 Shigeru Iwata
Shigeru Iwata Yoshiya Tanaka
Yoshiya Tanaka- First Department of Internal Medicine, University of Occupational and Environmental Health, Kitakyushu, Japan
Systemic lupus erythematosus (SLE) is an autoimmune disease that causes multiple organ damage in women of childbearing age and has a relapsing-remitting course. SLE is caused by the interaction between genetic and environmental factors, however, its underlying triggers remain unknown. Among the environmental factors, the involvement of infections as a trigger for SLE, especially those of viral etiology, has been widely reported. Human endogenous retroviruses (HERVs) may put patients at a genetic predisposition to SLE, while the Epstein-Barr virus (EBV) may play a role as an environmental factor that triggers the development of SLE. It has been suggested that EBV-infected B-cells may become resistant to apoptosis, resulting in the activation, proliferation, and antibody production of autoreactive B-cells, which cause tissue damage in SLE. However, the interaction between the virus and immune cells, as well as the impact of the virus on the differentiation and dysfunction of immune cells, remain unclear. In this review, we focus on the relationship between the development and pathogenesis of SLE and viral infections, as well as the mechanism of SLE exacerbation via activation of immune cells, such as B-cells, based on the latest findings.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease that causes multiple organ damage in women of childbearing age and has a relapsing-remitting course. In the pathogenesis of SLE, dysfunction of DNase I leads to impaired clearance of nucleic acids. Neutrophil extracellular traps (NETs) contain endogenous DNA released by neutrophils remaining in the body. As a result, plasmacytoid dendritic cells (pDCs) and myeloid dendritic cells (mDCs) are activated via Toll-like receptor (TLR) stimulation, leading to the production of large amounts of IFN-α. Dendritic cells induce helper T (Th) cell and B-cell differentiation as well as antibody production, either directly or through IFN-α production, resulting in progressive tissue damage by immune complex formation and deposition. Although it has been suggested that SLE is caused by the interaction of genetic and environmental factors, the underlying triggers of SLE remain unknown. Among the environmental factors, infections such as viruses, bacteria, parasites, and fungi play an important role in the development of autoimmune diseases. In particular, the involvement of viral infections in the development of SLE has been reported (1).
Viral infections have been implicated in the pathogenesis of autoimmune diseases through a variety of mechanisms, including structural or functional molecular mimicry/cross-reactivity, innate immune activation by IFN production, epigenetic factors, superantigen generation, bystander activation, regulation of apoptosis and clearance, and epitope spreading (2–14). However, the interactions between viruses and immune cells and the effects of viruses on the differentiation and dysfunction of immune cells are still unclear. In this review, we focus on the interaction of viruses with the host immune system and the mechanism of SLE exacerbation via activation of immune cells, mainly B-cells, based on the latest findings.
Regulation Mechanism of B-Cell Differentiation and Function in SLE Pathogenesis
B-cells play an important role in autoimmune diseases through the production of antibodies and cytokines, as well as antigen presentation. Under normal conditions, self-tolerance induction mechanisms are activated during B-cell differentiation to eliminate or inactivate self-reactive B-cells that express antigen receptors (BCRs) that recognize self-antigens. Self-tolerance induction mechanisms are known to include clonal loss, receptor editing, anergy, and lack of co-stimulation and regulation by regulatory cells. One and 2 occur mainly in the central nervous system, while 3 and 4 occur mainly in the periphery (15). However, in autoimmune diseases such as SLE, self-tolerance may be disrupted by environmental factors such as viruses, in addition to genetic predisposition, with activation, proliferation, class switching, and antibody production possibly induced by external factors other than B-cells, such as dendritic cells and Th cells, as well as by endogenous dysfunction of B-cells themselves.
B-cells are activated and differentiated by co-stimulatory molecular signals, such as B-cell receptors (BCR), CD40/CD40L, Toll-like receptor (TLR) 7 and 9, as well as cytokine signals, such as IL-4, IL-21, IFN-α, and IFN-γ. As a result, BCR signals through Syk, Btk, and PLC-γ, TLR signals through MyD88, TRAF6, and NFκB, and cytokine signals through JAK-STAT are activated, and the expression of transcription factors important for differentiation, such as BACH2, BCL6, XBP1, PRDM1, and IRF4 are induced, leading to plasmablast differentiation (16–18). We have reported that activated memory B-cells [IgD-CD27-double-negative (DN) memory, class-switched memory B-cells], and plasmablasts are deeply involved in the pathogenesis of SLE (19).
Th cells include a population of cells that play a particularly important role in B-cell differentiation. Follicular helper T cells (TFHs) are widely recognized. Recently, CXCR5−PD1hiCD4+ peripheral helper T cells (TPH) have been reported to increase in the synovial tissue of patients with rheumatoid arthritis (20). TPHs strongly express PD-1 and induce B-cell differentiation and antibody production in an IL-21-dependent manner (21). TFH cells express BCL6 and CXCR5, while TPH cells do not express CXCR5. TPH cells do express BLIMP1 and migrate to the peripheral tissue through the expression of chemokine receptors such as CCR2, CX3CR1, and CCR5. Caielli et al. also found that CXCR5−CXCR3+PD-1hiCD4+ helper T cells, which have a different ability to induce B-cell differentiation than either TFH or TPH, are increased in both the peripheral blood of SLE patients and the tubulointerstitial region of patients with proliferative lupus nephritis (22). This population exhibited a gene expression profile that was also different from that of TFH, and induced B-cell differentiation in an IL-21, CXCL13-independent, IL-3, IL-10-dependent manner, especially with succinate. The population was found to be a distinct subset from TFH and TPH and was proposed as Th10 cells.
The effector functions of CD8+ T cells in SLE patients, such as granzyme B and perforin production, are impaired (23). The disease is exacerbated when perforin is deleted in lupus-prone mice (24). In a murine model of graft vs. host disease, perforin- and FasL-dependent cytotoxicity by cytotoxic T lymphocytes (CTLs) is important for the regulation of autoreactive B-cells (25, 26). Enhancing CTL cytotoxicity suppresses B-cell autoreactivity and regulates SLE disease severity (27, 28). These results suggest that CTLs may be involved in the regulation of SLE.
