 John Puetz
John Puetz- Department of Pediatrics, Division of Pediatric Hematology/Oncology, Saint Louis University School of Medicine, St. Louis, MO, United States
After decades of investigation, gene therapy has received regulatory approval to treat hemophilia. However, since gene therapy investigations were initially conceived, other avenues of treatment have revolutionized the care of hemophilia. Emergent data is showing that gene therapy may not be as beneficial as hoped and more toxic than planned. At a minimum, a reassessment of risk/benefit estimate of gene therapy for hemophilia is needed.
Introduction
Over the past few years, multiple human gene therapy trials have successfully achieved hemostatic levels of factors VIII and IX using recombinant adeno-associated virus (rAAV) vectors (1, 2). As a culmination of decades of work, rAAV vectors have received regulatory approval for treatment of both hemophilia A and B. Unfortunately, over the past few years, data has emerged that now calls into question the wisdom of using rAAV vectors to treat hemophilia A and B. This manuscript will review current treatments of hemophilia, benefits of gene therapy, and the risks of gene therapy. Although other vectors are in development, since rAAV is the principal vector used for gene therapy in hemophilia, this manuscript will focus on rAAV toxicity. At a minimum, a reassessment of the risk/benefit estimate before routinely administering rAAV vectors to treat hemophilia seems prudent.
Current treatment of hemophilia
Hemophilia A and B are bleeding disorders in which patients bleed abnormally from trivial causes. Prior to modern treatment, patients suffered from recurrent abnormal bleeding leading to crippling, painful, arthropathies and premature death. Starting in the 1960’s and 70’s, factor replacement became available and was given in the home (3, 4). Bleeding episodes could finally be controlled. Patients underwent elective surgeries. However, joint disease and other complications persisted. Life expectancy improved but remained far from normal. Factor replacement led to alloimmunization (inhibitor) in some patients, which complicated treatment immensely. In the 1980s contamination of factor concentrates with HIV and hepatitis C devastated the hemophilia community (3, 4). In this environment, alternatives to episodic plasma derived factor replacement, including gene therapy, were proposed (5). Fortunately, advances in treatments in the 1990s started to turn the tide on the complications of hemophilia. Recombinant factor products were developed and improvements in plasma derived products eliminated the risk of HIV and hepatitis C transmission (4). Prophylactic infusion of factor reduced joint bleeding and other complications (6). Life expectancy improved further. However, the burden of treatment was significant. Prophylactic regimens required intravenous infusions at least twice weekly, and daily to achieve immune tolerance for those alloimmunized (6, 7). To facilitate the infusion, indwelling catheters were used. This led to infectious and thrombotic complications (8). The 2010s saw further improvements in treatment. Extended half-life products were developed that reduced the burden of prophylactic treatment (9, 10). Patients with hemophilia A and B are now treated with prophylactic regimens with once weekly (or less) infusions (9, 11). The factor VIII mimetic Emicizumab has revolutionized the treatment for hemophilia A (12–14). The annualized bleeding rate (ABR) for children with hemophilia A (alloimmunized or not) is approaching zero with subcutaneous injections of Emicizumab (13). This can be given as infrequently as once monthly (13, 14). The life expectancy of people with hemophilia is now approaching normal (15). Additional improvements in treatments are on the horizon. Treatments aimed at rebalancing hemostasis are nearing regulatory approval (14, 16, 17). Thus, a subcutaneous treatment for hemophilia B (alloimmunized or not) may soon be available. In diseases other than hemophilia, extended half-life strategies for monoclonal antibodies have been developed with therapeutic levels lasting 6 months (18). Similar technologies could be applied to hemophilia treatments, reducing the need for subcutaneous injections to 2–3 times a year.
The hope of gene therapy
While the life expectancy of people with hemophilia is near normal, it is not normal (15). Joint bleeding has been reduced, but not eliminated (6, 9–14). ABRs are approaching zero, but are not zero (9–14, 16, 17). The burden of treatment has been drastically reduced but persists. Treatments are not accessible to large populations of patients and where they are, patients do not always adhere to recommended treatments (19). The risk of transmitting infectious agents or other manufacturing mishaps remains (20). Emicizumab may increase the risk of thrombotic events (21). The financial costs of treatment remain substantial. In short, there remains room for improvement in the treatment of hemophilia. Current gene therapy regimens may fulfill that need. A one-time treatment with rAAV vectors has resulted in hemostatic levels for both factor VIII and IX (1, 2). Long term data is still needed, but current rAAV gene therapy may further reduce or eliminate joint disease and other bleeding complications. Although the financial cost of gene therapy is substantial, if hemostatic levels are sustained, the lifetime costs of treatment will be reduced (22). The most significant side effect of rAAV gene therapy for hemophilia during clinical trials has been mild liver inflammation, which has generally been controlled with steroids (1, 2). Although the specter of genotoxicity has been raised, none has been observed to date in human trials of rAAV for hemophilia. Thus, it has been claimed that it is time to “prepare the way” for hemophilia gene therapy (23). Or is it?
