 Yuxin Wang†
Yuxin Wang† Fengchao Zhao
Fengchao Zhao- Department of Orthopaedic Surgery, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
The mitochondrial calcium uniporter is an intensively investigated calcium channel, and its molecular components, structural features, and encoded genes have long been explored. Further studies have shown that the mitochondrial calcium unidirectional transporter (MCU) is a macromolecular complex related to intracellular and extracellular calcium regulation. Based on the current understanding, the MCU is crucial for maintaining cytosolic Ca2+ (cCa2+) homeostasis by modulating mitochondrial Ca2+ (mCa2+) uptake. The elevation of MCU-induced calcium levels is confirmed to be the main cause of mitochondrial reactive oxygen species (mROS) generation, which leads to disordered cellular metabolic patterns and cell death. In particular, in an I/R injury model, cancer cells, and adipocytes, MCU expression is maintained at high levels. As is well accepted, the AMPK/PGC-1α/SIRT3 pathway is believed to have an affinity for mROS formation and energy consumption. Therefore, we identified a link between MCU-related mROS formation and the AMPK/PGC-1α/SIRT3 signaling pathway in controlling cell metabolism and cell death, which may provide a new possibility of targeting the MCU to reverse relevant diseases.
Introduction
Previous studies have revealed that mitochondria regulate intracellular and extracellular calcium concentrations and signaling and are involved in a series of physiological and pathological processes, including energy metabolism, signaling regulation, smooth muscle contractility, cell proliferation, and cell death. Calcium permeation into cells is driven by the electrochemical gradient between the external space and matrix space (1). Mitochondrial calcium uptake is mediated by a highly selective calcium channel that is localized on the inner mitochondrial membrane called the MCU. Calcium has an extremely high affinity for the MCU, substantially exerting the properties of the MCU (2). Nevertheless, the precise and specific mechanism underlying the function of the MCU remains elusive. Many studies have attempted to illustrate the exact structure and function of the mitochondrial calcium uniporters. As experiments progress, it has been found that the uniporter is a complicated protein complex in humans that consists of four essential elements: MCU [located at the mitochondrial inner transmembrane for ion conduction that is inhibited by ruthenium red (RR) or Ru360] (3), MICU1 and MICU2 (two vital regulators that localize to the intermembrane space as gatekeepers) (4, 5), and EMRE (an essential membrane crossing subunit prompting the entry of calcium) (4). Ca2+ participates in various signaling pathways, inducing multiple cellular processes. Nevertheless, calcium uptake by mitochondria is attributed to three main functions: (1) maintenance of cellular metabolic homeostasis between the cytosol and mitochondria, (2) mediation of cCa2+ dynamics, and (3) modulation of various cell death pathways of apoptosis and necrosis (6–8). Ca2+ enters the mitochondrial matrix under large gradients across the membrane to take part in the tricarboxylic acid (TCA) cycle through dehydrogenase to regulate the generation of ATP, which is crucial to the responses required for energy, such as muscle contraction, exocytosis, biosynthesis, and neuronal signaling (6). Mitochondria attenuate cytosolic calcium through the transportation of calcium when there is a transient influx of Ca2+ from intracellular stores and extracellular sources. Moreover, it is also vital for the function under the current conditions of weak intracellular Ca2+ buffering (7). Cell death proceeds by constantly increasing mitochondrial free Ca2+, resulting in the overloading of Ca2+, which ultimately contributes to the intervention of ATP generation and the appearance of mROS (8). Taken together, the MCU functions as a calcium unidirectional transporter and has been demonstrated to affect multiple cell type activities, especially pathological processes. Many experiments have shown that the MCU modulates tumor metastasis (9), ischemia-reperfusion (I/R) injury (10), neuronal apoptosis (11), muscle atrophy (12), and abnormal adipocyte metabolism (13) via calcium transition. The AMP-activated protein kinase (AMPK)/peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α)/sirtuin-3 (SIRT3) signaling pathway, which is sensitive to the AMP/ATP ratio, is vital for energy metabolism (14). The formation of mROS also reflects the regulation of PGC-1α and SIRT3 on the expression levels of the MCU. In addition, mROS is thought to be the imperative step in triggering apoptosis and necrosis (15). Here, we analyzed the architecture and regulatory factors of MCU and revealed its vital role in the dynamic calcium balance. Moreover, the purpose of this review is to discuss how MCU-dependent mROS generation regulates cell metabolism and cell death, causing pathogenesis. We also explored the relationship between the MCU-mROS axis and the AMPK/PGC-1α/SIRT3 signaling pathway.
Structure of the Mitochondrial Calcium Uniporter
The Pore-Forming Subunits of MCU
The uniporter complex has been defined as an inward rectifying current that is universally expressed in all eukaryotes. However, in most fungi, genome sequence analysis indicates that the pore-forming MCU is the only component of the uniporter, which makes it an excellent heterologous expression system to reconstruct the MCU to exhibit uniporter properties in yeast. Based on this, investigators have found that an mCa2+ uptake response was evoked after reconstruction, suggesting that MCU is the pore-forming subunit of the uniporter complex (16, 17). Electrophysiological studies have demonstrated that the MCU, an ion channel, exhibits essential selectivity and high affinity for Ca2+, but it is sensitive to RR and its analog Ru360 (2). Computational analysis provides a hypothesis of ion channel topologies of the MCU pore-forming domain, which includes four identical subunits composed of two transmembrane helices (TM1 and TM2) separated by a conserved linker. The linker, facing the intermembrane space, is a short stretch of amino acids and contains a motif called “DIME (Asp-Ile-Met-Glu)”. In addition, the structure of the MCU comprises two coiled coils domain (CCD) and an N-terminal domain (NTD) (3, 18). In recent years, researchers have applied cryo-EM to further study the precise architecture of MCU. The overall structure is in accordance with the previously predicted hypothesis. MCU displays an exactly tetrameric architecture by means of EM under both high and low concentrations of calcium. TM1 and CC1 form a long and continuous helix at the periphery of the channel, while the TM2 helices line the central symmetry axis. There is a short helix that is defined as a junctional helix (JH), situated almost perpendicular to TM1. JH forms a junction between TM2 and CC2. In the CCD, CC1 and CC2 form a dimeric coiled coil, resulting in four dimeric coiled coils in the tetramer. Following the coiled-coil domain, the NTD, comprising six β-strands (β1–β6) and two α-helices (α1 and α2) that form the central core with two highly conserved leucine-rich loops (19), is directly connected to CC1 (20). The structure of the NTD forms the MCU oligomers, and deletion of the NTD results in a rapid decline in mCa2+ with complete cCa2+ dynamics (19). A residue of the NTD called the MCU-regulating acidic patch (MRAP) binds divalent cations to autoregulate Ca2+ uptake by matrix Ca2+ and Mg2+, occupying the MRAP domain, similar to a feedback mechanism (21). The strictly conserved sequence motif existing in all MCU homologs has been demonstrated to shape the selectivity filter of Ca2+. This sequence motif is located in the N-terminal region of TM2, with the carboxylate side chains of conserved acidic residues Asp and Glu from each protomer pointed into the central symmetry axis, forming two acidic rings along the channel pore. Asp is located at the intermembrane end of TM2 and its carboxylate ring coordinates hydrated Ca2+, while the diameter of the Glu carboxylate ring is too small for hydrated Ca2+ to pass through, suggesting that only dehydrated Ca2+ is allowed to pass through (17, 20, 22). Consequently, we have enough evidence to deduce that the Glu carboxylate ring is unsubstitutable for selective permeation.