Pathological Mechanism of SLE and Its Relationship to Viral Infection
Nucleic acids are involved in the pathogenesis of SLE as pathogen-associated molecular patterns (PAMPs) derived from viruses and bacteria (29–34), or damage-associated molecular patterns (DAMPs) of host origin (35–40). The immune system detects these molecules appropriately in the cytoplasm and extracellular space via pattern recognition receptors (PRRs). In the absence of infection, endogenous nucleic acids and PRRs are compartmentalized to prevent inappropriate activation of the immune system by these danger signals (41). In SLE, nucleic acids and proteins that bind to nucleic acids are the major self-antigens. The main PRRs involved in the pathogenesis of SLE are Toll-like receptor (TLR) 7 and TLR9, which recognize double-stranded RNA and DNA rich in hypomethylated CpGs, respectively (42, 43). In fact, Smith's antigen (Sm), a complex of ribonucleoprotein and non-histone nuclear RNA, as well as double-stranded DNA (dsDNA), are detected in patients with SLE (44). In SLE, PRR-mediated molecular mimicry/cross-reactivity between virus-derived nucleic acids (PAMPs) and host-derived nucleic acids (DAMPs) may lead to autoimmunity.
Overexpression of type 1 IFN-related genes, termed the “IFN signature,” has been observed in many SLE patients, and has been implicated in the etiology and pathogenesis of the disease (45). IFNα is involved in the pathogenesis and exacerbation of SLE through a variety of mechanisms, including direct and indirect effects on antigen-presenting cells, T cells, and B-cells. IFN-α is produced by plasmacytoid dendritic cells (pDCs) stimulated by viruses. IFN-α induces TLR7 expression in B-cells, and stimulation with IFN-α and TLR7 induces plasmablast differentiation (46). It has been suggested that type 1 IFN production induced by viral infection may promote these responses. Recently, the interaction between viruses and their ability to induce epigenetic modifications within the immune system has attracted attention. Epigenetic mechanisms allow the virus to create gene expression profiles that predispose the host to autoimmunity, such as altered DNA methylation in SLE susceptibility genes (e.g., IFN-related genes), histone modifications, and regulation by miRNAs (12–14).
During development of SLE, viruses produce superantigens that bind to T-cell receptor variable regions and major histocompatibility complex class II molecules. In addition, viruses promote the proliferation of autoreactive and memory T cells through a process of called “bystander activation,” which is T-cell receptor-independent and cytokine-dependent (2, 47). Viruses are involved in the survival of immune cells through the regulation of apoptosis and clearance. For example, it has been suggested that B-cells infected with EBV may become resistant to apoptosis. In addition, epitope spreading gradually leads to the production of antibodies against epitopes near the initial epitope, resulting in a diversity in antigen recognition by antibodies. As a result, self-reactive B-cells are activated, proliferate, and produce antibodies, leading to tissue damage. The impact of EBV on B-cell differentiation in the pathogenesis of SLE will be discussed in detail in Section 5.
Retroviruses, particularly human endogenous retroviruses (HERVs), have been implicated to cause genetic predisposition to SLE pathogenesis (48). In addition, viral infections with Epstein-Barr virus (EBV) may play a role as an environmental factor in triggering SLE pathogenesis (49). In particular, HERVs and EBVs involved in SLE pathogenesis are reviewed in the following sections.
Human Endogenous Retrovirus Infection as a Trigger for SLE
Human endogenous retroviruses (HERVs) have sequences similar to those found in the germline genomes of non-virus organisms (endogenous virus-like sequences). HERVs have been stably integrated into the host mammalian genome for 40 million years (50). HERVs are recognized as self-antigens, triggering the development of autoimmune diseases. HERVs are composed of three coding regions: gag, pol, and env. It also has a long terminal repeat (LTR) containing transcriptional regulatory sequences at both ends, as well as encodes proteases and reverse transcriptases. In the human genome, only about 1.5% of gene regions have the potential to be translated into proteins, but as much as 8% of the genome is encoded by fossilized retroviruses (51). Environmental factors (infections, estrogens, drugs, chemicals, ultraviolet radiation, etc.) can epigenetically reactivate the transcription of endogenous retroviral sequences. As many as 200 endogenous retroviral sequences have been reported, some of which (HRES-1, HERV-E 4-1, HERV-K10, HERV-K18 HERVs) are related to the development of SLE through: (1) autoantibody production by molecular mimicry, (2) stimulation of intracellular sensor molecules by viral nucleic acids, and (3) epigenetic regulation of host genes (52, 53).
Direct Stimulation of Intracellular Sensor Molecules by Viral Nucleic Acids
Binding of viral nucleic acids to intracellular sensor molecules induces the activation of a variety of pathways, all converging on a type 1 IFN response (54). Retroviruses are detected by TLRs in endosomes, which recognize dsRNA (TLR-3), ssRNA (TLR-7 and−8), and dsDNA (TLR-9) (55). In addition to TLRs in endosomes, retroviral nucleic acids are also sensitized by other PRRs in the cytoplasm (56). Although the nucleic acid sensing system is physiologically necessary to control viral infections, excessive activation may promote the progression of SLE by stimulating the production of pathogenic cytokines (57). The amount of mRNA from the HERV clone 4-1 gag region was significantly higher in SLE patients than in healthy subjects, and this amount was decreased with glucocorticoid treatment (48). However, there is no direct evidence of an association between HERV and TLRs in SLE. Further studies are warranted.
Autoantibody Production by Molecular Mimicry
HERV proteins cross-react with self-antigens (58). For example, HRES-1 encodes the nuclear autoantigen HRES-1/p28, which is targeted by cross-reactive antiviral antibodies (59). HRES-1 also encodes HRES/Rab4, which is integrated into chromosome 1q42, the SLE susceptibility locus, and is markedly overexpressed in T cells from patients with SLE (60). Overexpression of HRES-1/Rab 4 interferes with endosomal recycling of the CD3/TCRζ chain in SLE T cells, leading to decreased expression of the receptor and lowering of the threshold for T-cell activation (61, 62). In SLE CD4+ T-cells, hypomethylation of HERV-E 4-1 terminal repeats was observed, and HERV-E 4-1 mRNA expression was higher in SLE patients than in healthy controls, which was related to disease activity (63). Anti-HERV-K envelope antibodies have been identified in patients with SLE. The envelope glycoprotein 70 (gp70) and viral nucleoprotein complexes form a retroviral gp70-anti-gp70 immune complex upon TLR7 stimulation, and induce nephropathy in mouse models (64). Thus, there are a number of reports linking HERVs to the onset and pathogenesis of SLE, albeit indirectly.
