 Aoife M. O'Byrne
Aoife M. O'Byrne Tineke A. de Jong
Tineke A. de Jong Lisa G. M. van Baarsen
Lisa G. M. van Baarsen- 1Department of Rheumatology and Clinical Immunology, Amsterdam Institute for Infection and Immunity, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Experimental Immunology, Amsterdam Institute for Infection and Immunity, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 3Amsterdam Rheumatology and Immunology Center (ARC), Amsterdam, Netherlands
Rheumatoid arthritis (RA) is a chronic autoimmune disease of unknown etiology characterized by inflammation of the peripheral synovial joints leading to pannus formation and bone destruction. Rheumatoid Factor (RF) and anti-citrullinated protein antibodies (ACPA) are present years before clinical manifestations and are indicative of a break in tolerance that precedes chronic inflammation. The majority of studies investigating disease pathogenesis focus on the synovial joint as target site of inflammation while few studies explore the initial break in peripheral tolerance which occurs within secondary lymphoid organs such as lymph nodes. If explored during the earliest phases of RA, lymph node research may provide innovative drug targets for disease modulation or prevention. RA research largely centers on the role and origin of lymphocytes, such as pro-inflammatory T cells and macrophages that infiltrate the joint, as well as growing efforts to determine the role of stromal cells within the synovium. It is therefore important to explore these cell types also within the lymph node as a number of mouse studies suggest a prominent immunomodulatory role for lymph node stromal cells. Synovium and proximal peripheral lymph nodes should be investigated in conjunction with one another to gain understanding of the immunological processes driving RA progression from systemic autoimmunity toward synovial inflammation. This perspective seeks to provide an overview of current literature concerning the immunological changes present within lymph nodes and synovium during early RA. It will also propose areas that warrant further exploration with the aim to uncover novel targets to prevent disease progression.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease of unknown etiology that preferentially affects the peripheral joints. It is characterized by prolonged inflammation of the synovium which eventually leads to tissue destruction and pannus formation. Current research indicates that this chronic inflammation is driven by the infiltration of destructive pro-inflammatory lymphocytes into the joint leading to pro-inflammatory cytokine release and the initiation of an immune response. This response is perpetuated by the interaction of these destructive immune cells e.g. macrophages and stromal cells present within the synovium. Autoantibodies like rheumatoid factor (RF) and anti-citrullinated peptide antibodies (ACPA) are present in the peripheral blood of RA patients up to a decade before the clinical onset of synovitis and diagnosis (RA-risk individuals) (1–4). This highlights a break in immune tolerance which results in systemic autoimmunity years before diagnosis. Furthermore, prospective studies showed that depending on the risk profile only 30% of individuals with autoantibodies go on to develop arthritis (5, 6), suggesting that additional genetic and environmental factors influence disease onset.
The current paradigm highlights three key phases of arthritis development: the initial break in tolerance, the infiltration of immune cells into the synovial joints and finally established chronic synovitis leading to joint destruction (7). Although much is known about the chronic inflammatory process occurring in the synovium at the latter stages of disease, the changes that occur in the immune system during the earliest phases when there is break in tolerance but no apparent synovitis have yet to be fully elucidated. This break in tolerance is hypothesized to occur in secondary lymphoid organs such as lymph nodes (LNs). Currently, only a few human studies have explored this, while it may provide major insights into early RA pathogenesis and reveal mechanisms for restoring peripheral tolerance.
This review will outline our current knowledge of the immune cell interactions that occur in the LN of RA patients and how these may be linked to observations in the synovium (Figure 1). It will also postulate other immunological avenues that warrant future exploration to identify novel targets for treatment of early RA.
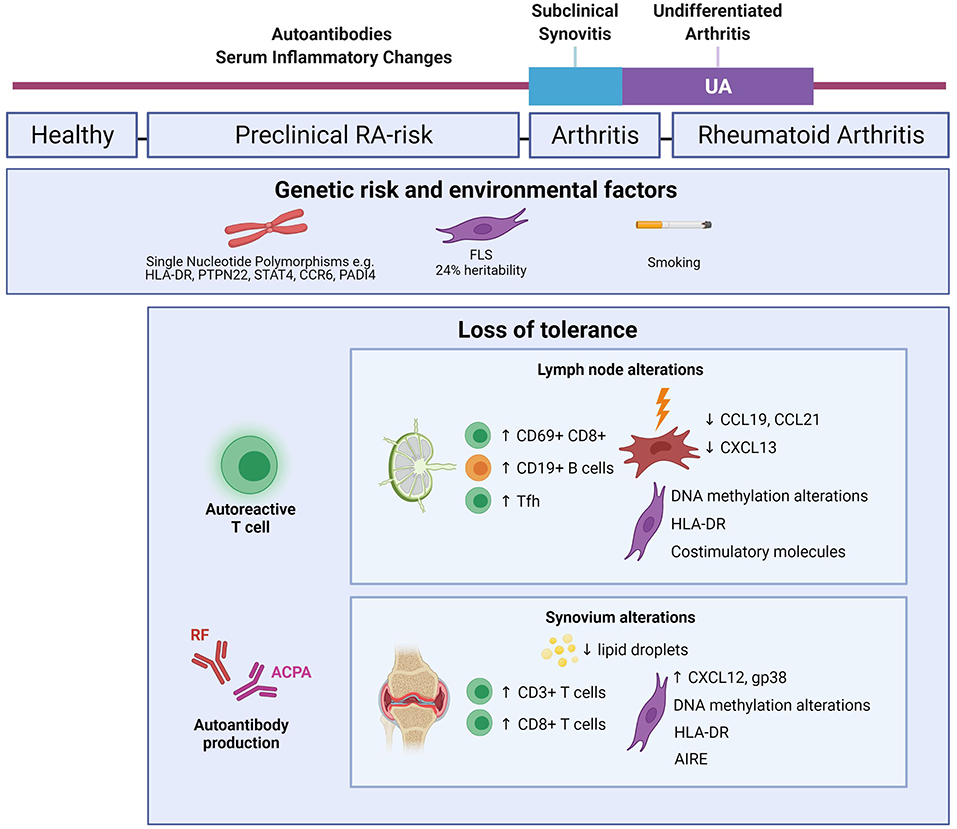
Figure 1. Mapping immune alterations during RA development. Schematic figure highlighting known changes within the immune system of RA-risk individuals and early RA. Genetic and environmental factors increase the likelihood of RA development in the healthy human population (5, 8). Depending on the risk profile, about 30% of RA-risk individuals go on to clinically develop RA (6). LN alterations are based on inguinal LN needle biopsy research comparing RA-risk individuals with healthy volunteers and early RA patients (9–13). Synovium alterations are based on synovial tissue biopsy research in RA-risk individuals and early RA patients (14–17, 57–59, 64, 65). Created with Biorender.com.
