 Alessio Cantore
Alessio Cantore Alessandro Fraldi
Alessandro Fraldi Vasco Meneghini
Vasco Meneghini Angela Gritti
Angela Gritti- 1San Raffaele Telethon Institute for Gene Therapy, Istituto di Ricovero e Cura a Carattere Scientifico San Raffaele Scientific Institute, Milan, Italy
- 2School of Medicine, Vita-Salute San Raffaele University, Milan, Italy
- 3CEINGE Biotecnologie Avanzate, Naples, Italy
- 4Department of Translational Medicine, University of Naples “Federico II”, Naples, Italy
In vivo genetic engineering has recently shown remarkable potential as a novel effective treatment for an ever-growing number of diseases, as also witnessed by the recent marketing authorization of several in vivo gene therapy products. In vivo genetic engineering comprises both viral vector-mediated gene transfer and the more recently developed genome/epigenome editing strategies, as long as they are directly administered to patients. Here we first review the most advanced in vivo gene therapies that are commercially available or in clinical development. We then highlight the major challenges to be overcome to fully and broadly exploit in vivo gene therapies as novel medicines, discussing some of the approaches that are being taken to address them, with a focus on the nervous system and liver taken as paradigmatic examples.
Introduction
Gene therapy (GT) has recently gained renewed interest and shown remarkable potential as a novel effective treatment for an ever-growing number of diseases, as also witnessed by the recent marketing authorization of several gene therapy products (1). In vivo genetic engineering, i.e., GT, involves the direct delivery of a GT medicinal product (GTMP) to patients either in situ in anatomically defined locations or systemically to reach organs or tissues such as central and peripheral nervous system (CNS, PNS), liver, muscles, and lungs. Emerging technologies for targeted gene editing are complementing the scope of conventional gene transfer, opening the way to precise gene correction that allows to silence, activate, or rewrite loci of interests in the genome. The GTMP may comprise a virus-derived or non-viral vehicle bearing a transgene expression cassette (gene transfer) or engineered site-specific nucleases or genetic/epigenetic modifiers with or without an exogenous DNA to be introduced into the host cells' genome (gene editing) (2–4). Short interfering RNAs (siRNAs) will not be considered here as GTMPs.
In vivo genetic engineering aims at genetically modifying somatic cells to: (i) treat genetic diseases, by adding functional genes (gene addition) or replacing dysfunctional ones (gene replacement), correcting or disrupting mutated disease-causing genes (gene subtraction) through pre-natal, post-natal or adult intervention; (ii) promote endogenous regeneration by delivering factors for tissue protection/engineering; (iii) tackle cancer by direct/indirect tumor cell elimination, including the use of oncolytic vectors (this will not be discussed here).
The most widely used delivery system for in vivo GT among viral vectors are adeno-associated viral (AAV) vectors (5). Lentiviral vectors (LV) are so far mostly used for ex vivo GT approaches, i.e., genetic engineering of cells in vitro and infusion of the modified cells back to patients, with only few examples related to in vivo delivery at the pre-clinical or early clinical stage (6, 7). Lipid nanoparticles (LNP) or chemical conjugates are used for small RNA delivery (8). Non-viral mediated delivery of genome editing components is generally at an earlier stage of development. The vast majority of current clinical trials rely on gene addition, only a few of them are based on gene editing strategies.
The availability of programmable nucleases, such as zinc-finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN) and, more recently, clustered regularly interspaced short palindromic repeat (CRISPR)–Cas-associated nucleases, has greatly expedited the progress of gene editing from concept to clinical practice (4, 9). Engineering of the Cas9 bacterial adaptive immunity response against phages allowed for the development of methods to generate sequence-specific modifications based on a single-guide RNA complementary to the target genomic sequence. In the last decade, CRISPR/Cas9 systems have been applied to genome and epigenome editing in order to disrupt genes, correct mutations, and silence disease-associated factors in different genetic and sporadic conditions. Genome editing has been predominantly performed ex vivo, however a few examples of in vivo gene editing exist in early-stage clinical trials.
Here we highlight the major hurdles currently limiting the full potential of in vivo genetic engineering (Figure 1) and review some possible solutions, with a focus on CNS and liver taken as paradigmatic examples.
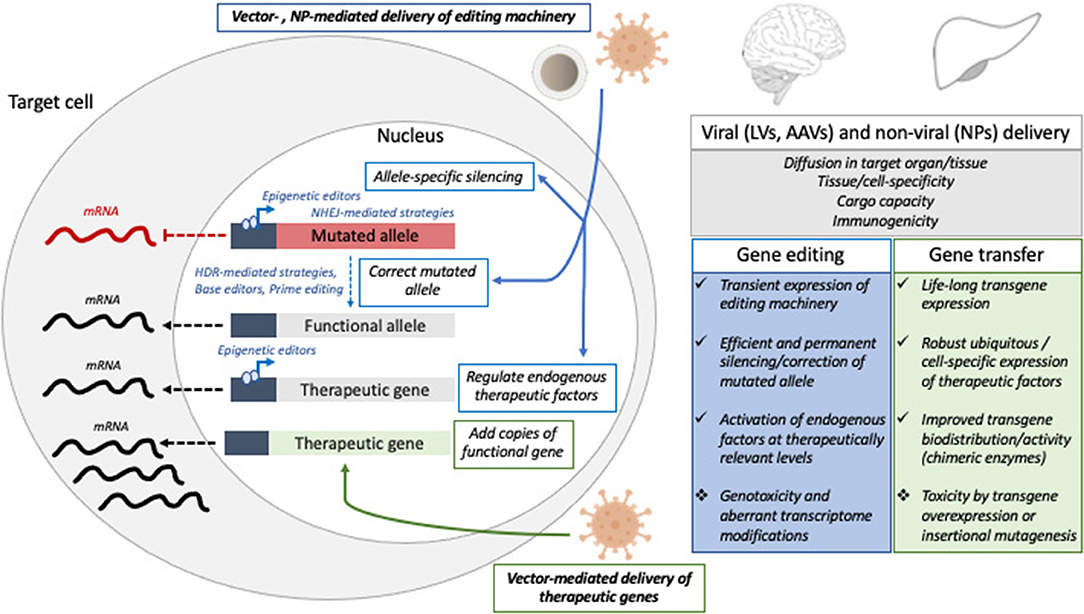
Figure 1. Schematic representation of gene editing and gene transfer approaches tested in pre-clinical and clinical settings to treat liver or CNS disorders, with a list of the major hurdles and challenges that might be addressed to improve the efficacy and safety of in vivo GTMP. NP, nanoparticles; LV, lentiviral vectors; AAV, adeno-associated viral vectors; NHEJ, non-homologous end joining; HDR, homology-directed repair.
