 Jerome Razanamahery1*
Jerome Razanamahery1* Sebastien Humbert2Jean-Francois Emile3Fleur Cohen-Aubart4Jean Fontan5Philippe Maksud6Sylvain Audia1Julien Haroche4
Sebastien Humbert2Jean-Francois Emile3Fleur Cohen-Aubart4Jean Fontan5Philippe Maksud6Sylvain Audia1Julien Haroche4- 1Internal Medicine Department and Clinical Immunology, Dijon University Hospital, Dijon, France
- 2Internal Medicine Department, Besancon University Hospital, Besançon, France
- 3Department of Pathology, Ambroise-Paré Hospital, Assistance-Publique Hopitaux de Paris, Paris, France
- 4Sorbonne Université, Assistance Publique Hôpitaux de Paris, Pitié-Salpêtrière Hospital, Internal Medicine Department 2, National Reference Center for Histiocytosis, Paris, France
- 5Department of Haematology, Besancon University Hospital, Besançon, France
- 6Department of Nuclear Medicine, Pitié-Salpêtrière Hospital, Paris, France
Immune thrombocytopenia (ITP) is a rare autoimmune-mediated condition characterized by isolated thrombocytopenia (<100 G/L) after exclusion of other causes. Mostly primary, it is associated with hematological malignancy, autoimmune disorders, or infection in 20% of patients. It is exceptionally described in patients with histiocytosis, mostly in children (seven patients in literature). We report a case of a 69-year-old man with ITP leading to the diagnosis of histiocytosis. At ITP's diagnosis, the patient had elevated gamma-globulins leading to computed tomography showing bilateral peri-renal infiltration. The biopsy showed enriched IgG-4 peri-renal Rosai Dorfman disease with MAP2K1 mutation, although peri-renal infiltration is highly suggestive of Erdheim-Chester disease. This overlapping association was described in men with mutation in MAP2K1 gene. Macrophages are implicated in the pathophysiology of ITP in multiple ways, notably by the phagocytosis of opsonized platelets and their function of antigen-presenting cells able to stimulate autoreactive T cells. Histiocytic cells derivate from monocyte-macrophage lineage. Activation of macrophages in active histiocytosis is responsible for consequential platelet destruction in ITP associated histiocytosis. Finally, this case highlights a rare presentation of ITP revealing histiocytosis, both being efficiently treated with rituximab.
Introduction
Immune thrombocytopenia (ITP) is a rare autoimmune disease characterized by isolated thrombocytopenia (<100 G/L) after exclusion of other causes (1). Although mostly primary, ITP is associated with chronic infections, connective tissue diseases, or hematological malignancies in 20% of patients (1). The association between histiocytosis and ITP is rare. It has been reported mostly in children (2–4); and in most cases, the diagnosis of both pathologies onset occurred several years apart.
Rosai-Dorfman disease (RDD) is a non-Langerhans cell histiocytosis characterized by histology demonstrating enlarged CD68+, CD1a−, and S100+ histiocytes with abundant lesions of emperipolesis (5). RDD locations are heterogeneous, but peri-nephric involvement is exceptional and usually consistent with Erdheim-Chester Disease (ECD). ECD is characterized by an iconic phenotype with long bone involvement, peri-nephric fat infiltration, and arteries' vascular sheathing (6). Histology usually demonstrates CD68+, CD1a−, and S100- histiocytes among fibrosis but is not as specific as in RDD. The first manifestations of both histiocytoses are heterogeneous but severe thrombocytopenia at diagnosis is uncommon.
Here, we report a case of ITP leading to RDD/ECD histiocytosis diagnosis successfully treated with Rituximab.