The Regulatory Subunits of MCU
It is generally accepted that the primary regulatory subunits centered around Ca2+ channel proteins are MICU1, MICU2, MCUR1, MCUb, and EMRE. MICU1, co-occurring with MCU in mammals but absent from most fungi, is a peripheral membrane protein with two EF-hand motifs that functions as a calcium-sensing regulator (23). MICU1 interacts with MCU through the D-ring of the DIME motif to constitute the conserved unit of a eukaryotic uniporter. It is not until cCa2+ rises above 3 μM that the gatekeeper MICU1 will activate the MCU channel by dissociating from MCU, making it adopt an open confirmation (24). To prove this hypothesis, genetic deletion of MICU1 was performed, and the results complemented the assumption that mCa2+ increase, even at low cCa2+, simultaneously caused mCa2+ overload (25). Moreover, when MICU1 was knocked down, the sensitivity of RR/Ru360 to MCU increased, while calcium uptake remarkably decreased, demonstrating that MICU1 likely competes with RR/Ru360 at the site of the pore-forming domain (5, 26).
MICU2 is a paralog of MICU1 and shares a similar function as a gatekeeper that assists MCU in taking up calcium from the cytoplasm. Silencing MICU2 by RNAi revealed a reduced rate of mCa2+ uptake, signifying that MICU2 is dispensable for mCa2+ uptake (27). Importantly, utilizing the transcription activator-like effector nuclease (TALEN) technology to knock out MICU1 and MICU2 in HEK293 T cells resulted in a steep Ca2+ uptake by MICU1 KO mitochondria due to the loss of gatekeeping. MICU2 KO mitochondria exhibited a decreased rate of Ca2+ influx (28). It follows that MICU2 may have a positive effect on the cooperation with MICU1 to achieve the function of gatekeeping. In addition to MICU1, heterooligomers of MICU1 and MICU2 also interact with MCU to control the gating and cooperative activation of the uniporter. However, the respective function and mechanism of each subunit in mediating the activity of the uniporter is complicated in the mammalian system because of differences ranging from the degree of gene silencing, tissue-specific protein composition (29, 30) from stoichiometry, and compensatory remodeling of the channel (25, 31).
In contrast, MCU regulator 1 (MCUR1), a positive regulator of the channel, exerted a blunt MCU current when the transmembrane voltage was clamped, indicating that MCUR1 acts directly on MCU to promote Ca2+ entry into the mitochondria instead of the reduced electrochemical driving force (32). Another laboratory finding showed that MCUR1 is an assembly factor of complex IV cytochrome oxidase, leading to an impaired oxidative phosphorylation in the absence of MCUR1 that is certainly a decreased driving force that decreases calcium uptake (33).
MCU proteome analysis revealed that EMRE, a 10-kilodalton protein with an EF hand domain, is ubiquitously expressed in all mammalian mitochondria. Experiments have demonstrated that a knockout of EMRE specifically decreases the uptake of calcium, while the appearance and proliferation of cells is not affected. Moreover, the abundance of other uniplex proteins and their mitochondrial localization also remain. What has changed is the association of MCU with MICU1 and MICU2. Hence, we can link the function of EMRE and MICU to calcium-sensing activity (4). Moreover, it has been proposed that EMRE senses the matrix calcium concentration with the acidic patch at its carboxy terminal to regulate MCU activity (34). However, in the study by Tsai et al., the interaction of MICU1 with MCU in the absence of EMRE was also displayed by coimmunoprecipitation (35).
Recently, MCUb, a paralog of the pore-forming subunit MCU, was investigated to determine whether it has a prominent influence on Ca2+ permeation. MCUb is conserved across all vertebrates and absent in species of plants, kinetoplastids, Nematoda, and Arthropoda, where MCU is present (18). The results of the present experiments suggest that MCUb is an important continuance of the uniplex and has a negative effect on Ca2+ uptake. MCUb dislocates the position of MCU to disconnect MCU from MICU1 and MICU2 to alter channel gating. Meanwhile, the presence of MCU and the expression of channel regulators MICU1 and MICU2 rapidly increases after MCUb depletion (36) (Figure 1).
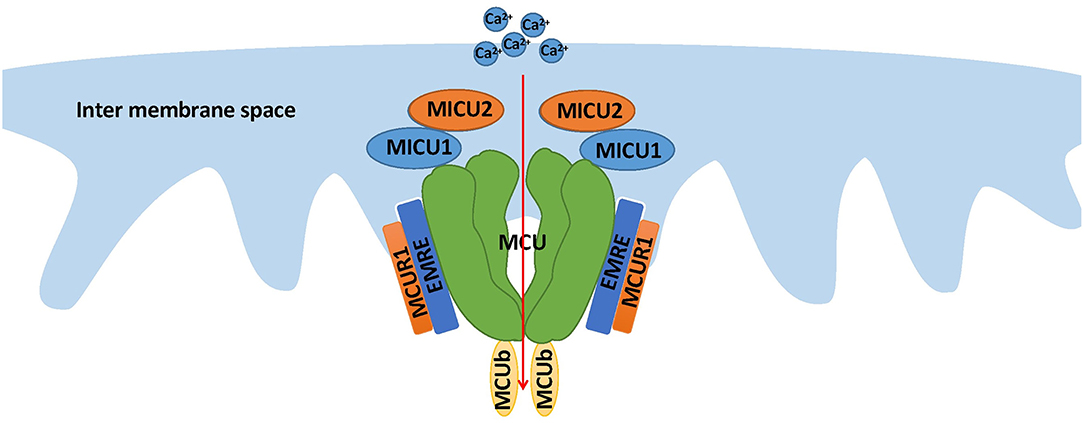
Figure 1. The structure of the MCU complex. This compound, localized at the inner membrane of the mitochondria, consists of the pore-forming subunit and regulator subunits, including MICU1, MICU2, EMRE, MCUR1, and MCUb. The most important function of MCU is mediating mitochondrial calcium uptake.
Modulation of MCU Expression
As demonstrated in experiments, cCa2+ signaling is influenced by both high and low cytosolic calcium concentrations (26, 36). MCU, a core ion channel transmitting Ca2+, may be an effective target to interrupt Ca2+ homeostasis and subsequently be involved in various pathological processes. In line with this finding, regulating the processes associated with MCU protein generation, including transcription, posttranscriptional modification, translation, and posttranslational modification (PTM), remarkably influence the expression level of proteins to change the interplay between cCa2+ and mCa2+. The Ca2+ regulated transcription factor CREB (cyclic adenosine monophosphate response element) binds to the MCU promoter. When cCa2+ decreases, phosphorylation of CREB is instantly initiated to alter transcription followed by the downregulation of MCU abundance and the decrease in mCa2+ (37). At the posttranscriptional level, microRNAs (miRNAs) are non-coding nucleotides and can modulate gene expression by combining with specific miRNAs to degrade target miRNAs or restrain translation (38). There have been valid findings linking miRNA-associated MCU expression with cell survival. The downregulation of MCU RNA and protein by miR-25 has been shown to protect tumor cells from cell death in colon cancer (39). Furthermore, subsequent studies utilized anti-miR25 and anti-miR138 in pulmonary arterial hypertension (PAH) patients whose MCU expression is downregulated, and the final conclusion revealed the predictable overexpression of the MCU protein (40). PTMs are composed of two predominant forms, namely, oxidation and phosphorylation (41). Previous studies confirmed the findings about the PTMs of MCU. In the NTD of MCU, there are conserved cysteines, which carry S-glutathionylation under oxidative stress to reconstruct NTD confirmation, which accelerates the sustained activity of MCU and a high rate of calcium uptake. In addition, phosphorylation is proposed to occur in the MCU PTM at sites S57 and S92 of the NTD in the presence of calmodulin kinase II (CaMKII). The implication from further studies revealed facilitated MCU current compared to the control, which inhibited the expression of CaMKII (19). These findings highlight the significance of regulating MCU expression and propose a novel route to interpose cell death and metabolic homeostasis (Figure 2).