Commercial and Clinical Stage Products
In vivo GT to the Nervous System
Currently, there are 3 commercial in vivo GT products and many more in clinical development (Table 1) (10). AAV vector-mediated gene replacement of a functional enzyme of the retinal pigment epithelium, or the regulatory protein survival of motor neuron is at the bases of Luxturna and Zolgensma, indicated for an inherited form of retinal blindness (Leber congenital amaurosis, LCA) or the genetic neurodegenerative disease spinal muscular atrophy, respectively (11, 12). Luxturna is administered in situ in the subretinal space, while Zolgensma is delivered systemically. In both cases, long-lasting therapeutic benefit has been shown, with remarkable recovery of vision and motor functions, respectively. Imlygic is an oncolytic vector indicated for melanoma (13).

Table 1. Commercial in vivo gene therapy products.
Encouraging results have also been shown for Duchenne muscular dystrophy by systemic delivery of AAV vectors expressing short forms of dystrophin in early-stage clinical trials (14). Systemic, intrathecal, and intraparenchymal administration of AAV vectors is under early clinical testing for several neurodegenerative diseases, both genetic early-onset (mucopolysaccharidoses (MPS), globoid leukodystrophy (GLD), Fabry disease, Canavan disease) and non-genetic adult-onset diseases [e.g., Parkinson disease (PD), Alzheimer Disease (AD)] (15). Clinical trials involving LV as delivery systems for in vivo GT are currently limited to PD, which benefits from intrastriatal injections of a LV coding for three genes essential for dopamine synthesis (16).
EDIT-101 is a gene-editing drug to treat LCA10 with Centrosomal Protein 290 (CEP290)-Related Retinal Degeneration (17). The approach is based on AAV-mediated single-dose subretinal delivery of a CRISPR-Cas9 system designed to excise the intronic IVS26 mutation in the photoreceptor CEP290 gene that causes abnormal splicing and termination of translation due to introduced cryptic exon. EDIT-101 recently entered clinical testing and enrolled 18 people with LCA10 (NCT03872479). To date, no study report has been published.
In vivo GT to the Liver
Systemic administration of AAV vectors expressing coagulation factor VIII or IX transgene in hepatocytes is in advanced phase of clinical testing as a treatment for the inherited coagulation disorder hemophilia and showed multi-year reconstitution of therapeutic amounts of the clotting factors, even though a decreasing trend in factor VIII activity has been reported (18–20). A similar strategy is under evaluation for some inherited liver metabolic diseases (such as familial hypercholesterolemia, hyperbilirubinemia, glycogen storage disease type-Ia, ornithine transcarbamilase deficiency) in earlier phase clinical trials (21).
NTLA-2001 is an in vivo gene-editing therapeutic that is designed to treat transtyrhetin (TTR)-related hereditary amyloidosis. Systemic administration of LNP delivering CRISPR/Cas9 RNA to the liver resulted in efficient disruption of TTR gene and subsequent reduction of the toxic misfolded TTR amyloid in 6 affected patients (22).
Despite these successes, several challenges remain to be addressed related to efficacy, safety and immunogenicity of in vivo GTMP, as well as manufacturing, regulatory aspects and sustainability, the latter not being the focus of this Mini Review. Below, we highlight the major challenges and elaborate on possible solutions to address some of them.
General Challenges Related to Efficacy and Safety of in vivo Genetic Engineering
The efficiency of the genetic engineering (viral gene transfer or genome editing), i.e., the actual quantity of genetically modified cells/genomic loci, may be limiting the efficacy of the procedure depending on the desired therapeutic effect. Tissue/cell-type specificity may be desirable or necessary according to different applications, yet hard to achieve. While the tropism of viral vectors can be controlled to a certain extent, it is currently more difficult to obtain specific targeting by non-viral delivery systems (23, 24). On the other hand, cargo capacity may be more limited for viral than non-viral mediated approaches. Despite a plethora of engineered transcriptional and post-transcriptional control elements available, the strength, cell-type specificity, physiological regulation, duration of transgene expression may all be difficult to control and switching expression on and off at will is yet to be achieved in the clinics (25, 26). For genome editing, efficient but transient expression of the editing machinery should be achieved. For non-monogenic diseases, the target genes to manipulate need to be defined. Ensuring the multi-year, ideally life-long durability of the therapeutic genetic modification is crucial in the context of genetic diseases and needs to rely on transgene integration or stability of the genomic edit in proliferating cells and/or in long-lived target cells; alternatively, safe re-administration of the GTMP has to be ensured (27–29).
Concerning the safety of the in vivo genetic engineering, the following risks need to be taken into consideration, carefully evaluated, and reduced to the minimum possible during the research and development phases: the acute responses to the delivery vehicles (30, 31), toxicities due to expression/overexpression of the transgene or other components of the GTMP, possible long-term adverse effects due to genomic insertions of vectors or other components of the GTMP (32), genotoxicity associated with off-target events, large deletion at the on-target loci, chromosomal rearrangements, and aberrant modifications of the transcriptome (33–36). Moreover, the effects of the GTMP on target cells' biology and functionality should be properly determined. Finally, the innate and adaptive immune responses to the delivery vehicle(s), the transgene product(s), including editing machineries of bacterial origin, and other components of the GTMP need to be assessed to avoid detrimental impacts on both the efficacy and safety of the procedure (37, 38).