Case Presentation
A 69-year-old man was referred for spontaneous epistaxis related to severe thrombocytopenia (7 G/L). He had a medical history of pancreatitis and took no medication. Physical examination showed isolated purpuric lesions of the lower limbs. Laboratory tests showed hemoglobin at 10.2 g/dL, white count cells at 5.36 G/L, platelets at 7 G/L without schistocytes on the blood smear. Coagulation tests were regular. Renal and liver functions were normal. Protein electrophoresis showed the protein level at 97 g/L (N: 60–80), albumin at 28.4 g/L (N > 40) and gamma globulins at 49.2 g/L (N: 8–13.5) without monoclonal gammopathy. Screening for infectious diseases (HIV/HBV/HCV) was negative. Immunological tests showed isolated antinuclear antibodies at 1/160 (N: <1/80) without antiphospholipid antibodies. Bone marrow aspiration showed no evidence for a myelodysplastic disorder nor for B9/B12 vitamin deficiency. The diagnosis of ITP was retained, and the patient received oral prednisone, 1 mg/kg/day for 4 weeks, resulting in clinical and biological response.
After the initial steroid course, the patient had a whole-body CT scan due to elevated gamma globulin level at ITP diagnosis. CT scan showed bilateral peri-nephric fat infiltration without other abnormalities (Figure 1A). At the biopsy time (3 months later), platelet count was 176 G/L, protein level at 86 g/L, albumin at 41 g/L, and gamma globulins at 23.1 g/L. The perinephric tissue biopsy showed infiltration with histiocytes, lymphocytes, and plasma cells rich in IgG-4 (130/high-power fields, IgG-4/IgG ratio: 25%; Figures 1B,B′). Histiocytes were CD68+, S100+, and CD1a-, with large nuclei and abundant lesions of emperipolesis (Figures 1C–E). Next-Generation Sequencing (NGS) on biopsy sample showed a c.395C>T variant on the MAP2K1 gene. Although the histology was characteristic of enriched IgG-4 RDD, the bilateral peri-nephric fat infiltration was more consistent with ECD, but the bone scintigraphy was normal, and heart and brain MRIs showed no histiocytic locations. Because histiocytosis was non-symptomatic, no specific treatment was started. At 1-year follow-up, the patient experienced an ITP relapse with purpuric lesions of the lower limbs and a platelet count at 30 G/L. Protein electrophoresis showed the protein level at 114 g/L, albumin at 22 g/L, and gamma globulins at 62.6 g/L, with IgG1 level at 54.7 g/L (N: 3.8–9.3) and IgG-4 at 11.36 g/L (N: 0.039–0.864) without monoclonal gammopathy. Immunological tests showed isolated antinuclear antibodies at 1/320 and positive anticardiolipin IgG without clinical signs suggestive of lupus. Bone marrow aspiration was consistent with peripheral thrombocytopenia, although NGS detected somatic variants in several genes (TET2, ASXL1, DNMT3A, JAK2). 18F-fluorodeoxyglucose PET-CT showed intense radiotracer uptake in peri-nephric fat fibrosis, mediastinal lymph nodes, and a low tracer uptake on the testis (Figure 1F). The patient received a first infusion of rituximab 1,000 mg for chronic ITP. Ten days later, he experienced a new flare with epistaxis and a platelet count at 15 G/L without response to intravenous immunoglobulins (IVIg, 1 g/kg/day, day 1 and 2) and prednisone (1 mg/kg/day). Salvage therapy with dexamethasone 40 mg for 4 days and vinblastine 10 mg allowed a biological response. A second pulse of rituximab 1,000 mg was performed 15 days after. After a two-year follow-up, the patient is still on prednisone (5 mg/day) and considered in clinical, biological (platelet count at 286 G/L, IgG1 at 5.32 g/L, and IgG-4 at 0.033 g/L) and metabolic (Figure 1F) remission.