The reality of gene therapy
While the hope of gene therapy is curative treatment, the reality is otherwise. As shown in Table 1, currently approved treatments are not curative (9, 11, 12, 24, 25). Even if not curative, a reduced burden of medication administration and a “bleed-free” mindset would still be beneficial (26). Although the clinical manifestations of hemophilia A and B are similar, they really are two different diseases. This is evident from the results of the phase 3 hemophilia gene therapy trials (24, 25). In both trials, hemostatic levels were achieved, however, there were substantial differences in durability and toxicity.
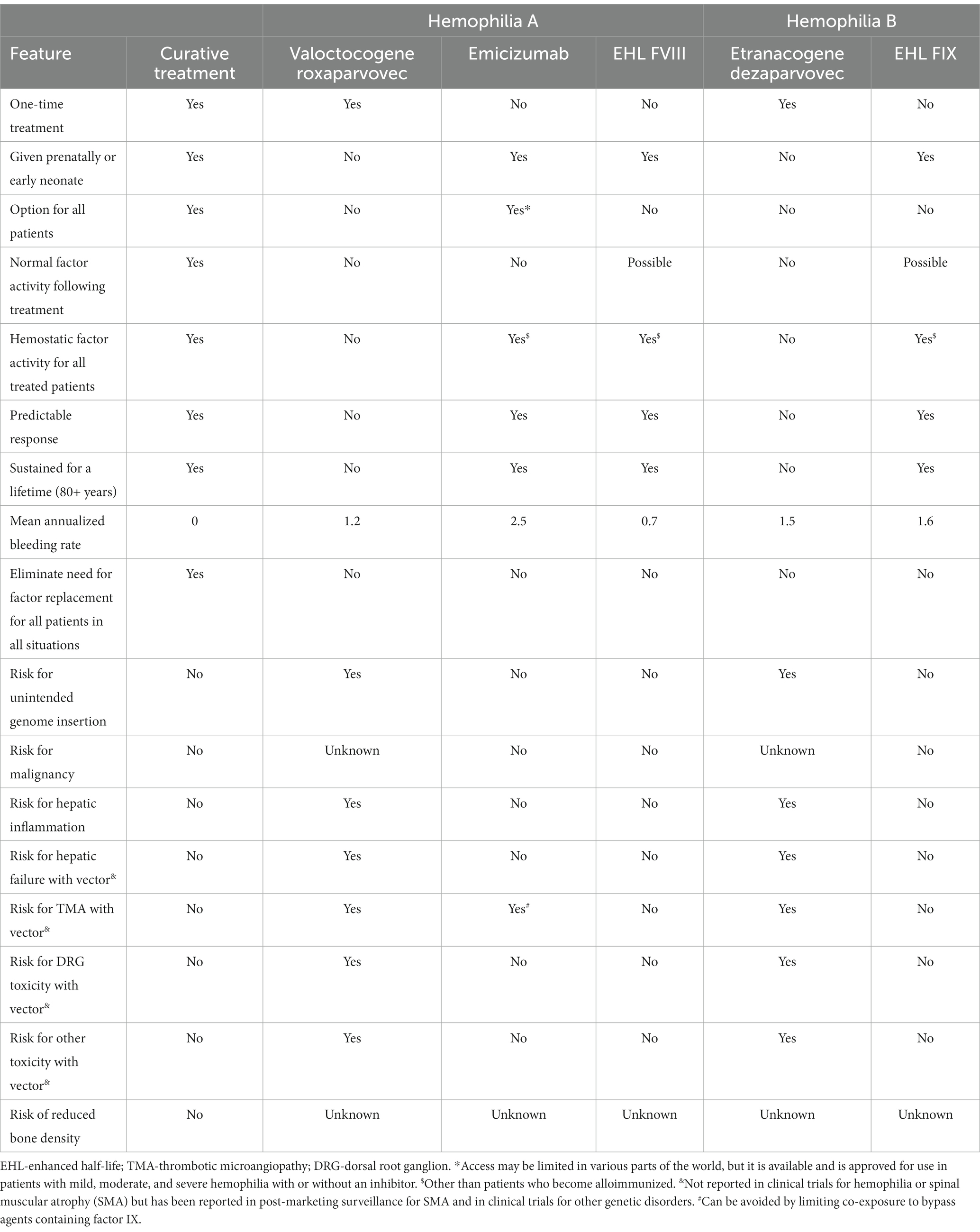
Table 1. Compares curative treatment, current gene therapy, and standard of care.