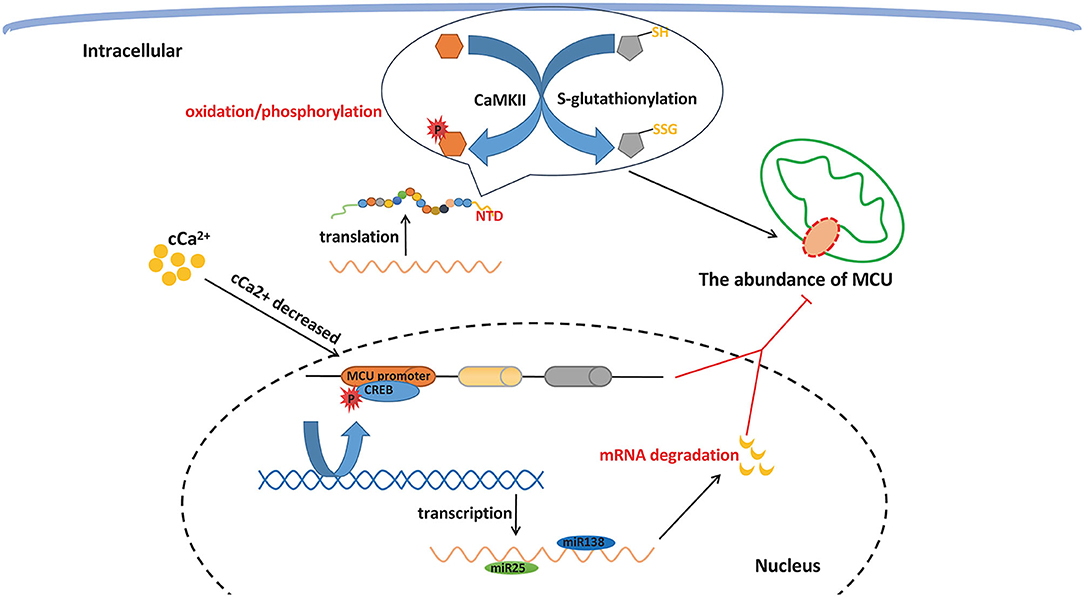
Figure 2. MCU expression regulation. MCU protein generation can be impacted at several molecular levels, including transcription, post transcriptional modification, translation, and post translational modification, which can remarkably change the interplay between cCa2+ and mCa2+ via influencing the expression level of protein.
MCU Is Involved in the Process of Metabolic Homeostasis AND Cell Death Via MROS
The Generation of mROS Under Elevated Mitochondrial Ca2+ Levels Induced by MCU
Mitochondria function in buffering the concentration of intracellular calcium. Mitochondria isolated from rat kidneys in the 1960s showed a potent ability to uptake calcium even at a low temperature (42). As studies have indicated, MCU, which displays a high affinity for calcium, was revealed to be the Ca2+ transportation protein localized in the IMM. Expression levels of MCU and calcium current were also found to be different in various tissues, including the heart, skeletal muscle, liver, and neurons. Fieni et al. (17) performed studies that provided evidence of a large density of MCU in neonatal cardiac tissue through patch-clamp experiments. The biochemical gel shift assay provided evidence that MCU is the only modality that senses mROS. When inflammation and oxidative stress increase mROS, modification of channels by mROS causes mCa2+ overload-mediated cell death. In turn, with changes in the extracellular microenvironment and the stimulation of hypoxia, the elevated mCa2+ taken up by MCU interrupts energy metabolism, causing the generation of mROS.
There are methods to induce the production of mROS with an increased mCa2+: (i) dehydrogenases that participate in the tricarboxylic acid cycle, including glycerol phosphate dehydrogenase (GPDH), pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (α-KGDH), are sensitive to mCa2+ (43). They cannot completely work without the existence of mCa2+. In contrast, the data show that an elevated mCa2+ stimulates the activity of these dehydrogenases and even the conductance of complexes I, III, IV, and V in OXPHOS (44). A large amount of O2 accelerates cellular metabolism, increasing the generation of mROS, which is a byproduct of the electron transport chain, although a low rate of O2 consumption has also been discovered to promote ROS production (45, 46). (ii) mtNOS, a constitutive nitric-oxide synthase (NOS) isoform, requires calcium to activate its function. Under high concentrations of mCa2+, mtNOS tends to decrease oxygen consumption via the competitive binding of nitric oxide to cytochrome oxidase. Reduced O2 consumption and the competitive binding of cytochrome oxidase hamper the conduction of electrons, which gives rise to a higher tendency of inducing complexes I and III to generate mROS (47). (iii) Opening of the mitochondrial permeability transition pore (mPTP) induced by MCU-dependent calcium overloading abruptly admits all solutes of molecular weight up to approximately 1,500 Da to permeate into the mitochondrial matrix, which results in mitochondrial depolarization, mitochondrial swelling, and rupture of the outer membrane, causing the release of cytochrome c (48–50). Intriguingly, opening of the mPTP can change the ionic strength within the intermembrane space, destroying the electrostatic interaction between cytochrome c and cardiolipin. The release of cytochrome c is triggered by the disruption of interaction, which blocks the activity of complex III and thus intercepts electron flow to enhance mROS production (46, 51). (iv) Studies have supported the hypothesis that Ca2+-cardiolipin (CL) interactions with cardiolipin headgroups mainly contribute to multiple membrane dysfunction. With regard to overloaded mCa2+, Ca2+-CL binding induces a sequence of chemical, dynamic events that favor the reorganization of membrane components, which causes the high rate generation of mROS at the ubiquinone level (52) (Figure 3).
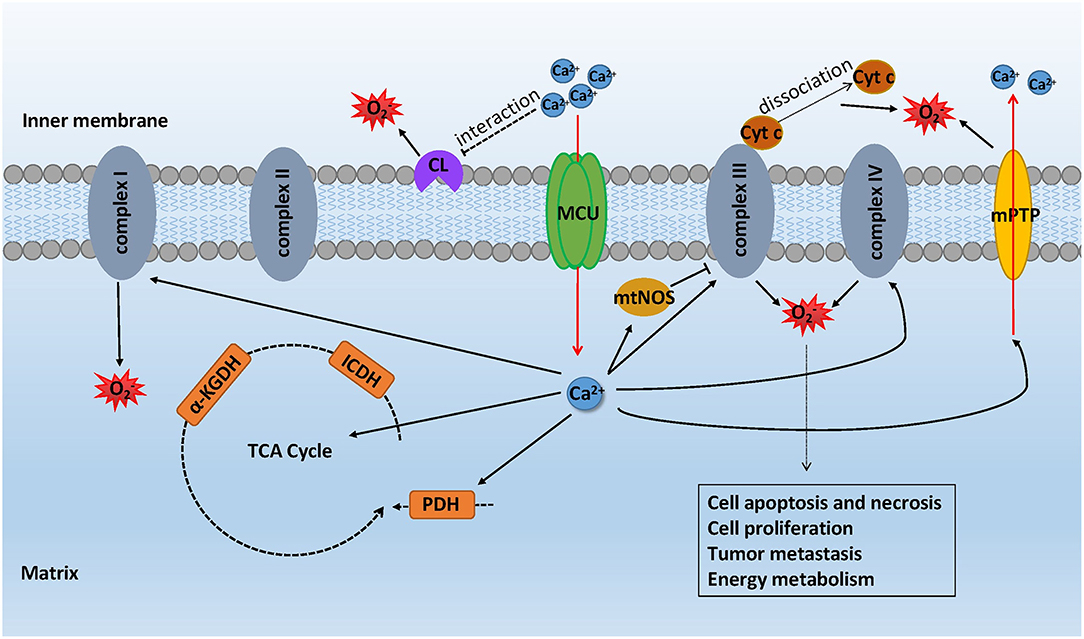
Figure 3. mROS formation mediated by MCU-dependent calcium uptake. With the stimulation of oxidative stress, the elevated mCa2+ took up by MCU interrupts energy metabolism causing the generation of mROS while the mROS formation can impact the cell metabolic function in turn. mCa2+ increases the activity of glycerol phosphate dehydrogenase (GPDH), pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (α-KGDH) in the TCA cycle, thus promoting mROS production. mtNOS, activated by mCa2+, hampers the conduction of electron, making complexes I and III generate mROS. mPTP opening results in the rupture of the outer membrane and thus causing the release of cytochrome c. Overloaded mCa2+ has an effect on Ca2+-CL binding, inducing the reorganization of membrane components, which causes the high-rate generation of mROS at the ubiquinone level.