Modifying the Transgene to Improve the Therapeutic Potential of in vivo GT
One of the main challenges in the clinical translation of in vivo GT is the difficulty in achieving and maintaining therapeutic amounts of the corrective gene in targeted tissues, avoiding the use of high dosage and/or repeated administration of the gene delivery vehicle (that, in most cases, is virus-derived), which is not only potentially toxic but also costly. Intra-vascular administration of GTMPs has been extensively tested in preclinical studies and is being exploited in clinical trials to treat the CNS as alternative approach to direct administration (via either intraparenchymal or intra cerebrospinal fluid injections), which in principle require lower amount of GMTPs but may represent an invasive approach. However, intra-vascular administration of GTMPs showed limited or no effectiveness on CNS pathology due to the impermeability of the blood-brain barrier (BBB) to large molecules (39). Therefore, this delivery route may require high doses of GTMPs, which may strongly reduce its clinical suitability. A possible strategy to overcome all these limitations is enhancing the therapeutic potential of the GTMP by modifying the expression cassette. Here, we give some examples on how this strategy can be applied to the treatment of inherited diseases due to enzymatic deficiency.
A way to modify the transgene expression cassette to enhance its therapeutic potential is adding specific peptides to generate chimeric enzymes with acquired capabilities. Lysosomal storage diseases (LSDs) are inherited metabolic conditions mostly caused by defective lysosomal hydrolases and often showing CNS involvement (40). The addition of heterologous signal peptides to soluble lysosomal enzymes has been showed to increase the secretion efficiency, thus improving enzyme bioavailability and tissue targeting upon in vivo GT in different models of LSDs, including MPS, GLD and Pompe diseases (41–44). In the case of Pompe disease, the liver directed administration of AAV encoding engineered secretable GAA (acid α-glucosidase) transgene in both mouse and non-human primate (NHP) animal models demonstrated improved efficacy associated to a clear dose advantage and reduced toxicity when compared to the native version of the GAA transgene. This approach is currently under clinical testing (NCT04093349). Furthermore, the fusion of the lysosomal hydrolase with specific protein domains capable to bind BBB receptors has been shown to allow active BBB crossing upon liver GT in preclinical LSD models. In these studies, the liver is converted into a factory for the engineered enzyme, which can cross the BBB and target the CNS upon secretion in the bloodstream (41, 45, 46). Interestingly, enzyme replacement therapy approaches based on the delivery of recombinant chimeric lysosomal enzyme fused to different BBB binding domains (BD) are under clinical evaluation for different MPS, thus supporting the potential clinical translation of GT protocols based on the viral mediated delivery of BBB-BD-modified enzymes.
An alternative way to enhance the therapeutic potential of the transgene is to use gain-of-function (GOF) mutants of the enzyme with increased activity and/or stability. Such “hyper functional” enzymes may be employed in in vivo gene transfer (as well as in enzyme replacement approaches) to produce a beneficial effect in targeted tissues at much lower doses and more efficiently compared to the respective WT enzymes. Naturally occurring GOF variants have already been used to treat liver diseases caused by inherited enzymatic defects. AAV vectors encoding a hyper-functional factor IX (FIX-Padua, R338L) has been explored for the treatment of hemophilia B. In dogs and mouse models of disease the use of such variant resulted in beneficial therapeutic effect and, at same time, allowed reducing the AAV vector dosage and, therefore, the risk of cellular immune response to vector capsid, which is one of the main complications of AAV GT for hemophilia B (18, 47, 48). In the case of lipoprotein lipase (LPL) deficiency (LPLD), an orphan disease associated with chylomicronemia, severe hypertriglyceridemia, metabolic complications, the use of AAV vectors encoding a GOF gene variant of LPL (S447X), showed efficacy in LPLD patients avoiding safety concerns related to immune response to AAV-capsid proteins (49). The possibility of generating GOF versions of enzymes “ad hoc” may greatly extend the possibility to apply safe GT protocols for the treatment of other metabolic diseases.
Ensuring Durability of Liver Gene Therapy for Monogenic Diseases
Gene therapy for monogenic diseases promises to be a once-in-a-lifetime treatment that could be delivered at young age and last life-long. The clinical success obtained by AAV vector-based liver GT in adults with hemophilia has raised the expectation to extend enrollment to pediatric patients to maximize the potential benefits for the patients and to broaden the indications to diseases that are more severe or lethal in childhood, such as inherited diseased of liver metabolism. Because AAV vectors do not actively integrate into the host cell genome, they are progressively diluted upon cell division in liver growth, thus challenging their use in pediatric patients. To address this issue, AAV re-dosing, integrating vectors and genome editing and are being explored.
The anti-vector immune response induced after the first administration indeed currently limits the efficiency of a second administration, thus efforts are underway to counteract the anti-AAV immune responses and allow effective re-administrations (50–52). LV integrate into the target cell chromatin and are maintained as cells divide, thus being suited for stable and potentially life-long transgene expression even following a single administration to newborn individuals. Systemic i.v. (intravenous) administration of LV has been shown to allow efficient and long-term gene transfer to the liver and achieve phenotypic correction of hemophilia in mice and dogs (53, 54). Allo-antigen free and phagocytosis-shielded LV have been generated, by high-level surface display of the phagocytosis inhibitor human CD47 (CD47hi) (55, 56). Following i.v. administration to NHP, these CD47hi LV provided amounts of circulating human coagulation factor transgene that would be therapeutic for hemophilia, the disease caused by the deficiency of one of these factors, without evidence of acute toxicity or genotoxicity. These LV are under development for clinical evaluation in hemophilia (57).
