-onset asthma: a distinct phenotype. J Allergy Clin Immunol. (2013) 132:336–41. doi: 10.1016/j.jaci.2013.04.052
38. Cline MG, Dodge R, Lebowitz MD, Burrows B. Determinants of percent pre- dicted FEVI in current asthmatics subjects. Chest. (1994) 106:1089–93. doi: 10.1378/chest.106.4.1089
39. Siroux V, Oryszcyant M-P, Paty E, Kauffmann F, Pison C, Vervloet D, et al. Relationships of allergic sensitization, total immunoglobulin E and blood eo- sinophils to asthma severity in children of the EGEA Study. Clin Exp Allergy. (2003) 33:746–51. doi: 10.1046/j.1365-2222.2003.01674.x
40. Liou A, Grubb J, Schechtman K, Hamilos DL. Causative and contributive factors to asthma severity and patterns of medications use in patients seeking specialized asthma care. Chest. (2003) 124:1781–8. doi: 10.1378/chest.124.5.1781
41. Fukutomi Y, Taniguchi M, Tsuburai T, Tanimoto H, Oshikata C, Ono E, et al. Obesity and aspirin intolerance are risk factors for difficult-to-treat asthma in Japanese non-atopic women. Clin Exp Allergy. (2012) 42:738–46. doi: 10.1111/j.1365-2222.2011.03880.x
42. Trivedi M, Denton E. Asthma in children and adults- what are the differences and what can they tell us about asthma? Front Pediatr. (2019) 7:256. doi: 10.3389/fped.2019.00256
43. Kuruvilla M, Lee F, Lee G. Understanding asthma phenotypes, endotypes and mechanisms of disease. Clin Rev Allergy Immunol. (2019) 56:219–33. doi: 10.1007/s12016-018-8712-1
44. Pakkasela J, Ilmarinen P, Honkamäki J, Tuomisto L, Andersén H, Piirilä P, et al. Age-specific incidence of allergic and non-allergic asthma. BMC Pulm Med. (2020) 20:9. doi: 10.1186/s12890-019-1040-2
45. Zoratti E, Havstad S, Wegienka G, Nocholas C, Bobbitt KR, Woodcroft KJ, et al. Differentiating asthma phenotypes in young adults through polyclonal cytokine profiles. Ann Allergy Asthma Immunol. (2014) 113:25–30. doi: 10.1016/j.anai.2014.04.013
46. Bafadhel M, McKenna S, Agbetile J, Fairs A, Desai D, Mistry V, et al. Aspergillus fumigatus during stable state and exacerbations of COPD. Eur Respir J. (2014) 43:64–71. doi: 10.1183/09031936.00162912
47. Jin J, Liu X, Sun Y. The prevalence of increased serum IgE and Aspergillus sensitization in patients with COPD and their association with symptoms and lung function. Respir Res. (2014) 15:130. doi: 10.1186/s12931-014-0130-1
48. Putcha N, Fawzy A, Matsui E, Liu M, Bowler R, Woodruff P, et al. Clinical phenotypes of atopy and asthma in COPD: a meta-analysis of SPIROMICS and COPDGene. Chest. (2020) 158:2333–45. doi: 10.1016/j.chest.2020.04.069
49. Tiew PY, Ko FWS, Pang SL, Matta SA, Sio YY, Poh ME, et al. Environmental fungal sensitisation associates with poorer clinical outcomes in COPD. Eur Respir J. (2020) 56:2000418. doi: 10.1183/13993003.00418-2020
50. Sherrill DL, Lebowitz MD, Halonen M, Barbee RA, Burrows B. Longitudinal evaluation of the association between pulmonary function and total serum IgE. Am J Respir Crit Care Med. (1995) 152:98–102. doi: 10.1164/ajrccm.152.1.7599870
51. Samaha H, Elsaid A, NasrEldin E. Total serum IgE level in COPD patients. Egypt J Chest Dis Tuber. (2015) 64:573–7. doi: 10.1016/j.ejcdt.2015.02.005
52. Jamieson DB, Matsui EC, Belli A, McCormack MC, Peng E, Pierre-Louis S, et al. Effects of allergic phenotype on respiratory symptoms and exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2013) 188:187–92. doi: 10.1164/rccm.201211-2103OC
53. Singh B, Arora S, Khanna V. Association of severity of COPD with IgE and interleukin-1 beta. Monaldi Arch Chest Dis. (2010) 73:86–7. doi: 10.4081/monaldi.2010.303
54. Sluiter HJ, Koeter GH, de Monchy JG, Postma DS, de Vries K, Orie NG. The Dutch hypothesis (chronic non-specific lung disease) revisited. Eur Respir J. (1991) 4:479–89.
