 Cameron D. Haydinger
Cameron D. Haydinger Lisia Barros Ferreira
Lisia Barros Ferreira Keryn A. Williams
Keryn A. Williams Justine R. Smith
Justine R. Smith- Flinders University College of Medicine and Public Health, Adelaide, SA, Australia
Macular edema is the pathological accumulation of fluid in the central retina. It is a complication of many retinal diseases, including diabetic retinopathy, retinal vascular occlusions and uveitis, among others. Macular edema causes decreased visual acuity and, when chronic or refractory, can cause severe and permanent visual impairment and blindness. In most instances, it develops due to dysregulation of the blood-retinal barrier which permits infiltration of the retinal tissue by proteins and other solutes that are normally retained in the blood. The increase in osmotic pressure in the tissue drives fluid accumulation. Current treatments include vascular endothelial growth factor blockers, corticosteroids, and non-steroidal anti-inflammatory drugs. These treatments target vasoactive and inflammatory mediators that cause disruption to the blood-retinal barrier. In this review, a clinical overview of macular edema is provided, mechanisms of disease are discussed, highlighting processes targeted by current treatments, and areas of opportunity for future research are identified.
1. Introduction
Macular edema is the pathological accumulation of fluid in the macula, the central region of the retina essential for high-acuity vision. It is a complication common to many ocular diseases. Fluid can accumulate diffusely in the central retina or within cysts usually localized in the inner nuclear layer or Henle’s fiber layer, or in the subretinal space, disrupting and distorting the retinal architecture and decreasing visual acuity (1, 2). In combination with the underlying pathology, the edema can progress to cause irreversible tissue damage with death of retinal cells, and permanent visual impairment. The common feature of most diseases that cause macular edema is dysfunction of the blood-retinal barrier (BRB), a set of structures that, in health, strictly regulates the passage of proteins, salts, metabolites and other solutes between the blood and the retinal tissue. Vasoactive growth factors and inflammatory cytokines, including vascular endothelial growth factor-A (VEGF) and tumor necrosis factor (TNF)-α, are upregulated in the ocular fluids in macular edema and have been implicated in disruption of the BRB. Dysregulated entry and accumulation of solutes in the retina disturb the balance of osmotic and hydrostatic forces, and lead to fluid entry when mechanisms maintaining fluid homeostasis are overcome. Current treatments target proteins and processes that are involved in angiogenesis, inflammation and blood-retinal barrier (BRB) dysfunction. This review provides an overview of clinical aspects of macular edema and discusses cellular and molecular mechanisms that are targeted by current treatments.
2. Clinical overview of macular edema
2.1. Associations and clinical features
Macular edema is a common feature of diverse eye diseases, such as diabetic retinopathy, retinal vascular occlusions, uveitis, post-cataract surgery inflammation (pseudophakic macular edema or Irvine-Gass syndrome), retinal dystrophies, drug reactions, intraocular tumors, serous central chorioretinopathy, radiation retinopathy and other retinal vascular abnormalities including retinal arterial macroaneurysms and retinal telangiectasia (3). Subretinal neovascular membranes observed in conditions such as age-related macular degeneration (AMD) are also associated with intraretinal and subretinal fluid accumulation (4).
Symptoms of macular edema include metamorphopsia, micropsia, blurred vision, a central scotoma, and reduction of contrast or color sensitivity. The clinical diagnosis of macular edema can be challenging in mild cases or when visualization of the fundus is impaired by poor pupillary dilation, cataract and other ocular media opacities. Therefore, fluorescein angiography and optical coherence tomography (OCT) are commonly employed to make the diagnosis. The latter is preferred due to its non-invasive nature, accurate measurements of retinal thickness and capacity for identifying other structural abnormalities, such as epiretinal membrane and vitreomacular traction, and it is the standard method for evaluating macular edema in clinical trials. Figure 1 illustrates the retinal changes that are observed by OCT in macular edema.
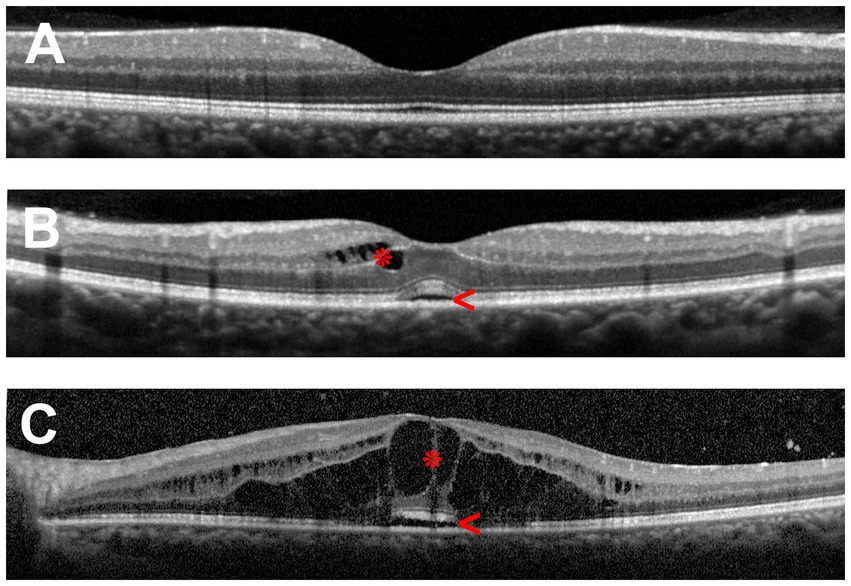
Figure 1. Optical coherence tomography (OCT) scans of the human macula in cross-section. (A) Healthy macula. (B) Macula of a patient with mild uveitic macular edema showing small numbers of cystic spaces in the inner retina (asterisk) and limited subretinal fluid (arrowhead). (C) Macula of a patient with severe uveitic macular edema demonstrating extensive retinal cystic changes (asterisk) and subretinal fluid (arrowhead).
The thickness of the retina is generally measured between the inner limiting membrane and Bruch’s membrane. By current spectral-domain OCT technology, the average and central (foveal) macular thicknesses in normal adult eyes are 334 and 226 μm, respectively, being thicker in males than in females (5). Three patterns of macular edema have been described using OCT: cystoid macular edema, diffuse macular edema, and subretinal detachment (6, 7). Diffuse retinal thickening is the main pattern in diabetic macular edema (4, 6), while in retinal vascular occlusions, accumulation of subretinal fluid is common (8). In uveitis, cystoid and diffuse macular edema are the typical forms of macular edema, whereas subretinal detachment is seen in less than one-third of cases, usually in combination with cystoid or diffuse edema (7, 9, 10).
2.2. Burden of disease
The distortion of the retinal architecture caused by fluid accumulation in the macula results in vision loss, and potentially irreversible visual impairment due to scarring when the condition becomes chronic. Macular edema is the main cause of vision loss in patients with diabetes mellitus (11), retinal vein occlusions (12, 13), and uveitis (14, 15). Populational studies have shown a mean prevalence of diabetic macular edema of around 6% in type 1 diabetes, and slightly higher in type 2 diabetes (11). Macular edema is extremely common in central retinal vein occlusions, while in branch retinal vein occlusion, macular edema occurs in 30% of cases (16, 17). Around one-third of patients with uveitis develop macular edema, being more frequent in intermediate uveitis and panuveitis, and less common in anterior uveitis (14, 15). The burden of macular edema not only relates to the decrease in quality of life experienced by patients, but also to the chronicity of the condition and the economic costs associated with treatment and frequent visits to medical centers (18–20). In uveitic macular edema in particular, the most affected population group is of working age, adding to the socioeconomic impact of the condition (21–23).
2.3. Current treatment
Treatment of macular edema depends on the underlying cause. The front-line therapy of macular edema associated with diabetic retinopathy or retinal vascular occlusions is anti-VEGF therapy, delivered into the eye by intravitreal injection and sometimes augmented with intravitreal corticosteroid and/or retinal laser photocoagulation (24–26). Ranibizumab, a recombinant humanized antibody fragment against VEGF, and aflibercept, a recombinant protein comprising the ligand-binding domains of VEGF receptors 1 and 2 fused to the Fc portion of immunoglobulin G1, are FDA-approved anti-VEGF drugs, while bevacizumab, a humanized monoclonal antibody against VEGF, is widely used off-label. As well as VEGF, aflibercept binds to placental growth factor (PlGF). The treatment of macular edema secondary to neovascular AMD also relies heavily on the use of anti-VEGF therapy (27). Newer anti-VEGF drugs, brolucizumab and faricimab, have recently been approved for the treatment of neovascular age-related macular degeneration, and faricimab is also approved for use in diabetic macular edema (28, 29). Potential complications of anti-VEGF therapy include infectious endophthalmitis, sterile intraocular inflammation, and retinal vasculitis particularly with brolucizumab (30, 31). However, a population-based cohort study did not find any increase in the risk of myocardial infarction or cerebrovascular accidents in patients treated for neovascular AMD (32). Among the intravitreal corticosteroid therapeutics, intravitreal injections of triamcinolone acetonide and the dexamethasone ‘Ozurdex’ implant may be given as adjunctive treatments.
In contrast, corticosteroids are the mainstay for managing macular edema in non-infectious uveitis. Corticosteroid eye drops may be used if macular edema is mild and associated with anterior uveitis. Among the topical corticosteroids, difluprednate has been shown to be most effective for treatment of uveitic macular edema (33, 34). Periocular triamcinolone acetonide injections and intravitreal injections of triamcinolone acetonide or the dexamethasone implant are used in unilateral or bilateral uveitis as monotherapy, or in combination with systemic immunomodulatory therapy to achieve a more rapid resolution of the edema (35–38). The most common complications of local corticosteroid treatments are elevated intraocular pressure and cataract. Systemic use of corticosteroids is indicated mainly for bilateral macular edema and should be used for a limited time due to the extensive list of side effects (39). A variety of conventional and biologic systemic immunomodulatory agents are used in the treatment of non-infectious uveitis, and a number may have activity against macular edema even in the absence of apparent intraocular inflammation (40). Intravitreal injections of VEGF agents, such as bevacizumab and ranibizumab, or methotrexate may be used in refractory uveitic macular edema or when corticosteroid therapeutics are contraindicated (41–43).
Other approaches have been used to treat macular edema. Non-steroidal anti-inflammatory drugs have an adjunct role in the treatment approach, and mostly are used for pseudophakic macular edema (44). Acetazolamide, an inhibitor of carbonic anhydrase (CA), is occasionally employed in cases of failed corticosteroid therapy, but sometimes is used as primary therapy for pseudophakic macular edema or macular edema associated with retinal dystrophies (44). Pars plana vitrectomy is mostly reserved for treating mechanical factors contributing to macular edema, such as epiretinal membranes and vitreoretinal traction. It is the last resort for the treatment of macular edema without mechanical factors, owing to its inherent risks, which include retinal breaks and detachment, vitreous hemorrhage, cataract, glaucoma and hypotony, and limited data on outcomes (44–47).