mROS Dysregulation Induced by MCU-Dependent Calcium Influx Causes Metabolic Disorders and Cell Death
As mitochondrial calcium is important for modulating mitochondrial activity, the change in MCU expression causes aberrant mCa2+ levels, which provokes the increasing production of mROS, as previously described. Obesity or age-related whitening and loss of brown adipose tissue (BAT) contribute to systemic metabolic dysfunction and the facilitation of energy storage, which has been demonstrated in a prior study (53). Wright et al. (13) reported that expression levels of the MCU complex were increased in mouse and human adipose tissue during obesity. In insulin-resistant (IR) adipocytes, MCU and its relevant components have been observed to be upregulated together with an increased mCa2+ uptake. Experiments have been performed on human IR adipocytes both in vivo and in vitro, which signified slightly different outcomes. The expression of MCU and MICU1 increased, while the other components, such as MICU2, were unaltered in vitro. In contrast, MCU, MICU1, and MICU2 all increased in the in vivo experiments. In conclusion, the level of MCU was upregulated both in vivo and in vitro. For this reason, the study further explored the relationship between inflammatory factors and the MCU. The data revealed that ad-MCU caused a decreased release of TNF-α and IL-6, indicating that markedly decreasing the MCU-induced calcium uptake remarkably influences inflammatory cytokine release. These results indicate a possible role for MCU in cell metabolism in adipocytes. However, the precise mechanism remains to be fully understood. In addition, several previous studies have reported the enhanced sensitivity of insulin in adipocytes under decreased intracellular calcium concentrations (54). Given the crucial modulatory function of calcium, with ER-mitochondria contact, ER calcium stores can be rapidly transferred to the mitochondria upon demand via MCU transmission (55). Gao et al. (56) performed experiments and discovered that in high-fat diet (HFD)-fed mice, total ROS levels assessed by dihydroethidium in frozen slices of BAT were remarkable increase. Meanwhile, oxygen consumption, carbon dioxide generation, energy expenditure (EE), and respiratory exchange ratio (RER) were also found to be reduced in HFD-fed mice. Moreover, the author observed a significantly elevated expression of the MCU and gatekeeper MICU1, but MCUb and MICU2 were not affected. Interestingly, MCU knockout by si-MCU completely blocked the uptake of mitochondrial calcium and dramatically reduced the excessive production of mROS, improving mitochondrial metabolic activity. In a study of idiopathic pulmonary fibrosis (IPF) subjects, researchers found that PGC-1α, which increases the enzymatic capacity for fatty acid oxidation (FAO) and abolishes glycolysis, was augmented in macrophages, whereas the effects of PGC-1α were diminished in a dominant-negative MCU model (57–59). These results may indicate the dysfunction of MCU, causing a disrupted aerobic respiratory function and an abnormal metabolism.
Intracellular calcium, acting as a second messenger, ubiquitously participates in all kinds of cellular biological events. Because of the importance of Ca2+ in signaling pathways, cCa2+ dynamics are strictly controlled. A significant quantity of data indicates that a small amount of calcium entering the mitochondria is beneficial to metabolic homeostasis, while a large amount of calcium is thought to induce cell death (60). Liao et al. (10, 43) studied the role of MCU in oxidative-induced cell death. HeLa cells treated with H2O2 generate massive mROS, arousing apoptosis, which was significantly inhibited by MCU overexpression. It was also discovered that MCU knockdown markedly suppressed mROS-induced apoptosis. To further confirm the importance of mCa2+, the researchers induced mutations, MCUD260A and MCUE263A, to make it lose the ability to absorb Ca2+. The expression of MCUD260A and MCUE263A was unable to achieve oxidative stress-induced apoptosis when treated with H2O2. Therefore, the mCa2+ uptake activity of MCU is essential for facilitating mROS-induced cell death. Accordingly, numerous cell death models, such as cardiac I/R injury and neuronal excitotoxicity, have validated the evidence of its negative effect in the I/R injury mouse model (10, 43). The abundance of MCU protein has been demonstrated to be increased, followed by a high concentration of mCa2+ in I/R injury. mROS production occurs after blood reenters into ischemic tissue, causing reperfusion injury. Sharply elevated mROS leads to the opening of the mPTP, which eventually provokes apoptosis and autophagy (60, 61). Proapoptotic factors, such as cytochrome c, are released into the cytosol by ruptured mitochondria, which can upregulate Bax and the Bax/Bcl-2 ratio and activate caspases and other apoptotic events, ultimately culminating in cell death (61). Therefore, the MCU-mROS-mPTP axis primarily regulates the cell death pathway. In addition, oxidative stress-stimulated formation of mROS alters the dynamic balance of mitochondrial fission and fusion under high levels of mCa2+. Such ruptured mitochondria trigger a sequence of cellular apoptosis events (62). Dynamin-related protein 1 (Drp1) and mitochondrial LC3II expression indicate the occurrence of fission and mitophagy, respectively (63). A previous investigation confirmed that MCU inhibition alleviates Drp1 accumulation and enhances mitochondrial LC3II expression. Additionally, atrophy type 1 (OPA1) expression, which is supposed to dominate mitochondrial fusion and is suppressed in I/R injury models, was recovered when MCU was repressed. Calpains, which are calcium-dependent thiol-proteases, are activated during I/R injury by calcium overload to phosphorylate Drp1. Nevertheless, the downregulation of OPA1 by siRNA transfection surprisingly abrogates the protective effects of calpain suppression on mitochondrial fission/fusion and mitophagy, indicating that MCU repression restrains calpain/OPA1-mediated mitochondrial fusion/mitophagy inhibition to shrink the myocardial infarction size and reduce levels of apoptosis (64).
In conclusion, high-level expression of MCU-induced mCa2+ overloading results in a disordered mitochondrial metabolism and cell death, which primarily depends on the generation of mROS.
The Role of MCU in Relevant Diseases
As a high level of calcium is reported to change the pattern of cellular energy metabolism from glycolysis to fatty acid oxidation and produce excessive mROS via MCU overexpression, the development of various diseases is closely associated with it (13).