In the hemophilia B study, 52 of the 54 subjects were able to achieve a mean factor IX activity above baseline of 36 IU/dL at 6 months and dropped minimally by 18 months to 34 IU/dL above baseline (25). Based on this, it has been estimated that >80% of subjects will maintain factor IX level > 2% at 25 years post treatment (27). The need for factor replacement was reduced when compared to the lead-in period. The ABR was also reduced when compared to the lead-in period. However, only 57% of the subjects were on an extended half-life product during the lead-in period. As shown in Table 1, the ABR following gene therapy showed no ABR reduction when compared to factor IX extended half-life studies (9, 25). The principal toxicity reported was elevation in liver transaminases. This occurred in 20% of subjects and was controlled with a course of steroids (median 11 weeks). The gene construct used for the trials was the gain of function factor IX Padua variant. This variant has never been used for human factor IX replacement prior to the gene therapy trials. Thus, the risk of inhibitor development is unknown. Extravascular factor IX binding to collagen type IV is also important for hemostatic activity (28). Also unknown is the factor IX Padua variant extravascular hemostatic activity in humans with hemophilia B.
For the hemophilia A trial, subjects achieved a median factor VIII activity of ~55 IU/dL at 6 months (24). Unfortunately, this was not durable. Within 2 years, roughly 5% of subjects had returned to prophylaxis. Within 5 years, factor VIII activity is estimated to drop to 6 IU/dL. Factor usage and the ABR fell compared to the lead-in period. However, none of the subjects were treated with Emicizumab or Efanesoctocog during the lead-in period (24). As shown in Table 1, the ABR following gene therapy was marginally better compared to clinical trial results of Emicizumab, and higher than trial results of Efanesoctocog (11, 12, 24). Again, the principal toxicity was elevation in liver transaminases. However, this was much more prevalent and recalcitrant to treatment compared to the hemophilia B study. Nearly 90% of hemophilia A subjects developed elevated transaminases. This was mostly treated with glucocorticoids alone for a median of 33 weeks. Roughly 20% of subjects received immunosuppression beyond glucocorticoids including tacrolimus and mycophenolate. The gene construct use was a b-domain deleted factor VIII, This variant has been used extensively for human factor VIII replacement. The dose of vector used during the hemophilia A trial was 3 times higher than the hemophilia B trial (6 × 1013 vg/kg vs. 2 ×1013 vg/kg) (24, 25). It is unknown to what extent this dosing difference may have contributed to the difference in toxicity.
Common to both studies was wide variability in response. Some subjects did not respond at all while others achieved supratherapeutic levels. For those subjects who did not respond or had an unsustained response, redosing with the same construct is not feasible. Potentially, patients can receive subsequent treatment with different vectors and constructs, but this needs to be investigated. Neither phase 3 study described treatment strategies used for surgical procedures. Based on the mean factor levels achieved, it is anticipated that a sizable proportion of subjects will still need factor replacement for surgical procedures.
In summary, the principal benefit of AAV gene therapy seen thus far is not a dramatic reduction in bleeding risk, but the need for frequent medication administration. Thus, gene therapy provides an added measure of convenience and potentially reduced cost. For hemophilia B, this benefit may outweigh the known risks described in the clinical trials. For hemophilia A, this is less clear.
However, there may be additional risk to hemophilia gene therapy with AAV vectors that were not evident in the clinical trials.
Additional risks of gene therapy
Hepatocellular carcinoma
The Adeno-associated virus was discovered in the 1960s and for decades was not thought to cause any human disease (29). For this reason, it was an ideal candidate for use as a gene therapy vector. However, Nault et al. reported wild type AAV (wAAV) insertions near the TERT promoter in human hepatocellular carcinoma (HCC) specimens (30). Additional wAAV insertions near 4 other oncogenes were found, and the insertions were clonal (30). These same oncogenes are targeted by hepatitis B, which is known to contribute to HCC (31). There remains debate as to whether the wAAV insertions are drivers of oncogenes or benign passengers (32). As far as hemophilia gene therapy goes, it really does not matter if wAAV causes HCC or not. wAAV is not infused into patients for gene therapy, rAAV is. The question for hemophilia gene therapy is does rAAV cause HCC?