3. Mechanisms of macular edema
3.1. Fluid forces driving macular edema
Macular edema is the outcome of the failure of mechanisms maintaining fluid homeostasis in the macula. The movement of fluid across capillaries throughout the body is classically described by Starling forces. A hydrostatic pressure gradient between the intravascular space and the tissue extracellular space tends to drive fluid into the tissue, and this is opposed by a colloid osmotic (oncotic) pressure gradient which tends to draw fluid out of the extracellular space into the blood vessels. A recent review by Cunha-Vaz discusses macular edema formation in the context of classic Starling forces (48). A relative increase in hydrostatic pressure in vessels versus the retinal tissue and a relative increase in oncotic pressure in the retinal tissue versus the vessels are the two forces that drive edema. Of these, macular edema in most diseases is thought to result from increased tissue oncotic pressure due to aberrant accumulation of solutes in the retinal extracellular space. The compliant nature of the retinal tissue, which can expand unimpeded into the vitreous, renders it particularly susceptible to edema as rises in tissue oncotic pressure readily incur increases in tissue volume. It should be noted that updated models of capillary filtration predict the tissue oncotic pressure to have limited effect on the rate of fluid flow across capillaries (49, 50). The relevance of these models to the retina is debatable, however, as in the absence of a known lymphatic system, the net movement of water between the retinal tissue and the blood must be zero or slightly outward due to metabolic water production, which counters the conjecture in these models that fluid flow is inward leading to a protected subglycocalyx space.
Tissues other than the macula are also susceptible to edema in unrelated conditions including pulmonary edema, cerebral edema and lymphedema. In all cases, the fundamental principle of imbalanced hydrostatic and osmotic forces applies. Pulmonary edema and lymphedema are quite distinct entities from macular edema, involving different barriers and unique underlying mechanisms. Cerebral edema, however, shares a degree of similarity with macular edema at the molecular level. It can be caused by disruption of the blood–brain barrier (BBB), the brain’s counterpart to the BRB, which is similar in its structural composition. Key molecules that disrupt both BRB and BBB integrity include VEGF (51) and matrix metalloproteinases (52), among others. The involvement of these factors in BRB disruption is discussed below. Shared mechanisms indicate the possibility of overlapping treatment opportunities.
It is unclear why the macula is uniquely vulnerable to edema as opposed to other regions of the retina. In their 2018 review, Daruich et al. described several hypotheses, including that disruption of junctional interactions between highly elongated Müller cells and photoreceptor axons in the perifoveal Henle’s layer may permit accumulation of protein, which is normally excluded from this region (2). They also proposed that a glymphatic system involving Müller glia may play a major role in retinal fluid and solute homeostasis, and this is discussed below. While the existence of such a system remains to be demonstrated, the unique cellular architecture of the fovea and perifovea likely challenges the mechanisms of solute clearance and fluid homeostasis that suffice to counter the abnormal accumulation of fluid in other regions of the retina. Mechanistic studies of the macula are difficult given that only humans and some primates possess a macula. Many studies of mechanisms underlying macular edema are performed in rodents or using cultured cells in vitro, and it is not apparent a priori which mechanisms will translate to the situation in the human macula.
The dysregulated entry of solutes to the retina occurs due to disruption of the BRB, and this is the main focus of most efforts to understand the mechanisms of this condition. An array of molecules and other insults have been described to modulate function of the BRB, and those selected for discussion in this review mainly relate to current treatments.
3.2. Cellular composition and structure of the blood-retinal barrier
The BRB is a group of structures in the eye that separates the blood from the neural retina and retinal extracellular space. There are two components: the inner BRB, and the outer BRB. The inner BRB is based around the retinal endothelial cells (REC) which line the retinal blood vessels, and the outer BRB is formed at the level of the retinal pigment epithelial (RPE) cells. As there can be non-selective bidirectional diffusion of solutes between the retinal extracellular space and the vitreous, barrier processes between the vitreous and blood vessels at the ciliary body might also be considered to form part of the BRB, although this region is not grouped in either the inner or outer BRB.
3.2.1. Structure of the inner blood retinal-barrier
The walls of the retinal vessels form the inner BRB. The luminal surface of retinal blood vessels is uninterruptedly lined by a squamous layer of RECs, and the membranes of adjacent RECs are held together by a continuous network of tight junctions. Figure 2A depicts the inner BRB structure. The RECs are not fenestrated, and they contain few pinocytic vesicles. The RECs and their junctions form a barrier that facilitates the regulated passage of solutes between the blood and the retina (53). Basement membrane completely covers the abluminal surface of the RECs. Evidence collected in vivo indicates the basement membrane does not directly constitute a barrier (53–55), although some evidence indicates it can impede diffusion of molecular tracers in vitro (56). Larger vessels have additional layers of smooth muscle, basement membrane, and adventitia, differing in structure depending on the size and type of vessel (57).
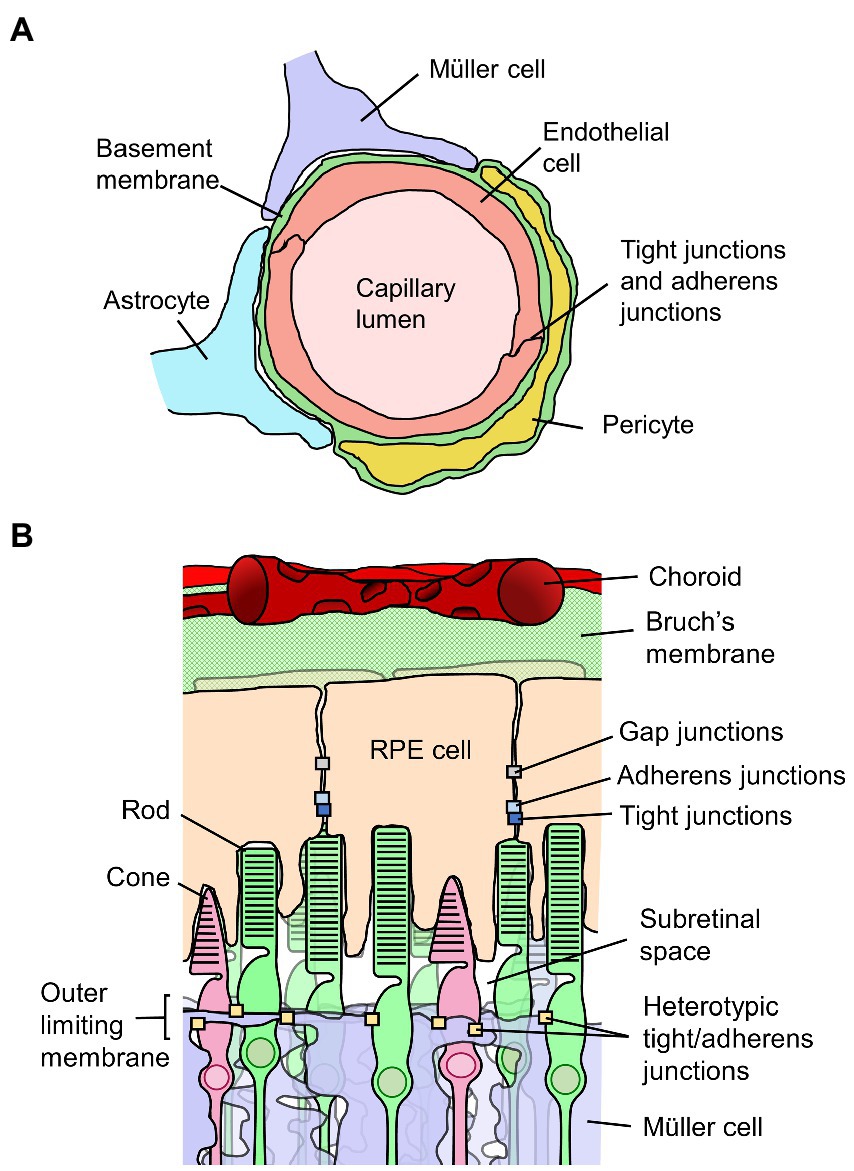
Figure 2. Diagrams of the inner and outer blood-retinal barriers (BRB). (A) Schematic cross-section of a retinal capillary indicating the location of junctions between retinal endothelial cells which form the inner BRB, along with surrounding cell types. (B) Diagram of the outer portion of the retina and choroid, indicating the junctions between retinal pigment epithelial (RPE) cells which form the outer BRB, along with surrounding structures.
Several cell types are situated in close proximity to the retinal vessels. Pericytes are embedded within the basement membrane of the abluminal wall of the vessels (57–59). In retinal capillaries, there is approximately a 1:1 ratio of endothelial cells to pericytes, meaning pericytes are abundant compared to most other capillary systems (58). Pericyte coverage of vessels is nonetheless discontinuous, so they do not form a barrier themselves. Müller glia, astrocytes and microglia directly border retinal vessels, separated by basement membrane from the RECs, pericytes or collagenous adventitia (60). Neurons are surrounded by Müller cells throughout the retina such that there is no direct contact between retinal vessels and neurons. Pericytes, astrocytes microglia and Müller cells are thought to contribute to the development of retinal vessels and the BRB (61–67). Whether these cell types are required to maintain the mature BRB is less clear and warrants further research. For example, depletion of pericytes from adult mice by induction of a genetically encoded diphtheria toxin gene did not induce BRB leakage (63), which countered the standing notion of the necessity of pericytes for BRB integrity (68). All of these cell types are, however, likely to be involved in a pathological or protective capacity in diseases affecting the BRB.
The supporting cell populations can influence fluid homeostasis via means other than direct effects on BRB integrity. As an example, Müller cells and astrocytes express the water channel aquaporin-4 (AQP4) in a distribution enriched at the endfeet bordering the vitreous and retinal vessels. Given its similar distribution to the Kir4.1 potassium channel, which plays a fundamental role in siphoning potassium from synapses to the blood and vitreous, AQP4 has been proposed as a mediator of ion-coupled water transport (69); however, this is uncertain with functional analysis indicating no interplay between Kir4.1 and AQP4 (70). Nevertheless, AQP4 is likely to be involved in retinal water homeostasis. In the brain, a so-called glymphatic system operates, whereby cerebrospinal fluid is circulated in the perivascular space between vessels and astrocyte endfeet rich in AQP4 (71). In the human macula, Daruich et al. observed channel-like structures outlined in the staining pattern of AQP4, particularly within Henle’s layer in the fovea, which is rich in AQP4 but absent of retinal vessels (2). They hypothesised that an AQP4-dependent fluid flow may exist to clear interstitial fluid from the central retina, with evidence of decreased AQP4 expression in the diabetic fovea. Direct evidence for this hypothesis is yet to be presented.
3.2.2. Structure of the outer blood-retinal barrier
The outer BRB is not situated at a vascular wall, but rather is formed by the retinal pigment epithelium, a monolayer of RPE cells situated behind the photoreceptors and immediately opposed to Bruch’s membrane which separates it from the vascular bed of the choriocapillaris (54, 72). Figure 2B depicts the outer BRB structure. In contrast to the inner BRB, the vessels of the choroid have thin and highly fenestrated endothelial cells, and do not form a selective barrier (53, 54, 73). As such, the epithelium is responsible for maintenance of the regulated environment required for retinal function. As applies to other epithelia, RPE cells are polarized and connected to one another laterally by tight junctions located at the apical side of their adjoining surfaces (72). Adherens junctions are present as well, located just basally of and partially overlapping the tight junctions (74). The RPE cells are also connected and electrically coupled via gap junctions (75). The polarized distributions of various membrane transporters in RPE cells facilitate the healthy functioning of the retina and are involved in retinal fluid homeostasis. Water injected into the subretinal space is predominantly cleared across the epithelium by mechanisms dependent on ion transport (76–78).