Epigenetic Regulation of Host Immune Genes
LTRs, which are often isolated sequences, are present in large numbers in the human genome, and regulate the transcription of adjacent genes. Some LTRs contain promoter/enhancer sequences that generally have little effect on gene transcription, thus preserving their regulatory activity. However, when the epigenetic mechanisms that normally regulate them are altered, their activity is pathologically increased. This epigenetic reset can occur in autoimmunity and cancer, resulting in LTR-dependent transcription of immune genes and reactivation of HERVs (52). MER 41, an LTR belonging to the HERV family, is a STAT1 binding site located near the IFN-γ-inducible absent in melanoma 2 (AIM2) gene and functions as its promoter (65). Javierre et al. reported the first high-throughput and candidate sequence analyses of DNA methylation to investigate the discordance of SLE in twins. SLE patients had decreased methylation of the AIM2 locus and increased AIM2 expression compared to healthy twins (66). Thus, HERV may be related to the epigenetic dysregulation of AIM2 in the pathogenesis of SLE.
EB Virus Infection as a Trigger for SLE
EBV is latent in memory B-cells (95% of the world population). After infection by saliva, it first infects pharyngeal epithelial cells, followed by quiescent B-cells, T cells, NK cells, and neutrophils (67). Primary infection in childhood is usually asymptomatic, but it can cause infectious mononucleosis in adolescents, and is also associated with Hodgkin's lymphoma and autoimmune diseases (47, 68–72). EBV is latent in the cells of diseased individuals, but can reactivate at any time, affecting innate and acquired immunity. A relationship between pediatric SLE and EBV-DNA in peripheral blood, as well as a relationship between adult SLE and history of EBV exposure, have been reported (73). In a recent study, higher levels of antibodies against early EBV infection were detected in patients with SLE than in healthy subjects, and EBV reactivation was correlated with lymphopenia (74). Seropositivity for anti-EBV antibodies is much higher in SLE (75–77). Two recent meta-analyses have confirmed a strong association between serum anti-EBV antibody (EBNA-1 and VCA IgG) positivity and SLE development, and that prior EBV infection is a prerequisite for SLE development, at least in some ethnic groups (78, 79). On many of these grounds, EBV, in particular, is considered to be one of the potent triggers for the development of SLE (80, 81).
Abnormalities in B-Cell Differentiation and Function Caused by EBV
It has been suggested that EBV-infected B-cells may become resistant to apoptosis. As a result, they induce the activation, proliferation, and antibody production of autoreactive B-cells, which damage tissues. EBV induces activation of naive B-cells similar to physiological signaling, such as the BCR signaling pathway via LMP (latent membrane protein) 2A (82) and CD40 stimulation via LMP1 (83). Cross-reactivity has been shown between the PPPGRRP sequence of EBNA-1 and PPPGMRPP, which may be the first epitope of the SmB antibody response, as well as between the GGSGSGPRHDGVRR sequence of EBNA-1 and TKYKQRHGWSHKD, which may be the first epitope of the anti-Ro antibody response (84). SLE-associated autoantibodies, such as anti-Sm antibodies, are commonly produced during infectious mononucleosis. Furthermore, EBV acts as a superantigen that stimulates a large number of T-cells, and exerts a number of mechanisms to induce autoimmunity.
Both lytic and latent EBV proteins elicit potent B- and T-cell responses. EBV induces high cytokine production by activated CD4 + cells, an increase in CD4 + EBV-specific cells producing IFN, and a decrease in CD8 + cell responses (85). The proliferation of EBV-infected B-cells is physiologically regulated by CD8 + cytotoxic T- cells (CTLs). However, in SLE, EBV-specific CD8 + CTLs are dysfunctional (86–88), blood EBV viral levels are chronically increased (89–91), and CTL responses by exhausted T-cells with increased PD-1 expression due to persistent EBV infection are reduced, possibly evading immune regulation (92). Sustained activation of the viral cycle increases the risk of both EBV-related malignant transformation and autoimmune disease in genetically predisposed individuals. Indeed, there is an inverse correlation between impaired function of EBV-specific CTLs and serum antibodies to soluble antigens (anti-EA/D antibodies), disease activity, and autoantibodies (86–88). Impaired EBV control by CTLs and persistent latent EBV infection are the major causes of widespread immunological changes that characterize SLE. Indeed, there are reports of 10- to 40-fold increases in EBV viral levels in peripheral blood B-cells and peripheral blood mononuclear cells from SLE patients compared to healthy controls, regardless of their receipt of immunosuppressive therapy (93–95).
The Relevance of Immunometabolism to B-Cell Differentiation via EBV Infection
Recently, “immunometabolism” has attracted much attention. Activation and differentiation of immune cells requires massive energy production, such as ATP, and biosynthesis of bioconstituents, such as nucleic acids and lipids, by metabolic transformation. It has been reported that cellular metabolic transformation to anabolism, including aerobic glycolysis, is important for immune cell activation, mainly in mice. Metabolic pathways involving glycolysis, oxidative phosphorylation, pentose phosphate circuitry, fatty acid β-oxidation, fatty acid synthesis, and amino acid metabolism (glutaminolysis) are involved in the production of products important for cell survival and growth, and are regulated by metabolic regulators such as mTOR, AMPK, HIF1α, and c-myc (96). Studies on the involvement of intracellular metabolism in B-cell activation have been reported using mouse models as follows: enhanced PI3K-Akt-mTORC1 signaling, glycolysis, and oxidative phosphorylation induce de novo lipid synthesis, which is important for B-cell proliferation and growth (97). In addition, the forced expression of mTORC1 promotes plasmablast differentiation. mTORC1 is activated in B-cells of a mouse model of lupus, while rapamycin inhibits B-cell proliferation and survival (98, 99). B-cell activating factor (BAFF) and its homolog a proliferation inducing ligand (APRIL) support B-cell differentiation and plasmablast differentiation. In vitro studies have shown that rapamycin inhibits BAFF-mediated proliferation and survival (99). Overexpression of BAFF increased lupus-like autoantibodies and enhanced glycolysis in B-cells in a transgenic mouse model (97). However, the role of B-cell metabolism in SLE, as well as its pathogenesis, remains unknown.
EBV acts on B-cell metabolism, especially in mitochondria and inhibits apoptosis. EBV encodes proteins that target mitochondria, including BHRF1 (BamHI-H right reading frame), BZLF1 (also known as Zebra protein), BALF1 (BamHI-A left frame transcript), LMP2A, and immediate-early Zta protein. BHRF1 accumulates in the outer mitochondrial membrane (OMM) of B-cells, preventing apoptosis, promoting the survival of EBV-infected cells, and viral replication. LMP2A induces mitochondrial fission in a Drp1-dependent manner (100, 101). Immediate early Zta protein blocks mitochondrial DNA replication and promotes viral replication (102).