LN Studies Reveal Immune Cell Activation During the Earliest Phases of RA
LNs are highly organized lymphoid structures situated throughout the human body that allow immune cell entry from surrounding tissues and blood to orchestrate a fast and effective immune response. They are the primary site of peripheral tolerance which aims to eliminate autoreactive T cells that escape central tolerance by exposing T cells residing in the LN to self-antigens. Studying human LNs during health and autoimmunity is challenging, because whole LNs are only obtained through surgery or autopsy and needle biopsies are too small to enable direct functional cellular analysis. The first study looking into LNs from RA patients suspected of lymphoma, only compared the cellular organization between LN and synovium in paired samples (17).
Our lab set up the infrastructure to study and compare LN needle biopsies from RA patients, RA-risk individuals and healthy volunteers (18). Initial cellular phenotyping by flow cytometry revealed that RA and RA-risk individuals have increased frequencies of CXCR3+CCR6−CCR4− Th1 cells (19), ILC1 (c-Kit-NKp44− ILCs) (20), memory CD8+ T cells (21), CD69+CD8+ T cells and more CD19+ B cells compared to healthy controls (9). Recent research suggests that CD69 expression may be indicative of a tissue resident memory T cell subset rather than an activated T cell subset (22). Evidence in mice suggests there is a circulating T effector population that is destined for a tissue resident phenotype (23, 24); however, the evidence for this in humans is undetermined. Of note, increased frequencies of CD69+CD8+ T cells were also observed in the blood of RA-risk individuals and RA patients (21). Whether the CD69+CD8+ T cells in LNs are tissue resident memory precursors that can later migrate and cause tissue inflammation within synovium warrants investigation.
T follicular helper cells (Tfh) are of particular interest as they can steer B cell activation and differentiation thereby potentially influencing autoantibody production. An increased frequency of both CD4+ and CD8+ Tfh could be detected in LN biopsies of RA-risk individuals and RA patients compared to healthy volunteers (10). Moreover, the augmented CD19+ B cell frequency in RA-risk individuals and RA patients correlated with Tfh frequency (9, 10). This may reflect germinal center activity resulting in autoantibody production within the LN due to break in tolerance, however, this needs further research.
Both dendritic cells (DCs) (25, 26) as well as lymph node stromal cells (LNSC) (27, 28) have the capacity to induce peripheral tolerance through presentation of peripheral tissue antigens (PTAs) to autoreactive T cells after which these cells undergo clonal deletion, differentiation into a regulatory T cell or become anergic. When studying DC subsets (CD1c+ myeloid DCs and CD304+ plasmacytoid DCs) in LN tissue it appeared that frequencies are comparable between RA-risk individuals and healthy controls, but increased in RA patients (29). This may suggest that these DCs are involved in sustaining inflammation during established RA, or that other DC subsets or other antigen presenting cells (APCs) are responsible for the initial break in tolerance. LNSC provide the structural integrity and framework for the important compartmentalization of lymphocytes within the LN (30). In recent years, mouse LNSC have been implicated more prominently in immune cell tolerance as they are able to delete self-reactive CD8+ T cells through PTA expression on major histocompatibility complex (MHC) I and on MHC II in the context of CD4+ T cells (27, 28, 31, 32). However, the presence of antibodies against self-antigens in early RA suggests this process is not always effective and provides impetus for examining LNSC in early RA. To investigate their functional capacities during RA development a human LNSC culture model has been developed (13). Transcriptome and methylome analysis of expanded LNSC highlighted key alterations in DNA methylation sites associated with antigen processing and presentation in RA-risk and RA patients when compared with healthy controls (11). Flow cytometry and qPCR data showed that all LNSC can express HLA-DR, co-stimulatory and co-inhibitory molecules (12), indicating that human LNSC are capable of modulating adaptive immunity. The expression of HLA-DR shows that human LNSC have the capacity to directly present PTAs while it was also shown that LNSC can express citrullinated antigens targeted by ACPAs (12). Whether such citrullinated antigens are presented as PTAs in LNSC and whether this process is altered in LNSC during systemic autoimmunity warrants further investigation.
LNSC produce key chemokines such as CCL19, CCL21 and CXCL13 to attract, retain and position lymphocytes within the LN and guide the interaction between T cells, B cells and APCs (33–35). Upon in vitro stimulation with TNFα and lymphotoxin α1β2, RA LNSC were significantly less capable to produce CCL19, CCL21 and CXCL13 (13). Furthermore, triggering TLR3 also showed lower induction of CCL19 in RA and RA-risk LNSC (36). Overall, these differences in RA-risk and RA LNSC may lead to imprecise localization of T cells upon LN entry and induce abnormal LNSC-T cell interactions leading to aberrant immune responses.