Unfortunately, the answer is yes, and here there is no debate (33). Pre-clinical Investigations of rAAV gene therapy date back over 2 decades. An early investigation of rAAV involved a mouse model of mucopolysaccharidosis VII (MPSVII). Neonatal mice with this disorder were successfully treated with rAAV. However, a small number developed HCC (34, 35). Because MPSVII is fatal in mice, it was unclear if the rAAV was causing the HCC or the underlying disease was. A follow-up study included a normal control group. These neonatal mice also developed HCC following rAAV (34). Investigation over the ensuing 2 decades showed that rAAV inserts and drives the RIAN locus, more specifically microRNA-341, in neonatal mice. This does not occur in adult mice. Humans do not have a RIAN ortholog, but dogs do (35). The combination of rAAV/neonatal mouse/RIAN locus are needed to induce HCC, or so it was thought (34).
Numerous trials of rAAV gene therapy for a variety of genetic disorders have been carried out in adult mice without the development of HCC (36). One difference in neonatal and adult mice is that neonatal mice have more proliferating cells, notably, hepatocytes. A recent investigation of rAAV gene therapy tested if rAAV could induce HCC in adult mice with liver injury (proliferating hepatocytes) (36). Investigators used either partial hepatectomy or fatty liver disease (high fat diet). Two rAAV vectors were used, one designed to drive the RIAN locus, and another vector control with a reporter gene. As expected, neither vector caused HCC above baseline in control adult mice. The rAAV vector designed to drive the RIAN locus caused HCC in both models of proliferating hepatocytes. Surprisingly, so did the control vector in adult mice with fatty liver disease (partial hepatectomy not tested) (36). In addition, a clinical trial of rAAV for phenylketonuria was placed on hold due to the development of HCC in a mouse model (2). Thus, rAAV can induce HCC in adult mice with proliferating hepatocytes. What about other animal models?
To date, HCC has not been observed in dogs or non-human primate rAAV gene therapy for hemophilia. However, clonally proliferating liver cells have been found (37). Nguyen et al. recently reported on a long term follow up of hemophilia A gene therapy in dogs using rAAV (37). Nine dogs were followed for up to 10 years. Liver samples were available in 6 of the dogs. rAAV integrations were found in 1,741 sites, and between 1 and 130 cells per integration. Preferential integrations near oncogenes in clonally proliferating cells were seen. The dogs were otherwise healthy. The authors did not report on the presence or absence of fatty liver disease. A second long term study of 8 hemophilic A dogs receiving rAAV gene therapy did not find clonal proliferation in the liver (38). This manuscript did not report on rAAV integrations, and additional analysis may be forthcoming. Long term data for hemophilia B gene therapy in dogs is also available (39, 40). Many canine gene therapy strategies for hemophilia B using rAAV targeted skeletal muscle and may not confer the same HCC risk as strategies targeting hepatocytes. There are no reports of HCC. However, detailed analysis for rAAV insertions and clonal proliferation have not been presented.
One subject in a human trial of rAAV for hemophilia B developed HCC (35). This subject had risk factors for developing HCC including hepatitis B and C. His liver specimen has been extensively evaluated and did not reveal genetic changes related to rAAV carcinogenesis but did have genetic changes typically found in HCC (35). No other human subjects have been reported with HCC following gene therapy for hemophilia. A recent report of liver biopsy results from 5 subjects who participated in a hemophilia A gene therapy trial did not show evidence of HCC or clonal proliferation (41). Notably, 4 of 5 subjects had liver steatosis, which apparently was subclinical.
In summary, emergent data has shown that rAAV can induce HCC in adult mice with liver disease (36). Long term studies of small number (<15) of hemophilia A dogs have shown clonal proliferation in the liver (37, 38). If rAAV causes HCC in a small proportion, or even 5%–10% of hemophilia A dogs, it could easily be missed by the current canine studies. To date, no studies have looked at the risk of HCC development of dogs or non-human primates with fatty liver disease following hemophilia AAV gene therapy. No humans have developed HCC or clonal proliferation caused by rAAV. Since the latency for the development of human HCC following rAAV may be decades, the risk of human HCC following rAAV cannot currently be estimated.
Genome integration
Another advantage of using rAAV as a vector is that it mostly remains episomal following insertion into a cell (42). Until recently, studies have suggested that 99+% of rAAV vector was episomal, and <1% integrates. Emergent data has shown a higher percentage of integrations. Up to 3% of rAAV may integrate into liver cells following gene therapy (42). Most hemophilia gene therapy protocols infuse 1014–1015 viral particles and target 1011 hepatocytes. Assuming a more conservative estimate of genome integrations of 0.1%, one could anticipate over 100 million integrations following gene therapy. Indeed, some experimental data confirms this notion (42). Therefore, there are a massive number of integrations following rAAV gene therapy. Most are not intact vectors. With 1011 hepatocytes in a human adult, each with a genome of 3 × 109 base pairs, most integrations are likely to land in an inactive genetic region if they integrate randomly. However, the above referenced dog hemophilia gene therapy study suggests that integrations are not random. They tend to occur near active genes, including oncogenes (37). One driver integration near the wrong oncogene at the wrong time could start the cell toward clonal proliferation and eventually, over years or decades, overt cancer.