Mrozek-Gorska et al. confirmed that EBV also causes significant changes in the cellular content of macromolecules such as proteins and RNA, as well as increased glucose uptake, enhanced glycolysis and mitochondrial functions, and the activation of germinal center-like differentiation programs only days after infection, with transcription factors and phenotypes similar to those of plasmablasts and early plasma cells (103). Wang et al. also confirmed that EBV modifies metabolic pathways to support rapid proliferation of B-cells, confirming that EBV induces mitochondrial remodeling and carbon monoxide (1C) metabolism. EBV-encoded EBNA2 and its target MYC enhance MTHFD2 expression, a central mitochondrial 1C enzyme, and plays a critical role in B-cell survival via nucleic acid synthesis, NADPH generation, and redox maintenance (104).
Immunometabolism in the Process of B-Cell Differentiation in SLE
EBV-induced expression of the latent protein LMP2A activates the PI3K/Akt/mTORC1 pathway (105, 106). However, the mechanism of activation and differentiation via mTOR in B-cells, and its involvement in the pathogenesis of SLE, remains unclear. We investigated the effects of IFN-α and a nucleic acid on the intracellular metabolism and differentiation of B-cells from SLE patients. p-mTOR expression levels were increased in B-cells from SLE patients compared to healthy controls and was correlated with the percentage of plasmablasts and disease activity (107). Activation of mTORC1 is regulated by various environmental signals, including amino acid levels. We found that methionine, an essential amino acid, is important for CpG- and IFN-α-induced plasmablast differentiation. In the presence of methionine, activation of mTORC1 induces the methyltransferase EZH2, which represses BACH2 expression via induction of H3K27me3 in the gene expression region of BACH2, consequently promoting BLIMP1 and XBP1 expression and plasmablast differentiation (Figure 1). B-cells from SLE patients overexpressed EZH2, which correlated with disease activity and autoantibody production (108) (Figure 1), and the expression level of DiOc6, which indicates hyperpolarization of the mitochondrial membrane, was higher in B-cells from SLE patients than in healthy subjects. In vitro stimulation of CD19 + cells with CpG and IFNα-induced plasmablast differentiation led to increased glycolysis, oxidative phosphorylation, and DiOc6 expression. The glutaminase inhibitor BPTES, which is a limited inhibitor of glutamine degradation, decreased DiOc6 expression, oxidative phosphorylation, ROS production, and ATP production, as well as suppressed plasmablast differentiation and antibody production. Metformin inhibited CpG- and IFNα-induced glutamine uptake, as well as suppressed plasmablast differentiation and antibody production (Figure 1) (109). In SLE patients, B-cells are activated and differentiated by viral and autoantigen-derived nucleic acids and IFN-α stimulation through the uptake of amino acids such as methionine and glutamine, which are related to the pathogenesis of SLE.
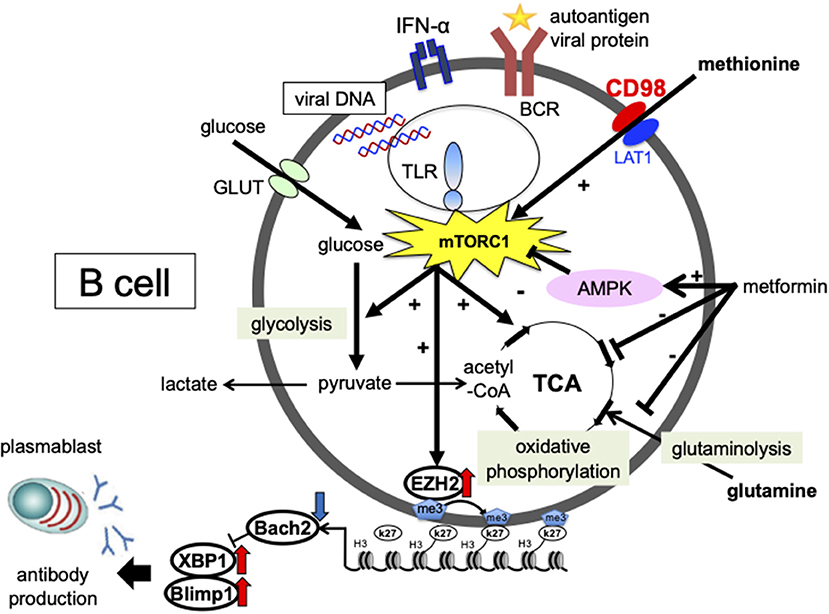
Figure 1. Involvement of cellular metabolism in the process of plasmablast differentiation in SLE. The glucose metabolic pathway consists of the glycolytic system, which converts glucose to pyruvate, and oxidative phosphorylation, which generates energy via the tricarboxylic acid (TCA) cycle. In the mitochondria, pyruvate is converted to acetyl-CoA, which enters the tricarboxylic acid cycle. Energy is also generated by glutamine metabolism. During anaerobic metabolism, pyruvate is converted into lactate. The essential amino acid methionine is strongly committed to plasmablast differentiation. In the presence of methionine, mTORC1 activation induces the expression of the methyltransferase enhancer zeste homolog 2 (EZH2). EZH2 induces H3K27me3 at BTB and CNC homolog 2 (BACH2) loci and suppresses BACH2 expression, leading to the induction of B lymphocyte-induced maturation protein-1 (BLIMP1) and X-box binding protein 1 (XBP1) expression and plasmablast differentiation. Metformin inhibited CpG- and IFN-α-induced glutamine uptake, mitochondrial function, and suppressed plasmablast differentiation. Enhanced cellular metabolism mediated by amino acids, such as methionine and glutamine, is important for plasmablast differentiation, which may be a potential therapeutic target for SLE.
Discussion
Viral infections are important environmental factors in the pathogenesis of SLE, and can trigger disease onset and relapse, as well as play an important role in altering clinical phenotypes. A wide range of viral-mediated immunomodulatory mechanisms have become increasingly evident. These mechanisms can profoundly alter the host immune system and interact to influence B-cell differentiation in SLE pathogenesis, such as the “immortalization” of autoreactive B-cells by EBV. Studies on the epigenetic regulation of IFN-responsive genes by reactivated HERVs have provided important insights into the missing link between environmental factors and genetic predisposition. In addition to hydroxychloroquine (HCQ), mycophenolate mofetil, anti-BAFF antibody belimumab, the anti-IFNAR antibody aniflorumab has been used for the treatment of SLE. Regarding the effect of HCQ on viral infections, HCQ has been shown to inhibit viral binding to receptors and entry into cells by inhibiting glycosylation and increasing pH during phagocytosis. However, in recent years, HCQ has not been recommended as a therapeutic agent for COVID-19 due to the lack of efficacy demonstrated for its effect on COVID-19 and many reports of concerns about side effects. In addition, aniflorumab is expected to be effective against SLE. However, there is also concern about the increased risk of viral infections because it suppresses the antiviral effects of type 1 IFN. Thus, a deeper understanding of the interaction between viral proteins and nucleic acids and the host's immune system is essential for the best use of therapeutic agents, such as TLR inhibitors, aniflorumab, and belimumab, as well as in considering the new concept of immunometabolism.