Site-specific integration of a corrective DNA in the genome remains an attractive therapeutic strategy for genetic diseases and represents an area of active investigation. The first report of successful in vivo genome editing in the liver in mice by ZFN dates back in 2011 by the K. High group, in collaboration with Sangamo Therapeutics (58, 59), an approach which has been later brought to early clinical testing in the context of Hunter's syndrome (60). The trial has been then closed and the results have not been published yet. In 2015, Barzel et al., reported a nuclease-free genome editing approach in the mouse liver, based on the spontaneous tendency of AAV vectors to integrate on a homology-dependent basis (61). This approach is being brought to early clinical testing in the context of the metabolic disease methyl malonic acidemia (https://investor.logicbio.com/news-releases/news-release-details/logicbio-therapeutics-announces-first-patient-dosed). Instead, Yin et al. reported in 2014 the first report of hepatocyte gene editing mediated by CRISPR/Cas9 for hereditary tyrosinemia type-I in mice (62). More recently, the advent of base editors has opened the possibility to perform single-base substitutions for therapeutic purposes (63). The availability of the highly efficient and transient LNP-based mRNA delivery system recently enabled nuclease-mediated or base-editor mediated genome editing in the liver of NHP and even humans (64, 65). Recently the results of the first clinical trial exploiting genome editing directed to the liver have been reported. These results showed high efficiency of gene disruption and evidence of therapeutic efficacy for the autosomal dominant disease TTR amyloidosis (22). The most advanced genome editing therapies remain so far mostly confined to gene subtraction approaches, however these encouraging results will fuel further progress toward more challenging gene correction approaches. Vector re-administration, integrating gene replacement and editing strategies have all advantages and disadvantages, thus extensive pre-clinical evaluations and risk/benefit assessments need to be conducted on an indication-per-indication basis.
Enhancing the Distribution and Cellular Selectivity of GTMPs to Improve in vivo CNS GT
The route of administration, the vector tropism, and the regulatory elements driving transgene expression are key determinants in defining the efficacy and safety of in vivo GT to treat CNS disorders.
In focal neurodegenerative disorders, intraparenchymal administration in the affected regions is well-tolerated and ensures a local distribution of the GTMP with low vector doses, thus reducing off-target effects in peripheral organs and immunogenicity (15). Convention enhanced delivery has been exploited to further increase the diffusion of the vector in the brain parenchyma by generating a pressure gradient in the infusion catheter leading to expansion of the extracellular space (66). The overall safety of intraparenchymal administration of AAV vectors has been shown in pediatric and adult patients affected by genetic (e.g., Canavan disease, Metachromatic Leukodystrophy, Batten's disease) and non-genetic (e.g., PD, AD) CNS disorders (66, 67). LV are alternative GT vehicles ensuring stable and robust expression of therapeutic transgenes in disease-bearing cells with negligible immune reactivity (68–72). The higher LV cargo capacity can be exploited to deliver multiple genes regulating metabolic processes that are hampered in genetic (i.e., GM2 gangliosidosis) (72) and sporadic (i.e., PD) diseases (70, 71). Indeed, the 8-year follow-up on ProSavin, a LV delivering key enzymes of the dopamine biosynthetic pathway, documented an improvement of the “off state” time in 8/15 treated PD patients, with GTMP-unrelated mild-to-moderate adverse events (16). Intrathecal or systemic administration can ensure a widespread biodistribution of viral vectors resulting in effective targeting of the spinal cord and in the rostro-caudal coverage of different brain regions (73). These approaches are better suited for the treatment of multifocal/diffuse neurodegenerative diseases (66, 67), including GM2 gangliosidosis (74). Still, they require higher vector doses and enhance targeting of off-target tissues, dorsal root ganglion pathology, and immune response against the GTMP (75, 76).
The selective delivery of GTMPs to the target cell populations/cell subtypes is necessary to improve both the efficacy and safety of GT. The efficiency of AAV vectors and LV in targeting different neuronal populations has been proven in rodents and NHP (15). The higher tropism of LVs for oligodendrocytes (69, 77–79), astrocytes (80) and microglia (81, 82) defines these vectors as a good candidate for gene transfer in glial populations. Recently, AAV hybrid serotypes and AAV variants generated by directed evolution or structural mutagenesis have been selected for their enhanced transduction efficiency in macroglia cells (83–85). In particular, systemic administration of the AAV9 variant AAV-F showed high proficiency for astrocyte transduction and a CNS distribution similar to the BBB-crossing AAV9.PHP.B variant (86), suggesting their potential use for less invasive targeting of cells involved in neuroinflammation processes.
The cell specificity of GTMPs could be enhanced by the inclusion of lineage-specific regulatory elements in the transgenic constructs. The size of cell-type specific promoters has been shortened to fit AAV cargo capacity and tested in pre-clinical models, resulting in upstream regulatory elements able to enhance and/or restrict transgene expression in neurons (e.g., NSE, CaMKII and Syn1 promoters) (87), astrocytes (e.g., gfaABC (1)D promoter) (84), oligodendrocytes (e.g., Mag promoter) (88), or microglia and brain-infiltrating macrophages (e.g., F4/80 and CD68) (89, 90). De-targeting strategies based on endogenous microRNAs selectively expressed in off-target cell populations could further increase cell-specific transgene expression (81, 82), decrease the targeting of off-target cells/tissues (91, 92), and mitigate immune responses (93). The multiplexing of different microRNA de-targeting strategies favors the refinement of the post-translational regulation of transgene expression.