Other cancers
In addition to the above-mentioned HCC following rAAV gene therapy for hemophilia, 3 additional cases of cancer have been reported in humans, and one in a dog (43–45). As with the HCC, the other cancers (tonsillar carcinoma, salivary gland carcinoma, leukemia) have been thoroughly investigated and shown not to be caused by rAAV (43–45). It is unknown if immunosuppression received during clinical trials may have contributed to oncogenesis. To date, several hundred subjects have participated in clinical trials involving rAAV for hemophilia with reported observation periods lasting several years. There is easily over 1,000 person years of observation. Thus 4 reports of human cancer may not be unexpected. However, a proper epidemiological investigation seems indicated.
Unfolded protein response
The target cell for hemophilia gene therapy using rAAV is the hepatocyte. Although factor VIII is made in the liver, it is not made in the hepatocyte (46). Accordingly, hemophilia A gene therapy targets a cell that does not typically produce factor VIII. Factor IX is naturally made in the hepatocyte. Factor VIII is a large protein with complex folding. Misfolded factor VIII protein can lead to cellular toxicity via the unfolded protein response (UPR) (47). This has been shown to occur when non-native cells are driven to express factor VIII. This occurs both in vitro (Chinese hamster ovary cells) and in vivo (mouse hepatocytes) for factor VIII (47, 48). Factor IX expression is stable for years following rAAV gene therapy in animal models and humans (1, 2). While factor VIII expression has been stable following rAAV gene therapy in animals, this has not been the case in humans when therapeutic levels are achieved. Several clinical trials of AAV gene therapy for hemophilia A have shown that hemostatic levels are not sustained (24, 49). The etiology for the falling levels remains unclear. An immune response has been investigated and does not clearly seem to be the cause (35). UPR could provide another explanation. However, as above, liver biopsies from human subjects following hemophilia A gene therapy failed to demonstrate evidence for UPR at the time of biopsies (41). Biopsies were performed 2.6–4.1 years following infusion of rAAV. So cellular toxicity/loss from UPR occurring prior to this would have been missed.
Although an immune response and UPR have been independently proposed as explanations for falling factor VIII expression following AAV gene therapy, an investigation by Butterfield et al. suggests that they may not be mutually exclusive (50). In addition, this study also suggested that translational shutdown (related to UPR and immune response) rather than loss of hepatocytes could lead to falling factor VIII expression following AAV gene therapy.
Concerns about UPR and HCC have also been raised. Kapelanski-Lamoureux et al. have investigated UPR and HCC risk in mice (51). In their study, all mice fed a high fat diet following receipt of a B-domain deleted factor VIII gene therapy vector (non-AAV) via hydrodynamic tail vein injection developed liver tumors. This happened less so with a factor VIII variant vector less prone to misfolding and not at all with a control vector. This suggests that factor VIII misfolding in mice fed a high fight diet contributes to the development of HCC independent of viral vector integration.
Spinal muscular atrophy
One of the first rAAV gene therapy treatments to achieve regulatory approval was for the treatment of spinal muscular atrophy (SMA). This is a degenerative neuromuscular disorder that results in early death in those affected with severe (infantile) forms. rAAV gene therapy for this disorder has met with widespread success (52). While affected infants treated with rAAV gene therapy are not normal, they are achieving developmental milestones with an extended lifespan. Like hemophilia rAAV trials, the principal toxicity seen during the SMA trials was mild liver inflammation that is controlled with steroids (53). Now that over 3,000 infants have received gene therapy for this disorder, rare side effects not seen during the clinical trials are being observed (52). At least 9 cases of thrombotic microangiography (TMA) have been reported in the medical literature following rAAV gene therapy for SMA, one of which was fatal (54). Thirty (two fatalities) cases of TMA are reported in the U.S. (United States) Food and Drug Administration Adverse Event Reporting System (FAERS) (55). It is unknown if there is any overlap between cases reported in FAERS and the medical literature. The manufacturer of SMA rAAV has reported two cases of hepatotoxicity leading to fatalities (56). FAERS reports 120 cases of hepatobiliary disorders and 8 fatalities (55). One should interpret the FAERS data with caution, as the cause of death is not listed, only “Reactions.” Duplicate reports may also be present. Therefore, patients who died from causes unrelated to rAAV may be included in this database. Table 2 shows the number of reported relevant “Reactions” and deaths in the FAERS database for regulatory approved medications for SMA (55–59). A comparison of clinical trial results suggests that the event free survival rate is similar between Onasemnogene abeparvovec and Risdiplam, and higher than Nusinersen (59). Post-marketing surveillance from the FAERS database also shows higher mortality reporting for Nusinersen. TMA and hepatobiliary disease following treatment for SMA seems to be relatively unique to Onasemnogene abeparvovec. Use of rAAV in other clinical trials has also resulted in hepatotoxicity related fatalities (54). Because SMA and hemophilia are different diseases, different age groups were treated, and different doses used, similar toxicities may not occur in hemophilia patients following regulatory approval for rAAV gene therapy. However, the potential for rare, serious, and potentially fatal toxicities should be included in any risk/benefit calculation.