The outer limiting membrane must also be considered when discussing the outer BRB. It is not a true basement membrane but is formed by tight-like and adherens junctions among Müller cells and the cell bodies of photoreceptors (2). The pore size of the outer limiting membrane is small, restricting the passage of large molecules including albumin (79), so the membrane may be an important factor in macular edema development or in the type or location of edema. For instance, it may play a key role in the development or localization of subretinal edema.
Although the choroid sits outside the site of the outer BRB, together with Bruch’s membrane, it may still influence fluid homeostasis across the barrier. For instance, the hydraulic conductivity of Bruch’s membrane decreases with age (80), which is a possible factor in the accumulation of subretinal fluid in exudative age-related macular degeneration (81). Changes to the choroid are also likely to affect processes of retinal fluid absorption across the RPE cells. There are choroidal changes in diabetes, including choroidal vascular loss, choroidal neovascularization, and altered subfoveal choroidal thickness, which are reported to correlate with edema (82, 83). There is conflicting data on the subfoveal thickness changes, which is discussed in a report by Lutty (84). A precise mechanistic link between these changes and formation of retinal edema is yet unclear. Choriocapillaris loss may decrease the capacity for local fluid clearance or induce local hypoxia and promote expression of VEGF (85), with subsequent neovascularization and elevated vascular permeability. Serum leakage resulting from neovascularization may influence RPE cell transport processes and osmotic balance.
3.2.3. Molecular composition of the intercellular junctions of the blood-retinal barrier
The key structures responsible for paracellular BRB integrity are the junctional interactions between the RECs or RPE cells. Junctional complexes connect adjacent cells completely and greatly limit the passage of solutes through the gaps between cells. Generally considered most crucial for this function are the tight junctions (zonulae occludens). Tight junctions are formed by the adherent interactions of specific protein complexes on adjacent cells. These complexes comprise various transmembrane and intracellular proteins, and their composition varies depending on the cell type or context. In general, the extracellular regions of transmembrane proteins adhere in a homo- or heterotypic manner to tight junction proteins on adjacent cells. Common transmembrane proteins include occludin and other MARVEL domain proteins, claudins, and JAMs. Occludin is present at tight junctions in both RECs and in RPE cells. Claudin −1, −2 and − 5 are the predominant claudins at REC tight junctions (86). Claudin-19 is the predominant claudin at RPE tight junctions, with claudin-3 and -10 also expressed (87, 88). The cytoplasmic regions of the transmembrane proteins contact intracellular proteins which link the tight junctions to the actin cytoskeleton, and which participate in cell signaling. Common intracellular proteins include the zonula occludens (ZO) proteins, ZO-1, −2 and − 3. Tight junctions are dynamic, with certain proteins diffusing within the junction or undergoing exchange between cytosolic and membrane-localised pools (89). In addition to restricting paracellular passage of solutes, tight junctions contribute to processes of cell polarity, especially in epithelial cells, where they are located at the boundary between apical and basolateral plasma membrane domains (90), although mature tight junctions are not necessary for polarization (91).
Adherens junctions (zonulae adherens) are another structural component that joins adjacent cells in the inner and outer BRBs. These junctions are also formed by interactions of like proteins between two cells. The nectin family and cadherin family are major adhesion proteins at adherens junctions. Vascular endothelial (VE)-cadherin is the main player in endothelial cells (92). In RPE cells, it has been reported that placental (P)-cadherin is most highly expressed – at least at the mRNA level – which differs from most epithelia, as epithelial (E)-cadherin usually predominates (93). The cytoplasmic regions of cadherins interact with members of the catenin family, p120-, α- and β-catenin which link to and locally modulate the actin cytoskeleton, and participate in cell signaling and transcriptional control (94). The nectins link to the actin cytoskeleton via afadin. While adherens junctions are not classically regarded as key mediators of the barrier function in barrier epithelia and endothelia, there is functionally important interplay between adherens junctions and tight junctions (95). In fact, intravitreal injection of an anti-VE-cadherin antibody in rat eyes reportedly increased vascular permeability by nearly five-fold, demonstrating a direct importance of adherens junctions in BRB function (96).
The composition of tight junctions and adherens junctions and roles of the proteins involved in these cell–cell contacts have been reviewed by others (see references (72, 94, 97, 98)). Here, mechanisms of disruption of the junctions, as well as disruption of the transcellular route of BRB dysfunction are discussed as related to macular edema.
3.3. Blood-retinal barrier dysfunction: routes of solute transit and molecular mediators
Dysfunction of the BRB underlies development of macular edema. Loss of selectivity of the exchange of molecules between the blood and retinal tissue at the level of either the inner or outer BRB permits entry of proteins and other solutes to the retinal tissue. Leakage is usually evident well prior to macular edema formation. It is likely that edema eventuates when the severity of leakage overcomes solute and fluid clearance mechanisms (99). The rate of clearance of solutes from the subretinal space is inversely proportional to their size (100). Injection of variously sized, tagged dextran tracers into patients with uveitic macular edema showed rates of leakage roughly proportional to the size of the tracer. Sizes up to 20 kDa leaked into the macula, but 150 kDa did not (101). These data indicate that, while infiltration of larger solutes likely poses a greater challenge for clearance, complete BRB breakdown permitting non-selective entry of the largest solutes is not necessary for edema to form.
Solutes can enter the retina across the dysfunctional BRB in two ways: paracellularly or transcellularly. The paracellular route involves passage of solutes via the gaps between cells due to disruption of the junctional contacts. The transcellular route involves increased vesicular transport of solutes across cells and/or increased passive diffusion due to elevated membrane permeability. The routes of breakdown may differ between diseases, and both routes may be involved (102).
The widespread nature of BRB leakage in many diseases suggests that soluble factors likely play a role. Many growth factors and inflammatory cytokines have been found elevated in the intraocular fluids of patients with diseases complicated by macular edema, and there is evidence that several act to disrupt BRB integrity. These molecules can drive BRB breakdown in multiple ways. They may act directly on the RECs or RPE cells to increase permeability by affecting the expression or regulation of junctional molecules (103, 104); they may act on other retinal cell types to further induce their own expression or that of other inflammatory mediators, facilitating a persistent inflammatory state; or they may attract and promote adhesion of leukocytes to the retinal vasculature (105, 106) which then induce BRB breakdown directly or further release inflammatory mediators (107, 108). The relative importance of various inflammatory and vasoactive molecules, their cellular sources, and the precise sequence of events leading to BRB breakdown and macular edema is not yet clear, and likely differs between diseases and individual patients. A selection of mediators of BRB dysfunction of relevance to current macular edema treatments is discussed below.
3.3.1. Vascular endothelial growth factor-A and placental growth factor
Vascular endothelial growth factor-A is a major mediator of pathological vascular changes in several ocular diseases. The presence of VEGF is higher in the ocular fluids of patients with diabetic retinopathy and central retinal vein occlusion than in healthy controls (109). It is upregulated in experimental autoimmune uveoretinitis (110). Macular edema in retinal vascular occlusion is driven by VEGF in many but not necessarily all cases (111). Intravitreal levels of VEGF and IL-6 correlate with the severity of macular edema and the size of the non-perfusion area (112, 113). This is consistent with the observations that VEGF expression is induced by hypoxia in vitro in various retinal cell types, and hypoxia-conditioned medium stimulates proliferation of RECs (114).
Vascular endothelial growth factor-A increases permeability of bovine REC monolayers in vitro (115). Mechanistically, VEGF activates protein kinase C (PKC)β which directly phosphorylates occludin, targeting it for ubiquitination (116, 117). This alters trafficking of occludin and its normal localization at tight junctions (116). Inhibition of PKC, transfection of a dominant negative PKC, or mutation of the phosphorylated residue of occludin counters VEGF-induced permeation of bovine REC monolayers (115, 118). Occludin content is lower in retinas of diabetic rats, and phosphorylation of occludin is elevated (118, 119). Intravitreal injection of VEGF induces fluorescein leakage from retinal vessels in rats in vivo, and this is almost completely abolished by inhibition of PKCβ (120). However, phase 3 studies of a PKCβ inhibitor showed limited benefit in treatment of diabetic retinopathy (121). There is a wealth of evidence supporting the mechanism of VEGF-induced alteration of occludin distribution leading to functional changes in tight junctions in vitro. When assessed at the ultrastructural level, however, REC tight junctions often appear non-leaky in models of diabetes, indicating other mechanisms of BRB breakdown are also likely at play (122–124). Intravitreal injection of VEGF in cynomolgus monkeys increased vascular permeability without opening tight junctions (125). Instead, there was a stark change in the distribution of pinocytic vesicles in RECs, said to indicate an increase in vesicular transport of macromolecules from the vascular lumen to the retinal extracellular space. It is possible that non-responsiveness to VEGF blockers may also occur where outer BRB breakdown is apparent. Both VEGF and anti-VEGF treatments are reported to have minimal effects on RPE cell tight junctions in vitro, meaning other mechanisms may need to be targeted (87). Outer BRB breakdown may also limit the proposed ability of the epithelium to remove VEGF from the subretinal space (87).
In endothelial cells, adherens junction components including VE-cadherin, p120- and β-catenin, and plakoglobin undergo tyrosine phosphorylation in response to VEGF which coincides with increased permeability (126). A more recent study showed that VEGF induces activation of the small GTPase, Rac, then p21-activated kinase (PAK), which phosphorylates a specific serine residue on VE-cadherin leading to its internalization, disassembly of the adherens junction, and increased permeability (127).
Placental growth factor is an angiogenic growth factor in the VEGF family, which may also be involved in promoting BRB permeability. It is greatly upregulated in retinae of Akita diabetic mice, and its knockout in this model protects against retinal capillary leakage and degeneration (128). The PlGF gene deletion not only abrogated the decrease in expression of ZO-1 and VE-cadherin in diabetic mice, but elevated their expression relative to non-diabetic controls. Expression of these junctional molecules correlated with expression of two putative regulators, sonic hedgehog and angiopoetin-1, the latter of which has been shown to protect against BRB breakdown (129). Hypoxia-inducible factor (HIF)-1α and VEGF expression were upregulated in Akita mice, and this was countered by PlGF knockout.
Clearly, VEGF plays a major role in driving progression of retinal diseases and macular edema, which is irrefutably evidenced by the broad utility of anti-VEGF therapies. Targeting VEGF with intravitreal injections of monoclonal antibodies or high-affinity decoy receptors are effective means of treating most diseases complicated by macular edema, including wet age-related macular degeneration (130), diabetic macula edema (131, 132), and macular edema caused by CRVO (133), BRVO (134) and uveitis (135). However, there are concerns of ocular and systemic toxicity in targeting VEGF, and long-term frequent reinjection is required (136–138). Also, the fact that there are cases resistant to anti-VEGF treatment suggests other pathways may also be involved in these diseases.