Nanoparticles (NPs) delivering large-size Cas9 nucleases, genome or epigenome modifiers are the ideal GTMP vehicle to ensure effective on-target editing by transient and safer expression of the editing machinery. Intraparenchymal injection of CRISPR/Cas9-loaded NPs have been tested in animal models to treat focal neurodegenerative disorders, such as Fragile X syndrome (94) and AD (95). The limited distribution and rapid clearance of NPs hamper their application in multifocal neurodegenerative diseases, for which multiple site administration or NP functionalization to increase cell-specific uptake and the BBB crossing are required to ensure CNS distribution upon systemic injection (96). Future in vivo validation of NP platforms to deliver GTMPs in the brain of large animals is a crucial step in the long path toward their clinical applications.
Conclusion
In vivo genetic engineering has experienced considerable progress in the last decade and a few landmark studies have convincingly shown that somatic genetic modification for therapeutic purposes can be safely achieved in humans. These new advanced therapies remain highly complex, only partially understood, difficult and costly to develop. Yet, they hold tremendous therapeutic potential and promise to revolutionize medicine. We have highlighted some of the many challenges that still need to be addressed and some avenues that are being explored for broader exploitation and effective introduction of these therapies into clinical practice. To achieve this goal, technical advances need to be accompanied by a continuous dialogue and cooperation between academia, biotechnology and pharmaceutical companies, regulators, policy makers and a civil society with high education and trust in science.
Author Contributions
AC, AF, VM, and AG: wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Italian Ministry of Health (GR-2019-12368956) to AC; Cure Sanfilippo Foundation and Sanfilippo Children's Foundation (Joint grant 2021-2023), Fondazione Telethon (Tigem Core grant 2016-2020) to AF; Fondazione Telethon (Tiget Core Grant 2016-2021, grant D2), European Leukodystrophy Association (ELA 2019-015I2), European Joint Program on Rare Diseases (EJPRD; project NG4Leuko) to AG; EU Marie Curie fellowship (895111 ASTRO-EDITING), Italian Ministry of Health (GR-2019-12368930), ELA (ELA 2020-010I2) to VM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AAV, adeno-associated viral; AD, Alzheimer Disease; BBB, blood-brain barrier; CNS, central nervous system; CRISPR, clustered regularly interspaced short palindromic repeat; GAA, acid α-glucosidase a pag. 6; GLD, globoid leukodystrophy; GOF, gain-of-function; GT, gene therapy; GTMP, gene therapy medicinal product; i.v., intravenous; LCA, Leber congenital amaurosis; LNP, lipid nanoparticles; LPL, lipoprotein lipase; LPLD, LPL deficiency; LSD, lysosomal storage diseases; LV, lentiviral vectors; MPS, mucopolysaccharidoses; NHP, non-human primate; PD, Parkinson's disease; PNS, peripheral nervous system; TALEN, transcription activator-like effector nucleases; TTR, transthyretin; ZFN, zinc-finger nucleases.
References
1. Alhakamy NA, Curiel DT, Berkland CJ. The era of gene therapy: From preclinical development to clinical application. Drug Discov Today. (2021) 26:1602–19. doi: 10.1016/j.drudis.2021.03.021
2. Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. (2018) 359:4672. doi: 10.1126/science.aan4672
4. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. (2020) 38:824–44. doi: 10.1038/s41587-020-0561-9
5. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther. (2021) 29:464–88. doi: 10.1016/j.ymthe.2020.12.007
6. Naldini L. Gene therapy returns to centre stage. Nature. (2015) 526:351–60. doi: 10.1038/nature15818
7. Poletti V, Mavilio F. Designing lentiviral vectors for gene therapy of genetic diseases. Viruses. (2021) 13:1526. doi: 10.3390/v13081526
8. Akinc A, Maier MA, Manoharan M, Fitzgerald K, Jayaraman M, Barros S, et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat Nanotechnol. (2019) 14:1084–7. doi: 10.1038/s41565-019-0591-y
9. Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther. (2020) 5:1. doi: 10.1038/s41392-019-0089-y
10. Jensen TL, Gotzsche CR, Woldbye DPD. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. (2021) 14:695937. doi: 10.3389/fnmol.2021.695937
11. Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. (2017) 390:849–60. doi: 10.1016/S0140-6736(17)31868-8
12. Lowes LP, Alfano LN, Arnold WD, Shell R, Prior TW, McColly M, et al. Impact of age and motor function in a phase 1/2A study of infants with SMA type 1 receiving single-dose gene replacement therapy. Pediatr Neurol. (2019) 98:39–45. doi: 10.1016/j.pediatrneurol.2019.05.005
13. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. (2015) 33:2780–8. doi: 10.1200/JCO.2014.58.3377
14. Mackenzie SJ, Nicolau S, Connolly AM, Mendell JR. Therapeutic approaches for duchenne muscular dystrophy: old and new. Semin Pediatr Neurol. (2021) 37:100877. doi: 10.1016/j.spen.2021.100877
15. Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. (2018) 17:767. doi: 10.1038/nrd.2018.158