55. Stoll P, Bahker A, Ulrich M, Bratke K, Garbe K, Christian Virchow J, et al. The dendritic cell high-affinity IgE receptor is overexpressed in both asthma and severe COPD. Clin Exp Allergy. (2016) 46:575–83. doi: 10.1111/cea.12664
56. Omenaas E, Bakke P, Elsayed S, Hanoa R, Gulsvik A. Total and specific serum IgE levels in adults: relationship to sex, age and environmental factors. Clin Exp Allergy. (1994) 24:530–9. doi: 10.1111/j.1365-2222.1994.tb00950.x
57. Sigurs NA. cohort of children hospitalised with acute RSV bronchiolitis: impact on later respiratory disease. Paediatr Respir Rev. (2002) 3:177–83. doi: 10.1016/S1526-0542(02)00191-4
58. Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. (2000) 161:1501–7. doi: 10.1164/ajrccm.161.5.9906076
59. Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. (2008) 178:667–72. doi: 10.1164/rccm.200802-309OC
60. Illi S, von Mutius E, Lau S, Bergmann R, Niggemann B, Sommerfeld C, et al. Early childhood infectious diseases and the development of asthma up to school age: a birth cohort study. BMJ. (2001) 322:390–5. doi: 10.1136/bmj.322.7283.390
61. Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. (2005) 171:137–41. doi: 10.1164/rccm.200406-730OC
62. Smith-Norowitz TA, Mandal M, Joks R, Norowitz LT, Weaver D, Durkin HG, et al. IgE anti-respiratory syncytial virus antibodies detected in serum of pediatric patients with asthma. Hum Immunol. (2015) 76:519–24. doi: 10.1016/j.humimm.2015.06.002
63. Smith-Norowitz TA, Kusonruksa M, Wong D, Norowitz MM, Joks R, Durkin HG, et al. Long-term persistence of IgE anti-influenza A HIN1 virus antibodies in serum of children and adults following influenza A vaccination with subsequent H1N1 infection: a case study. J Inflamm Res. (2012) 5:111–6. doi: 10.2147/JIR.S34152
64. Welliver RC. Respiratory syncytial virus and other respiratory viruses. Pediatr Infect Dis J. (2003) 22(Suppl. 2):S6–10; discussion S-2. doi: 10.1097/00006454-200302001-00002
65. Tam JS, Jackson WT, Hunter D, Proud D, Grayson MH. Rhinovirus specific IgE can be detected in human sera. J Allergy Clin Immunol. (2013) 132:1241–3. doi: 10.1016/j.jaci.2013.07.011
66. Busse WW, Morgan WJ, Gergen PJ, Mitchell HE, Gern JE, Liu AH, et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N Engl J Med. (2011) 364:1005–15. doi: 10.1056/NEJMoa1009705
67. Teach SJ, Gill MA, Togias A, Sorkness CA, Arbes SJ Jr, Calatroni A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol. (2015) 136:1476–85. doi: 10.1016/j.jaci.2015.09.008