A remarkable feature observed in both RA and RA-risk LN T cells and LNSC is the diminished capacity to produce cytokines or chemokines upon in vitro stimulation which may indicate an exhausted phenotype (13, 19, 21, 36). T cell exhaustion is the result of repeated antigen stimulation which leads to increased inhibitory receptor expression such as PD1 and low effector function (37). Although extensively explored in chronic viral infection and cancer, the role of exhaustion in autoimmunity is as yet unclear. Future work to determine whether this reduction in cytokine production in LN T cells is a result of exhaustion is ongoing. Exhausted T cells are the target of immunotherapy such as anti-PD1 treatment. Recent studies of tumor draining LNs suggest they may harbor a reservoir of exhausted T cells that, if targeted, may improve therapeutic efficacy (38, 39). In the context of cancer, targeting an exhausted T cell population within the LN can allow for beneficial unleashing of the immune system. It is interesting to postulate whether exhaustion is induced in autoimmunity and how this may contribute to disease flare or remission. The increase in CD69+CD8+ T cells within RA and RA-risk LNs could also point toward a pre-exhausted memory phenotype. A recent human study identified a CD69+CXCR5+CXCR4+TCF1+ population in healthy LNs that had reduced expression of effector molecules and showed similarities to the mouse CD8+ T cell subset found to respond to checkpoint blockade therapy in a number of chronic infection mouse models (40). The CD69+CD8+ T cells increased in RA LNs may be an expansion of this newly identified human population which is possibly exhausted.
Overall, LN biopsy studies in RA-risk individuals provide a great model to explore the earliest break in tolerance which occurs in seropositive RA. Although, no LN data is currently available on the difference between RA-risk individuals that developed RA and those who did not, a number of changes observed in RA patients were also present in a proportion of RA-risk individuals. This may suggest a continuum that contributes to disease progression; however, follow up studies are needed to confirm this. Exploring if and how these early differences are reflected in the chronic inflammatory environment of the synovium provides great insight into early RA pathogenesis and may provide novel targets for preventive intervention.
Insights From Synovium During Early RA Development
Several large cohort studies of RA synovium from treatment naïve patients have unearthed cellular heterogeneity. This enables pathology-based stratification which can be associated with clinical treatment response. The three main pathotypes described are diffuse myeloid, lympho-myeloid and pauci-immune representing myeloid dominance, lymphoid dominance and stromal dominance respectively (41–44). Analysis of synovial tissue biopsies not only allows for disease stratification and exploring how chronic inflammation persists in RA but also how it is initiated following loss of tolerance, presumably instigated in lymphoid organs. Synovial biopsies of autoantibody positive RA-risk individuals in almost all cases lack B cells and plasma cells (14) and do not yet display overt immune cell infiltration when compared to synovium from healthy controls (15), suggesting that synovial inflammation is likely to occur closer to clinical arthritis. The presence of CD3+ T cells in RA-risk synovium has been associated with subsequent arthritis development (14). Especially the combined presence of synovial CD8+ T cells with ACPA positivity increased the risk of RA development. This data suggest an early role for T cells in arthritis development. How these synovial T cells relate to the increased frequency of CD69+CD8+ T cells observed in the LNs of RA-risk individuals is worthy of exploration.
The origin of T cells in the synovium and how and where they have been activated is still largely unknown. Part of them may reflect activated resident T cells but probably the majority have been activated in peripheral lymphoid organs after which they then migrate to the synovium, as changes in the T cell compartment can be found in peripheral blood of early RA and RA-risk individuals (7, 45, 46). As previously highlighted, our work on LN-derived T cells suggest that there may be increased Tfh cells in RA patients (10) as well as a possibly exhausted population in RA-risk individuals (19, 21) which are both characterized by increased PD1 expression. Whether these changes are mirrored within the synovium is unclear. A number of studies have reported high PD1 expression on T cells within RA synovial tissue (47–49). Mass cytometry analysis of synovial tissue uncovered an expanded PD1highCXCR5−CD4+ T cell population that were akin to Tfh cells as they exhibited increased IL21, CXCL13, IFNγ and IL10 production (49). PD1+CXCR5− memory CD4+ T cells were enriched within synovial tissue and fluid compared to their CXCR5+ memory CD4+ T cell counterpart; however, the phenotype of this population was not explored further (49). It was concluded that exhaustion was not present due to the population's ability to produce IL21 and CXCL13 (49). A larger, more in depth study is required to further characterize this PD1highCXCR5− population and to determine at what phase of RA it emerges. Similar studies in juvenile idiopathic arthritis suggest that synovial T cells are effector populations and not exhausted (50); however, this has not been confirmed in the case of RA.
Not much is known about the role of CD8+ Tfh cells in RA, although the frequency of this population was also increased in RA LN (10). PD1highCD8+ T cells producing IL21 and also exhibiting increased CD28, ICOS, CD69 and HLA-DR expression have been observed in the synovial fluid and peripheral blood of RA patients (51) which like in the findings of Rao et al. (49) did also not express CXCR5. Whether this subset originates from the same CD69+CD8+ T cells found upregulated in the LN of RA-risk and early RA patients is of great interest but difficult to elucidate.
The possible interactions of these Tfh cells present within the synovium with B cells is interesting in relation to autoantibody production; however, B cells and plasma cells were not detectable by immunohistochemistry in synovial biopsies of RA-risk individuals (14, 15). This is despite the presence of autoantibodies and increased B cell frequencies in the lymph nodes of these individuals (9, 10). Furthermore, no significant changes have been observed in B cell frequency in the synovium of seropositive early RA compared to their seronegative counterpart (52) which further negates a role for B cells in autoantibody production within the synovium. Lymphoid neogenesis, relating to the aggregation of T and B cell lymphocytes, is observed in the synovium of a proportion of RA patients; however, its presence is associated with the degree of synovitis and not linked to clinical outcome or autoantibody status (53). This suggests these lymphoid aggregates are a result of chronic inflammation rather than a consequence of initial loss in peripheral tolerance.
It is clear from these studies that a population of T cells within the synovium that bear resemblance to Tfh cells and can provide B cell help are present in RA synovium. However, how this population arises and where these cells received their activation and differentiation cues is still contentious. Determining at what stage of RA pathogenesis this cell population arises will provide much insight into their functional role. Synovial cues directing the attraction, retention and activation of T cells may originate from altered resident synovial fibroblasts (FLS).