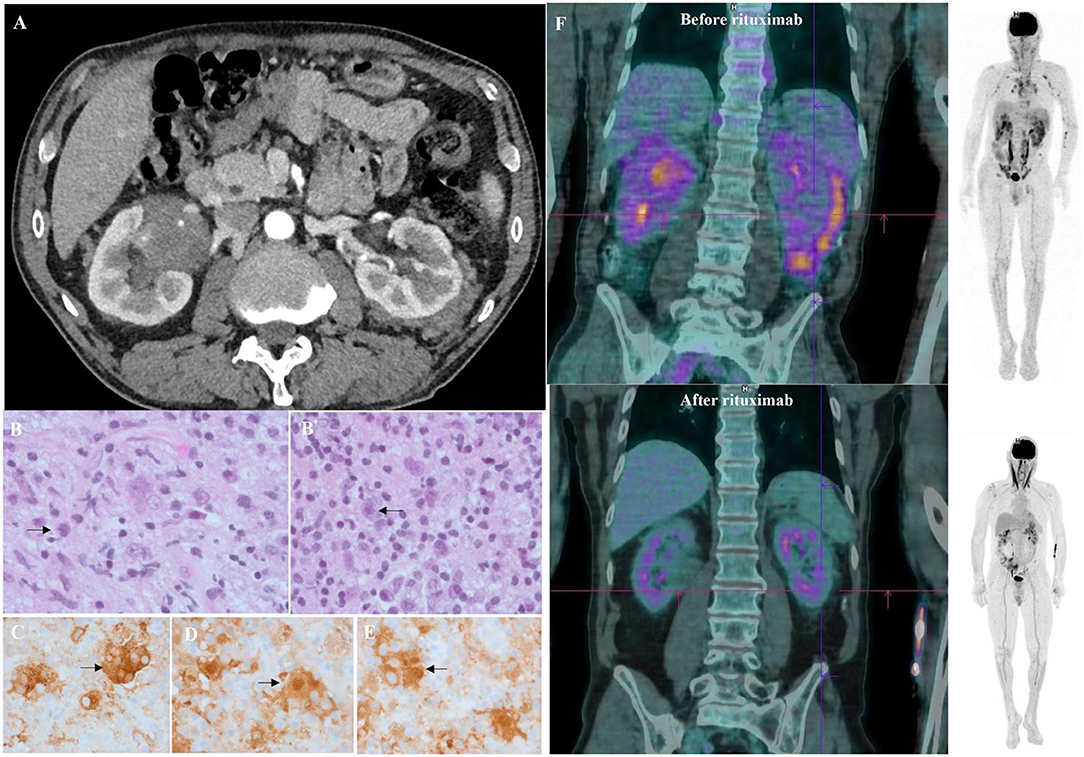
Figure 1. Clinical, radiological, and histological features of mixed Erdheim Chester Disease (ECD)/Rosai Dorfman Disease (RDD) histiocytosis. (A) Axial computed tomography (CT) of the patient demonstrating infiltration of peri-nephric fat defined as “hairy-kidney,” highly suggestive of ECD after 4 weeks of corticosteroids for ITP. (B) Tissue guided biopsy of perinephric lesions consistent with Rosai-Dorfman disease histology as shown by multinucleated histiocytes with large nuclei, abundant cytoplasm and lesions of emperipolesis (arrows). Haematoxylin and eosin staining (HES), original magnification ×400. Biopsy was performed 3 months after ITP diagnosis. (B′) Same samples showing perinephric lesions consistent with Rosai-Dorfman disease histology as shown by multinucleated histiocytes with large nuclei, abundant cytoplasm, and lesions of emperipolesis (arrows). Haematoxylin and eosin staining (HES), original magnification ×100. Biopsy was performed 3 months after ITP diagnosis. (C) Sample samples showing tissue infiltration with multinucleated histiocytes with CD163 expression on immunostaining (HES; immunohistochemistry, ×400) consistent with Rosai-Dorfman disease. (D) Same sample showing a strong expression of S100 protein (brown staining) by the multinucleated histiocytes (HES; immunochemistry, ×400) consistent with Rosai-Dorfman disease. (E) Same sample showing a strong expression of S100 protein (brown staining) by the multinucleated histiocytes (HES; immunochemistry, ×400) consistent with Rosai-Dorfman disease. (F) Sagittal 18FDG-PET CT-scan showing radiotracer uptake before (upper image) and after (lower image) rituximab infusions. An intense radiotracer uptake on “hairy-kidney” and a low tracer uptake on the testis was observed before treatment.