68. van Rijt LS, van Kessel CH, Boogaard I, Lambrecht BN. Respiratory viral infections and asthma pathogenesis: a critical role for dendritic cells? J Clin Virol. (2005) 34:161–9. doi: 10.1016/j.jcv.2005.07.002
69. Steinke JW, Borish L. Immune responses in rhinovirus-induced asthma exacerbations. Curr Allergy Asthma Rep. (2016) 16:78. doi: 10.1007/s11882-016-0661-2
70. Kay AB. Allergy and allergic diseases. First of two parts. N Engl J Med. (2001) 344:30–7. doi: 10.1056/NEJM200101043440106
71. Grayson MH, Cheung D, Rohlfing MM, Kitchens R, Spiegel DE, Tucker J, et al. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med. (2007) 204:2759–69. doi: 10.1084/jem.20070360
72. Khan SH, Park SS, Sirajuddin IA, Grayson MH. Respiratory virus and asthma: the role of immunoglobulin E. Clin Ther. (2008) 30:1017–24. doi: 10.1016/j.clinthera.2008.06.002
73. Alexeyev OA, Ahlm C, Billheden J, Settergren B, Wadell G, Juto P. Elevated levels of total and Puumala virus-specific immunoglobulin E in the Scandinavian type of hemorrhagic fever with renal syndrome. Clin Diagn Lab Immunol. (1994) 1:269–72. doi: 10.1128/cdli.1.3.269-272.1994
74. Calenoff E, Zhao JC, Derlacki EL, Harrison WH, Selmeczi K, Dutra JC, et al. Patients with Meniere's disease possess IgE reacting with herpes family viruses. Arch Otolaryngol Head Neck Surg. (1995) 121:861–4. doi: 10.1001/archotol.1995.01890080029005
75. Koraka P, Murgue B, Deparis X, Setiati TE, Suharti C, van Gorp EC, et al. Elevated levels of total and dengue virus-specific immunoglobulin E in patients with varying disease severity. J Med Virol. (2003) 70:91–8. doi: 10.1002/jmv.10358
76. Las Heras J, Swanson VL. Sudden death of an infant with rhinovirus infection complicating bronchial asthma: case report. Pediatr Pathol. (1983) 1:319–23. doi: 10.3109/15513818309040669
77. Rager KJ, Langland JO, Jacobs BL, Proud D, Marsh DG, Imani F. Activation of antiviral protein kinase leads to immunoglobulin E class switching in human B cells. J Virol. (1998) 72:1171–6. doi: 10.1128/JVI.72.2.1171-1176.1998
78. Skoner DP, Doyle WJ, Tanner EP, Kiss J, Fireman P. Effect of rhinovirus 39 (RV-39) infection on immune and inflammatory parameters in allergic and non-allergic subjects. Clin Exp Allergy. (1995) 25:561–7. doi: 10.1111/j.1365-2222.1995.tb01095.x
79. Votava M, Bartosova D, Krchnakova A, Crhova K, Kubinova L. Diagnostic importance of heterophile antibodies and immunoglobulins IgA, IgE, IgM and low-avidity IgG against Epstein-Barr virus capsid antigen in children. Acta Virol. (1996) 40:99–101.