Fibroblasts in the synovium of patients with established RA have been studied for decades showing that these cells invade the cartilage and are responsible for tissue degradation and bone erosion [reviewed in (54)]. During destructive joint inflammation, FLS have an activated phenotype and secrete several immunomodulatory factors (55, 56). Epigenome and methylome analysis performed by several labs showed that FLS from RA patients undergo DNA methylation changes that can be linked to disease development (8, 57–59). A recent, elegantly designed functional genomics atlas study demonstrated that FLS account for up to 24% of RA heritability providing evidence of a causal role for FLS in RA development (8). This is in line with a prospective study which compared synovial biopsies of RA-risk individuals who later developed RA after follow up, with those who did not. Using immunohistochemistry analyses no overt immune cell infiltration was found in the synovium of RA-risk individuals who later developed disease (14, 15) while gene expression profiling points toward an activated stromal cell gene signature with increased podoplanin and CXCL12 levels and decreased lipid droplets (16). These studies provide strong evidence for a causal role for FLS in driving disease pathogenesis at an early stage.
Fibroblasts are known to exhibit immunomodulatory capacities when interacting with nearby or incoming immune cells and several studies have investigated their potential as antigen-presenting cells (60, 61). Similar to stromal cells in the LN, FLS create a pro-survival and anti-apoptotic microenvironment in the synovium by secreting cytokines and chemokines that support the survival of immune cells supporting inflammation (62, 63). AIRE was recently identified as a cytokine-induced RA-risk gene in RA FLS (64). RNA sequencing revealed that AIRE did not induce PTAs in FLS, but stimulates pro-inflammatory cytokine and chemokine secretion associated with RA development. AIRE expression was hardly detected on mRNA level in unstimulated RA FLS, but largely increased and detected at protein level after TNFα and IL1β stimulation (64). Single-cell RNA sequencing on RA FLS showed a putative subpopulation of CD90+HLA-DRhigh FLS expanded in the sublining of RA synovium. Additionally, inflammatory mediators such as IL6 could be linked to this FLS subset (65). The HLA-DRhigh FLS subpopulation was shown to express genes related to MHC-II presentation and IFNγ signaling (65), suggesting that FLS have the capacity to present antigens and interact with immune cells directly. It will be of interest to investigate whether this HLA-DRhigh FLS subset is expanded already in RA-risk individuals and to study its potential capacity to regulate peripheral tolerance.
Future Research Avenues
Detailed analysis of tissues involved in initial triggering of RA are fundamental for finding predictive biomarkers for disease progression and for the discovery of novel drug targets to aid future preventive treatment strategies (Figure 2). As said, we propose that the first signs of RA can be identified in lymphoid organs as they are the epicenter of immune activation in which APCs, including stromal cells, can trigger T-cell mediated B-cell activation, thereby initiating an inflammatory response and autoantibody production. Studying tissue samples derived from lymphoid organs of individuals at risk of developing RA will provide crucial insights into the earliest stages of disease pathogenesis. Presently, within the field of rheumatology, only a few studies have been performed on human LN biopsies while this biopsy procedure is frequently performed in routine clinical care in the field of oncology. In addition, our observational study investigating the perspectives of study participants, indicated that inguinal LN biopsy sampling is well-tolerated, safe and provides sufficient material for further molecular and cellular analyses (Fiechter et al.)1. Accordingly, available data provides a strong case for the application of this research tool in order to identify novel biomarkers and drug targets in individuals at risk of developing RA.
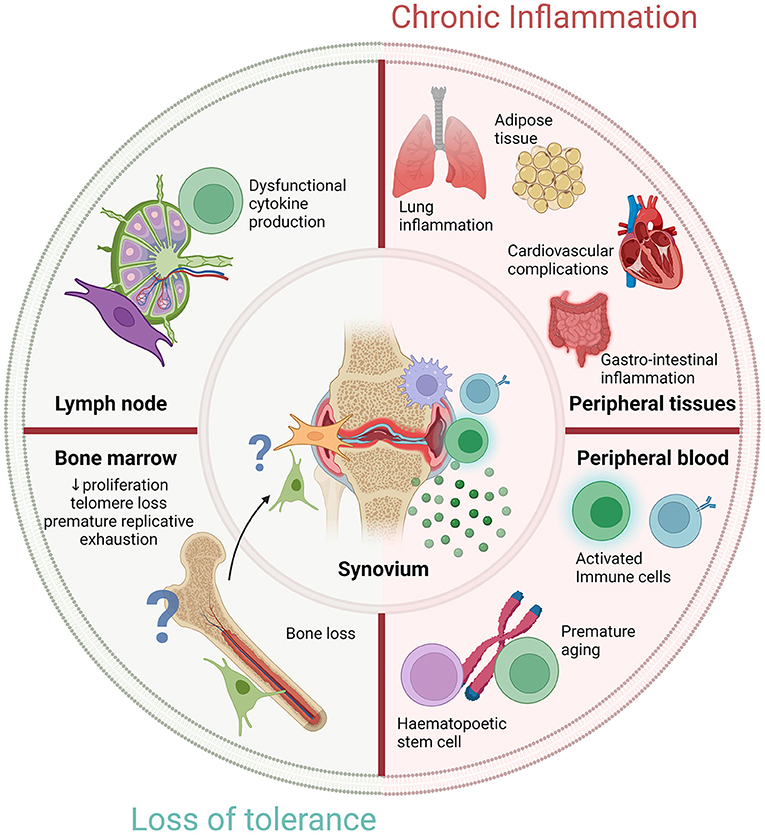
Figure 2. Novel targets to restore cellular fitness before RA development. Diagram representing novel targets of either chronic inflammation or loss of tolerance to prevent RA development in RA-risk individuals. Outer circle shows four critical areas that show significant changes in early RA. Lymph node and bone marrow alterations are observed, potentially due to a loss of tolerance in RA which warrant further investigation. Timely normalization of cellular fitness within lymphoid organs or other peripheral tissues could possibly prevent RA development in RA-risk individuals. Peripheral blood research may identify predictive biomarkers for early diagnosis or disease prognosis. Chronic inflammation is observed in distinct locations outside the synovium but whether these are the consequence of a loss of tolerance or caused by inflammation is unknown. The locations of the outer circle culminate in the chronic inflammatory environment observed within the synovium but how exactly they contribute warrants further examination. Created with Biorender.com.