Discussion
We report the first presentation of ITP leading to the diagnosis of histiocytosis. RDD location in peri-nephric space is rare (7), usually described as a solitary nodule or hilar mass or hypodense subcapsular infiltration. Bilateral peri-nephric fat infiltration is highly suggestive of ECD, raising the hypothesis of an overlapping histiocytosis, which was already described in men with testicular involvement and gain of function variant of MAP2K1 gene (8). In this cohort, no patient had elevated IgG-4 plasmocytes on tissue biopsy or profound thrombocytopenia suggestive for ITP. The occurrence of somatic variants in bone marrow consecutive to clonal haematopoiesis of indeterminate potential (CHIP) is frequently reported in ECD patients (9), reinforcing the hypothesis of an “overlapping histiocytosis.”
Immunological conditions (i.e., autoimmune haemolytic anemia, IgG-4-related disease, systemic lupus erythematosus) have been reported in about 10% of patients with RDD (5, 10), even more frequently in a cohort of ECD patients (11). ITP has rarely been reported in patients with histiocytosis. In most cases, diagnosis of ITP and histiocytosis onset occurred several years apart (Table 1).
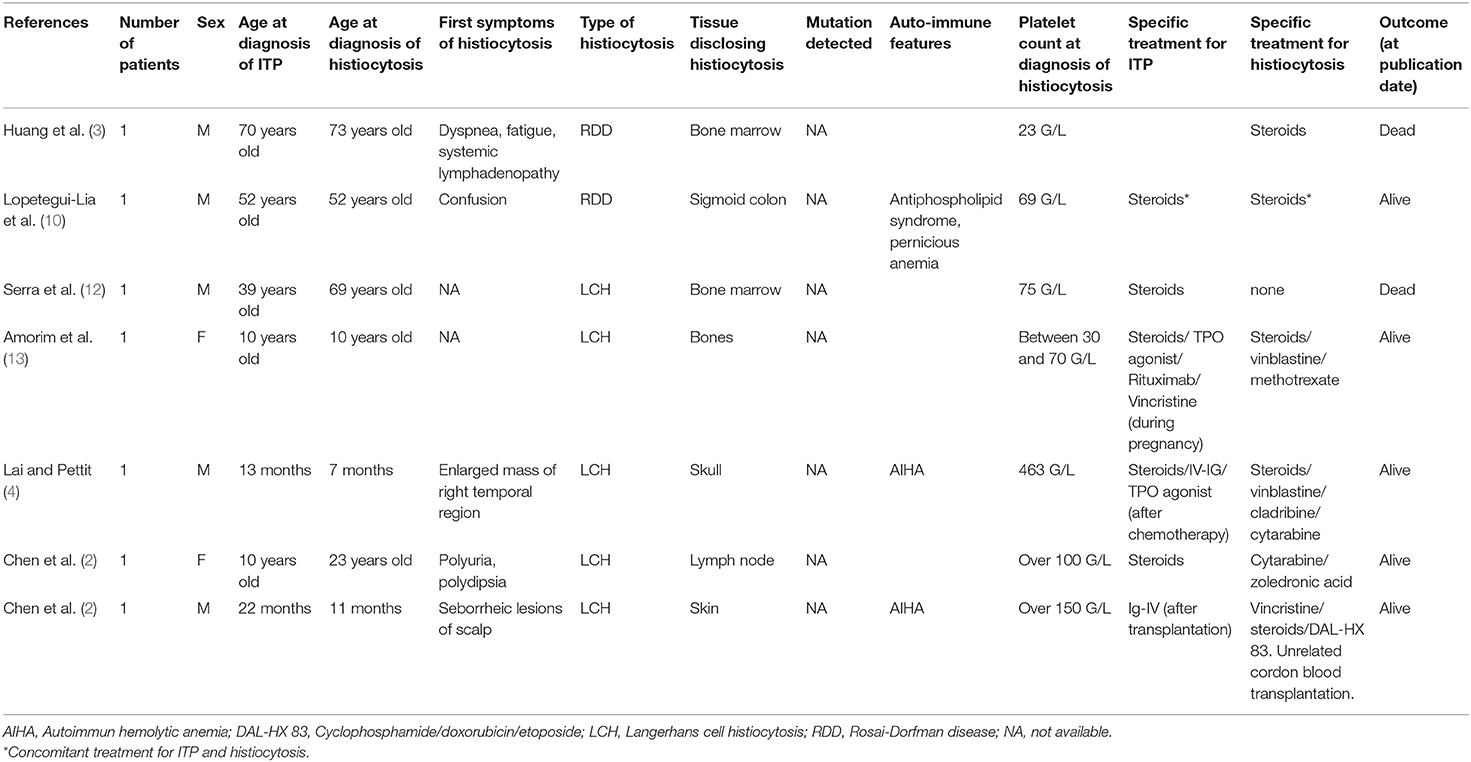
Table 1. Characteristics of previously reported patients with ITP and histiocytosis.
The pathophysiology of ITP is complex, macrophages playing a crucial role by phagocyting opsonized platelets in the spleen (14). Macrophages also participate in the stimulation of autoreactive T cells, among which T-follicular helper cells are essential for the stimulation and differentiation of autoreactive B-cell lymphocytes that produce antiplatelet antibodies (15).
A recent study highlighted an increased expression of M2-macrophage markers (CD163, CX3CR1) in the peripheral blood of ITP patients suggesting their potential immunomodulatory role in ITP pathogenesis (16).