80. Welliver RC, Sun M, Rinaldo D, Ogra PL. Predictive value of respiratory syncytial virus-specific IgE responses for recurrent wheezing following bronchiolitis. J Pediatr. (1986) 109:776–80. doi: 10.1016/S0022-3476(86)80692-8
81. English K, Brady C, Corcoran P, Cassidy JP, Mahon BP. Inflammation of the respiratory tract is associated with CCL28 and CCR10 expression in a murine model of allergic asthma. Immunol Lett. (2006) 103:92–100. doi: 10.1016/j.imlet.2005.09.011
82. Grayson MH, Ramos MS, Rohlfing MM, Kitchens R, Wang HD, Gould A, et al. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol. (2007) 179:1438–48. doi: 10.4049/jimmunol.179.3.1438
83. John AE, Thomas MS, Berlin AA, Lukacs NW. Temporal production of CCL28 corresponds to eosinophil accumulation and airway hyperreactivity in allergic airway inflammation. Am J Pathol. (2005) 166:345–53. doi: 10.1016/S0002-9440(10)62258-4
84. Pan J, Kunkel EJ, Gosslar U, Lazarus N, Langdon P, Broadwell K, et al. A novel chemokine ligand for CCR10 and CCR3 expressed by epithelial cells in mucosal tissues. J Immunol. (2000) 165:2943–9. doi: 10.4049/jimmunol.165.6.2943
85. Wang W, Soto H, Oldham ER, Buchanan ME, Homey B, Catron D, et al. Identification of a novel chemokine (CCL28), which binds CCR10 (GPR2). J Biol Chem. (2000) 275:22313–23. doi: 10.1074/jbc.M001461200
86. Sigurs N. Epidemiologic and clinical evidence of a respiratory syncytial virus-reactive airway disease link. Am J Respir Crit Care Med. (2001) 163:S2–6. doi: 10.1164/ajrccm.163.supplement_1.2011109
87. Welliver RC, Duffy L. The relationship of RSV-specific immunoglobulin E antibody responses in infancy, recurrent wheezing, and pulmonary function at age 7-8 years. Pediatr Pulmonol. (1993) 15:19–27. doi: 10.1002/ppul.1950150104
88. Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B, Bjorksten B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics. (1995) 95:500–5.
89. Busse WW, Lemanske RF Jr, Gern JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet. (2010) 376:826–34. doi: 10.1016/S0140-6736(10)61380-3
90. Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ. (1995) 310:1225–9. doi: 10.1136/bmj.310.6989.1225
91. Soto-Quiros M, Avila L, Platts-Mills TA, Hunt JF, Erdman DD, Carper H, et al. High titers of IgE antibody to dust mite allergen and risk for wheezing among asthmatic children infected with rhinovirus. J Allergy Clin Immunol. (2012) 129:1499–505 e5. doi: 10.1016/j.jaci.2012.03.040
92. Papadopoulos NG, Christodoulou I, Rohde G, Agache I, Almqvist C, Bruno A, et al. Viruses and bacteria in acute asthma exacerbations–a GA(2) LEN-DARE systematic review. Allergy. (2011) 66:458–68. doi: 10.1111/j.1398-9995.2010.02505.x
93. Denlinger LC, Sorkness RL, Lee WM, Evans MD, Wolff MJ, Mathur SK, et al. Lower airway rhinovirus burden and the seasonal risk of asthma exacerbation. Am J Respir Crit Care Med. (2011) 184:1007–14. doi: 10.1164/rccm.201103-0585OC
94. Lemanske RF Jr, Jackson DJ, Gangnon RE, Evans MD, Li Z, Shult PA, et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol. (2005) 116:571–7. doi: 10.1016/j.jaci.2005.06.024
95. Lee KK, Hegele RG, Manfreda J, Wooldrage K, Becker AB, Ferguson AC, et al. Relationship of early childhood viral exposures to respiratory symptoms, onset of possible asthma and atopy in high risk children: the Canadian Asthma Primary Prevention Study. Pediatr Pulmonol. (2007) 42:290–7. doi: 10.1002/ppul.20578