A so far unexplored lymphoid organ in RA-risk individuals is the bone marrow (BM), while already decades ago it has been postulated that RA may be a BM disorder (66). A more recent micro-CT study has shown bone loss in ACPA positive healthy individuals without clinical signs of arthritis (67). Of interest, animal labeling studies have shown migration of fibroblastic cells from the BM to the synovium at the onset of arthritis (68). Besides the production of hematopoietic stem cells, the BM is highly important for the development of mesenchymal stromal cells (MSC). There is compelling evidence that BM-MSC are instrumental for effective hematopoiesis and have important immunomodulatory and regenerative capacities (69–71). Accordingly, clinical trials have been initiated to examine the therapeutic properties of MSC in many different diseases, including RA (72). However, the inconsistent findings reported, reflect that the understanding of MSC biology, especially in the context of autoimmunity, is limited. A few studies have investigated BM-MSC in patients with RA (73–75). These studies show that RA BM-MSC are normal in frequency and have a normal differentiation capacity (73). However, RA BM-MSC exhibit reduced proliferation, telomere loss, and premature replicative exhaustion (73). Moreover, BM-MSC from RA patients are less supportive for hematopoietic stem and progenitor cell (HSPC) survival (74). Currently, it is unknown whether these BM-MSC abnormalities are caused by inflammation or take place already before disease onset in RA-risk individuals.
Increased immune cell aging, determined by measuring the shortening of telomeres, has been observed in several chronic inflammatory diseases, including RA (76–78). Immune cells from 30 year-old RA patients show a biological cellular age of a 50 year-old healthy individual (79). Of note, in RA this accelerated aging is already observed in HSPC (80); however, it is unknown whether premature aging is an intrinsic defect present already before onset of disease or a secondary effect due to inflammatory signals or treatment. It is unclear whether this accelerated aging originates in lymphoid organs and is also observed in tissue resident cells like MSC which will have a major influence on MSC-mediated cell survival and immunomodulation.
Many of the questions still outstanding regarding RA pathogenesis are those of disease origin and cellular contribution. We postulate that RA-risk individuals can be studied as a model to investigate the earliest phases of systemic autoimmunity wherein self-tolerance is lost potentially due to accelerated cellular aging affecting adaptive immune responses. It is important to unravel whether accelerated or premature aging can already be detected in preclinical phases of RA by studying cellular characteristics of senescence in lymphocytes as well as stromal cells isolated from synovial, LN and BM tissue biopsies obtained from RA-risk individuals. If present, such research will lead to the discovery of novel targets to restore cellular fitness before RA onset. Research focusing on detailed analyses of human tissue samples obtained during the earliest preclinical phases of RA are highly challenging but will provide great advancement toward the ultimate goal of disease prevention.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
LB received an AMC Fellowship and funding from a ZonMw VIDI project (91718371) and from the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement No. 847551 (ARCAID).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
RA, rheumatoid arthritis; RA-risk, individuals at risk of developing rheumatoid arthritis; LN, lymph node; LNSC, lymph node stromal cells; FLS, synovial fibroblasts; BM, bone marrow; BM-MSC, bone marrow stromal cells; PTA, peripheral tissue antigens; DC, dendritic cell; APC, antigen presenting cell; Tfh, T follicular helper; IL21, Interleukin 21; RF, rheumatoid factor; ACPA, anti-citrullinated peptide antibodies.
Footnotes
1. ^Fiechter RH, Bolt JW, van de Sande MGH, Aalbers CJ, Landewé RBM, Maas M, et al. Ultrasound-guided lymph node biopsy sampling to study the immunopathogenesis of rheumatoid arthritis: a well-tolerated valuable research tool.
References
1. Gerlag DM, Raza K, van Baarsen LGM, Brouwer E, Buckley CD, Burmester GR, et al. EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann Rheum Dis. (2012) 71:638–41. doi: 10.1136/annrheumdis-2011-200990
2. Jørgensen KT, Wiik A, Pedersen M, Hedegaard CJ, Vestergaard BF, Gislefoss RE, et al. Cytokines, autoantibodies and viral antibodies in premorbid and postdiagnostic sera from patients with rheumatoid arthritis: case-control study nested in a cohort of Norwegian blood donors. Ann Rheum Dis. (2008) 67:860–6. doi: 10.1136/ard.2007.073825