Author Contributions
SI wrote the manuscript. YT reviewed and edited the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by JSPS KAKENHI grant number #JP16K09928 and the University of Occupational and Environmental Health, Japan, through the UOEH Grant for Advanced Research (#H29-903 and #H30-905).
Conflict of Interest
YT received research grants from Mitsubishi-Tanabe, Takeda, Daiichi-Sankyo, Chugai, Bristol-Myers, MSD, Astellas, Abbvie, and Eisai.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tanaka Y. State-of-the-art treatment of systemic lupus erythematosus. Int J Rheum Dis. (2020) 23:465–71. doi: 10.1111/1756-185X.13817
2. Pan Q, Liu Z, Liao S, Ye L, Lu X, Chen X, et al. Current mechanistic insights into the role of infection in systemic lupus erythematosus. Biomed Pharmacother. (2019) 117:109122. doi: 10.1016/j.biopha.2019.109122
3. Kanduc D, Shoenfeld Y. From anti-EBV immune responses to the EBV diseasome via cross-reactivity. Glob Med Genet. (2020) 7:51–63. doi: 10.1055/s-0040-1715641
4. Dreyfus DH, Farina A, Farina GA. Molecular mimicry, genetic homology, and gene sharing proteomic “molecular fingerprints” using an EBV (Epstein-Barr Virus)-derived microarray as a potential diagnostic method in autoimmune disease. Immunol Res. (2018) 66:686–95. doi: 10.1007/s12026-018-9045-0
5. Tu J, Wang X, Geng G, Xue X, Lin X, Zhu Z, et al. The possible effect of B-cell epitopes of epstein-barr virus early antigen, membrane antigen, latent membrane protein-1, and−2A on. Systemic Lupus Erythematosus Front Immunol. (2018) 9:187. doi: 10.3389/fimmu.2018.00187
6. Migliorini P, Baldini C, Rocchi V, Bombardieri S. Anti-Sm and anti-RNP antibodies. Autoimmunity. (2005) 38:47–54. doi: 10.1080/08916930400022715
7. Cornaby C, Gibbons L, Mayhew V, Sloan C, Welling A, Poole BD. B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett. (2015) 163:56–68. doi: 10.1016/j.imlet.2014.11.001
8. Pacheco Y, Acosta-Ampudia Y, Monsalve DM, Chang C, Gershwin ME, Anaya JM. Bystander activation and autoimmunity. J Autoimmun. (2019) 103:102301. doi: 10.1016/j.jaut.2019.06.012
9. Barrat FJ, Su L. A pathogenic role of plasmacytoid dendritic cells in autoimmunity and chronic viral infection. J Exp Med. (2019) 216:1974–85. doi: 10.1084/jem.20181359
10. Fitzsimmons L, Kelly GL. EBV and apoptosis: the viral master regulator of cell fate? Viruses. (2017) 9:339. doi: 10.3390/v9110339
11. Zan H, Tat C, Casali P. MicroRNAs in lupus. Autoimmunity. (2014) 47:272–85. doi: 10.3109/08916934.2014.915955
12. Lanata CM, Chung SA, Criswell LA. DNA methylation 101: what is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci Med. (2018) 5:e000285. doi: 10.1136/lupus-2018-000285
13. Wang Z, Chang C, Peng M, Lu Q. Translating epigenetics into clinic: focus on lupus. Clin Epigenetics. (2017) 9:78. doi: 10.1186/s13148-017-0378-7
14. Long H, Yin H, Wang L, Gershwin ME, Lu Q. The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J Autoimmun. (2016) 74:118–38. doi: 10.1016/j.jaut.2016.06.020
15. Goodnow CC, Sprent J, Barbara Fazekas de St Groth BF, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. (2005) 435:590–7. doi: 10.1038/nature03724
16. Iwata S, Yamaoka K, Niiro H, Nakano K, Wang SP, Akashi K, et al. Amplification of Toll-like receptor-mediated signaling through spleen tyrosine kinase in human B-cell activation. J Allergy Clin Immunol. (2012) 129:1594–601. doi: 10.1016/j.jaci.2012.03.014
17. Kurosaki T, Shinohara H, Baba Y. B cell signaling and fate decision. Annu Rev Immunol. (2010) 28:21–55. doi: 10.1146/annurev.immunol.021908.132541
18. Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, et al. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus. erythematosus. Ann Rheum Dis. (2010) 69:305–8. doi: 10.1136/ard.2008.096495
19. Iwata S, Tanaka Y. B-cell subsets, signaling and their roles in secretion of autoantibodies. Lupus. (2016) 25:850–6. doi: 10.1177/0961203316643172
20. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. (2017) 542:110–14. doi: 10.1038/nature20810
21. Bocharnikov AV, Keegan J, Wacleche VS, Cao Y, Fonseka CY, Wang G, et al. PD1hiCXCR5− T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight. (2019) 4:130062. doi: 10.1172/jci.insight.130062
22. Caielli S, Veiga DT Balasubramanian P, Athale S, Domic B, Murat E, et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med. (2019) 25:75–81. doi: 10.1038/s41591-018-0254-9
23. Comte D, Karampetsou MP, Yoshida N, Kis-Toth K, Kyttaris VC, Tsokos GC, et al. Signaling lymphocytic activation molecule family member 7 engagement restores defective effector CD8+ T cell function in systemic lupus erythematosus. Arthrit Rheumatol. (2017) 69:1035–44. doi: 10.1002/art.40038
24. Peng SL, Moslehi J, Robert ME, Craft J. Perforin protects against autoimmunity in lupus-prone mice. J Immunol. (1998) 160:652–60.