96. Kristjansson S, Bjarnarson SP, Wennergren G, Palsdottir AH, Arnadottir T, Haraldsson A, et al. Respiratory syncytial virus and other respiratory viruses during the first 3 months of life promote a local TH2-like response. J Allergy Clin Immunol. (2005) 116:805–11. doi: 10.1016/j.jaci.2005.07.012
97. Carroll KN, Wu P, Gebretsadik T, Griffin MR, Dupont WD, Mitchel EF, et al. Season of infant bronchiolitis and estimates of subsequent risk and burden of early childhood asthma. J Allergy Clin Immunol. (2009) 123:964–6. doi: 10.1016/j.jaci.2008.12.011
98. Wu P, Dupont WD, Griffin MR, Carroll KN, Mitchel EF, Gebretsadik T, et al. Evidence of a causal role of winter virus infection during infancy in early childhood asthma. Am J Respir Crit Care Med. (2008) 178:1123–9. doi: 10.1164/rccm.200804-579OC
99. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. (2008) 14:633–40. doi: 10.1038/nm1770
100. Proud D, Chow CW. Role of viral infections in asthma and chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2006) 35:513–8. doi: 10.1165/rcmb.2006-0199TR
101. Papi A, Luppi F, Franco F, Fabbri LM. Pathophysiology of exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. (2006) 3:245–51. doi: 10.1513/pats.200512-125SF
102. Aaron SD, Donaldson GC, Whitmore GA, Hurst JR, Ramsay T, Wedzicha JA. Time course and pattern of COPD exacerbation onset. Thorax. (2012) 67:238–43. doi: 10.1136/thoraxjnl-2011-200768
103. Hurst JR, Donaldson GC, Wilkinson TM, Perera WR, Wedzicha JA. Epidemiological relationships between the common cold and exacerbation frequency in COPD. Eur Respir J. (2005) 26:846–52. doi: 10.1183/09031936.05.00043405
104. Johnston NW, Olsson M, Edsbacker S, Gerhardsson de Verdier M, Gustafson P, McCrae C, et al. Colds as predictors of the onset and severity of COPD exacerbations. Int J Chron Obstruct Pulmon Dis. (2017) 12:839–48. doi: 10.2147/COPD.S127146
105. Stolz D, Papakonstantinou E, Grize L, Schilter D, Strobel W, Louis R, et al. Time-course of upper respiratory tract viral infection and COPD exacerbation. Eur Respir J. (2019) 54:1900407. doi: 10.1183/13993003.00407-2019
106. Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet. (2014) 384:691–702. doi: 10.1016/S0140-6736(14)61136-3
107. FitzGerald G, Boonen S, Compston JE, Pfeilschifter J, LaCroix AZ, Hosmer DW Jr, et al. Differing risk profiles for individual fracture sites: evidence from the Global Longitudinal Study of Osteoporosis in Women (GLOW). J Bone Miner Res. (2012) 27:1907–15. doi: 10.1002/jbmr.1652
108. Legatzki A, Rosler B, von Mutius E. Microbiome diversity and asthma and allergy risk. Curr Allergy Asthma Rep. (2014) 14:466. doi: 10.1007/s11882-014-0466-0
109. Ohta K, Ichinose M, Nagase H, Yamaguchi M, Sugiura H, Tohda Y, et al. Japanese guideline for adult asthma 2014. Allergol Int. (2014) 63:293–333. doi: 10.2332/allergolint.14-RAI-0766
110. Iikura M, Hojo M, Koketsu R, Watanabe S, Sato A, Chino H, et al. The importance of bacterial and viral infections associated with adult asthma exacerbations in clinical practice. PLoS ONE. (2015) 10:e0123584. doi: 10.1371/journal.pone.0123584
111. Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. (1997) 10:505–20. doi: 10.1128/CMR.10.3.505
112. Balaban N, Rasooly A. Staphylococcal enterotoxins. Int J Food Microbiol. (2000) 61:1–10. doi: 10.1016/S0168-1605(00)00377-9
113. Al-Daccak R, Mehindate K, Damdoumi F, Etongue-Mayer P, Nilsson H, Antonsson P, et al. Staphylococcal enterotoxin D is a promiscuous superantigen offering multiple modes of interactions with the MHC class II receptors. J Immunol. (1998) 160:225–32.