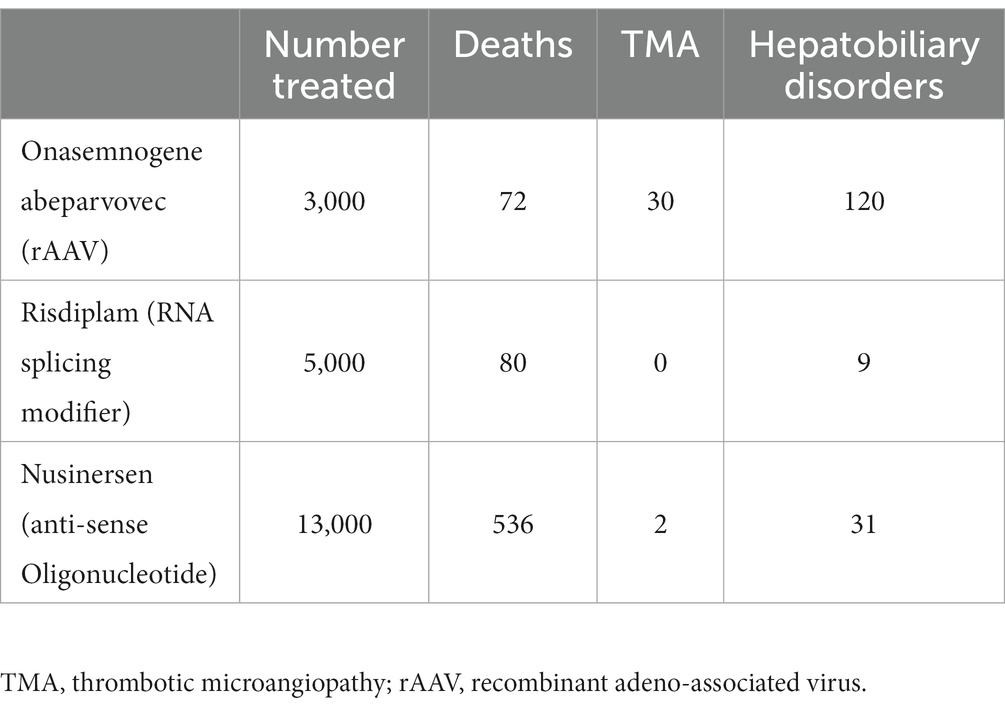
Table 2. Reactions and deaths reported in the Food and Drug Administration Adverse Event Reporting System for medications approved to treat spinal muscular atrophy.
Dorsal root ganglion
Dorsal root ganglion (DRG) pathology has been commonly found in non-human primate gene therapy using rAAV (60). This has been found in a variety of vectors for a variety of diseases. It occurs more commonly with central nervous system administration but is also seen following intravenous administration of rAAV. Immunosuppression does not seem to ameliorate the toxicity. It is not seen with non-expressing vectors (61). Accordingly, the toxicity seems related to recombinant protein expression. Fortunately, the toxicity is mild and short lived, and importantly, does not seem to cause obvious symptoms in the non-human primates. DRG toxicity has been reported in human trials of rAAV, but not for hemophilia (1, 2, 54, 62). However, it does not appear that subjects enrolled in hemophilia rAAV gene therapy trials were carefully evaluated for DRG toxicity, or if they were, this data is not reported in published manuscripts (1, 2, 24, 25).
Conclusion
The past 2–3 decades have seen tremendous advances in the treatment of hemophilia. Unlike many other genetic diseases, hemophilia patients can expect a near normal lifespan with controllable morbidities. The burden of treatment remains but is becoming lighter. Thus, any new treatments are tasked with demonstrating superior efficacy (reduced bleeding risk, medication administration, morbidity, mortality, costs, etc.) to current and near future treatments with little to no added risk. In other words, gene therapy must have “flawlessly safety” (63). Gene therapy for hemophilia using rAAV does not appear to meet those requirements.