3. Nielen MMJ, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MHMT, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. (2004) 50:380–6. doi: 10.1002/art.20018
4. Rantapää-Dahlqvist S, de Jong BAW, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. (2003) 48:2741–9. doi: 10.1002/art.11223
5. de Hair MJH, Landewé RBM, van de Sande MGH, van Schaardenburg D, van Baarsen LGM, Gerlag DM, et al. Smoking and overweight determine the likelihood of developing rheumatoid arthritis. Ann Rheum Dis. (2013) 72:1654–8. doi: 10.1136/annrheumdis-2012-202254
6. van de Stadt LA, van der Horst AR, de Koning MHMT, Bos WH, Wolbink GJ, van de Stadt RJ, et al. The extent of the anti-citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann Rheum Dis. (2011) 70:128–33. doi: 10.1136/ard.2010.132662
7. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol. (2021) 22:10–8. doi: 10.1038/s41590-020-00816-x
8. Ge X, Frank-Bertoncelj M, Klein K, McGovern A, Kuret T, Houtman M, et al. Functional genomics atlas of synovial fibroblasts defining rheumatoid arthritis heritability. Genome Biol. (2021) 22:247. doi: 10.1186/s13059-021-02460-6
9. van Baarsen LGM, de Hair MJH, Ramwadhdoebe TH, Zijlstra IJAJ, Maas M, Gerlag DM, et al. The cellular composition of lymph nodes in the earliest phase of inflammatory arthritis. Ann Rheum Dis. (2013) 72:1420–4. doi: 10.1136/annrheumdis-2012-202990
10. Anang DC, Ramwadhdoebe TH, Hahnlein J, van Kuijk B, Smits N, van Lienden KP, et al. Increased frequency of CD4+ and CD8+ follicular helper T cells in human lymph node biopsies during the earliest stages of rheumatoid arthritis. bioRxiv [Preprint]. (2021). doi: 10.1101/2021.11.10.467883
11. Karouzakis E, Hähnlein J, Grasso C, Semmelink JF, Tak PP, Gerlag DM, et al. Molecular characterization of human lymph node stromal cells during the earliest phases of rheumatoid arthritis. Front Immunol. (2019) 10:1863. doi: 10.3389/fimmu.2019.01863
12. Hähnlein JS, Nadafi R, Jong TA de, Semmelink JF, Remmerswaal EBM, Safy M, et al. Human lymph node stromal cells have the machinery to regulate peripheral tolerance during health and rheumatoid arthritis. Int J Mol Sci. (2020) 21:5713. doi: 10.3390/ijms21165713
13. Hähnlein JS, Nadafi R, de Jong T, Ramwadhdoebe TH, Semmelink JF, Maijer KI, et al. Impaired lymph node stromal cell function during the earliest phases of rheumatoid arthritis. Arthritis Res Ther. (2018) 20:35. doi: 10.1186/s13075-018-1529-8
14. De Hair MJH, Van De Sande MGH, Ramwadhdoebe TH, Hansson M, Landewé R, Van Der Leij C, et al. Features of the synovium of individuals at risk of developing rheumatoid arthritis: implications for understanding preclinical rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:513–22. doi: 10.1002/art.38273
15. Van De Sande MGH, De Hair MJH, Van Der Leij C, Klarenbeek PL, Bos WH, Smith MD, et al. Different stages of rheumatoid arthritis: features of the synovium in the preclinical phase. Ann Rheum Dis. (2011) 70:772–7. doi: 10.1136/ard.2010.139527
16. van Baarsen LGM, de Jong TA, de Hair MJH, Semmelink JF, Choi IY, Gerlag DM, et al. Synovial gene signatures associated with the development of rheumatoid arthritis in at risk individuals: a prospective study. bioRxiv [Preprint]. (2021). doi: 10.1101/2021.04.27.440770
17. Page G, Miossec P. Paired synovium and lymph nodes from rheumatoid arthritis patients differ in dendritic cell and chemokine expression. J Pathol. (2004) 204:28–38. doi: 10.1002/path.1607
18. de Hair MJH, Zijlstra IAJ, Boumans MJH, van de Sande MGH, Maas M, Gerlag DM, et al. Hunting for the pathogenesis of rheumatoid arthritis: core-needle biopsy of inguinal lymph nodes as a new research tool. Ann Rheum Dis. (2012) 71:1911–2. doi: 10.1136/annrheumdis-2012-201540
19. Ramwadhdoebe TH, Hähnlein J, Maijer KI, van Boven LJ, Gerlag DM, Tak PP, et al. Lymph node biopsy analysis reveals an altered immunoregulatory balance already during the at-risk phase of autoantibody positive rheumatoid arthritis. Eur J Immunol. (2016) 46:2812–21. doi: 10.1002/eji.201646393
20. Rodríguez-Carrio J, Hähnlein JS, Ramwadhdoebe TH, Semmelink JF, Choi IY, van Lienden KP, et al. Brief report: altered innate lymphoid cell subsets in human lymph node biopsy specimens obtained during the at-risk and earliest phases of rheumatoid arthritis. Arthritis Rheumatol (Hoboken, NJ). (2017) 69:70–6. doi: 10.1002/art.39811
21. Ramwadhdoebe TH, Hähnlein J, van Kuijk BJ, Choi IY, van Boven LJ, Gerlag DM, et al. Human lymph-node CD8(+) T cells display an altered phenotype during systemic autoimmunity. Clin Transl Immunol. (2016) 5:e67. doi: 10.1038/cti.2016.8
22. Kumar B V, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
23. Kok L, Masopust D, Schumacher TN. The precursors of CD8+ tissue resident memory T cells: from lymphoid organs to infected tissues. Nat Rev Immunol. (2021). doi: 10.1038/s41577-021-00590-3.[Epub ahead of print].