ROS production has been well demonstrated to play a crucial role in many types of cancer (65, 66). The primary source of ROS is the mitochondrial electron transport chain (ETC) in most mammalian cells. The generation of ROS, which is mostly dependent on NADH/NAD, is correlated with cellular metabolism. ROS-high tumor sphere (RH-TS) cells were found to have higher intramitochondrial superoxide levels than normal cells. The intramitochondrial superoxide level measured by flow cytometry suggested that endogenous ROS in RH-TS cells were largely generated in mitochondria. With the generation of mROS, the biosynthesis of fatty acids in RH-TS cells was reduced, while the breakdown of fatty acids increased (67). Liu et al. (68) reported that MCU is instrumental for the growth of colorectal cancer (CRC). RT-qPCR and Western blotting analysis showed that the expression levels of both the MCU protein and mRNA were markedly upregulated in the majority of malignant colorectal tissue compared to the adjacent normal tissue. Immunohistochemical and Kaplan-Meier analyses also validated these results. More importantly, studies indicated that treatment with H2O2 and ROS scavengers reversed the effects of MCU knockdown and MCU overexpression in vivo. Taken together, these findings reveal that MCU-mediated calcium uptake stimulates mitochondrial biogenesis to expedite CRC growth via excessive ROS generation. Similarly, in hepatocellular carcinoma (HCC) cells, the upregulation of MCU and the downregulation of MICU1 led to an increased basal mCa2+ compared to the control cells. It is well known that the electron transport chain is substantially strengthened by increased calcium entering the mitochondria, generating additional mROS. In particular, mCa2+ uptake increases NAD+ conversion into NADH and downregulates the deacetylase activity of SIRT3, inactivating SOD2, which subsequently loses the capacity to eliminate mROS. As a result, HCC proliferation and cell migration and invasion are enhanced via ROS in response to high expression levels of MCU (69). Beyond these, Arvizo et al. (35) probed novel roles of MCU in ovarian cancer. Tosatto et al. (70) found that triple-negative breast cancer (TNBC), the most aggressive breast carcinoma subtype, is also influenced by MCU in terms of tumor size, lung metastasis, and lymph node infiltration by creating an MCU deletion in an MDA-MB-231 cell model with CRISPR/Cas9 nuclease RNA-guided genome editing technology.
In addition, MCU is involved in cardiac I/R injury and neurodegenerative disease. The cardiac microvasculature is particularly susceptible to the deleterious effects of I/R injury, which are mediated by dysregulated intracellular calcium. Li et al. (71) reported that sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) recycles calcium from the cytosol back to the endoplasmic reticulum, preventing I/R-induced luminal stenosis, vascular wall edema, endothelial barrier integrity, and erythrocyte morphological changes. Furthermore, they found that SERCA overexpression suppressed MCU expression and simultaneously attenuated intracellular calcium overloading. Applying a calcium activator or using an MCU agonist remarkably induced endothelial necroptosis in vitro and abolished the protective effects of SERCA overexpression in a reperfused heart tissue in vivo. Signal transducer and activator of transcription 3 (STAT3) in intermittent hypobaric hypoxia (IHH) has been demonstrated to convey cardio-protection against I/R injury. Wu et al. (72) further studied its mechanism in a post-ischemic myocardial cell model. These cardioprotective effects were abrogated by the STAT3 inhibitor AG490, which opens the MCU. Colocalization of STAT3 and MCU was also observed in rat I/R cardiomyocytes. The results of this study ultimately revealed that activated STAT3 interacts with the NTD of MCU to alleviate mitochondrial calcium overload, protecting against cardiac I/R. Enlightened by the examination of MCU in I/R injury of cardiomyocytes, MCU-mediated neuron apoptosis and necrosis, which crucially account for neurodegenerative and neural ischemic disease, have been universally probed to identify a novel therapeutic target. Veronica et al. (73) have investigated whether that mCa2+ overload is sufficient to decide the neural cell fate by overexpressing MCU both in an exosomatic model of mouse primary cortical neurons and an endosomatic model of injecting MCU-coding adenoviral particles into the mouse brain cortex. In a rat cerebral ischemia model, MCU was observed to be directly phosphorylated by activated proline-rich tyrosine kinase 2 (Pyk2) and aroused Ca2+ overloading and mROS accumulation, thus leading to neuronal apoptosis and ischemic stroke. Inhibiting the phosphorylation of MCU with Pyk2 antagonist can prevent mCa2+ overload, mitochondrial injury, proapoptotic protein release, and cell death (74). In addition, Maria et al. (75) and Smijin et al. (76) reported the relevance of hereditary spastic paraplegia (HSP7) and Parkinson's disease (PD) to MCU function, respectively. The researchers found that the mitochondrial matrix ATPase associated with diverse cellular activities (m-AAA) protease plays an important role in regulating the function of mitochondria. Protein synthesis and respiration in mitochondria are damaged after inhibiting m-AAA (77). In particular, m-AAA depletion impairs MCU assembly, leading to degradation of the EMRE subunit, which induces the massive entry of mCa2+ into the Purkinje cells and ultimately evokes HSP7. Dopaminergic neurons with hybridization and immunohistochemical labels were rescued after the deletion of MCU in the model of PD (76, 78). Alzheimer's disease (AD) is a progressive neurodegenerative disorder with cognitive dysfunction in an individual. The experiments found the blockade of MCU exhibits neuroprotective activity of AD animal models, which was induced by intracerebroventricular injection of streptozotocin (ICV-STZ). In addition, the experimental rats were treated with the MCU blocker, RR. The results indicated that RR attenuated ICV-STZ induced memory-related behavioral abnormalities and decreased level of acetylcholine and choline acetyltransferase activity. The percentage of apoptotic cells in ICV-STZ challenged rat brain regions was also alleviated by RR (79).
The Relationship Between the MCU-MROS AXIS And the AMPK/PGC-1α/SIRT3 Signaling Pathway
As AMPK/PGC-1α is an intracellular energy sensor and modulator, it is activated in response to energy depletion and high demands for energy supply. SIRT3 that served as the downstream target of AMPK-PGC-1α signaling plays a key role in the regulation of mitochondrial biogenesis and oxidative stress. Most studies have proved that this pathway is related to ischemia-reperfusion injury, neurodegenerative diseases, and abnormal glucose metabolism in diabetes (80, 81). It has a high degree of overlap with the diseases involved in the MCU-mROS axis.