3.3.2. Tumor necrosis factor-α and interleukin-1β
Tumor necrosis factor-α is a master inflammatory cytokine which has been shown to increase barrier permeability of REC monolayers in vitro (139). When injected into the vitreous in rabbits, TNF-α increased the proportion of REC tight junctions that appeared open, and albumin was detected within these junctions, indicating leakiness (140). Larger vesicles also stained positively for albumin in RECs in treated eyes, but not controls. These results indicate breakdown of both transcellular and paracellular pathways. Interestingly, 24 h after injection, TNF-α-induced tight junction changes had reversed, a promising sign for a potential treatment target. When injected into the vitreous in rats, it also induced barrier leakage, and the effect of injection recovered after 7 days (141). Huang et al. used two mouse models of diabetes to assess the pathological role of TNF-α (142). Vascular leakage late in disease was completely prevented by TNF-α knockout. Interestingly, these observations did not correlate with the expression of TNF-α in the control diabetic mice, as TNF-α expression was not elevated in mid or late diabetic retinae. Instead, it was proposed that early expression of TNF-α might initiate a chain of events leading to increased apoptosis of retinal cells and BRB breakdown in later stages. In bovine REC monolayers, TNF-α treatment decreased expression of ZO-1 and claudin-5 but increased expression of occludin (139). It disrupted the normal border staining pattern of all three. The increase in barrier permeability induced by TNF-α was prevented by dexamethasone or by an inhibitor of PKCζ. The PKCζ inhibitor also prevented BRB disruption induced by intravitreal injection of TNF-α in rats in vivo.
Treatment of human fetal RPE cells in vitro with TNF-α decreased resistance of monolayers by 80% (143). Interestingly, in a particular serum-free medium, this effect was only observed when TNF-α was applied to the apical side of the monolayer. A clear molecular mechanism for TNF-α-mediated barrier disruption was elusive, with no changes observed in levels of several junctional molecules tested, but it was suggested that TNF-α may induce changes in tight junction tortuosity.
Anti-TNF-α monoclonal antibodies are sometimes used in the treatment of non-infectious uveitis (144). They show high efficacy, although corticosteroids and conventional immunosuppressive drugs are generally the first-line treatment (145). Tumor necrosis factor-α has essential roles as an infection-fighting agent, so patients require close monitoring due to risk of developing infections, and increased risk of cancer may also be a concern. Some data suggest TNF inhibitors used for treatment of non-ocular diseases could even worsen or increase the risk of developing uveitis, a point which will require larger-scale studies to resolve (146).
Along with TNF-α, another master inflammatory cytokine which is of increasing interest is IL-1β. Similar to TNF-α, when injected into the rabbit vitreous, IL-1β increases the number of open, leaky tight junctions (140). It also increases the permeability of REC monolayers in vitro (139). In streptozotocin-diabetic rats, retinal IL-1β expression increased between 1.5 and 4.5 months of diabetes (147). IL-1β mRNA is induced by high glucose in bovine RECs in vitro (147). It induces its own expression in RECs, Müller cells and brain astrocytes in vitro, which is prevented by inhibition of protein kinase C (147). The effects of IL-1β may be due to elevated oxidative stress, decreased expression of occludin, ZO-1 and claudin-5, and altered distribution of VE-cadherin (148). Interleukin-1β may be a promising target for new therapeutics: a receptor antagonist (anakinra), a decoy receptor (rilonacept), and a neutralizing antibody (canakinumab) are already in clinical use for other indications.
3.3.3. Interleukin-6
Interleukin-6 is a cytokine intimately linked with inflammatory and autoimmune disease, and is a promising target for treatment of non-infectious uveitis (149). Circumstantial evidence implicates IL-6 in diseases leading to macular edema. It is elevated in patients with uveitis (150). It is elevated in diabetic macular edema (151) and correlates with macular thickness (152). IL-6 is also upregulated in patients with central retinal vein occlusion, especially those with ischemia (112). Its expression often correlates with that of VEGF.
Experimental evidence for the role of IL-6 in disease is less clear at present. IL-6 did not induce noticeable barrier permeability to mannitol when injected into the vitreous in rats (141). In vitro, IL-6 treatment disrupted the barrier formed by the ARPE-19 RPE cell line, as evidenced by increased permeability to 40 kDa dextran and decreased electrical resistance (153). Distribution of ZO-1 at the borders was disrupted, suggesting tight junction dysfunction may be involved. In contrast, IL-6 had no effect on the permeability of human REC monolayers (153). It can, however, reportedly induce permeability of non-REC monolayers in vitro (104), although in some cases the effect requires exogenous addition of IL-6 receptor (154). Together, these data suggest IL-6 may be especially active on RPE cells in inducing permeability.
As opposed to or in addition to acting directly on the barrier cells, IL-6 may act via less direct means, promoting inflammation to indirectly facilitate disease progression. Knockout of IL-6 or treatment with anti-IL-6 or anti-IL-6 receptor antibody ameliorates mouse experimental autoimmune uveoretinitis, a model of human non-infectious uveitis (155–157). A major mechanism underlying the effectiveness of IL-6 blockade in this model is the prevention of differentiation and expansion of T helper 17 cells (155, 156). Clinical trials of biologic drugs that target IL-6 signaling – such as tocilizumab and sarilumab – for macular edema are ongoing, with initial positive results (149, 158).
3.3.4. Matrix metalloproteinases
Growth factors and inflammatory cytokines are not the only targets of treatments for macular edema. Corticosteroids provide effective treatment for macular edema resulting from various diseases, including diabetes mellitus, central and branch retinal vein occlusions, and uveitis (159–161). These drugs are very broad-acting and have potent anti-inflammatory effects. One of many mechanisms by which they may act is the suppression of matrix metalloproteinase (MMP) expression and activity (162). The MMPs are a family of endopeptidases which degrade extracellular matrix and basement membranes among other macromolecules, and they have been suggested to play a role in progression of diabetic retinopathy (163).
Matrix metalloproteinases, MMP2, MMP9 and MMP14, are upregulated in the retinal vessels at the RNA level in streptozotocin-diabetic rats (96, 164). Retinal vascular permeability is induced in this model, and this could be prevented by intraperitoneal injection of a broad spectrum MMP inhibitor. Mechanistically, MMP inhibition prevented the loss of surface staining of VE-cadherin on RECs. The elevated presence of advanced glycation end products was proposed to underly the heightened activity of MMPs in the diabetic model. MMPs may play a role in modulating both inner and outer BRB integrity, with treatment of the ARPE19 RPE cell line, as well as bovine RECs, with recombinant MMP2 or MMP9 decreasing electrical resistance of the monolayers (164). In this study, the mechanism was proposed to be degradation of occludin and tight junction alterations (164).
3.3.5. Carbonic anhydrase
Carbonic anhydrase inhibitors, including acetazolamide, have a history of use for treating macular edema, especially that caused by retinitis pigmentosa and diabetic retinopathy (165–167). Isoforms CA-I and CA-II were found to be elevated in the vitreous of patients with proliferative diabetic retinopathy (168). Injection of CA into rat vitreous induced vessel leakage which was prevented by co-injection of CA inhibitors. It is possible that the elevated intravitreal CA derives from intraocular hemorrhage, as injection of lysed red blood cells recapitulated vessel leakage, and this can be prevented by CA inhibition. The effect of CA on BRB permeability was found to be mediated by the kallikrein-kinin system. Notably, injection of VEGF resulted in a similar magnitude increase in vascular permeability as injection of CA, VEGF did not act via the same pathway, and the effect of CA and VEGF together was additive, suggesting dual treatment could prove useful. In addition to the above mechanism, CA inhibitors also act on membrane-bound CA on RPE cells and increase resorption of subretinal fluid across the epithelium (169–171). The precise mechanism by which this works is an area of continuing research. A recent mathematical model of ion transport across the RPE cell barrier predicted this behavior (78).
3.4. Dysfunction of the transcellular arm of the blood-retinal barrier
As is evident from this discussion, molecular studies of BRB breakdown frequently focus on changes in expression and localization of junctional proteins, which are key players in paracellular BRB integrity. There is ample evidence that many molecules elevated in retinal diseases affect paracellular integrity. However, the ultrastructural studies that have been performed have indicated transcellular routes of BRB dysfunction are likely equally important as paracellular dysfunction. There are several means of transcellular passage (92), including simple diffusion – which is minimal for hydrophilic molecules – ion channels for salts and small molecules, specific transporters for amino acids and metabolites, and small vesicles termed caveolae, which when dysregulated can facilitate non-selective entry of proteins and solutes. Small vesicles are scarce in RECs compared to non-barrier endothelia, and this is reportedly a key feature that contributes to the inner BRB (172). Electron microscopy shows that vesicles are predominantly located at the abluminal side of the RECs, which has been suggested to indicate most transport occurs in the retina-to-blood direction (125). Caveolae are the best studied means of trans-endothelial vesicular transport across the BRB, reviewed by Klassen et al. (92). Caveolin-1 is the predominant protein constituent of caveolae. Both its knockout (173), and its overexpression (174) have been reported to disrupt BRB function. Caveolin-1 and caveolae are likely multifunctional and are only beginning to be understood in the context of the BRB.
Based on the ultrastructural localization of albumin in animal models of diabetes and in diabetic human retinas, tight junctions between RECs or RPE cells rarely appear disrupted, with albumin leakage instead suggested to be predominantly transcellular via vesicles or via diffusion through hyperpermeable plasma membranes due to degenerative changes in RECs or RPE cells (122–124, 175). In a rat model of experimental autoimmune uveoretinitis, leakage of albumin into the retinal tissue and presence of albumin in REC vesicles and RPE vacuoles was evident prior to ultrastructural evidence of disruption of tight junctions (140). Tight junctions did subsequently open and also permit passage of albumin. Tight junctions also appear to remain intact in the face of acute ischemic or chemical injury despite significant damage to RECs and transcellular leakage of tracers, although leakage in this setting is likely due to gross damage to the RECs rather than dysregulated vesicle transport (55). The potential for the transcellular route of BRB transit to account for vessel leakiness is also apparent in the process of inner BRB development. Newly sprouting vessels in later stages of development are leaky to tracers, but possess functional tight junctions (176). Leakage occurs through RECs via vesicles, and the fully functional BRB only forms upon suppression of transcytosis (176).
It is not yet clear which route of breakdown is responsible for the greatest portion of solute entry during BRB dysfunction, and the situation in the human macula may differ to animal models and to peripheral regions of the retina. Paracellular and transcellular routes may both be involved in many cases of macular edema.
4. Conclusion
Current treatments for macular edema show effectiveness in some, but not all situations. The continued use of broad acting treatments such as corticosteroids as first-line agents in spite of significant side effects highlights the incomplete understanding of the mechanisms underlying disease and the need for continuing research. Ongoing efforts to understand mechanisms governing regulation of junctional complex molecules in response to growth factors and inflammatory cytokines may clarify the contexts in which these mediators are involved. Major holes in current knowledge also include the reason why the macula is uniquely vulnerable to edema, as well as the mechanisms underlying dysfunction of transcellular integrity. Dysfunction of the BRB has a diverse spectrum of causes, in addition to those presented here. Though macular edema may be a common outcome of BRB dysfunction, the disparate causes of BRB dysfunction may require a battery of options such that treatment may be tailored to the cause of each case.
Author contributions
KW and JS conceptualized the review and reviewed and edited the manuscript draft. CH extensively reviewed the literature and drafted Sections 1. Introduction, 3. Mechanisms of macular edema, and 4. Conclusion. LF extensively reviewed the literature and drafted Section 2. Clinical overview of macular edema and Figure 1. All authors read and approved the final manuscript.
Funding
This work was supported by a Research Grant from Macular Disease Foundation Australia.