Regarding histiocytosis ontogeny, cells originate from the fetal liver (17), the yolk sac (18), but mainly from the bone marrow (19, 20). Cells deriving from common bone marrow myeloid precursors give rise to blood monocytes who infiltrate several tissues and differentiate into dendritic cells and macrophages. Those monocytes can virtually infiltrate any tissues. Once in tissues, macrophages lose their migration ability (21) and become less sensitive to cell death signals (22). Those modifications are mandatory for tissue macrophage-homing and transformation into histiocytes (23).
In active histiocytosis, tissue macrophages from the same origins can be simultaneously activated and cause synchronous damages. Correlation between ITP and histiocytosis activity might be explained by macrophages activation into tissues resulting in platelet destruction.
The hypermetabolism of histiocytosis locations on PET-CT scan during ITP flare might support this postulate.
Regarding treatments, steroids associated or not with IVIg depending on the haemorrhagic score, are the first-line therapy in newly diagnosed ITP. Second-line therapies such as thrombopoietin receptor agonists, rituximab are used in persistent ITP (>3 months) while splenectomy is dedicated to chronic ITP (>12 months). Salvage therapy with vinca-alkaloids has shown efficacy in refractory and life-threatening situations (1). In our case, rituximab was favored for persistent ITP because of its established efficiency in ITP (24), RDD (25), and IgG-4-related disease (26). Because of a relapse of ITP that was refractory to steroids and IVIg, vinblastine was proposed to rapidly increase platelet count before the response to rituximab was obtained. Finally, remission of both ITP and IgG-4-related features associated with RDD occurred 6 weeks after the last rituximab infusion. The complete therapeutic sequence has been reported in Figure 2.