Long term data with rAAV gene therapy is lacking. Although indirect evidence points toward the possibility of normalizing the lifespan of some hemophilia patients, that remains to be proven. Hemophilia gene therapy may reduce the risk of hemophilic arthropathy that develops during adulthood. Because rAAV gene therapy will not be given to children, it will have no impact on joint disease that develops prior to adulthood. The lifetime risk of joint disease in people in the general population is well over 50% (64). Thus, even if it is shown that gene therapy reduces the risk of hemophilic arthropathy, large numbers of hemophilia patients will still develop joint disease. The inconvenience of monthly subcutaneous injections currently or soon to be available for hemophilia remains. However, in comparison to most chronic diseases in which daily administration of medication is needed, the burden of hemophilia treatment is no longer onerous. Hence, the benefits of hemophilia gene therapy with rAAV are marginal.
It is often claimed that the risk of hemophilia gene therapy with rAAV is “low,” but what exactly is meant by “low” is not defined (2, 35, 62). If the latency for development of HCC with rAAV is like that of hepatitis B and C, we will not know the true risk for HCC following rAAV gene therapy for several decades. Clinical trials are not powered to detect rare complications. Therefore, the risk of hemophilia gene therapy with rAAV cannot currently be quantified. It is not zero and is potentially catastrophic. In addition, a proper epidemiological investigation of the growing number of reports of cancer following rAAV gene therapy for hemophilia is needed. Also needed are investigations of DRG toxicities in hemophilia patients following rAAV gene therapy. The reason(s) for falling factor VIII activity following rAAV gene therapy remain unresolved and need clarification before rAAV is widely used for this disorder.
Gene therapy for hemophilia was first proposed when the complications of this disorder were substantial. In the decades it has taken for rAAV gene therapy to become successful, gains in the standard of care have become revolutionary. Any new treatment that is “low” risk may be too high of a risk for hemophilia patients. All new treatment requires an assessment of risks and benefits. Despite the rare fatal risks due to rAAV for SMA, the benefits still outweigh the risks for this disorder. Based on the data presented here, that does not seem to be the case for hemophilia gene therapy with rAAV.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the author, without undue reservation.
Author contributions
JP: Writing – original draft, Conceptualization, Investigation.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
This manuscript was originally posted on the Authorea preprint server (65).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Leebeck, F, and Miesbach, W. Gene therapy for hemophilia: a review on clinical benefit, limitations, and remaining issues. Blood. (2021) 138:923–31. doi: 10.1182/blood.2019003777
2. Nathwani, A. Gene therapy for hemophilia. Hematology. (2022) 2022:569–78. doi: 10.1182/hematology.2022000388
4. Mannucci, P, and Tuddenham, E. The Hemophilias – from Royal Genes to gene therapy. N Engl J Med. (2001) 344:1773–9. doi: 10.1056/NEJM200106073442307
5. Anson, D, Hock, R, Austen, D, Smith, K, Brownlee, G, Verma, I, et al. Towards gene therapy for Hemophilia B. Mol Biol Med. (1987) 4:11–20.
6. Manco-Johnson, M, Abshire, T, Shapiro, A, Riske, B, Hacker, M, Kilcoyne, R, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe Hemophilia. N Engl J Med. (2007) 357:535–44. doi: 10.1056/NEJMoa067659
7. Di Michele, D. Immune tolerance induction in haemophilia: evidence and the way forward. J Thromb Haemost. (2011) 9:216–25. doi: 10.1111/j.1538-7836.2011.04349.x
8. Ljung, R. The risk associated with indwelling catheters in children with haemophilia. Br J Haematol. (2007) 138:580–6. doi: 10.1111/j.1365-2141.2007.06703.x