Acknowledgments
The authors wish to thank Janet Matthews for administrative support in preparing this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Spaide, RF. Retinal vascular cystoid macular edema: review and new theory. Retina. (2016) 36:1823–42. doi: 10.1097/IAE.0000000000001158
2. Daruich, A, Matet, A, Moulin, A, Kowalczuk, L, Nicolas, M, Sellam, A, et al. Mechanisms of macular edema: beyond the surface. Prog Retin Eye Res. (2018) 63:20–68. doi: 10.1016/j.preteyeres.2017.10.006
3. Tranos, PG, Wickremasinghe, SS, Stangos, NT, Topouzis, F, Tsinopoulos, I, and Pavesio, CE. Macular edema. Surv Ophthalmol. (2004) 49:470–90. doi: 10.1016/S0039-6257(04)00109-2
4. Trichonas, G, and Kaiser, PK. Optical coherence tomography imaging of macular oedema. Br J Ophthalmol. (2014) 98:ii24. doi: 10.1136/bjophthalmol-2014-305305
5. Myers, CE, Klein, BEK, Meuer, SM, Swift, MK, Chandler, CS, Huang, Y, et al. Retinal thickness measured by spectral-domain optical coherence tomography in eyes without retinal abnormalities: the beaver dam eye study. Am J Ophthalmol. (2015) 159:445–56.e1. doi: 10.1016/j.ajo.2014.11.025
6. Otani, T, Kishi, S, and Maruyama, Y. Patterns of diabetic macular edema with optical coherence tomography. Am J Ophthalmol. (1999) 127:688–93. doi: 10.1016/S0002-9394(99)00033-1
7. Markomichelakis, NN, Halkiadakis, I, Pantelia, E, Peponis, V, Patelis, A, Theodossiadis, P, et al. Patterns of macular edema in patients with uveitis: qualitative and quantitative assessment using optical coherence tomography. Ophthalmology. (2004) 111:946–53. doi: 10.1016/j.ophtha.2003.08.037
8. Spaide, RF, Lee, JK, Klancnik, JK Jr, and Gross, NE. Optical coherence tomography of branch retinal vein occlusion. Retina. (2003) 23:343–7. doi: 10.1097/00006982-200306000-00009
9. Grajewski, RS, Boelke, AC, Adler, W, Meyer, S, Caramoy, A, Kirchhof, B, et al. Spectral-domain optical coherence tomography findings of the macula in 500 consecutive patients with uveitis. Eye. (2016) 30:1415–23. doi: 10.1038/eye.2016.133
10. Iannetti, L, Accorinti, M, Liverani, M, Caggiano, C, Abdulaziz, R, and Pivetti-Pezzi, P. Optical coherence tomography for classification and clinical evaluation of macular edema in patients with uveitis. Ocul Immunol Inflamm. (2008) 16:155–60. doi: 10.1080/09273940802187466
11. Lee, R, Wong, TY, and Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. (2015) 2:17. doi: 10.1186/s40662-015-0026-2
12. McIntosh, RL, Rogers, SL, Lim, L, Cheung, N, Wang, JJ, Mitchell, P, et al. Natural history of central retinal vein occlusion: an evidence-based systematic review. Ophthalmology. (2010) 117:1113–23.e15. doi: 10.1016/j.ophtha.2010.01.060
13. Rogers, SL, McIntosh, RL, Lim, L, Mitchell, P, Cheung, N, Kowalski, JW, et al. Natural history of branch retinal vein occlusion: an evidence-based systematic review. Ophthalmology. (2010) 117:1094–101.e5. doi: 10.1016/j.ophtha.2010.01.058
14. Rothova, A, Suttorp-van Schulten, MS, Frits Treffers, W, and Kijlstra, A. Causes and frequency of blindness in patients with intraocular inflammatory disease. Br J Ophthalmol. (1996) 80:332–6. doi: 10.1136/bjo.80.4.332
15. Lardenoye, CW, van Kooij, B, and Rothova, A. Impact of macular edema on visual acuity in uveitis. Ophthalmology. (2006) 113:1446–9. doi: 10.1016/j.ophtha.2006.03.027
16. Flaxel, CJ, Adelman, RA, Bailey, ST, Fawzi, A, Lim, JI, Vemulakonda, GA, et al. Retinal vein occlusions preferred practice pattern. Ophthalmology. (2020) 127:P288–320. doi: 10.1016/j.ophtha.2019.09.029
17. Zhou, JQ, Xu, L, Wang, S, Wang, YX, You, QS, Tu, Y, et al. The 10-year incidence and risk factors of retinal vein occlusion: the Beijing eye study. Ophthalmology. (2013) 120:803–8. doi: 10.1016/j.ophtha.2012.09.033
18. Hariprasad, SM, Mieler, WF, Grassi, M, Green, JL, Jager, RD, and Miller, L. Vision-related quality of life in patients with diabetic macular oedema. Br J Ophthalmol. (2008) 92:89–92. doi: 10.1136/bjo.2007.122416
19. Jeon, HL, Lee, H, Yoon, D, Lee, Y, Kim, JH, Jee, D, et al. Burden of diabetic macular oedema in patients receiving antivascular endothelial growth factor therapy in South Korea: a healthcare resource use and cost analysis. BMJ Open. (2020) 10:e042484. doi: 10.1136/bmjopen-2020-042484
20. Lanzetta, P, Nuys, KV, Tran, I, Gallagher, M, Colman, S, and Lakdawalla, D. The economic burden of diabetic macular edema from a U.S. private payer perspective. Investig Ophthalmol Vis Sci. (2011)
21. Silva, LMP, Arantes, TE, Casaroli-Marano, R, Vaz, T, Belfort, R Jr, and Muccioli, C. Quality of life and psychological aspects in patients with visual impairment secondary to uveitis: a clinical study in a tertiary care hospital in Brazil. Ocul Immunol Inflamm. (2019) 27:99–107. doi: 10.1080/09273948.2017.1370551
22. Kempen, JH, Altaweel, MM, Holbrook, JT, Jabs, DA, and Sugar, EA. The multicenter uveitis steroid treatment trial: rationale, design, and baseline characteristics. Am J Ophthalmol. (2010) 149:550–61.e10. doi: 10.1016/j.ajo.2009.11.019
23. Schiffman, RM, Jacobsen, G, and Whitcup, SM. Visual functioning and general health status in patients with uveitis. Arch Ophthalmol. (2001) 119:841–9. doi: 10.1001/archopht.119.6.841
24. Ehlers, JP, Yeh, S, Maguire, MG, Smith, JR, Mruthyunjaya, P, Jain, N, et al. Intravitreal pharmacotherapies for diabetic macular edema: a report by the American Academy of ophthalmology. Ophthalmology. (2022) 129:88–99. doi: 10.1016/j.ophtha.2021.07.009
25. Ehlers, JP, Kim, SJ, Yeh, S, Thorne, JE, Mruthyunjaya, P, Schoenberger, SD, et al. Therapies for macular edema associated with branch retinal vein occlusion: a report by the American Academy of ophthalmology. Ophthalmology. (2017) 124:1412–23. doi: 10.1016/j.ophtha.2017.03.060
26. Yeh, S, Kim, SJ, Ho, AC, Schoenberger, SD, Bakri, SJ, Ehlers, JP, et al. Therapies for macular edema associated with central retinal vein occlusion: a report by the American Academy of ophthalmology. Ophthalmology. (2015) 122:769–78. doi: 10.1016/j.ophtha.2014.10.013
27. Flaxel, CJ, Adelman, RA, Bailey, ST, Fawzi, A, Lim, JI, Vemulakonda, GA, et al. Age-related macular degeneration preferred practice pattern. Ophthalmology. (2020) 127:P1–P65. doi: 10.1016/j.ophtha.2019.09.024
28. Dugel, PU, Koh, A, Ogura, Y, Jaffe, GJ, Schmidt-Erfurth, U, Brown, DM, et al. HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology. (2020) 127:72–84. doi: 10.1016/j.ophtha.2019.04.017
29. Sharma, A, Kumar, N, Kuppermann, BD, Bandello, F, and Loewenstein, A. Faricimab: expanding horizon beyond VEGF. Eye. (2020) 34:802–4. doi: 10.1038/s41433-019-0670-1
30. Cox, JT, Eliott, D, and Sobrin, L. Inflammatory complications of intravitreal anti-VEGF injections. J Clin Med. (2021) 10:981. doi: 10.3390/jcm10050981
31. Levin, AM, Chaya, CJ, Kahook, MY, and Wirostko, BM. Intraocular pressure elevation following intravitreal anti-VEGF injections: short- and long-term considerations. J Glaucoma. (2021) 30:1019–26. doi: 10.1097/IJG.0000000000001894
32. Dalvin, LA, Starr, MR, AbouChehade, JE, Damento, GM, Garcia, M, Shah, SM, et al. Association of intravitreal anti-vascular endothelial growth factor therapy with risk of stroke, myocardial infarction, and death in patients with exudative age-related macular degeneration. JAMA Ophthalmol. (2019) 137:483–90. doi: 10.1001/jamaophthalmol.2018.6891
33. Schallhorn, JM, Niemeyer, KM, Browne, EN, Chhetri, P, and Acharya, NR. Difluprednate for the treatment of uveitic cystoid macular edema. Am J Ophthalmol. (2018) 191:14–22. doi: 10.1016/j.ajo.2018.03.027
34. Symes, RJ, and Forooghian, F. Topical difluprednate monotherapy for uveitic macular edema. Can J Ophthalmol. (2016) 51:47–9. doi: 10.1016/j.jcjo.2015.10.007
35. Leder, HA, Jabs, DA, Galor, A, Dunn, JP, and Thorne, JE. Periocular triamcinolone acetonide injections for cystoid macular edema complicating noninfectious uveitis. Am J Ophthalmol. (2011) 152:441–8.e2. doi: 10.1016/j.ajo.2011.02.009
36. Shin, JY, and Yu, HG. Intravitreal triamcinolone injection for uveitic macular edema: a randomized clinical study. Ocul Immunol Inflamm. (2015) 23:430–6. doi: 10.3109/09273948.2015.1025982
37. Pohlmann, D, Vom Brocke, GA, Winterhalter, S, Steurer, T, Thees, S, and Pleyer, U. Dexamethasone inserts in noninfectious uveitis: a single-center experience. Ophthalmology. (2018) 125:1088–99. doi: 10.1016/j.ophtha.2017.12.038
38. Cao, JH, Mulvahill, M, Zhang, L, Joondeph, BC, and Dacey, MS. Dexamethasone intravitreal implant in the treatment of persistent uveitic macular edema in the absence of active inflammation. Ophthalmology. (2014) 121:1871–6. doi: 10.1016/j.ophtha.2014.04.012
39. Foster, CS, Kothari, S, Anesi, SD, Vitale, AT, Chu, D, Metzinger, JL, et al. The ocular immunology and uveitis foundation preferred practice patterns of uveitis management. Surv Ophthalmol. (2016) 61:1–17. doi: 10.1016/j.survophthal.2015.07.001
40. Radosavljevic, A, Agarwal, M, Bodaghi, B, Smith, JR, and Zierhut, M. Medical therapy of uveitic macular edema: biologic agents. Ocul Immunol Inflamm. (2020) 28:1239–50. doi: 10.1080/09273948.2019.1709648
41. Bae, JH, Lee, CS, and Lee, SC. Efficacy and safety of intravitreal bevacizumab compared with intravitreal and posterior sub-tenon triamcinolone acetonide for treatment of uveitic cystoid macular edema. Retina. (2011) 31:111–8. doi: 10.1097/IAE.0b013e3181e378af
42. Cervantes-Castaneda, RA, Giuliari, GP, Gallagher, MJ, Yilmaz, T, MacDonell, RE, Quinones, K, et al. Intravitreal bevacizumab in refractory uveitic macular edema: one-year follow-up. Eur J Ophthalmol. (2009) 19:622–9. doi: 10.1177/112067210901900417
43. Taylor, SR, Habot-Wilner, Z, Pacheco, P, and Lightman, SL. Intraocular methotrexate in the treatment of uveitis and uveitic cystoid macular edema. Ophthalmology. (2009) 116:797–801. doi: 10.1016/j.ophtha.2008.10.033
44. Pepple, KL, Nguyen, MH, Pakzad-Vaezi, K, Williamson, K, Odell, N, Lee, C, et al. Response of inflammatory cystoid macular edema to treatment using oral acetazolamide. Retina. (2019) 39:948–55. doi: 10.1097/IAE.0000000000002044