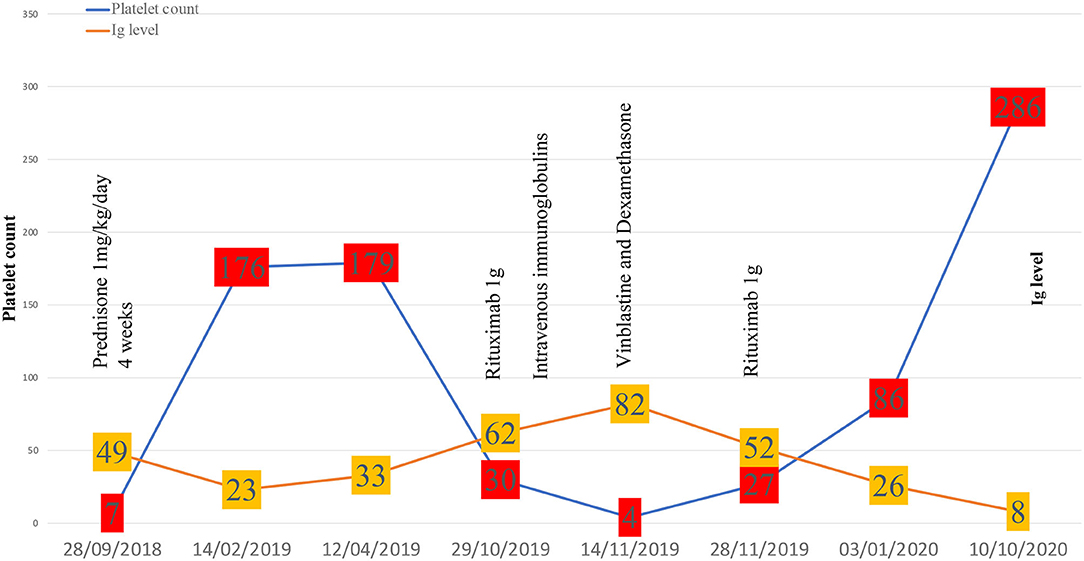
Figure 2. Evolution of immune thrombocytopenia.
In both pathologies, the long-term objective is to maintain remission out of treatment. Low-dose steroids are recommended for “auto-immune features” related to RDD (5, 25), but there is no consensus regarding maintenance therapy duration after remission. After 1 year of remission, steroids could be interrupted if the patient fulfills clinical, biological, and metabolic remission criteria. Surveillance with platelet count, Ig-G4 ratio at least every 6 months, and 18FDG-PET every year are useful to assess disease activity. In case of relapse of ITP, a new infusion of rituximab could be of interest if circulating B cells are detectable, as previously shown (27).
If not, treatment with azathioprine can be an interesting option for both ITP and RDD with efficacy reported in both pathologies (28, 29). Salvage therapy with dexamethasone and vinblastine could be a good option in severe bleeding. As MEK-inhibitors have shown a dramatic efficacy in histiocytic disorder (30, 31), Cobimetinib would be the best option for RDD life-threatening involvement. Interestingly, targeted therapies (BRAF and MEK-inhibitors) does not worsen auto-immunity in histiocytosis (11). Furthermore, severe thrombocytopenia has rarely been reported as a frequently associated side effect.
Although this association is not usual, ITP is more likely to be associated with histiocytosis rather than being primary, both being efficiently treated with rituximab.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JR collected the data and wrote the initial draft. J-FE confirmed histological features of RDD and determined the mutational status. All authors provided clinical, radiographic, histological data of the patient, participated in editing, and approved the final version of the manuscript.