25. Soloviova K, Puliaiev M, Puliaev R, Irina Puliaeva I, Via CS. Both perforin and FasL are required for optimal CD8 T cell control of autoreactive B cells and autoantibody production in parent-into-F1 lupus mice. Clin Immunol. (2018) 194:34–42. doi: 10.1016/j.clim.2018.06.007
26. Shustov A, Luzina I, Nguyen P, Papadimitriou JC, Handwerger B, Elkon KB, et al. Role of perforin in controlling B-cell hyperactivity and humoral autoimmunity. J Clin Invest. (2000) 106:R39–47. doi: 10.1172/JCI8876
27. Nguyen V, Rus H, Chen C, Rus V. CTL-promoting effects of IL-21 counteract murine lupus in the parent → F1 graft-versus-host disease model. J Immunol. (2016) 196:1529–40. doi: 10.4049/jimmunol.1501824
28. Puliaev R, Puliaeva I, Welniak LA, Ryan AE, Haas M, Murphy WJ, et al. CTL-promoting effects of CD40 stimulation outweigh B cell-stimulatory effects resulting in B cell elimination and disease improvement in a murine model of lupus. J Immunol. (2008) 181:47–61. doi: 10.4049/jimmunol.181.1.47
29. Medzhitov R. Approaching the asymptote: 20 years later. Immunity. (2009) 30:766–75. doi: 10.1016/j.immuni.2009.06.004
30. Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete. interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci USA. (1996) 93:2879–83. doi: 10.1073/pnas.93.7.2879
31. Sparwasser T, Koch ES, Vabulas RM, Heeg K, Lipford GB, Ellwart JW, et al. Bacterial DNA and immunostimulatory CpG oligonucleotides. trigger maturation and activation of murine dendritic cells. Eur J Immunol. (1998) 28:2045–54. doi: 10.1002/(SICI)1521-4141(199806)28:06<2045::AID-IMMU2045>3.0.CO
32. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature. (2000) 408:740–5. doi: 10.1038/35047123
33. Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via. species- specific CpG motif recognition. Proc Natl Acad Sci USA. (2001) 98:9237–42. doi: 10.1073/pnas.161293498
34. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9- mediated recognition of herpes simplex virus-2 by plasmacytoid. dendritic cells. J Exp Med. (2003) 198:513–20. doi: 10.1084/jem.20030162
35. Matzinger P. The danger model: a renewed sense of self. Science. (2002) 296:301–5. doi: 10.1126/science.1071059
36. Gallo PM, Gallucci S. The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol. (2013) 4:138. doi: 10.3389/fimmu.2013.00138
37. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
38. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. doi: 10.1084/jem.20151876
39. Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. (2018) 560:238–42. doi: 10.1038/s41586-018-0363-0
40. Malathi K, Dong B, Gale Jr M, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. (2007) 448:816–19. doi: 10.1038/nature06042
41. Gallucci S, Maffei ME. DNA sensing across the tree of life. Trends Immunol. (2017) 38:719–32. doi: 10.1016/j.it.2017.07.012
42. Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, et al. Critical role of TLR7 in the acceleration of systemic. lupus erythematosus in TLR9-deficient mice. J Autoimmun. (2010) 34:339–48. doi: 10.1016/j.jaut.2009.11.001
43. Gies V, Schickel JN, Jung S, Joublin A, Glauzy S, Knapp AM, et al. Impaired TLR9 responses in B cells from patients with systemic lupus. erythematosus. JCI Insight. (2018) 3:96795. doi: 10.1172/jci.insight.96795
44. Didier K, Bolko L, Giusti D, Toquet S, Robbins A, Antonicelli F, et al. Autoantibodies associated with connective tissue diseases: what. Meaning for clinicians? Front Immunol. (2018) 9:541. doi: 10.3389/fimmu.2018.00541
45. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 17:711–23. doi: 10.1084/jem.20021553
46. Marshak-RothsteinA. Toll-likereceptorsinsystemicautoimmunedisease. Nature Rev Immunol. (2006) 6:823–35. doi: 10.1038/nri1957
47. Rigante D, Mazzoni MBS, Esposito S. The cryptic interplay between systemic lupus erythematosus and infections. Autoimmun Rev. (2014) 13:96–102. doi: 10.1016/j.autrev.2013.09.004
48. Ogasawara H, Naito T, Kaneko H, Hishikawa T, Sekigawa I, Hashimoto H, et al. Quantitative analyses of messenger RNA of human endogenous retrovirus in SLE patients. J Rheumatol. (2001) 28:533–8.
49. James JA, Neas BR, Moser KL, Hall T, Bruner GR, Sestak AL, et al. Systemic lupus erythematosus in adults is associated with previous Epstein-Barr virus exposure. Arthrit Rheum. (2001) 44:1122–6. doi: 10.1002/1529-013144:5<1122::AID–ANR193>3.0.CO;2-D
50. Belshaw R, Pereira V, Katzourakis A, Talbot G, Pačes J, Burt A, et al. Long-term reinfection of the human genome by endogenous retroviruses. Proc Natl Acad Sci USA. (2004) 101:4894–99. doi: 10.1073/pnas.0307800101
51. Weiss RA. The discovery of endogenous retroviruses. Retrovirology. (2006) 3:67. doi: 10.1186/1742-4690-3-67
52. Talotta R, Atzeni F, Laska MJ. Retroviruses in the pathogenesis of systemic lupus erythematosus: are they potential therapeutic targets? Autoimmunity. (2020) 53:177–91. doi: 10.1080/08916934.2020.1755962
53. Greenig M. HERVs, immunity and autoimmunity: understanding the connection. Peer J. (2019) 7:e6711. doi: 10.7717/peerj.6711
54. Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular nucleic acid detection in autoimmunity. Annu Rev Immunol. (2017) 35:313–36. doi: 10.1146/annurev-immunol-051116-052331
55. Browne EP. The role of toll like receptors in retroviral infection. Microorganisms. (2020) 8:1787. doi: 10.3390/microorganisms8111787
56. Barral PM, Sarkar D, Su Z, Barber GN, DeSalle R, Racaniello VR, et al. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: key regulators of innate immunity. Pharmacol Ther. (2009) 124:219–234. doi: 10.1016/j.pharmthera.2009.06.012
57. Shehab M, Sherri N, Hussein H, Salloum N, Rahal EA. Endosomal toll-like receptors mediate enhancement of interleukin-17a production triggered by epstein-barr virus DNA in mice. J Virol. (2019) 93:e00987–19. doi: 10.1128/JVI.00987-19
58. Chen CJ, Lin KH, Lin SC, Tsai WC, Yen JH, Chang SJ, et al. High prevalence of immunoglobulin a antibody against epstein-barr virus capsid antigen in adult patients with lupus with disease flare: case control studies. J Rheumatol. (2005) 32:44–7.