45. Tranos, P, Scott, R, Zambarakji, H, Ayliffe, W, Pavesio, C, and Charteris, DG. The effect of pars plana vitrectomy on cystoid macular oedema associated with chronic uveitis: a randomised, controlled pilot study. Br J Ophthalmol. (2006) 90:1107–10. doi: 10.1136/bjo.2006.092965
46. Gutfleisch, M, Spital, G, Mingels, A, Pauleikhoff, D, Lommatzsch, A, and Heiligenhaus, A. Pars plana vitrectomy with intravitreal triamcinolone: effect on uveitic cystoid macular oedema and treatment limitations. Br J Ophthalmol. (2007) 91:345–8. doi: 10.1136/bjo.2006.101675
47. Schilling, H, Heiligenhaus, A, Laube, T, Bornfeld, N, and Jurklies, B. Long-term effect of acetazolamide treatment of patients with uveitic chronic cystoid macular edema is limited by persisting inflammation. Retina. (2005) 25:182–8. doi: 10.1097/00006982-200502000-00011
48. Cunha-Vaz, J. The blood-retinal barrier in the management of retinal disease: EURETINA award lecture. Ophthalmologica. (2017) 237:1–10. doi: 10.1159/000455809
49. Adamson, RH, Lenz, JF, Zhang, X, Adamson, GN, Weinbaum, S, and Curry, FE. Oncotic pressures opposing filtration across non-fenestrated rat microvessels. J Physiol. (2004) 557:889–907. doi: 10.1113/jphysiol.2003.058255
50. Woodcock, TE, and Woodcock, TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth. (2012) 108:384–94. doi: 10.1093/bja/aer515
51. van Bruggen, N, Thibodeaux, H, Palmer, JT, Lee, WP, Fu, L, Cairns, B, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. (1999) 104:1613–20. doi: 10.1172/JCI8218
52. Shigemori, Y, Katayama, Y, Mori, T, Maeda, T, and Kawamata, T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir Suppl. (2006) 96:130–3. doi: 10.1007/3-211-30714-1_29
53. Cunha-Vaz, JG, Shakib, M, and Ashton, N. Studies on the permeability of the blood-retinal barrier. I. on the existence, development, and site of a blood-retinal barrier. Br J Ophthalmol (1966) 50:441–53. doi: 10.1136/bjo.50.8.441, 453
54. Cunha-Vaz, JG. The blood-retinal barriers. Doc Ophthalmol. (1976) 41:287–327. doi: 10.1007/BF00146764
55. Cunha-Vaz, JG, and Shakib, M. Ultrastructural mechanisms of breakdown of the blood-retina barrier. J Pathol Bacteriol. (1967) 93:645–52. doi: 10.1002/path.1700930225
56. Chronopoulos, A, Trudeau, K, Roy, S, Huang, H, Vinores, SA, and Roy, S. High glucose-induced altered basement membrane composition and structure increases trans-endothelial permeability: implications for diabetic retinopathy. Curr Eye Res. (2011) 36:747–53. doi: 10.3109/02713683.2011.585735
57. Hogan, MJ, and Feeney, L. The ultrastructure of the retinal blood vessels. I. the large vessels. J Ultrastruct Res. (1963) 39:10–28. doi: 10.1016/s0022-5320(63)80033-7
58. Hogan, MJ, and Feeney, L. The ultrastructure of the retinal vessels. II. The small vessels. J Ultrastruct Res. (1963) 49:29–46. doi: 10.1016/s0022-5320(63)80035-0
59. Ishikawa, T. Fine structure of retinal vessels in man and the macaque monkey. Investig Ophthalmol. (1963) 2:1–15.
60. Hogan, MJ, and Feeney, L. The ultrastructure of the retinal vessels. III. Vascular-glial relationships. J Ultrastruct Res. (1963) 49:47–64. doi: 10.1016/s0022-5320(63)80035-0
61. Kim, JH, Kim, JH, Yu, YS, Kim, DH, and Kim, KW. Recruitment of pericytes and astrocytes is closely related to the formation of tight junction in developing retinal vessels. J Neurosci Res. (2009) 87:653–9. doi: 10.1002/jnr.21884
62. Choi, YK, Kim, JH, Kim, WJ, Lee, HY, Park, JA, Lee, SW, et al. AKAP12 regulates human blood-retinal barrier formation by downregulation of hypoxia-inducible factor-1alpha. J Neurosci. (2007) 27:4472–81. doi: 10.1523/JNEUROSCI.5368-06.2007
63. Park, DY, Lee, J, Kim, J, Kim, K, Hong, S, Han, S, et al. Plastic roles of pericytes in the blood-retinal barrier. Nat Commun. (2017) 8:15296. doi: 10.1038/ncomms15296
64. Yao, H, Wang, T, Deng, J, Liu, D, Li, X, and Deng, J. The development of blood-retinal barrier during the interaction of astrocytes with vascular wall cells. Neural Regen Res. (2014) 9:1047–54. doi: 10.4103/1673-5374.133169
65. Gardner, TW, Lieth, E, Khin, SA, Barber, AJ, Bonsall, DJ, Lesher, T, et al. Astrocytes increase barrier properties and ZO-1 expression in retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. (1997) 38:2423–7.
66. Checchin, D, Sennlaub, F, Levavasseur, E, Leduc, M, and Chemtob, S. Potential role of microglia in retinal blood vessel formation. Invest Ophthalmol Vis Sci. (2006) 47:3595–602. doi: 10.1167/iovs.05-1522
67. Tout, S, Chan-Ling, T, Holländer, H, and Stone, J. The role of Müller cells in the formation of the blood-retinal barrier. Neuroscience. (1993) 55:291–301. doi: 10.1016/0306-4522(93)90473-S
68. Santos, GSP, Prazeres, P, Mintz, A, and Birbrair, A. Role of pericytes in the retina. Eye. (2018) 32:483–6. doi: 10.1038/eye.2017.220
69. Nagelhus, EA, Mathiisen, TM, and Ottersen, OP. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience. (2004) 129:905–13. doi: 10.1016/j.neuroscience.2004.08.053
70. Ruiz-Ederra, J, Zhang, H, and Verkman, AS. Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 potassium channels in retinal Müller cells. J Biol Chem. (2007) 282:21866–72. doi: 10.1074/jbc.M703236200
71. Iliff, JJ, Wang, M, Liao, Y, Plogg, BA, Peng, W, Gundersen, GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. (2012) 4:147ra11. doi: 10.1126/scitranslmed.3003748
72. Naylor, A, Hopkins, A, Hudson, N, and Campbell, M. Tight junctions of the outer blood retina barrier. Int J Mol Sci. (2019) 21:211. doi: 10.3390/ijms21010211
73. Törnquist, P, Alm, A, and Bill, A. Permeability of ocular vessels and transport across the blood-retinal-barrier. Eye. (1990) 4:303–9. doi: 10.1038/eye.1990.41
74. Rizzolo, LJ. Polarity and the development of the outer blood-retinal barrier. Histol Histopathol. (1997) 12:1057–67.
75. Fadjukov, J, Wienbar, S, Hakanen, S, Aho, V, Vihinen-Ranta, M, Ihalainen TO, et al. Gap junctions and connexin hemichannels both contribute to the electrical properties of retinal pigment epithelium. J Gen Physiol. (2022) 154:e202112916. doi: 10.1085/jgp.202112916
76. Negi, A, and Marmor, MF. Quantitative estimation of metabolic transport of subretinal fluid. Invest Ophthalmol Vis Sci. (1986) 27:1564–8.
77. Negi, A, and Marmor, MF. Mechanisms of subretinal fluid resorption in the cat eye. Invest Ophthalmol Vis Sci. (1986) 27:1560–3.
78. Dvoriashyna, M, Foss, AJE, Gaffney, EA, and Repetto, R. Fluid and solute transport across the retinal pigment epithelium: a theoretical model. J R Soc Interface. (2020) 17:20190735. doi: 10.1098/rsif.2019.0735
79. Bunt-Milam, AH, Saari, JC, Klock, IB, and Garwin, GG. Zonulae adherentes pore size in the external limiting membrane of the rabbit retina. Invest Ophthalmol Vis Sci. (1985) 26:1377–80.
80. Fisher, RF. The influence of age on some ocular basement membranes. Eye. (1987) 1:184–9. doi: 10.1038/eye.1987.35
81. Soubrane, G. Macular edema of choroidal origin. Dev Ophthalmol. (2017) 58:202–19. doi: 10.1159/000455282
82. Kim, JT, Lee, DH, Joe, SG, Kim, JG, and Yoon, YH. Changes in choroidal thickness in relation to the severity of retinopathy and macular edema in type 2 diabetic patients. Invest Ophthalmol Vis Sci. (2013) 54:3378–84. doi: 10.1167/iovs.12-11503
83. Amjad, R, Lee, CA, Farooqi, HMU, Khan, H, and Paeng, DG. Choroidal thickness in different patterns of diabetic macular edema. J Clin Med. (2022) 11:6169. doi: 10.3390/jcm11206169
85. Kliffen, M, Sharma, HS, Mooy, CM, Kerkvliet, S, and de Jong, PT. Increased expression of angiogenic growth factors in age-related maculopathy. Br J Ophthalmol. (1997) 81:154–62. doi: 10.1136/bjo.81.2.154
86. Luo, Y, Xiao, W, Zhu, X, Mao, Y, Liu, X, Chen, X, et al. Differential expression of claudins in retinas during normal development and the angiogenesis of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. (2011) 52:7556–64. doi: 10.1167/iovs.11-7185
87. Peng, S, Adelman, RA, and Rizzolo, LJ. Minimal effects of VEGF and anti-VEGF drugs on the permeability or selectivity of RPE tight junctions. Invest Ophthalmol Vis Sci. (2010) 51:3216–25. doi: 10.1167/iovs.09-4162
88. Peng, S, Rao, VS, Adelman, RA, and Rizzolo, LJ. Claudin-19 and the barrier properties of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. (2011) 52:1392–403. doi: 10.1167/iovs.10-5984
89. Shen, L, Weber, CR, and Turner, JR. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J Cell Biol. (2008) 181:683–95. doi: 10.1083/jcb.200711165
90. Rodriguez-Boulan, E, and Macara, IG. Organization and execution of the epithelial polarity programme. Nat Rev Mol Cell Biol. (2014) 15:225–42. doi: 10.1038/nrm3775
91. Baas, AF, Kuipers, J, van der Wel, NN, Batlle, E, Koerten, HK, Peters, PJ, et al. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cells. (2004) 116:457–66. doi: 10.1016/S0092-8674(04)00114-X
92. Klaassen, I, Van Noorden, CJ, and Schlingemann, RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res. (2013) 34:19–48. doi: 10.1016/j.preteyeres.2013.02.001
93. Yang, X, Chung, JY, Rai, U, and Esumi, N. Cadherins in the retinal pigment epithelium (RPE) revisited: P-cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PLoS One. (2018) 13:e0191279. doi: 10.1371/journal.pone.0191279
94. Hartsock, A, and Nelson, WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. (2008) 1778:660–9. doi: 10.1016/j.bbamem.2007.07.012
95. Campbell, HK, Maiers, JL, and DeMali, KA. Interplay between tight junctions & adherens junctions. Exp Cell Res. (2017) 358:39–44. doi: 10.1016/j.yexcr.2017.03.061
96. Navaratna, D, McGuire, PG, Menicucci, G, and Das, A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes. (2007) 56:2380–7. doi: 10.2337/db06-1694
97. Zihni, C, Mills, C, Matter, K, and Balda, MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. (2016) 17:564–80. doi: 10.1038/nrm.2016.80
98. Díaz-Coránguez, M, Ramos, C, and Antonetti, DA. The inner blood-retinal barrier: cellular basis and development. Vis Res. (2017) 139:123–37. doi: 10.1016/j.visres.2017.05.009
99. Sander, B, Larsen, M, Moldow, B, and Lund-Andersen, H. Diabetic macular edema: passive and active transport of fluorescein through the blood-retina barrier. Invest Ophthalmol Vis Sci. (2001) 42:433–8.