16. Palfi S, Gurruchaga JM, Lepetit H, Howard K, Ralph GS, Mason S, et al. Long-term follow-up of a Phase I/II Study of ProSavin, a lentiviral vector gene therapy for Parkinson's disease. Hum Gene Ther Clin Dev. (2018) 29:148–55. doi: 10.1089/humc.2018.081
17. Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. (2019) 25:229–33. doi: 10.1038/s41591-018-0327-9
18. George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. (2017) 377:2215–27. doi: 10.1056/NEJMoa1708538
19. Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N Engl J Med. (2020) 382:29–40. doi: 10.1056/NEJMoa1908490
20. Batty P, Lillicrap D. Hemophilia gene therapy: approaching the first licensed product. Hemasphere. (2021) 5:e540. doi: 10.1097/HS9.0000000000000540
21. Zabaleta N, Hommel M, Salas D, Gonzalez-Aseguinolaza G. Genetic-based approaches to inherited metabolic liver diseases. Hum Gene Ther. (2019) 30:1190–203. doi: 10.1089/hum.2019.140
22. Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. (2021) 385:493–502. doi: 10.1056/NEJMoa2107454
23. Domenger C, Grimm D. Next-generation AAV vectors-do not judge a virus (only) by its cover. Hum Mol Genet. (2019) 28:R3–R14. doi: 10.1093/hmg/ddz148
24. Duverge A, Negroni M. Pseudotyping lentiviral vectors: when the clothes make the virus. Viruses. (2020) 12:1311. doi: 10.3390/v12111311
25. Merlin S, Follenzi A. Transcriptional targeting and MicroRNA regulation of lentiviral vectors. Mol Ther Methods Clin Dev. (2019) 12:223–32. doi: 10.1016/j.omtm.2018.12.013
26. Lipinski DM. A comparison of inducible gene expression platforms: implications for recombinant adeno-associated virus (rAAV) vector-mediated ocular gene therapy. Adv Exp Med Biol. (2019) 1185:79–83. doi: 10.1007/978-3-030-27378-1_13
27. Pierce GF, Iorio A. Past, present and future of haemophilia gene therapy: From vectors and transgenes to known and unknown outcomes. Haemophilia. (2018) 24(suppl. 6):60–7. doi: 10.1111/hae.13489
28. Teboul L, Herault Y, Wells S, Qasim W, Pavlovic G. Variability in genome editing outcomes: challenges for research reproducibility and clinical safety. Mol Ther. (2020) 28:1422–31. doi: 10.1016/j.ymthe.2020.03.015
29. Kumar SR, Markusic DM, Biswas M, High KA, Herzog RW. Clinical development of gene therapy: results and lessons from recent successes. Mol Ther Methods Clin Dev. (2016) 3:16034. doi: 10.1038/mtm.2016.34
30. Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. (2018) 29:285–98. doi: 10.1089/hum.2018.015
31. Wilson JM, Flotte TR. Moving forward after two deaths in a gene therapy trial of myotubular myopathy. Hum Gene Ther. (2020) 31:695–6. doi: 10.1089/hum.2020.182
32. Biasco L, Rothe M, Buning H, Schambach A. Analyzing the genotoxicity of retroviral vectors in hematopoietic cell gene therapy. Mol Ther Methods Clin Dev. (2018) 8:21–30. doi: 10.1016/j.omtm.2017.10.002
33. Yee JK. Off-target effects of engineered nucleases. FEBS J. (2016) 283:3239–48. doi: 10.1111/febs.13760
34. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. (2018) 36:765–71. doi: 10.1038/nbt.4192
35. Zhou C, Sun Y, Yan R, Liu Y, Zuo E, Gu C, et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. (2019) 571:275–8. doi: 10.1038/s41586-019-1314-0
36. Grunewald J, Zhou R, Garcia SP, Iyer S, Lareau CA, Aryee MJ, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. (2019) 569:433–7. doi: 10.1038/s41586-019-1161-z
37. Shirley JL, de Jong YP, Terhorst C, Herzog RW. Immune responses to viral gene therapy vectors. Mol Ther. (2020) 28:709–22. doi: 10.1016/j.ymthe.2020.01.001
38. Mehta A, Merkel OM. Immunogenicity of Cas9 protein. J Pharm Sci. (2020) 109:62–7. doi: 10.1016/j.xphs.2019.10.003
39. Pardridge WM. Drug and gene delivery to the brain: the vascular route. Neuron. (2002) 36:555–8. doi: 10.1016/S0896-6273(02)01054-1
40. Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. (2018) 4:27. doi: 10.1038/s41572-018-0025-4
41. Sorrentino NC, D'Orsi L, Sambri I, Nusco E, Monaco C, Spampanato C, et al. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol Med. (2013) 5:675–90. doi: 10.1002/emmm.201202083
42. Sorrentino NC, Cacace V, De Risi M, Maffia V, Strollo S, Tedesco N, et al. Enhancing the therapeutic potential of sulfamidase for the treatment of mucopolysaccharidosis IIIA. Mol Ther Methods Clin Dev. (2019) 15:333–42. doi: 10.1016/j.omtm.2019.10.009
43. Puzzo F, Colella P, Biferi MG, Bali D, Paulk NK, Vidal P, et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci Transl Med. (2017) 9:6375. doi: 10.1126/scitranslmed.aam6375
44. Ricca A, Cascino F, Morena F, Martino S, Gritti A. In vitro validation of chimeric beta-galactosylceramidase enzymes with improved enzymatic activity and increased secretion. Front Mol Biosci. (2020) 7:167. doi: 10.3389/fmolb.2020.00167
45. Spencer BJ, Verma IM. Targeted delivery of proteins across the blood-brain barrier. Proc Natl Acad Sci USA. (2007) 104:7594–9. doi: 10.1073/pnas.0702170104
46. Pan X, Sands SA, Yue Y, Zhang K, LeVine SM, Duan D. An engineered galactosylceramidase construct improves AAV gene therapy for krabbe disease in twitcher mice. Hum Gene Ther. (2019) 30:1039–51. doi: 10.1089/hum.2019.008
47. Crudele JM, Finn JD, Siner JI, Martin NB, Niemeyer GP, Zhou S, et al. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. (2015) 125:1553–61. doi: 10.1182/blood-2014-07-588194