59. Perl A, Nagy G, Koncz A, Gergely P, Fernandez D, Doherty E, et al. Molecular mimicry and immunomodulation by the HRES-1 endogenous retrovirus in SLE. Autoimmunity. (2008) 41:287–97. doi: 10.1080/08916930802024764
60. Godavarthy A, Kelly R, Jimah J, Beckford M, Caza T, Fernandez D, et al. Lupus-associated endogenous retroviral LTR polymorphism and epigenetic imprinting promote HRES-1/RAB4 expression and MTOR activation. JCI Insight. (2020) 5:e134010. doi: 10.1172/jci.insight.134010
61. Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4- regulated lysosomal degradation. J Immunol. (2009) 182:2063–73. doi: 10.4049/jimmunol.0803600
62. Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res. (2014) 2014:419029. doi: 10.1155/2014/419029
63. Wu Z, Mei X, Zhao D, Sun Y, Song J, Pan W, et al. DNA methylation modulates HERV-E expression in CD4+ T cells from systemic lupus erythematosus patients. J Dermatol Sci. (2015) 77:110–16. doi: 10.1016/j.jdermsci.2014.12.004
64. Baudino L, Yoshinobu K, Morito N, Santiago-Raber ML, Izui S. Role of endogenous retroviruses in murine SLE. Autoimmun Rev. (2010) 10:27–34. doi: 10.1016/j.autrev.2010.07.012
65. Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. (2016) 351:1083–7. doi: 10.1126/science.aad5497
66. Biola M, Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. (2010) 20:170–9. doi: 10.1101/gr.100289.109
67. Cohen J. Primary immunodeficiencies associated with EBV disease. Curr Top Microbiol Immunol. (2015) 390:241–65. doi: 10.1007/978-3-319-22822-8_10
68. Esposito S, Bosis S, Semino M, Rigante D. Infections and systemic lupus erythematosus. Eur J Clin Microbiol Infect Dis. (2014) 33:1467–75. doi: 10.1007/s10096-014-2098-7
69. Nelson P, Rylance P, Roden D, Trela M, Tugnet N. Viruses as potential pathogenic agents in systemic lupus erythematosus. Lupus. (2014) 23:596–605. doi: 10.1177/0961203314531637
70. Rigante D, Esposito S. Infections and systemic lupus erythematosus: binding or sparring partners? Int J Mol Sci. (2015) 16:17331–43. doi: 10.3390/ijms160817331
71. Illescas-Montes R, Corona-Castro CC, Melguizo-Rodríguez L, Ruiz C, Costela-Ruiz VJ. Infectious processes and systemic lupus erythematosus. Immunology. (2019) 158:153–60. doi: 10.1111/imm.13103
72. Lossius A, Johansen JN, Torkildsen Ø, Vartdal F, Holmy T. Epstein-barr virus in systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis—association and causation. Viruses. (2012) 4:3701–30. doi: 10.3390/v4123701
73. James J, Harley JB, Scofield RH. Role of epstein–barr virus in systemic lupus erythematosus. Curr Opin Rheumatol. (2006) 18:462–7. doi: 10.1097/01.bor.0000240355.37927.94
74. Rasmussen NS, Draborg AH, Nielsen CT, Jacobsen S, Houen G. Antibodies to early EBV, CMV, and HHV6 antigens in systemic lupus erythematosus patients. Scand J Rheumatol. (2015) 44:143–49. doi: 10.3109/03009742.2014.973061
75. James JA, Neas BR, Moser KL, Hall T, Bruner GR, Sestak AL, et al. Systemic lupus erythematosus in adults is associated with previous epstein-barr virus exposure. Arthrit Rheum. (2001) 44:1122–26. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D
76. Vista ES, Weisman MH, Ishimori ML, Chen H, Bourn RL, Bruner BF, et al. Strong viral associations with SLE among filipinos. Lupus Sci Med. (2017) 4:e000214. doi: 10.1136/lupus-2017-000214
77. Cui J, Yan W, Xu S, Wang Q, Zhang W, Liu W, et al. Anti-epstein-barr virus antibodies in beijing during 2013-2017: what we have found in the different patients. PLoS ONE. (2018) 13:e0193171. doi: 10.1371/journal.pone.0193171
78. Hanlon P, Avenell A, Aucott L, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between epstein-barr virus and systemic lupus erythematosus. Arthritis Res Ther. (2014) 16:R3. doi: 10.1186/ar4429
79. Li ZX, Zeng S, Wu HX. The risk of systemic lupus erythematosus associated with epstein-barr virus infection: a systematic review and meta-analysis. Clin Exp Med. (2019) 19:23–36. doi: 10.1007/s10238-018-0535-0
80. Caza T, Oaks Z, Perl A. Interplay of infections, autoimmunity, and immunosuppression in systemic lupus erythematosus. Int Rev Immunol. (2014) 33:330–63. doi: 10.3109/08830185.2013.863305
81. Kamen DL. Environmental influences on systemic lupus erythematosus expression. Rheum Dis Clin North Am. (2014) 40:401–12. doi: 10.1016/j.rdc.2014.05.003
82. Mancao CW, Hammerschmidt W. Epstein-Barr virus latent membrane protein 2A is a B- cell receptor mimic and essential for B-cell survival. Blood. (2007) 110:3715–21. doi: 10.1182/blood-2007-05-090142
83. Kieser A, Sterz R. The latent membrane protein 1 (LMP1). Curr Top Microbiol Immunol. (2015) 391:119–49. doi: 10.1007/978-3-319-22834-1_4
84. McClain MT, Heilen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med. (2005) 11:85–9. doi: 10.1038/nm1167
85. Zandman-Goddard G, Shoenfeld Y. Infections and SLE. Autoimmunity. (2005) 38:473–85. doi: 10.1080/08916930500285352
86. Draborg AH, Jacobsen S, Westergaard M, Mortensen S, Larsen JL, Houen G, et al. Reduced response to epstein-barr virus antigens by T-cells in systemic lupus erythematosus patients. Lupus Sci Med. (2014) 1:e000015. doi: 10.1136/lupus-2014-000015
87. Draborg AH, Sandhu N, Larsen N, Larsen JL, Jacobsen S, Houen G. Impaired cytokine responses to epstein-barr virus antigens in systemic lupus erythematosus patients. J Immunol Res. (2016) 2016:6473204. doi: 10.1155/2016/6473204
88. Draborg A, Izarzugaza JMG, Houen G. How compelling are the data for epstein-barr virus being a trigger for systemic lupus and other autoimmune diseases? Curr Opin Rheumatol. (2016) 28:398–404. doi: 10.1097/BOR.0000000000000289
89. McClain MT, Poole BD, Bruner BF, Kaufman KM, Harley JB, James JA. An altered immune response to epstein-barr nuclear antigen 1 in pediatric systemic lupus erythematosus. Arthrit Rheum. (2006) 54:360–68. doi: 10.1002/art.21682
90. Tsokos GC, Magrath IT, Balow JE. Epstein-barr virus induces normal B cell responses but defective suppressor T cell responses in patients with systemic lupus erythematosus. J Immunol. (1983) 131:1797–801.