100. Marmor, MF, Negi, A, and Maurice, DM. Kinetics of macromolecules injected into the subretinal space. Exp Eye Res. (1985) 40:687–96. doi: 10.1016/0014-4835(85)90138-1
101. Atkinson, EG, Jones, S, Ellis, BA, Dumonde, DC, and Graham, E. Molecular size of retinal vascular leakage determined by FITC-dextran angiography in patients with posterior uveitis. Eye. (1991) 5:440–6. doi: 10.1038/eye.1991.71
102. Klaassen, I, Hughes, JM, Vogels, IM, Schalkwijk, CG, Van Noorden, CJ, and Schlingemann, RO. Altered expression of genes related to blood-retina barrier disruption in streptozotocin-induced diabetes. Exp Eye Res. (2009) 89:4–15. doi: 10.1016/j.exer.2009.01.006
103. Stamatovic, SM, Keep, RF, Kunkel, SL, and Andjelkovic, AV. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via rho and rho kinase. J Cell Sci. (2003) 116:4615–28. doi: 10.1242/jcs.00755
104. Maruo, N, Morita, I, Shirao, M, and Murota, S. IL-6 increases endothelial permeability in vitro. Endocrinology. (1992) 131:710–4. doi: 10.1210/endo.131.2.1639018
105. Miyamoto, K, Khosrof, S, Bursell, SE, Rohan, R, Murata, T, Clermont, AC, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. (1999) 96:10836–41. doi: 10.1073/pnas.96.19.10836
106. Rangasamy, S, McGuire, PG, Franco Nitta, C, Monickaraj, F, Oruganti, SR, and Das, A. Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One. (2014) 9:e108508. doi: 10.1371/journal.pone.0108508
107. Joussen, AM, Poulaki, V, Mitsiades, N, Cai, WY, Suzuma, I, Pak, J, et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J. (2003) 17:76–8. doi: 10.1096/fj.02-0157fje
108. Joussen, AM, Poulaki, V, Le, ML, Koizumi, K, Esser, C, Janicki, H, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. (2004) 18:1450–2. doi: 10.1096/fj.03-1476fje
109. Aiello, LP, Avery, RL, Arrigg, PG, Keyt, BA, Jampel, HD, Shah, ST, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. (1994) 331:1480–7. doi: 10.1056/NEJM199412013312203
110. Vinores, SA, Chan, CC, Vinores, MA, Matteson, DM, Chen, YS, Klein, DA, et al. Increased vascular endothelial growth factor (VEGF) and transforming growth factor beta (TGFbeta) in experimental autoimmune uveoretinitis: upregulation of VEGF without neovascularization. J Neuroimmunol. (1998) 89:43–50. doi: 10.1016/S0165-5728(98)00075-7
111. Noma, H, Yasuda, K, and Shimura, M. Cytokines and the pathogenesis of macular edema in branch retinal vein occlusion. J Ophthalmol. (2019) 2019:5185128. doi: 10.1155/2019/5185128
112. Noma, H, Funatsu, H, Mimura, T, Harino, S, and Hori, S. Vitreous levels of interleukin-6 and vascular endothelial growth factor in macular edema with central retinal vein occlusion. Ophthalmology. (2009) 116:87–93. doi: 10.1016/j.ophtha.2008.09.034
113. Noma, H, Minamoto, A, Funatsu, H, Tsukamoto, H, Nakano, K, Yamashita, H, et al. Intravitreal levels of vascular endothelial growth factor and interleukin-6 are correlated with macular edema in branch retinal vein occlusion. Graefes Arch Clin Exp Ophthalmol. (2006) 244:309–15. doi: 10.1007/s00417-004-1087-4
114. Aiello, LP, Northrup, JM, Keyt, BA, Takagi, H, and Iwamoto, MA. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol. (1995) 113:1538–44. doi: 10.1001/archopht.1995.01100120068012
115. Murakami, T, Felinski, EA, and Antonetti, DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. (2009) 284:21036–46. doi: 10.1074/jbc.M109.016766
116. Murakami, T, Frey, T, Lin, C, and Antonetti, DA. Protein kinase cβ phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes. (2012) 61:1573–83. doi: 10.2337/db11-1367
117. Antonetti, DA, Barber, AJ, Hollinger, LA, Wolpert, EB, and Gardner, TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. (1999) 274:23463–7. doi: 10.1074/jbc.274.33.23463
118. Harhaj, NS, Felinski, EA, Wolpert, EB, Sundstrom, JM, Gardner, TW, and Antonetti, DA. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci. (2006) 47:5106–15. doi: 10.1167/iovs.06-0322
119. Antonetti, DA, Barber, AJ, Khin, S, Lieth, E, Tarbell, JM, and Gardner, TW. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group Diabet. (1998) 47:1953–9. doi: 10.2337/diabetes.47.12.1953
120. Aiello, LP, Bursell, SE, Clermont, A, Duh, E, Ishii, H, Takagi, C, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. (1997) 46:1473–80. doi: 10.2337/diab.46.9.1473
121. Sheetz, MJ, Aiello, LP, Davis, MD, Danis, R, Bek, T, Cunha-Vaz, J, et al. The effect of the oral PKC β inhibitor ruboxistaurin on vision loss in two phase 3 studies. Invest Ophthalmol Vis Sci. (2013) 54:1750–7. doi: 10.1167/iovs.12-11055
122. Vinores, SA, Van Niel, E, Swerdloff, JL, and Campochiaro, PA. Electron microscopic immunocytochemical demonstration of blood-retinal barrier breakdown in human diabetics and its association with aldose reductase in retinal vascular endothelium and retinal pigment epithelium. Histochem J. (1993) 25:648–63. doi: 10.1007/BF00157879
123. Vinores, SA, Van Niel, E, Swerdloff, JL, and Campochiaro, PA. Electron microscopic immunocytochemical evidence for the mechanism of blood-retinal barrier breakdown in galactosemic rats and its association with aldose reductase expression and inhibition. Exp Eye Res. (1993) 57:723–35. doi: 10.1006/exer.1993.1180
124. Vinores, SA, Derevjanik, NL, Mahlow, J, Berkowitz, BA, and Wilson, CA. Electron microscopic evidence for the mechanism of blood-retinal barrier breakdown in diabetic rabbits: comparison with magnetic resonance imaging. Pathol Res Pract. (1998) 194:497–505. doi: 10.1016/S0344-0338(98)80118-0
125. Hofman, P, Blaauwgeers, HG, Tolentino, MJ, Adamis, AP, Nunes Cardozo, BJ, Vrensen, GF, et al. VEGF-A induced hyperpermeability of blood-retinal barrier endothelium in vivo is predominantly associated with pinocytotic vesicular transport and not with formation of fenestrations. Vascular endothelial growth factor-a. Curr Eye Res. (2000) 21:637–45. doi: 10.1076/0271-3683(200008)2121-VFT637
126. Esser, S, Lampugnani, MG, Corada, M, Dejana, E, and Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. (1998) 111:1853–65. doi: 10.1242/jcs.111.13.1853
127. Gavard, J, and Gutkind, JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. (2006) 8:1223–34. doi: 10.1038/ncb1486
128. Huang, H, He, J, Johnson, D, Wei, Y, Liu, Y, Wang, S, et al. Deletion of placental growth factor prevents diabetic retinopathy and is associated with Akt activation and HIF1α-VEGF pathway inhibition. Diabetes. (2015) 64:200–12. doi: 10.2337/db14-0016
129. Nambu, H, Nambu, R, Oshima, Y, Hackett, SF, Okoye, G, Wiegand, S, et al. Angiopoietin 1 inhibits ocular neovascularization and breakdown of the blood-retinal barrier. Gene Ther. (2004) 11:865–73. doi: 10.1038/sj.gt.3302230
130. Heier, JS, Brown, DM, Chong, V, Korobelnik, JF, Kaiser, PK, Nguyen, QD, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. (2012) 119:2537–48. doi: 10.1016/j.ophtha.2012.09.006
131. Nguyen, QD, Brown, DM, Marcus, DM, Boyer, DS, Patel, S, Feiner, L, et al. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology. (2012) 119:789–801. doi: 10.1016/j.ophtha.2011.12.039
132. Korobelnik, JF, Do, DV, Schmidt-Erfurth, U, Boyer, DS, Holz, FG, Heier, JS, et al. Intravitreal aflibercept for diabetic macular edema. Ophthalmology. (2014) 121:2247–54. doi: 10.1016/j.ophtha.2014.05.006
133. Brown, DM, Campochiaro, PA, Singh, RP, Li, Z, Gray, S, Saroj, N, et al. Ranibizumab for macular edema following central retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology. (2010) 117:1124–33.e1. doi: 10.1016/j.ophtha.2010.02.022
134. Campochiaro, PA, Heier, JS, Feiner, L, Gray, S, Saroj, N, Rundle, AC, et al. Ranibizumab for macular edema following branch retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology. (2010) 117:1102–12.e1. doi: 10.1016/j.ophtha.2010.02.021
135. Mackensen, F, Heinz, C, Becker, MD, and Heiligenhaus, A. Intravitreal bevacizumab (avastin) as a treatment for refractory macular edema in patients with uveitis: a pilot study. Retina. (2008) 28:41–5. doi: 10.1097/IAE.0b013e318156db75
136. Nguyen, QD, Shah, SM, Khwaja, AA, Channa, R, Hatef, E, Do, DV, et al. Two-year outcomes of the ranibizumab for edema of the mAcula in diabetes (READ-2) study. Ophthalmology. (2010) 117:2146–51. doi: 10.1016/j.ophtha.2010.08.016
137. Campochiaro, PA, Hafiz, G, Channa, R, Shah, SM, Nguyen, QD, Ying, H, et al. Antagonism of vascular endothelial growth factor for macular edema caused by retinal vein occlusions: two-year outcomes. Ophthalmology. (2010) 117:2387–94.e1-5. doi: 10.1016/j.ophtha.2010.03.060
138. Saint-Geniez, M, Maharaj, AS, Walshe, TE, Tucker, BA, Sekiyama, E, Kurihara, T, et al. Endogenous VEGF is required for visual function: evidence for a survival role on Müller cells and photoreceptors. PLoS One. (2008) 3:e3554. doi: 10.1371/journal.pone.0003554
139. Aveleira, CA, Lin, CM, Abcouwer, SF, Ambrósio, AF, and Antonetti, DA. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. (2010) 59:2872–82. doi: 10.2337/db09-1606