114. Choi YW, Kotzin B, Herron L, Callahan J, Marrack P, Kappler J. Interaction of Staphylococcus aureus toxin “superantigens” with human T cells. Proc Natl Acad Sci U S A. (1989) 86:8941–5. doi: 10.1073/pnas.86.22.8941
115. Pinchuk IV, Beswick EJ, Reyes VE. Staphylococcal enterotoxins. Toxins (Basel). (2010) 2:2177–97. doi: 10.3390/toxins2082177
116. Huvenne W, Hellings PW, Bachert C. Role of staphylococcal superantigens in airway disease. Int Arch Allergy Immunol. (2013) 161:304–14. doi: 10.1159/000350329
117. Bachert C, Zhang N. Chronic rhinosinusitis and asthma: novel understanding of the role of IgE ‘above atopy'. J Intern Med. (2012) 272:133–43. doi: 10.1111/j.1365-2796.2012.02559.x
118. Bachert C, Zhang N, Holtappels G, De Lobel L, van Cauwenberge P, Liu S, et al. Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J Allergy Clin Immunol. (2010) 126:962–8, 8e1-6. doi: 10.1016/j.jaci.2010.07.007
119. Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. (2015) 42:756–66. doi: 10.1016/j.immuni.2015.03.014
120. Reginald K, Westritschnig K, Werfel T, Heratizadeh A, Novak N, Focke-Tejkl M, et al. Immunoglobulin E antibody reactivity to bacterial antigens in atopic dermatitis patients. Clin Exp Allergy. (2011) 41:357–69. doi: 10.1111/j.1365-2222.2010.03655.x
121. Sachse F, Becker K, von Eiff C, Metze D, Rudack C. Staphylococcus aureus invades the epithelium in nasal polyposis and induces IL-6 in nasal epithelial cells in vitro. Allergy. (2010) 65:1430–7. doi: 10.1111/j.1398-9995.2010.02381.x
122. Tomassen P, Vandeplas G, Van Zele T, Cardell LO, Arebro J, Olze H, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. (2016) 137:1449–56.e4. doi: 10.1016/j.jaci.2015.12.1324
123. Broker BM, Mrochen D, Peton V. The T cell response to Staphylococcus aureus. Pathogens. (2016) 5:31. doi: 10.3390/pathogens5010031
124. Cho SH, Strickland I, Tomkinson A, Fehringer AP, Gelfand EW, Leung DY. Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J Invest Dermatol. (2001) 116:658–63. doi: 10.1046/j.0022-202x.2001.01331.x
125. Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature. (2013) 503:397–401. doi: 10.1038/nature12655
126. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol. (2015) 13:529–43. doi: 10.1038/nrmicro3521
127. Teufelberger AR, Nordengrun M, Braun H, Maes T, De Grove K, Holtappels G, et al. The IL-33/ST2 axis is crucial in type 2 airway responses induced by Staphylococcus aureus-derived serine protease-like protein D. J Allergy Clin Immunol. (2018) 141:549–59.e7. doi: 10.1016/j.jaci.2017.05.004
128. Clementsen P, Larsen FO, Milman N, Svok PS, Norn S. Haemophilus influenzae release histamine and enhance histamine release from human bronchoalveolar cells. Acta Pathol Microbiol Scand. (1995) 103:806–12. doi: 10.1111/j.1699-0463.1995.tb01438.x
129. Kjaergard LL, Larsen FO, Norn S, Clementsen P, Skov PS, Permin H. Basophil-bound IgE and serum IgE directed against Haemophilus influenzae and Streptococcus pneumoniae in patients with chronic bronchitis during acute exacerbations. Acta Pathol Microbiol Scand. (1996) 104:61–7. doi: 10.1111/j.1699-0463.1996.tb00687.x
130. Norn S, Jensen L, Kjaergard LL, Permin H, Skov PS, Espersen F. Bacteria-induced IgE-mediated histamine release: examination of patients with chronic bronchitis (CB) during acute exacerbations. Agents Actions. (1994) 41:C22–3. doi: 10.1007/BF02007749
131. Otczyk D, Clancy R, Cripps A Haemophilus influenzae and smoking-related obstructive airway disease. Int J COPD. (2011) 6:345–51. doi: 10.2147/COPD.S19359