Previous works have provided evidence that decreased SIRT3 expression is the primary feature of BAT whitening. Unlike SIRT1, which primarily exists in the nucleus, SIRT3 preferentially localizes in the mitochondria (82). SIRT3 knockout (KO) or si-Sirt3 significantly promoted MCU expression via increased acetylation of histone lysine 27 on the MCU promoter. Moreover, with a decrease in H3K27ac, MCU expression was remarkably reduced. These conspicuous phenomena demonstrate that acetylation of H3K27 plays a key role in the histone modification status to control the transcription of MCU (56). It is well accepted that SIRT3 is an NAD+-dependent mitochondrial deacetylase that deacetylates SOD2 to eliminate the formation of mROS. MCU-mediated mCa2+ uptake downregulates the NAD+/NADH ratio, which diminishes SOD2 deacetylation activity. Furthermore, the activity of SIRT3 and SOD2 drastically reversed MCU overexpression-induced HCC cell migration and invasion (69). Interestingly, SIRT3 not only modulates the metabolic enzymes of mitochondria but also directly regulates mitochondrial morphology dynamics by targeting OPA1 during stress (83). Additionally, Kong et al. (84) reported that SIRT3 was required for PGC-1α-essential induction of ROS-detoxifying enzymes and several constituents of the respiratory chain, such as glutathione peroxidase-1, ATP synthase 5c, and cytochrome c. The data show that the significantly increased deacetylase activity of SIRT3 is consistent with MCU knockdown, whereas MCU overexpression induces the reverse condition (69). In mouse adipose tissue, knockout of SIRT3 increased mitochondrial oxidative stress, resulting in accelerated obesity and metabolic syndrome (85). Capsaicin was proven to be significant in increasing BAT activity and aerobic respiratory function. SIRT3 mediated the promotional effects of capsaicin on BAT metabolism and ROS generation by enhancing MCU activity. The inhibitory effect of capsaicin on driving mitochondrial calcium overload in BAT was impaired after SIRT3 KO. Likewise, mitochondrial ROS were elevated in cells treated with si-Sirt3, compared to WT cells. Gao et al. (56) indicated that SIRT3 alleviates mCa2+ overload by inhibiting MCU expression. Nevertheless, both overexpression and knockdown of MCU failed to alter the expression of SIRT3, which indicates that MCU is a downstream target of SIRT3. In addition, researchers constructed a cardiac I/R injury model via coronary artery ligation and reperfusion, which showed that mice lacking cyclophilin D exhibited a reduced infarct size. Hafner et al. (86) discovered that SIRT3 deacetylates cyclophilin D on lysine 166 to interrupt the opening of the mPTP. Cardiac myocytes from mice lacking SIRT3 exhibit an increased mitochondrial swelling with age due to ascending mPTP opening, eventually contributing to cell death. Experimental results demonstrate that in both muscle cells and hepatocytes, PGC-1α strongly stimulated mouse SIRT3 gene expression by activating the SIRT3 promoter, indicating that SIRT3 works as a downstream target gene of PGC-1α (84). Activation of AMPK is a sensor of energy metabolism that increases mitochondrial biogenesis, glucose uptake, and fatty acid oxidation by upregulating gene expression in these pathways. It has also been found that activation of AMPK inevitably induces PGC-1α gene expression. In turn, the effects of AMPK on energy metabolism are almost entirely dependent on the PGC-1α protein. AMPK directly phosphorylates PGC-1α to initiate downstream cellular events (87). However, experiments have shown that loss of SIRT3 impacts the phosphorylation of AMPK and PGC-1α expression in skeletal muscle. Accordingly, SIRT3 upregulation strengthened the downstream activation of PGC-1α via a higher phosphorylation level of AMPK compared to the control group (88). These results suggest that SIRT3 and AMPK form a positive-feedback loop, acting as a link between the energy metabolism of cells and mitochondrial function.
Increasing studies have investigated whether mitochondria act as cCa2+ buffers. The rapid accumulation of mCa2+ occurs when cCa2+ increases. Consistently, different patterns of mCa2+ elevation cause contrary results. Investigations have observed that transient mCa2+ accelerates mitochondrial bioenergetics and activates the energy metabolism cycle, while calcium overload increases mROS levels and evokes mitophagy (89–91). Analogously, with a transiently elevated cCa2+, AMPK phosphorylation is significantly activated, but AMPK activity is blocked by sustained high intracellular Ca2+ levels. There may be a close connection between calcium-modulated mitochondrial bioenergetics and the AMPK activated state (92, 93). This phenomenon may provide a defense mechanism to prevent cells from undergoing apoptosis and autophagy under acute stress (Figure 4).
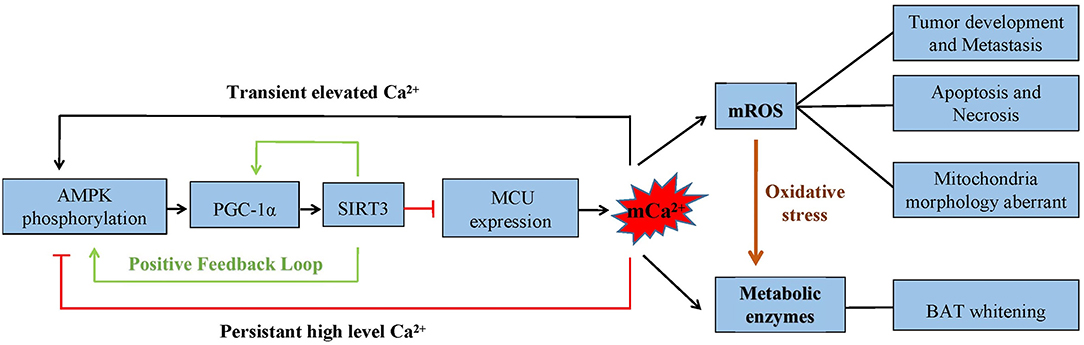
Figure 4. The AMPK/PGC-1α/SIRT3 signaling pathway inhibits the expression of MCU. The capability of mCa2+ uptake is decreased, which inhibits mROS generation. Cell metabolism, apoptosis, and necrosis are significantly affected by mCa2+-dependent mROS production. Also, the crosstalk between AMPK/PGC-1α and SIRT3 forms a positive feedback loop. The phosphorylation of AMPK is found to be different under a transient and persistent elevation of calcium.
Perspective
Calcium has been reported to change the pattern of cellular energy metabolism from glycolysis to fatty acid oxidation, which is commonly presumed to affect cellular function and even pathogenesis (13). Mitochondria have consistently played an irreplaceable role in maintaining Ca2+ homeostasis, which is largely dependent on the MCU. With the increase in the understanding of MCU, a spectrum of experiments has been performed to reveal the effects of MCU in a myriad of physiological and pathological processes. However, depending on the cell type, an abnormal expression of MCU has distinct effects (9). In neurodegeneration, I/R injury of cardiac and neurologic tissue, skeletal muscle metabolism, cancer cell metastasis, and cell death, mitochondria control Ca2+ signaling as a common final pathway in controlling mROS production and organelle structure. Although the AMPK/PGC-1α/SIRT3 pathway has been proven to be crucial in governing the cellular metabolic state and cell death, integral mitochondrial calcium signaling networks remain unknown but has become vital for our understanding of a series of therapeutic targets in various diseases.
Author Contributions
FZ: provided the idea. YW: contributed to editing the manuscript. YW and XL: conceived the study, supervised the research and wrote the paper. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by A Project of Scientific Research Fund of Zhejiang Provincial Education Department (188310-542126/015). No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this manuscript.
References
1. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. (2003) 4:517–29. doi: 10.1038/nrm1155
2. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. (2004) 427:360–4. doi: 10.1038/nature02246
3. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. (2011) 476:341–5. doi: 10.1038/nature10234
4. Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. (2013) 342:1379–82. doi: 10.1126/science.1242993
5. Paillard M, Csordás G, Huang KT, Várnai P, Joseph SK, Hajnóczky G. MICU1 interacts with the d-ring of the MCU pore to control Its Ca(2+) flux and sensitivity to Ru360. Mol Cell. (2018) 72:778–85.e3. doi: 10.1016/j.molcel.2018.09.008
6. Glancy B Balaban RS Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. (2012) 51:2959–73. doi: 10.1021/bi2018909
7. Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. (1997) 137:633–48. doi: 10.1083/jcb.137.3.633
8. Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. (1998) 1366:177–96. doi: 10.1016/S0005-2728(98)00112-1
9. Vultur A, Gibhardt CS, Stanisz H, Bogeski I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch. (2018) 470:1149–63. doi: 10.1007/s00424-018-2162-8
10. de J García-Rivas G, Carvajal K, Correa F, Zazueta C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br J Pharmacol. (2006) 149:829–37. doi: 10.1038/sj.bjp.0706932
11. Nichols M, Pavlov EV, Robertson GS. Tamoxifen-induced knockdown of the mitochondrial calcium uniporter in Thy1-expressing neurons protects mice from hypoxic/ischemic brain injury. Cell Death Dis. (2018) 9:606. doi: 10.1038/s41419-018-0607-9
12. Zampieri S, Mammucari C, Romanello V, Barberi L, Pietrangelo L, Fusella A, et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol Rep. (2016) 4. doi: 10.14814/phy2.13005
13. Wright LE, Vecellio Reane D, Milan G, Terrin A, Di Bello G, Belligoli A, et al. Increased mitochondrial calcium uniporter in adipocytes underlies mitochondrial alterations associated with insulin resistance. Am J Physiol Endocrinol Metab. (2017) 313:E641–50. doi: 10.1152/ajpendo.00143.2016
14. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
15. Fu B, Zhao J, Peng W, Wu H, Zhang Y. Zhang, Resveratrol rescues cadmium-induced mitochondrial injury by enhancing transcriptional regulation of PGC-1α and SOD2 via the Sirt3/FoxO3a pathway in TCMK-1 cells. Biochem Biophys Res Commun. (2017) 486:198–204. doi: 10.1016/j.bbrc.2017.03.027
16. Kovács-Bogdán E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, et al. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci USA. (2014) 111:8985–90. doi: 10.1073/pnas.1400514111
17. Nguyen NX, Armache JP, Lee C, Yang Y, Zeng W, Mootha VK, et al. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature. (2018) 559:570–4. doi: 10.1038/s41586-018-0333-6
18. Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. Embo j. (2013) 32:2362–76. doi: 10.1038/emboj.2013.157
19. Lee Y, Min CK, Kim TG, Song HK, Lim Y, Kim D, et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. (2015) 16:1318–33. doi: 10.15252/embr.201540436
20. Yoo J, Wu M, Yin Y, Herzik MA, Lander GC, Lee SY. Cryo-EM structure of a mitochondrial calcium uniporter. Science. (2018) 361:506–11.