Conflict of Interest
FC-A and JH are investigators (FC-A being the PI) of an academic study on the efficacy of cobimetinib for treating histiocytoses.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lambert MP, Gernsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood. (2017) 129:2829–35. doi: 10.1182/blood-2017-03-754119
2. Chen R-L, Wu P-L, Hsu Y-H, Kuo P-L. Evans syndrome after unrelated cord blood transplantation for disseminated Langerhans cell histiocytosis in a child. J Pediatr Hematol Oncol. (2007) 29:348–50. doi: 10.1097/MPH.0b013e3180556467
3. Huang Q, Chang KL, Weiss LM. Extranodal Rosai-Dorfman disease involving the bone marrow: a case report. Am J Surg Pathol. (2006) 30:1189–92. doi: 10.1097/01.pas.0000209846.52046.62
4. Lai CY, Pettit T. Immune thrombocytopenia in a child with refractory langerhans cell histiocytosis following cladribine containing therapy. J Pediatr Hematol Oncol. (2019) 41:620–3. doi: 10.1097/MPH.0000000000001243
5. Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile J-F, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. (2018) 131:2877–90. doi: 10.1182/blood-2018-03-839753
6. Haroche J, Cohen Aubart F, Amoura Z. Erdheim-Chester disease. Blood. (2020) doi: 10.1182/blood.2019002766
7. Brown WE, Coakley FV, Heaney M. Renal involvement by Rosai-Dorfman disease: CT findings. Abdom Imaging. (2002) 27:214–6. doi: 10.1007/s00261-001-0061-4
8. Razanamahery J, Diamond EL, Cohen-Aubart F, Plate K-H, Charlotte F, Hélias-Rodzewicz Z, et al. Erdheim-Chester disease with concomitant Rosai-Dorfman like lesions: a distinct entity mainly driven by MAP2K1. Haematologica. (2020) 105:e5–8. doi: 10.3324/haematol.2019.216937
9. Cohen Aubart F, Roos-Weil D, Armand M, Marceau-Renaut A, Emile J-F, Duployez N, et al. High frequency of clonal hematopoiesis in Erdheim-Chester disease. Blood. (2020) 137:485–92. doi: 10.1182/blood.2020005101
10. Lopetegui-Lia N, Asad SD, Jafri SI, Harrison JS. Autoimmune diseases and Rosai-Dorfman disease coexist more commonly than expected: two case reports. Am J Case Rep. (2019) 20:770–2. doi: 10.12659/AJCR.915627
11. Roeser A, Cohen-Aubart F, Breillat P, Myara M, Emile JF, Charlotte F, et al. Autoimmunity associated with Erdheim-Chester disease improves with BRAF/MEK inhibitors. Haematologica. (2019) 104:e502–5. doi: 10.3324/haematol.2018.214007
12. Serra JT, Silva R, Aldomiro F, Duarte FP. Bone marrow langerhans cell histiocytosis in association with Kasabach-Merritt syndrome: The difficulty of a differential diagnosis. Case Rep Oncol. (2020) 13:408–13. doi: 10.1159/000506539
13. Amorim de JG, Abecasis MR, Rodrigues FMNL. Refractory severe thrombocytopenia during pregnancy: how to manage. Rev Bras Ginecol Obstet. (2018) 40:803–7. doi: 10.1055/s-0038-1675186
14. Norris PAA, Segel GB, Burack WR, Sachs U, Lissenberg-Thunnissen S, Vidarsson G, et al. FcγRI and FcγRIII on splenic macrophages mediate phagocytosis of anti-glycoprotein IIb/IIIa autoantibody-opsonized platelets in immune thrombocytopenia. Haematologica. (2020) 106:250–4. doi: 10.3324/haematol.2020.248385
15. Audia S, Rossato M, Santegoets K, Spijkers S, Wichers C, Bekker C, et al. Splenic TFH expansion participates in B-cell differentiation and antiplatelet-antibody production during immune thrombocytopenia. Blood. (2014) 124:2858–66. doi: 10.1182/blood-2014-03-563445
16. Shao X, Wu B, Chen P, Zhan Y, Li F, Hua F, et al. The role of M2 macrophage in primary immune thrombocytopenia. Blood. (2019) 134:2355. doi: 10.1182/blood-2019-129667
17. Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. (2020) 135:1319–31. doi: 10.1182/blood.2019000934
18. Haroche J, Cohen-Aubart F, Emile J-F, Donadieu J, Amoura Z. Neurodegeneration in histiocytoses might start in utero. Lancet Neurol. (2017) 16:953–4. doi: 10.1016/S1474-4422(17)30373-3
19. Durham BH, Roos-Weil D, Baillou C, Cohen-Aubart F, Yoshimi A, Miyara M, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. (2017) 130:176–80. doi: 10.1182/blood-2016-12-757377
20. Milne P, Bigley V, Bacon CM, Néel A, McGovern N, Bomken S, et al. Hematopoietic origin of langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. (2017) 130:167–75. doi: 10.1182/blood-2016-12-757823
21. Berres M-L, Allen CE, Merad M. Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol. (2013) 120:127–61. doi: 10.1016/B978-0-12-417028-5.00005-3
22. Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. (2005) 436:720–4. doi: 10.1038/nature03890
23. Hogstad B, Berres M-L, Chakraborty R, Denoyelle C, Kuilman T, van der Horst C, et al. RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in langerhans cell histiocytosis lesions. J Exp Med. (2018) 215:319–36. doi: 10.1084/jem.20161881
24. Khellaf M, Charles-Nelson A, Fain O, Terriou L, Viallard J-F, Cheze S, et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: results from a prospective registry including 248 patients. Blood. (2014) 124:3228–36. doi: 10.1182/blood-2014-06-582346
25. Goyal G, Ravindran A, Young JR, Shah M, Bennani N, Patnaik M, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. (2019) 105:348–57. doi: 10.3324/haematol.2019.219626
26. Zhang W, Stone JH. Management of IgG4-related disease. Lancet Rheumatol. (2019) 1:e55–65. doi: 10.1016/S2665-9913(19)30017-7
27. Deshayes S, Khellaf M, Zarour A, Layese R, Fain O, Terriou L, et al. Long-term safety and efficacy of rituximab in 248 adults with immune thrombocytopenia: results at 5 years from the French prospective registry ITP-ritux. Am J Hematol. (2019) 94:1314–24. doi: 10.1002/ajh.25632
28. Le Guenno G, Galicier L, Uro-Coste E, Petitcolin V, Rieu V, Ruivard M. Successful treatment with azathioprine of relapsing Rosai-Dorfman disease of the central nervous system. J Neurosurg. (2012) 117:486–9. doi: 10.3171/2012.5.JNS12148
29. Provan D, Arnold DM, Bussel JB, Chong B, Cooper N, Gernsheimer T, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. (2019) 3:3780–817. doi: 10.1182/bloodadvances.2019000812
30. Diamond EL, Durham BH, Ulaner GA, Drill E, Buthorn J, Ki M, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. (2019) 567:521–4. doi: 10.1038/s41586-019-1012-y
Keywords: immune thrombocytopenia, histiocytosis, Rosai Dorfman disease, Erdheim Chester disease, rituximab
Citation: Razanamahery J, Humbert S, Emile J-F, Cohen-Aubart F, Fontan J, Maksud P, Audia S and Haroche J (2021) Immune Thrombocytopenia Revealing Enriched IgG-4 Peri-Renal Rosai-Dorfman Disease Successfully Treated with Rituximab: A Case Report and Literature Review. Front. Med. 8:678456. doi: 10.3389/fmed.2021.678456
Received: 09 March 2021; Accepted: 13 May 2021;
Published: 16 June 2021.
Edited by:
Lalitha Nayak, Case Western Reserve University, United StatesReviewed by:
Roland Schroers, Ruhr University Bochum, GermanyEleonora Timperi, Institut Curie, France
Copyright © 2021 Razanamahery, Humbert, Emile, Cohen-Aubart, Fontan, Maksud, Audia and Haroche. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome Razanamahery, cmF6YW5hbWFoZXJ5Lmplcm9tZUBob3RtYWlsLmZy