132. Roth M, Zhong J, Zumkeller C. S'Ng C T, Goulet S, Tamm M. The role of IgE-receptors in IgE-dependent airway smooth muscle cell remodelling. PLoS ONE. (2013) 8:e56015. doi: 10.1371/journal.pone.0056015
133. Roth M, Zhao F, Zhong J, Lardinois D, Tamm M. Serum IgE induced airway smooth muscle cell remodeling is independent of allergens and is prevented by omalizumab. PLoS ONE. (2015) 10:e0136549. doi: 10.1371/journal.pone.0136549
134. Fang L, Wang X, Sun Q, Papakonstantinou E, S'ng C, Tamm M, et al. IgE downregulates PTEN through MicroRNA-21-5p and stimulates airway smooth muscle cell remodeling. Int J Mol Sci. (2019) 20:875. doi: 10.3390/ijms20040875
135. Jones R, Noble P, Elliot J, James A. Airway remodeling in COPD: it's not asthma! Respirology. (2016) 21:1347–56. doi: 10.1111/resp.12841
136. Woolcock AJ, Anderson SD, Peat JK, Du Toit JI, Zhang YG, Smith CM, et al. Characteristics of bronchial hyperresponsiveness in chronic obstructive pulmonary disease and in asthma. Am Rev Respir Dis. (1991) 143:1438–43. doi: 10.1164/ajrccm/143.6.1438
137. Ramsdell JW, Nachtwey FJ, Moser KM. Bronchial hyperreactivity in chronic obstructive bronchitis. Am Rev Resp Dis. (1982) 126:829–32.
138. van Schayck CP, Dompeling E, Molema J, Folgering H, van Grunsven PM, van Weel C. Does bronchial hyperresponsiveness precede or follow airway obstruction in asthma or COPD? Neth J Med. (1994) 45:145–53.
139. Rijcken B, Schouten JP, Weiss ST, Speizer FE, van der Lende R. The relationship of nonspecific bronchial responsiveness to respiratory symptoms in a random population sample. Am Rev Respir Dis. (1987) 136:62–8. doi: 10.1164/ajrccm/136.1.62
140. Stolz D, Anderson SD, Gysin C, Miedinger D, Surber C, Tamm M, et al. Airway reactivity to inhaled mannitol in cigarette smokers: a longitudinal study. Respir Med. (2007) 101:1470–6. doi: 10.1016/j.rmed.2007.01.008
141. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO global initiative for chronic obstructive lung disease (GOLD) workshop summary. Am J Respir Crit Care Med. (2001) 163:1256–76. doi: 10.1164/ajrccm.163.5.2101039
142. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med. (1968) 278:1355–60. doi: 10.1056/NEJM196806202782501
143. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Eng J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
144. Papakonstantinou E, Savic S, Siebeneichler A, Strobel W, Jones PW, Tamm M, et al. pilot study to test the feasibility of histological characterisation of asthma-COPD overlap. Eur Respir J. (2019) 53:1801941. doi: 10.1183/13993003.01941-2018
145. Shamji MH, Thomsen I, Layhadi JA, Kappen J, Holtappels G, Sahiner U, et al. Broad IgG repertoire in patients with chronic rhinosinusitis with nasal polyps regulates proinflammatory IgE responses. J Allergy Clin Immunol. (2019) 143:2086–94.e2. doi: 10.1016/j.jaci.2019.02.001
Keywords: COPD, asthma, IgE, atopy, allergy
Citation: Karakioulaki M, Papakonstantinou E, Goulas A and Stolz D (2021) The Role of Atopy in COPD and Asthma. Front. Med. 8:674742. doi: 10.3389/fmed.2021.674742
Received: 01 March 2021; Accepted: 26 July 2021;
Published: 18 August 2021.
Edited by:
Luigino Calzetta, University of Parma, ItalyReviewed by:
Chin Kook Rhee, The Catholic University of Korea, South KoreaKonstantinos Bartziokas, University Hospital of Ioannina, Greece
Copyright © 2021 Karakioulaki, Papakonstantinou, Goulas and Stolz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daiana Stolz, ZGFpYW5hLnN0b2x6QHVzYi5jaA==