21. Lee SK, Shanmughapriya S, Mok MC, Dong Z, Tomar D, Carvalho E, et al. Structural insights into mitochondrial calcium uniporter regulation by divalent cations. Cell Chem Biol. (2016) 23:1157–1169. doi: 10.1016/j.chembiol.2016.07.012
22. Fan C, Fan M, Orlando BJ, Fastman NM, Zhang J, Xu Y, et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature. (2018) 559:575–9. doi: 10.1038/s41586-018-0330-9
23. Bick AG, Calvo SE, Mootha VK. Evolutionary diversity of the mitochondrial calcium uniporter. Science. (2012) 336:886. doi: 10.1126/science.1214977
24. Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. (2012) 151:630–44. doi: 10.1016/j.cell.2012.10.011
25. Liu JC, Liu J, Holmström KM, Menazza S, Parks RJ, Fergusson MM, et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. (2016) 16:1561–73. doi: 10.1016/j.celrep.2016.07.011
26. Wettmarshausen J, Goh V, Huang KT, Arduino DM, Tripathi U, Leimpek A, et al. MICU1 confers protection from MCU-dependent manganese toxicity. Cell Rep. (2018) 25:1425–35.e7. doi: 10.1016/j.celrep.2018.10.037
27. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE. (2013) 8:e55785. doi: 10.1371/journal.pone.0055785
28. Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. (2014) 15:299–307. doi: 10.1002/embr.201337946
29. Murgia M, Rizzuto R. Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell Calcium. (2015) 58:11–7. doi: 10.1016/j.ceca.2014.11.001
30. Reane DV, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, et al. A MICU1 splice variant confers high sensitivity to the mitochondrial Ca(2+) uptake machinery of skeletal muscle. Mol Cell. (2016) 64:760–73. doi: 10.1016/j.molcel.2016.10.001
31. Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, et al. Tissue-specific mitochondrial decoding of cytoplasmic Ca(2+) signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. (2017) 18:2291–300. doi: 10.1016/j.celrep.2017.02.032
32. Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, et al. MCUR1 Is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. (2016) 15:1673–85. doi: 10.1016/j.celrep.2016.04.050
33. Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. (2015) 21:109–16. doi: 10.1016/j.cmet.2014.12.004
34. Vais H, Mallilankaraman K, Mak DO, Hoff H, Payne R, Tanis JE, et al. EMRE is a matrix Ca(2+) sensor that governs gatekeeping of the mitochondrial Ca(2+) uniporter. Cell Rep. (2016) 14:403–10. doi: 10.1016/j.celrep.2015.12.054
35. Tsai MF, Phillips CB, Ranaghan M, Tsai CW, Wu Y, Williams C, et al. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife. (2016) 5:e15545. doi: 10.7554/eLife.15545
36. Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, et al. MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation. (2019) 140:1720–33. doi: 10.1161/CIRCULATIONAHA.118.037968
37. Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal. (2015) 8:ra23. doi: 10.1126/scisignal.2005673
38. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. (2008) 9:102–14. doi: 10.1038/nrg2290
39. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. (2013) 23:58–63. doi: 10.1016/j.cub.2012.11.026
40. Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, et al. MicroRNA-138 and MicroRNA-25 down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am J Respir Crit Care Med. (2017) 195:515–29. doi: 10.1164/rccm.201604-0814OC
41. Theillet FX, Smet-Nocca C, Liokatis S, Thongwichian R, Kosten J, Yoon MK, et al. Cell signaling, post-translational protein modifications and NMR spectroscopy. J Biomol NMR. (2012) 54:217–36. doi: 10.1007/s10858-012-9674-x
42. Vasington FD, Murphy JV. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem. (1962) 237:2670–7. doi: 10.1016/S0021-9258(19)73805-8
43. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. (2009) 1787:1309–16. doi: 10.1016/j.bbabio.2009.01.005
44. Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. (2013) 52:2793–809. doi: 10.1021/bi3015983
45. Neretti N, Wang PY, Brodsky AS, Nyguyen HH, White KP, Rogina B, et al. Long-lived Indy induces reduced mitochondrial reactive oxygen species production and oxidative damage. Proc Natl Acad Sci USA. (2009) 106:2277–82. doi: 10.1073/pnas.0812484106
46. Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. (2002) 99:1259–63. doi: 10.1073/pnas.241655498
47. Solien J, Haynes V, Giulivi C. Differential requirements of calcium for oxoglutarate dehydrogenase and mitochondrial nitric-oxide synthase under hypoxia: impact on the regulation of mitochondrial oxygen consumption. Comp Biochem Physiol A Mol Integr Physiol. (2005) 142:111–7. doi: 10.1016/j.cbpb.2005.05.004
48. Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. (2003) 304:463–70. doi: 10.1016/S0006-291X(03)00618-1
49. Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. (1976) 251:5069–77. doi: 10.1016/S0021-9258(17)33220-9
50. Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. (2009) 46:821–31. doi: 10.1016/j.yjmcc.2009.02.021
51. Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. (1985) 237:408–14. doi: 10.1016/0003-9861(85)90293-0
52. Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol. (1990) 52:451–66. doi: 10.1146/annurev.ph.52.030190.002315
53. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. (2011) 11:85–97. doi: 10.1038/nri2921
54. Zemel MB. Insulin resistance vs. hyperinsulinemia in hypertension: insulin regulation of Ca2+ transport and Ca(2+)-regulation of insulin sensitivity. J Nutr. (1995) 125:1738s−43s. doi: 10.1093/jn/125.suppl_6.1738S
55. Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, et al. Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta. (2009) 1787:1342–51. doi: 10.1016/j.bbabio.2009.03.015
56. Gao P, Jiang Y, Wu H, Sun F, Li Y, He H, et al. Inhibition of mitochondrial calcium overload by SIRT3 prevents obesity- or age-related whitening of brown adipose tissue. Diabetes. (2020) 69:165–80. doi: 10.2337/db19-0526
57. Bhoumik A, Takahashi S, Breitweiser W, Shiloh Y, Jones N, Ronai ZE. ATM–dependent phosphorylation of ATF2 is required for the DNA damage response. Mol Cell. (2005) 18:577–87. doi: 10.1016/j.molcel.2005.04.015
58. Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, et al. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. (2005) 280:19587–93. doi: 10.1074/jbc.M408862200
59. Fuchs SY, Tappin I, Ronai Z. Stability of the ATF2 transcription factor is regulated by phosphorylation and dephosphorylation. J Biol Chem. (2000) 275:12560–4. doi: 10.1074/jbc.275.17.12560
60. Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. (2009) 1787:1395–401. doi: 10.1016/j.bbabio.2009.06.009
61. Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, et al. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. (2002) 4:769–81. doi: 10.1089/152308602760598918