48. Nathwani AC, Davidoff AM, Tuddenham EGD. Advances in gene therapy for hemophilia. Hum Gene Ther. (2017) 28:1004–12. doi: 10.1089/hum.2017.167
49. Gaudet D, Methot J, Dery S, Brisson D, Essiembre C, Tremblay G, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. (2013) 20:361–9. doi: 10.1038/gt.2012.43
50. Meliani A, Boisgerault F, Hardet R, Marmier S, Collaud F, Ronzitti G, et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat Commun. (2018) 9:4098. doi: 10.1038/s41467-018-06621-3
51. Leborgne C, Barbon E, Alexander JM, Hanby H, Delignat S, Cohen DM, et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med. (2020) 26:1096–101. doi: 10.1038/s41591-020-0911-7
52. Ilyinskii PO, Michaud AM, Roy CJ, Rizzo GL, Elkins SL, Capela T, et al. Enhancement of liver-directed transgene expression at initial and repeat doses of AAV vectors admixed with ImmTOR nanoparticles. Sci Adv. (2021) 7:321. doi: 10.1126/sciadv.abd0321
53. Cantore A, Nair N, Della Valle P, Di Matteo M, Matrai J, Sanvito F, et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood. (2012) 120:4517–20. doi: 10.1182/blood-2012-05-432591
54. Cantore A, Ranzani M, Bartholomae CC, Volpin M, Valle PD, Sanvito F, et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med. (2015) 7:277ra28. doi: 10.1126/scitranslmed.aaa1405
55. Milani M, Annoni A, Bartolaccini S, Biffi M, Russo F, Di Tomaso T, et al. Genome editing for scalable production of alloantigen-free lentiviral vectors for in vivo gene therapy. EMBO Mol Med. (2017) doi: 10.15252/emmm.201708148
56. Milani M, Annoni A, Moalli F, Liu T, Cesana D, Calabria A, et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci Transl Med. (2019) 11:493. doi: 10.1126/scitranslmed.aav7325
57. Cantore A, Naldini L. WFH State-of-the-art paper 2020: In vivo lentiviral vector gene therapy for haemophilia. Haemophilia. (2021) 27:122–5. doi: 10.1111/hae.14056
58. Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. (2011) 475:217–21. doi: 10.1038/nature10177
59. Anguela XM, Sharma R, Doyon Y, Miller JC, Li H, Haurigot V, et al. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood. (2013) 122:3283–7. doi: 10.1182/blood-2013-04-497354
61. Barzel A, Paulk NK, Shi Y, Huang Y, Chu K, Zhang F, et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. (2015) 517:360–4. doi: 10.1038/nature13864
62. Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. (2016) 34:328–33. doi: 10.1038/nbt.3471
63. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. (2018) 19:770–88. doi: 10.1038/s41576-018-0059-1
64. Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. (2021) 593:429–34. doi: 10.1038/s41586-021-03534-y
65. Rothgangl T, Dennis MK, Lin PJC, Oka R, Witzigmann D, Villiger L, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat Biotechnol. (2021) 39:949–57. doi: 10.1038/s41587-021-00933-4
66. Lonser RR, Akhter AS, Zabek M, Elder JB, Bankiewicz KS. Direct convective delivery of adeno-associated virus gene therapy for treatment of neurological disorders. J Neurosurg. (2020) 134:1751–63. doi: 10.3171/2020.4.JNS20701
67. Uchitel J, Kantor B, Smith EC, Mikati MA. Viral-mediated gene replacement therapy in the developing central nervous system: current status and future directions. Pediatr Neurol. (2020) 110:5–19. doi: 10.1016/j.pediatrneurol.2020.04.010
68. Dissen GA, Lomniczi A, Neff TL, Hobbs TR, Kohama SG, Kroenke CD, et al. In vivo manipulation of gene expression in non-human primates using lentiviral vectors as delivery vehicles. Methods. (2009) 49:70–7. doi: 10.1016/j.ymeth.2009.06.004
69. Meneghini V, Lattanzi A, Tiradani L, Bravo G, Morena F, Sanvito F, et al. Pervasive supply of therapeutic lysosomal enzymes in the CNS of normal and Krabbe-affected non-human primates by intracerebral lentiviral gene therapy. EMBO Mol Med. (2016) 8:489–510. doi: 10.15252/emmm.201505850
70. Jarraya B, Boulet S, Ralph GS, Jan C, Bonvento G, Azzouz M, et al. Dopamine gene therapy for Parkinson's disease in a nonhuman primate without associated dyskinesia. Sci Transl Med. (2009) 1:2ra4. doi: 10.1126/scitranslmed.3000130
71. Badin RA, Binley K, Van Camp N, Jan C, Gourlay J, Robert C, et al. Gene therapy for Parkinson's disease: preclinical evaluation of optimally configured TH:CH1 fusion for maximal dopamine synthesis. Mol Ther Methods Clin Dev. (2019) 14:206–16. doi: 10.1016/j.omtm.2019.07.002
72. Ornaghi F, Sala D, Tedeschi F, Maffia MC, Bazzucchi M, Morena F, et al. Novel bicistronic lentiviral vectors correct beta-Hexosaminidase deficiency in neural and hematopoietic stem cells and progeny: implications for in vivo and ex vivo gene therapy of GM2 gangliosidosis. Neurobiol Dis. (2020) 134:104667. doi: 10.1016/j.nbd.2019.104667
73. Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. (2016) 34:204–9. doi: 10.1038/nbt.3440
74. Lahey HG, Webber CJ, Golebiowski D, Izzo CM, Horn E, Taghian T, et al. Pronounced therapeutic benefit of a single bidirectional AAV vector administered systemically in sandhoff mice. Mol Ther. (2020) 28:2150–60. doi: 10.1016/j.ymthe.2020.06.021
75. Hordeaux J, Buza EL, Dyer C, Goode T, Mitchell TW, Richman L, et al. Adeno-associated virus-induced dorsal root ganglion pathology. Hum Gene Ther. (2020) 31:808–18. doi: 10.1089/hum.2020.167