91. Kang I, Quan T, Nolasco H, Park SH, Hong MS, Crouch J, et al. Defective control of latent epstein-barr virus infection in systemic lupus erythematosus. J Immunol. (2004) 172:1287–9. doi: 10.4049/jimmunol.172.2.1287
92. Larsen M, Sauce D, Deback C, Arnaud L, Mathian A, Miyara M, et al. Exhausted cytotoxic control of epstein-barr virus in human lupus. PLoS Pathog. (2011) 7:e1002328. doi: 10.1371/journal.ppat.1002328
93. Gross AJ, Hochberg D, Rand WM, Thorley-Lawson DA. EBV and systemic lupus erythematosus: a new perspective. J Immunol. (2005) 174:6599–607. doi: 10.4049/jimmunol.174.11.6599
94. Moon UY, Park SJ, Oh ST, Kim WU, Park SH, Lee SH, et al Patients with systemic lupus erythematosus have abnormally elevated epstein-barr virus load in blood. Arthrit Res Ther. (2004) 6:R295–302. doi: 10.1186/ar1181
95. Yu SF, Wu HC, Tsai WC, Yen JH, Chiang W, You CY, et al. Detecting epstein-barr virus DNA from peripheral blood mononuclear cells in adult patients with systemic lupus erythematosus in Taiwan. Med Microbiol Immunol. (2005) 194:115–20. doi: 10.1007/s00430-004-0230-5
96. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
97. Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. (2014) 192:3626–36. doi: 10.4049/jimmunol.1302062
98. Wu T, Qin X, Kurepa Z, Kumar KR, Liu K, Kanta H, et al. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus, J Clin Invest. (2007) 117:2186–96. doi: 10.1172/JCI30398
99. Zeng Q, Zhang H, Qin J, Xu Z, Gui L, Liu B, et al. Rapamycin inhibits BAFF-stimulated cell proliferation and survival by suppressing mTOR-mediated PP2A-Erk 1/2 signaling pathway in normal and neoplastic B-lymphoid cells. Cell Mol Life Sci. (2015) 72:4867–84. doi: 10.1007/s00018-015-1976-1
100. Cavallari I, Scattolin G, Silic-Benussi M, Raimondi V, D'Agostino DM, Ciminale V. Mitochondrial proteins coded by human tumor viruses. Front Microbiol. (2018) 9:81. doi: 10.3389/fmicb.2018.00081
101. Pal AD, Basak NP, Banerjee AS, Banerjee S. Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis. (2014) 35:1592–601. doi: 10.1093/carcin/bgu069
102. Wiedmer A, Wang P, Zhou J, Rennekamp AJ, Tiranti V, Zeviani M, et al. Epstein-barr virus immediate-early protein Zta Co-Opts mitochondrial single-stranded DNA binding protein to promote viral and inhibit mitochondrial DNA replication. J Virol. (2008) 82:4647–55. doi: 10.1128/JVI.02198-07
103. Mrozek-Gorska P, Buschle A, Pich D, Schwarzmayr T, Fechtner R, Sciaaldone A, et al. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc Natl Acad Sci USA. (2019) 116:16046–55. doi: 10.1073/pnas.1901314116
104. Wang LW, Shen H, Nobre L, Ersing I, Paulo JA, Trudeau S, et al. Epstein-Barr-Virus-Induced one-carbon metabolism drives B cell transformation. Cell Metab. (2019) 30:539–55.e11. doi: 10.1016/j.cmet.2019.06.003
105. Portis T, Longnecker R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene. (2004) 23:8619–28. doi: 10.1038/sj.onc.1207905
106. Moody CA, Scott RS, Amirghahari N, Nathan CA, Young LS, Dawson CW, et al. Modulation of the cell growth regulator mTOR by epstein-barr virus-encoded LMP2A. J Virol. (2005) 79:5499–506. doi: 10.1128/JVI.79.9.5499-5506.2005
107. Torigoe M, Iwata S, Nakayamada S, Sakata K, Zhang M, Hajime M, et al. Metabolic reprogramming commits differentiation of human CD27 + IgD + B Cells to plasmablasts or CD27−IgD− cells. J Immunol. (2017) 199:425–34. doi: 10.4049/jimmunol.1601908
108. Zhang M, Iwata S, Hajime M, Ohkubo N, Todoroki Y, Miyata, et al. Methionine commits cells to differentiate into plasmablasts through epigenetic regulation of BTB and CNC Homolog 2 by the methyltransferase EZH2. Arthrit Rheumatol. (2020) 72:1143–53. doi: 10.1002/art.41208
109. Sumikawa MH, Iwata S, Zhang M, Miyata H, Ueno M, Todoroki Y, et al. An enhanced mitochondrial function through glutamine metabolism in plasmablast differentiation in systemic lupus erythematosus. Rheumatology. (2021). doi: 10.1093/rheumatology/keab824. [Epub ahead of print].
Glossary
SLE; Systemic lupus erythematosus
HERVs; Human Endogenous Retroviruses
EBV; Epstein-Barr virus
pDCs; Plasmacytoid dendritic cells
mDCs; Myeloid dendritic cells
TLR; Toll-like receptor
Th; T helper
IFN; interferon
BCRs; B cell receptors
IL; interleukin
TFH; Follicular helper T cells
TPH; Peripheral helper T cells
CTLs; Cytotoxic T cells
PAMPs; Pathogen-associated molecular pattern molecules
DAMPs; damage-associated molecular patterns
PRRs; pattern-recognition receptors
dsDNA; double-stranded DNA
LTR; long terminal repeat
AIM2; absent in melanoma 2
LMP; latent membrane protein
BAFF; B Cell Activating Factor
OMM; outer mitochondrial membrane
HCQ; Hydroxychloroquine
IFNAR; interferon-α/β receptor
Keywords: SLE, viral infection, human endogenous retroviruses, Epstein-Barr virus, B cell, immunometabolism
Citation: Iwata S and Tanaka Y (2022) Association of Viral Infection With the Development and Pathogenesis of Systemic Lupus Erythematosus. Front. Med. 9:849120. doi: 10.3389/fmed.2022.849120
Received: 05 January 2022; Accepted: 03 February 2022;
Published: 25 February 2022.
Edited by:
Jixin Zhong, Huazhong University of Science and Technology, ChinaReviewed by:
Zheng Wei, Yale University, United StatesMichelle Swanson-Mungerson, Midwestern University, United States
Copyright © 2022 Iwata and Tanaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshiya Tanaka, dGFuYWthQG1lZC51b2VoLXUuYWMuanA=