140. Luna, JD, Chan, CC, Derevjanik, NL, Mahlow, J, Chiu, C, Peng, B, et al. Blood-retinal barrier (BRB) breakdown in experimental autoimmune uveoretinitis: comparison with vascular endothelial growth factor, tumor necrosis factor alpha, and interleukin-1beta-mediated breakdown. J Neurosci Res. (1997) 49:268–80. doi: 10.1002/(SICI)1097-4547(19970801)49:3<268::AID-JNR2>3.0.CO;2-A
141. Bamforth, SD, Lightman, S, and Greenwood, J. The effect of TNF-alpha and IL-6 on the permeability of the rat blood-retinal barrier in vivo. Acta Neuropathol. (1996) 91:624–32. doi: 10.1007/s004010050476
142. Huang, H, Gandhi, JK, Zhong, X, Wei, Y, Gong, J, Duh, EJ, et al. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest Ophthalmol Vis Sci. (2011) 52:1336–44. doi: 10.1167/iovs.10-5768
143. Peng, S, Gan, G, Rao, VS, Adelman, RA, and Rizzolo, LJ. Effects of proinflammatory cytokines on the claudin-19 rich tight junctions of human retinal pigment epithelium. Invest Ophthalmol Vis Sci. (2012) 53:5016–28. doi: 10.1167/iovs.11-8311
144. Jiang, Q, Li, Z, Tao, T, Duan, R, Wang, X, and Su, W. TNF-α in uveitis: from bench to clinic. Front Pharmacol. (2021) 12:740057. doi: 10.3389/fphar.2021.740057
145. Leal, I, Rodrigues, FB, Sousa, DC, Duarte, GS, Romão, VC, Marques-Neves, C, et al. Anti-TNF drugs for chronic uveitis in adults- a systematic review and meta-analysis of randomized controlled trials. Front Med. (2019) 6:104. doi: 10.3389/fmed.2019.00104
146. Nicolela Susanna, F, and Pavesio, C. A review of ocular adverse events of biological anti-TNF drugs. J Ophthalmic Inflamm Infect. (2020) 10:11. doi: 10.1186/s12348-020-00202-6
147. Liu, Y, Biarnés Costa, M, and Gerhardinger, C. IL-1β is upregulated in the diabetic retina and retinal vessels: cell-specific effect of high glucose and IL-1β autostimulation. PLoS One. (2012) 7:e36949. doi: 10.1371/journal.pone.0036949
148. Palenski, TL, Sorenson, CM, and Sheibani, N. Inflammatory cytokine-specific alterations in retinal endothelial cell function. Microvasc Res. (2013) 89:57–69. doi: 10.1016/j.mvr.2013.06.007
149. Mesquida, M, Leszczynska, A, Llorenç, V, and Adán, A. Interleukin-6 blockade in ocular inflammatory diseases. Clin Exp Immunol. (2014) 176:301–9. doi: 10.1111/cei.12295
150. van Kooij, B, Rothova, A, Rijkers, GT, and de Groot-Mijnes, JD. Distinct cytokine and chemokine profiles in the aqueous of patients with uveitis and cystoid macular edema. Am J Ophthalmol. (2006) 142:192–4. doi: 10.1016/j.ajo.2006.02.052
151. Funatsu, H, Yamashita, H, Noma, H, Mimura, T, Yamashita, T, and Hori, S. Increased levels of vascular endothelial growth factor and interleukin-6 in the aqueous humor of diabetics with macular edema. Am J Ophthalmol. (2002) 133:70–7. doi: 10.1016/S0002-9394(01)01269-7
152. Oh, IK, Kim, SW, Oh, J, Lee, TS, and Huh, K. Inflammatory and angiogenic factors in the aqueous humor and the relationship to diabetic retinopathy. Curr Eye Res. (2010) 35:1116–27. doi: 10.3109/02713683.2010.510257
153. Mesquida, M, Drawnel, F, Lait, PJ, Copland, DA, Stimpson, ML, Llorenç, V, et al. Modelling macular edema: the effect of IL-6 and IL-6R blockade on human blood-retinal barrier integrity in vitro. Transl Vis Sci Technol. (2019) 8:32. doi: 10.1167/tvst.8.5.32
154. Alsaffar, H, Martino, N, Garrett, JP, and Adam, AP. Interleukin-6 promotes a sustained loss of endothelial barrier function via Janus kinase-mediated STAT3 phosphorylation and de novo protein synthesis. Am J Phys Cell Physiol. (2018) 314:C589–602. doi: 10.1152/ajpcell.00235.2017
155. Yoshimura, T, Sonoda, KH, Ohguro, N, Ohsugi, Y, Ishibashi, T, Cua, DJ, et al. Involvement of Th17 cells and the effect of anti-IL-6 therapy in autoimmune uveitis. Rheumatology. (2009) 48:347–54. doi: 10.1093/rheumatology/ken489
156. Hohki, S, Ohguro, N, Haruta, H, Nakai, K, Terabe, F, Serada, S, et al. Blockade of interleukin-6 signaling suppresses experimental autoimmune uveoretinitis by the inhibition of inflammatory Th17 responses. Exp Eye Res. (2010) 91:162–70. doi: 10.1016/j.exer.2010.04.009
157. Tode, J, Richert, E, Koinzer, S, Klettner, A, Pickhinke, U, Garbers, C, et al. Intravitreal injection of anti-interleukin (IL)-6 antibody attenuates experimental autoimmune uveitis in mice. Cytokine. (2017) 96:8–15. doi: 10.1016/j.cyto.2017.02.023
158. Sadiq, MA, Hassan, M, Afridi, R, Halim, MS, Do, DV, Sepah, YJ, et al. Posterior segment inflammatory outcomes assessed using fluorescein angiography in the STOP-UVEITIS study. Int J Retina Vitreous. (2020) 6:47. doi: 10.1186/s40942-020-00245-w
159. Haller, JA, Bandello, F, Belfort, R Jr, Blumenkranz, MS, Gillies, M, Heier, J, et al. Randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with macular edema due to retinal vein occlusion. Ophthalmology. (2010) 117:1134–46.e3. doi: 10.1016/j.ophtha.2010.03.032
160. Yeh, S, Khurana, RN, Shah, M, Henry, CR, Wang, RC, Kissner, JM, et al. Efficacy and safety of suprachoroidal CLS-TA for macular edema secondary to noninfectious uveitis: phase 3 randomized trial. Ophthalmology. (2020) 127:948–55. doi: 10.1016/j.ophtha.2020.01.006
161. Silva, PS, Sun, JK, and Aiello, LP. Role of steroids in the management of diabetic macular edema and proliferative diabetic retinopathy. Semin Ophthalmol. (2009) 24:93–9. doi: 10.1080/08820530902800355
162. De Paiva, CS, Corrales, RM, Villarreal, AL, Farley, WJ, Li, DQ, Stern, ME, et al. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Exp Eye Res. (2006) 83:526–35. doi: 10.1016/j.exer.2006.02.004
163. Mohammad, G, and Siddiquei, MM. Role of matrix metalloproteinase-2 and -9 in the development of diabetic retinopathy. J Ocul Biol Dis Infor. (2012) 5:1–8. doi: 10.1007/s12177-012-9091-0
164. Giebel, SJ, Menicucci, G, McGuire, PG, and Das, A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Investig. (2005) 85:597–607. doi: 10.1038/labinvest.3700251
165. Grover, S, Fishman, GA, Fiscella, RG, and Adelman, AE. Efficacy of dorzolamide hydrochloride in the management of chronic cystoid macular edema in patients with retinitis pigmentosa. Retina. (1997) 17:222–31. doi: 10.1097/00006982-199717030-00009
166. Fishman, GA, Gilbert, LD, Fiscella, RG, Kimura, AE, and Jampol, LM. Acetazolamide for treatment of chronic macular edema in retinitis pigmentosa. Arch Ophthalmol. (1989) 107:1445–52. doi: 10.1001/archopht.1989.01070020519031
167. Giusti, C, Forte, R, Vingolo, EM, and Gargiulo, P. Is acetazolamide effective in the treatment of diabetic macular edema? A pilot study Int Ophthalmol. (2001) 24:79–88. doi: 10.1023/A:1016354517304
168. Gao, BB, Clermont, A, Rook, S, Fonda, SJ, Srinivasan, VJ, Wojtkowski, M, et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med. (2007) 13:181–8. doi: 10.1038/nm1534
169. Wolfensberger, TJ, Dmitriev, AV, and Govardovskii, VI. Inhibition of membrane-bound carbonic anhydrase decreases subretinal pH and volume. Doc Ophthalmol. (1999) 97:261–71. doi: 10.1023/A:1002496223131
170. Wolfensberger, TJ, Chiang, RK, Takeuchi, A, and Marmor, MF. Inhibition of membrane-bound carbonic anhydrase enhances subretinal fluid absorption and retinal adhesiveness. Graefes Arch Clin Exp Ophthalmol. (2000) 238:76–80. doi: 10.1007/s004170050013
171. Wolfensberger, TJ, Mahieu, I, Jarvis-Evans, J, Boulton, M, Carter, ND, Nógrádi, A, et al. Membrane-bound carbonic anhydrase in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. (1994) 35:3401–7.
172. Sagaties, MJ, Raviola, G, Schaeffer, S, and Miller, C. The structural basis of the inner blood-retina barrier in the eye of Macaca mulatta. Invest Ophthalmol Vis Sci. (1987) 28:2000–14.
173. Gu, X, Fliesler, SJ, Zhao, YY, Stallcup, WB, Cohen, AW, and Elliott, MH. Loss of caveolin-1 causes blood-retinal barrier breakdown, venous enlargement, and mural cell alteration. Am J Pathol. (2014) 184:541–55. doi: 10.1016/j.ajpath.2013.10.022
174. Tian, XF, Xia, XB, Xu, HZ, Xiong, SQ, and Jiang, J. Caveolin-1 expression regulates blood-retinal barrier permeability and retinal neovascularization in oxygen-induced retinopathy. Clin Exp Ophthalmol. (2012) 40:e58–66. doi: 10.1111/j.1442-9071.2011.02656.x
175. Vinores, SA, Derevjanik, NL, Ozaki, H, Okamoto, N, and Campochiaro, PA. Cellular mechanisms of blood-retinal barrier dysfunction in macular edema. Doc Ophthalmol. (1999) 97:217–28. doi: 10.1023/A:1002136712070
Keywords: human, retina, macula, macular edema, diabetic retinopathy, retinal vein occlusion, uveitis
Citation: Haydinger CD, Ferreira LB, Williams KA and Smith JR (2023) Mechanisms of macular edema. Front. Med. 10:1128811. doi: 10.3389/fmed.2023.1128811
Edited by:
Georgios Panos, Nottingham University Hospitals NHS Trust, United KingdomCopyright © 2023 Haydinger, Ferreira, Williams and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justine R. Smith, anVzdGluZS5zbWl0aEBmbGluZGVycy5lZHUuYXU=