24. Kok L, Dijkgraaf FE, Urbanus J, Bresser K, Vredevoogd DW, Cardoso RF, et al. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med. (2020) 217:e20191711. doi: 10.1084/jem.20191711
25. Audiger C, Rahman MJ, Yun TJ, Tarbell K V, Lesage S. The importance of dendritic cells in maintaining immune tolerance. J Immunol. (2017) 198:2223–31. doi: 10.4049/jimmunol.1601629
26. Osorio F, Fuentes C, López MN, Salazar-Onfray F, González FE. Role of dendritic cells in the induction of lymphocyte tolerance. Front Immunol. (2015) 6:535. doi: 10.3389/fimmu.2015.00535
27. Nadafi R, Gago de Graça C, Keuning ED, Koning JJ, de Kivit S, Konijn T, et al. Lymph node stromal cells generate antigen-specific regulatory T cells and control autoreactive T and B cell responses. Cell Rep. (2020) 30:4110–23.e4. doi: 10.1016/j.celrep.2020.03.007
28. Baptista AP, Roozendaal R, Reijmers RM, Koning JJ, Unger WW, Greuter M, et al. Lymph node stromal cells constrain immunity via MHC class II self-antigen presentation. Elife. (2014) 3:e04433. doi: 10.7554/eLife.04433
29. Ramwadhdoebe TH, Ramos MI, Maijer KI, van Lienden KP, Maas M, Gerlag DM, et al. Myeloid dendritic cells are enriched in lymph node tissue of early rheumatoid arthritis patients but not in at risk individuals. Cells. (2019) 8:756. doi: 10.3390/cells8070756
30. Harlé G, Kowalski C, Garnier L, Hugues S. Lymph node stromal cells: Mapmakers of t cell immunity. Int J Mol Sci MDPI AG. (2020) 21:1–18. doi: 10.3390/ijms21207785
31. Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, et al. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med. (2010) 207:689–97. doi: 10.1084/jem.20092642
32. Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A, et al. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med. (2010) 207:681–8. doi: 10.1084/jem.20092465
33. Lian J, Luster AD. Orchestrates the generation of adaptive immune responses. Curr Opin Cell Biol. (2015) 36:1–6. doi: 10.1016/j.ceb.2015.05.003
34. Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, et al. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. (2007) 8:1076–85. doi: 10.1038/ni1499
35. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. (2007) 8:1255–65. doi: 10.1038/ni1513
36. Hähnlein JS, Ramwadhdoebe TH, Semmelink JF, Choi IY, Berger FH, Maas M, et al. Distinctive expression of T cell guiding molecules in human autoimmune lymph node stromal cells upon TLR3 triggering. Sci Rep. (2018) 8:1736. doi: 10.1038/s41598-018-19951-5
37. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
38. van Pul KM, Fransen MF, van de Ven R, de Gruijl TD. Immunotherapy goes local: the central role of lymph nodes in driving tumor infiltration and efficacy. Front Immunol. (2021) 12:643291. doi: 10.3389/fimmu.2021.643291
39. Fransen MF, Schoonderwoerd M, Knopf P, Camps MGM, Hawinkels LJAC, Kneilling M, et al. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight. (2018) 3:e124507. doi: 10.1172/jci.insight.124507
40. Miron M, Kumar B V, Meng W, Granot T, Carpenter DJ, Senda T, et al. Human lymph nodes maintain TCF-1(hi) memory t cells with high functional potential and clonal diversity throughout life. J Immunol. (2018) 201:2132–40. doi: 10.4049/jimmunol.1800716
41. Humby F, Lewis M, Ramamoorthi N, Hackney JA, Barnes MR, Bombardieri M, et al. Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients. Ann Rheum Dis. (2019) 78:761–72. doi: 10.1136/annrheumdis-2018-214539
42. Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol. (2013) 25:334–44. doi: 10.1097/BOR.0b013e32835fd8eb
43. Lewis MJ, Barnes MR, Blighe K, Goldmann K, Rana S, Hackney JA, et al. Molecular portraits of early rheumatoid arthritis identify clinical and treatment response phenotypes. Cell Rep. (2019) 28:2455–70.e5. doi: 10.1016/j.celrep.2019.07.091
44. Dennis GJ, Holweg CTJ, Kummerfeld SK, Choy DF, Setiadi AF, Hackney JA, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther. (2014) 16:R90. doi: 10.1186/ar4555
45. Ponchel F, Burska AN, Hunt L, Gul H, Rabin T, Parmar R, et al. T-cell subset abnormalities predict progression along the Inflammatory Arthritis disease continuum: implications for management. Sci Rep. (2020) 10:3669. doi: 10.1038/s41598-020-60314-w
46. Lübbers J, van Beers-Tas MH, Vosslamber S, Turk SA, de Ridder S, Mantel E, et al. Changes in peripheral blood lymphocyte subsets during arthritis development in arthralgia patients. Arthritis Res Ther. (2016) 18:205. doi: 10.1186/s13075-016-1102-2
47. Guo Y, Walsh AM, Canavan M, Wechalekar MD, Cole S, Yin X, et al. Immune checkpoint inhibitor PD-1 pathway is down-regulated in synovium at various stages of rheumatoid arthritis disease progression. PLoS One. (2018) 13:e0192704. doi: 10.1371/journal.pone.0192704
48. Canavan M, Floudas A, Veale DJ, Fearon U. The PD-1:PD-L1 axis in Inflammatory Arthritis. BMC Rheumatol. (2021) 5:1. doi: 10.1186/s41927-020-00171-2
49. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. (2017) 542:110–4. doi: 10.1038/nature20810
50. Petrelli A, Mijnheer G, Hoytema van Konijnenburg DP, van der Wal MM, Giovannone B, Mocholi E, et al. PD-1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. J Clin Invest. (2018) 128:4669–81. doi: 10.1172/JCI96107
51. Higashioka K, Yoshimura M, Sakuragi T, Ayano M, Kimoto Y, Mitoma H, et al. Human PD-1(hi)CD8(+) T Cells Are a Cellular Source of IL-21 in Rheumatoid Arthritis. Front Immunol. (2021) 12:654623. doi: 10.3389/fimmu.2021.654623
52. van de Sande MGH, de Hair MJH, Schuller Y, van de Sande GPM, Wijbrandts CA, Dinant HJ, et al. The features of the synovium in early rheumatoid arthritis according to the 2010 ACR/EULAR classification criteria. PLoS ONE. (2012) 7:e36668. doi: 10.1371/journal.pone.0036668