62. Yang J, Guo W, Wang J, Yang X, Zhang Z, Zhao Z. T-2 toxin-induced oxidative stress leads to imbalance of mitochondrial fission and fusion to activate cellular apoptosis in the human liver 7702 cell line. Toxins. (2020) 12:43. doi: 10.3390/toxins12010043
63. Tangmansakulchai K, Abubakar Z, Kitiyanant N, Suwanjang W, Leepiyasakulchai C, Govitrapong P, et al. Calpastatin overexpression reduces oxidative stress-induced mitochondrial impairment and cell death in human neuroblastoma SH-SY5Y cells by decreasing calpain and calcineurin activation, induction of mitochondrial fission and destruction of mitochondrial fusion. Mitochondrion. (2016) 30:151–61. doi: 10.1016/j.mito.2016.07.009
64. Guan L, Che Z, Meng X, Yu Y, Li M, Yu Z, et al. MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy Inhibition. J Cell Mol Med. (2019) 23:7830–7843. doi: 10.1111/jcmm.14662
65. Yang HL, Thiyagarajan V, Shen PC, Mathew DC, Lin KY, Liao JW, et al. Anti-EMT properties of CoQ0 attributed to PI3K/AKT/NFKB/MMP-9 signaling pathway through ROS-mediated apoptosis. J Exp Clin Cancer Res. (2019) 38:186. doi: 10.1186/s13046-019-1196-x
66. Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. (2016) 12:999–1014. doi: 10.1080/15548627.2016.1166318
67. Wang C, Shao L, Pan C, Ye J, Ding Z, Wu J, et al. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial-mesenchymal transition. Stem Cell Res Ther. (2019) 10:175. doi: 10.1186/s13287-019-1265-2
68. Liu Y, Jin M, Wang Y, Zhu J, Tan R, Zhao J, et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct Target Ther. (2020) 5:59. doi: 10.1038/s41392-020-0155-5
69. Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, et al. MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene. (2017) 36:5897–909. doi: 10.1038/onc.2017.167
70. Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol Med. (2016) 8:569–85. doi: 10.15252/emmm.201606255
71. Li C, Ma Q, Toan S, Wang J, Zhou H, Liang J. Liang, SERCA overexpression reduces reperfusion-mediated cardiac microvascular damage through inhibition of the calcium/MCU/mPTP/necroptosis signaling pathways. Redox Biol. (2020) 36:101659. doi: 10.1016/j.redox.2020.101659
72. Wu L, Tan JL, Chen ZY, Huang G. Cardioprotection of post-ischemic moderate ROS against ischemia/reperfusion via STAT3-induced the inhibition of MCU opening. Basic Res Cardiol. (2019) 114:39. doi: 10.1007/s00395-019-0747-9
73. Granatiero V, Pacifici M, Raffaello A, De Stefani D, Rizzuto R. Overexpression of mitochondrial calcium uniporter causes neuronal death. Oxid Med Cell Longev. (2019) 2019:1681254. doi: 10.1155/2019/1681254
74. Zhang K, Yan J, Wang L, Tian X, Zhang T, Guo L, et al. The Pyk2/MCU pathway in the rat middle cerebral artery occlusion model of ischemic stroke. Neurosci Res. (2018) 131:52–62. doi: 10.1016/j.neures.2017.09.002
75. Patron M, Sprenger HG, Langer T. Langer, m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res. (2018) 28:296–306. doi: 10.1038/cr.2018.17
76. Soman SK, Bazała M, Keatinge M, Bandmann O, Kuznicki J. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson's disease. Biol Open. (2019) 8:bio044347. doi: 10.1242/bio.044347
77. Gerdes F, Tatsuta T, Langer T. Mitochondrial AAA proteases—towards a molecular understanding of membrane-bound proteolytic machines. Biochim Biophys Acta. (2012) 1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015
78. Soman S, Keatinge M, Moein M, Da Costa M, Mortiboys H, Skupin A, et al. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1(-/-) zebrafish. Eur J Neurosci. (2017) 45:528–35. doi: 10.1111/ejn.13473
79. Sharma Y, Garabadu D. Ruthenium red, mitochondrial calcium uniporter inhibitor, attenuates cognitive deficits in STZ-ICV challenged experimental animals. Brain Res Bull. (2020) 164:121–35. doi: 10.1016/j.brainresbull.2020.08.020
80. Yu L, Gong B, Duan W, Fan C, Zhang J, Li Z, et al. Wang, Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK-PGC-1α-SIRT3 signaling. Sci Rep. (2017) 7:41337. doi: 10.1038/srep41337
81. Cardoso SM, Correia SC, Carvalho C, Moreira PI. Mitochondria in Alzheimer's Disease and Diabetes-Associated Neurodegeneration: License to Heal!. Handb Exp Pharmacol. (2017) 240:281–308. doi: 10.1007/164_2017_3
82. Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. (2005) 280:13560–7. doi: 10.1074/jbc.M414670200
83. Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, et al. Gupta, SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. (2014) 34:807–19. doi: 10.1128/MCB.01483-13
84. Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, et al. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE. (2010) 5:e11707. doi: 10.1371/journal.pone.0011707
85. Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. (2011) 44:177–90. doi: 10.1016/j.molcel.2011.07.019
86. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. (2010) 2:914–23. doi: 10.18632/aging.100252
87. Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. (2007) 104:12017–22. doi: 10.1073/pnas.0705070104
88. Palacios OM, Carmona JJ, Michan S, Chen KY, Manabe Y, Ward III JL, et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging. (2009) 1:771–83. doi: 10.18632/aging.100075
89. Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, et al. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy. (2013) 9:1677–86. doi: 10.4161/auto.24795
90. Ikeda K, Maretich P, Kajimura S. The common and distinct features of brown and beige adipocytes. Trends Endocrinol Metab. (2018) 29:191–200. doi: 10.1016/j.tem.2018.01.001
91. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. (2012) 13:566–78. doi: 10.1038/nrm3412
92. Park S, Scheffler TL, Gerrard DE. Chronic high cytosolic calcium decreases AICAR-induced AMPK activity via calcium/calmodulin activated protein kinase II signaling cascade. Cell Calcium. (2011) 50:73–83. doi: 10.1016/j.ceca.2011.05.009
Keywords: mitochondrial calcium uniporter, mitochondrial reactive oxygen species, metabolism, AMPK/PGC-1α/SIRT3, cell death
Citation: Wang Y, Li X and Zhao F (2021) MCU-Dependent mROS Generation Regulates Cell Metabolism and Cell Death Modulated by the AMPK/PGC-1α/SIRT3 Signaling Pathway. Front. Med. 8:674986. doi: 10.3389/fmed.2021.674986
Received: 02 March 2021; Accepted: 14 June 2021;
Published: 09 July 2021.
Edited by:
Federica Saponaro, University of Pisa, ItalyReviewed by:
Serenella Anzilotti, Institute of Research and Medical Care (IRCCS) SDN, ItalyMichela Rossi, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2021 Wang, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengchao Zhao, emhhb2ZlbmdjaGFvQHpqdS5lZHUuY24=
†These authors have contributed equally to this work