76. Ballon DJ, Rosenberg JB, Fung EK, Nikolopoulou A, Kothari P, De BP, et al. Quantitative whole-body imaging of I-124-labeled adeno-associated viral vector biodistribution in nonhuman primates. Hum Gene Ther. (2020) 31:1237–59. doi: 10.1089/hum.2020.116
77. Lattanzi A, Neri M, Maderna C, di Girolamo I, Martino S, Orlacchio A, et al. Widespread enzymatic correction of CNS tissues by a single intracerebral injection of therapeutic lentiviral vector in leukodystrophy mouse models. Hum Mol Genet. (2010) 19:2208–27. doi: 10.1093/hmg/ddq099
78. Lattanzi A, Salvagno C, Maderna C, Benedicenti F, Morena F, Kulik W, et al. Therapeutic benefit of lentiviral-mediated neonatal intracerebral gene therapy in a mouse model of globoid cell leukodystrophy. Hum Mol Genet. (2014) 23:3250–68. doi: 10.1093/hmg/ddu034
79. Ricca A, Rufo N, Ungari S, Morena F, Martino S, Kulik W, et al. Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum Mol Genet. (2015) 24:3372–89. doi: 10.1093/hmg/ddv086
80. Delzor A, Escartin C, Deglon N. Lentiviral vectors: a powerful tool to target astrocytes in vivo. Curr Drug Targets. (2013) 14:1336–46. doi: 10.2174/13894501113146660213
81. Akerblom M, Sachdeva R, Quintino L, Wettergren EE, Chapman KZ, Manfre G, et al. Visualization and genetic modification of resident brain microglia using lentiviral vectors regulated by microRNA-9. Nat Commun. (2013) 4:1770. doi: 10.1038/ncomms2801
82. Clayton K, Delpech JC, Herron S, Iwahara N, Ericsson M, Saito T, et al. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol Neurodegener. (2021) 16:18. doi: 10.1186/s13024-021-00440-9
83. Koerber JT, Klimczak R, Jang JH, Dalkara D, Flannery JG, Schaffer DV. Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol Ther. (2009) 17:2088–95. doi: 10.1038/mt.2009.184
84. Hammond SL, Leek AN, Richman EH, Tjalkens RB. Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS ONE. (2017) 12:e0188830. doi: 10.1371/journal.pone.0188830
85. Powell SK, Khan N, Parker CL, Samulski RJ, Matsushima G, Gray SJ, et al. Characterization of a novel adeno-associated viral vector with preferential oligodendrocyte tropism. Gene Ther. (2016) 23:807–14. doi: 10.1038/gt.2016.62
86. Hanlon KS, Meltzer JC, Buzhdygan T, Cheng MJ, Sena-Esteves M, Bennett RE, et al. Selection of an efficient AAV vector for robust CNS transgene expression. Mol Ther Methods Clin Dev. (2019) 15:320–32. doi: 10.1016/j.omtm.2019.10.007
87. Radhiyanti PT, Konno A, Matsuzaki Y, Hirai H. Comparative study of neuron-specific promoters in mouse brain transduced by intravenously administered AAV-PHP.eB. Neurosci Lett. (2021) 756:135956. doi: 10.1016/j.neulet.2021.135956
88. von Jonquieres G, Frohlich D, Klugmann CB, Wen X, Harasta AE, Ramkumar R, et al. Recombinant human myelin-associated glycoprotein promoter drives selective AAV-mediated transgene expression in oligodendrocytes. Front Mol Neurosci. (2016) 9:13. doi: 10.3389/fnmol.2016.00013
89. Cucchiarini M, Ren XL, Perides G, Terwilliger EF. Selective gene expression in brain microglia mediated via adeno-associated virus type 2 and type 5 vectors. Gene Ther. (2003) 10:657–67. doi: 10.1038/sj.gt.3301925
90. Rosario AM, Cruz PE, Ceballos-Diaz C, Strickland MR, Siemienski Z, Pardo M, et al. Microglia-specific targeting by novel capsid-modified AAV6 vectors. Mol Ther Methods Clin Dev. (2016) 3:16026. doi: 10.1038/mtm.2016.26
91. Taschenberger G, Tereshchenko J, Kugler S. A MicroRNA124 target sequence restores astrocyte specificity of gfaABC1D-driven transgene expression in AAV-mediated gene transfer. Mol Ther Nucleic Acids. (2017) 8:13–25. doi: 10.1016/j.omtn.2017.03.009
92. Xie J, Xie Q, Zhang H, Ameres SL, Hung JH, Su Q, et al. MicroRNA-regulated, systemically delivered rAAV9: a step closer to CNS-restricted transgene expression. Mol Ther. (2011) 19:526–35. doi: 10.1038/mt.2010.279
93. Majowicz A, Maczuga P, Kwikkers KL, van der Marel S, van Logtenstein R, Petry H, et al. Mir-142-3p target sequences reduce transgene-directed immunogenicity following intramuscular adeno-associated virus 1 vector-mediated gene delivery. J Gene Med. (2013) 15:219–32. doi: 10.1002/jgm.2712
94. Lee B, Lee K, Panda S, Gonzales-Rojas R, Chong A, Bugay V, et al. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat Biomed Eng. (2018) 2:497–507. doi: 10.1038/s41551-018-0252-8
95. Park H, Oh J, Shim G, Cho B, Chang Y, Kim S, et al. In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer's disease. Nat Neurosci. (2019) 22:524–8. doi: 10.1038/s41593-019-0352-0
Keywords: gene therapy, liver, central nervous system, gene editing, translational medicine
Citation: Cantore A, Fraldi A, Meneghini V and Gritti A (2022) In vivo Gene Therapy to the Liver and Nervous System: Promises and Challenges. Front. Med. 8:774618. doi: 10.3389/fmed.2021.774618
Received: 12 September 2021; Accepted: 16 December 2021;
Published: 18 January 2022.
Edited by:
Nuno M. Neves, University of Minho, PortugalReviewed by:
Haihui Pan, George Washington University, United StatesHai Bac Tran, University of Adelaide, Australia
Copyright © 2022 Cantore, Fraldi, Meneghini and Gritti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessio Cantore, Y2FudG9yZS5hbGVzc2lvQGhzci5pdA==