53. Van De Sande MGH, Thurlings RM, Boumans MJH, Wijbrandts CA, Modesti MG, Gerlag DM, et al. Presence of lymphocyte aggregates in the synovium of patients with early arthritis in relationship to diagnosis and outcome: Is it a constant feature over time? Ann Rheum Dis. (2011) 70:700–3. doi: 10.1136/ard.2010.139287
54. Mousavi MJ, Karami J, Aslani S, Tahmasebi MN, Vaziri AS, Jamshidi A, et al. Transformation of fibroblast-like synoviocytes in rheumatoid arthritis; from a friend to foe. Autoimmun Highlights. (2021) 12:3. doi: 10.1186/s13317-020-00145-x
55. Turner JD, Filer A. The role of the synovial fibroblast in rheumatoid arthritis pathogenesis. Curr Opin Rheumatol. (2015) 27:175–82. doi: 10.1097/BOR.0000000000000148
56. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. (2013) 9:24–33. doi: 10.1038/nrrheum.2012.190
57. Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol. (2013) 190:1297–303. doi: 10.4049/jimmunol.1202572
58. Ai R, Hammaker D, Boyle DL, Morgan R, Walsh AM, Fan S, et al. Joint-specific DNA methylation and transcriptome signatures in rheumatoid arthritis identify distinct pathogenic processes. Nat Commun. (2016) 7:11849. doi: 10.1038/ncomms11849
59. Ai R, Laragione T, Hammaker D, Boyle DL, Wildberg A, Maeshima K, et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat Commun. (2018) 9:1921. doi: 10.1038/s41467-018-04310-9
60. Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. (2007) 56:1497–506. doi: 10.1002/art.22573
61. Yoshitomi H. Regulation of immune responses and chronic inflammation by fibroblast-like synoviocytes. Front Immunol. (2019) 10:1–8. doi: 10.3389/fimmu.2019.01395
62. Filer A, Ward LSC, Kemble S, Davies CS, Munir H, Rogers R, et al. Identification of a transitional fibroblast function in very early rheumatoid arthritis. Ann Rheum Dis. (2017) 76:2105–12. doi: 10.1136/annrheumdis-2017-211286
63. Juarez M, McGettrick HM, Scheel-Toellner D, Yeo L, Spengler J, de Paz B, et al. DKK1 expression by synovial fibroblasts in very early rheumatoid arthritis associates with lymphocyte adhesion in an in vitro flow co-culture system. Arthritis Res Ther. (2016) 18:14. doi: 10.1186/s13075-016-0915-3
64. Bergström B, Lundqvist C, Vasileiadis GK, Carlsten H, Ekwall O, Ekwall A-KH. The rheumatoid arthritis risk gene AIRE is induced by cytokines in fibroblast-like synoviocytes and augments the pro-inflammatory response. Front Immunol. (2019) 10:1384. doi: 10.3389/fimmu.2019.01384
65. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. (2019) 20:928–42. doi: 10.1038/s41590-019-0378-1
66. Berthelot JM, Bataille R, Maugars Y, Prost A. Rheumatoid arthritis as a bone marrow disorder. Semin Arthritis Rheum. (1996) 26:505–14. doi: 10.1016/S0049-0172(96)80039-4
67. Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F, et al. Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann Rheum Dis. (2014) 73:854–60. doi: 10.1136/annrheumdis-2012-202958
68. Nakagawa S, Toritsuka Y, Wakitani S, Denno K, Tomita T, Owaki H, et al. Bone marrow stromal cells contribute to synovial cell proliferation in rats with collagen induced arthritis. J Rheumatol. (1996) 23:2098–103.
69. Shi Y, Hu G, Su J, Li W, Chen Q, Shou P, et al. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell Res. (2010) 20:510–8. doi: 10.1038/cr.2010.44
70. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. (2012) 12:383–96. doi: 10.1038/nri3209
71. Frenette PS, Pinho S, Lucas D, Scheiermann C. Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu Rev Immunol. (2013) 31:285–316. doi: 10.1146/annurev-immunol-032712-095919
72. Munir H, Ward LSC, McGettrick HM. Mesenchymal stem cells as endogenous regulators of inflammation. Adv Exp Med Biol. (2018) 1060:73–98. doi: 10.1007/978-3-319-78127-3_5
73. Kastrinaki M-C, Sidiropoulos P, Roche S, Ringe J, Lehmann S, Kritikos H, et al. Functional, molecular and proteomic characterisation of bone marrow mesenchymal stem cells in rheumatoid arthritis. Ann Rheum Dis. (2008) 67:741–9. doi: 10.1136/ard.2007.076174
74. Papadaki HA, Kritikos HD, Gemetzi C, Koutala H, Marsh JCW, Boumpas DT, et al. Bone marrow progenitor cell reserve and function and stromal cell function are defective in rheumatoid arthritis: evidence for a tumor necrosis factor alpha-mediated effect. Blood. (2002) 99:1610–9. doi: 10.1182/blood.V99.5.1610
75. Papadaki HA, Kritikos HD, Valatas V, Boumpas DT, Eliopoulos GD. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: improvement following anti-tumor necrosis factor-alpha antibody therapy. Blood. (2002) 100:474–82. doi: 10.1182/blood-2002-01-0136
76. Bauer ME. Accelerated immunosenescence in rheumatoid arthritis: impact on clinical progression. Immun Ageing. (2020) 17:6. doi: 10.1186/s12979-020-00178-w
77. Chalan P, van den Berg A, Kroesen B-J, Brouwer L, Boots A. Rheumatoid arthritis, immunosenescence and the hallmarks of aging. Curr Aging Sci. (2015) 8:131–46. doi: 10.2174/1874609808666150727110744
78. Goronzy JJ, Weyand CM. Mechanisms underlying T cell ageing. Nat Rev Immunol. (2019) 19:573–83. doi: 10.1038/s41577-019-0180-1
79. Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM, et al. cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. (2000) 97:9203–8. doi: 10.1073/pnas.97.16.9203
Keywords: rheumatoid arthritis, lymph node, synovium, tolerance, fibroblasts, T cells, pre-RA
Citation: O'Byrne AM, de Jong TA and van Baarsen LGM (2022) Bridging Insights From Lymph Node and Synovium Studies in Early Rheumatoid Arthritis. Front. Med. 8:820232. doi: 10.3389/fmed.2021.820232
Received: 22 November 2021; Accepted: 21 December 2021;
Published: 14 January 2022.
Edited by:
Aurélie Najm, University of Glasgow, United KingdomReviewed by:
Martin Helmut Stradner, Medical University of Graz, AustriaCopyright © 2022 O'Byrne, de Jong and van Baarsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa G. M. van Baarsen, ZS5nLnZhbmJhYXJzZW5AYW1zdGVyZGFtdW1jLm5s
†These authors have contributed equally to this work and share first authorship