9. Santagostino, E, Martinowitz, U, Lissitchkov, T, Pan-Petesch, P, Hanabusa, H, Oldenburg, J, et al. Long-acting recombinant coagulation factor IX albumin fusion protein (rIX-FP) in hemophilia B: results of a phase 3 trial. Blood. (2016) 127:1761–9. doi: 10.1182/blood-2015-09-669234
10. Mahlangu, J, Powell, J, Ragni, M, Chowdary, P, Josephson, N, Pabinger, I, et al. Phase 3 study of recombinant factor VIII fc fusion protein in severe hemophilia A. Blood. (2014) 123:317–25. doi: 10.1182/blood-2013-10-529974
11. Von Drygalski, A, Chowdary, P, Kulkarni, R, Susen, S, Konkle, B, Oldenburg, J, et al. Efanesoctocog alfa prophylaxis for patients with severe hemophilia A. N Engl J Med. (2023) 388:310–8. doi: 10.1056/NEJMoa2209226
12. Mahlangu, J, and Oldenburg, J. Paz-Priel, Negrier C, Niggli H, Mancuso M et al. Emicizumab prophylaxis in patients who have Hemophilia without inhibitors. N Engl J Med. (2018) 379:811–22. doi: 10.1056/NEJMoa1803550
13. Young, G, Liesner, R, Chang, T, Sidonio, R, Oldenburg, J, Jimenez-Yuste, V, et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. (2019) 134:2127–38. doi: 10.1182/blood.2019001869
14. Nogami, K, and Shima, M. Current and future therapies for haemophilia—beyond factor replacement therapies. Br J Haematol. (2023) 200:23–34. doi: 10.1111/bjh.18379
15. Hassan, S, Monahan, R, Mauser-Bunschoten, E, van Vulpen, L, Eikenboom, J, Beckers, E, et al. Mortality, life expectancy, and cause of death of persons with hemophilia in the Netherlands 2001-2018. J Thromb Haemost. (2021) 19:645–53. doi: 10.1111/jth.15182
16. Frei-Jones, M, Cepo, K, d’Oiron, R, Sim Goh, A, Mathias, M, and Odgaard-Jensen, J. Subcutaneous Concizumab prophylaxis in patients with Hemophilia A or B with inhibitors: efficacy and safety results by Hemophilia subtype from phase 3 explorer 7 trial. Blood. (2022) 140:466–8. doi: 10.1182/blood-2022-166522
17. Young, G, Srivastava, A, Kavakli, K, Ross, C, Sathar, J, Tran, H, et al. Efficacy and safety of Fitusiran prophylaxis, a siRNA therapeutic, in a multicenter phase 3 study (ATLAS-INH) in people with Hemophilia A or B, with inhibitors (PwHI). Blood. (2021) 138:4. doi: 10.1182/blood-2021-150273
18. Mahhitt, L, Dagan, R, Yuan, Y, Baca Cots, M, Bosheva, M, Nadhi, S, et al. Nirsevimab for prevention of RSV in healthy late-preterm and term infants. N Engl J Med. (2022) 386:837–46. doi: 10.1056/NEJMoa2110275
19. Dover, S, Blanchette, V, Wrathall, D, Pullenayegum, E, Kazandjian, D, Song, B, et al. Hemophilia prophylaxis adherence and bleeding using a tailored, frequency-escalated approach: the Canadian Hemophilia primary prophylaxis study. Res Pract Thromb Haemost. (2020) 4:318–25. doi: 10.1002/rth2.12301
20. National Hemophilia Foundation. Stimate Recall. (2023) Available at: https://www.hemophilia.org/news/stimate-recall-update (Accessed 30 June 2023).
21. Abbattista, M, Ciavarella, A, Noone, D, and Peyvandi, F. Hemorrhagic and thrombotic adverse events associated with Emicizumab and extended half-life factor VIII replacement drugs: EudraVigilance data of 2021. J Thromb Haemost. (2023) 21:5460552. doi: 10.1016/j.jth.2023.01.010
22. Bolous, N, Chen, Y, Wang, H, Davidoff, A, Devidas, M, Jacobs, T, et al. The cost-effectiveness of gene therapy for severe hemophilia B: a microsimulation study from the United States perspective. Blood. (2021) 138:1677–90. doi: 10.1182/blood.2021010864
23. Thornburg, C. Prepare the way for Hemophilia A gene therapy. N Engl J Med. (2022) 386:1081–2. doi: 10.1056/NEJMe2200878
24. Mahlangu, J, Kaczmarek, R, von Drygalski, A, Shapriro, S, Chou, C, Ozelo, M, et al. Two-year outcomes of Valoctocogene Roxaparvovec therapy for hemophilia A. N Engl J Med. (2023) 388:694–705. doi: 10.1056/NEJMoa2211075
25. Pipe, S, Leebek, F, Recht, M, Key, N, Castaman, G, Miesbach, S, et al. Gene therapy with Etranacogene Dezaparvovec for hemophilia B. N Engl J Med. (2023) 388:706–18. doi: 10.1056/NEJMoa2211644
26. Pierce, G, and Herzog, R. Two gene therapies for hemophilia available: now what? Mol Ther. (2023) 31:919–20. doi: 10.1016/j.ymthe.2023.03.001
27. Shah, J, Kim, H, Sivamurthy, K, Monahan, P, and Fries, H. Comprehensive analysis and prediction of long-term durability of factor IX activity following etranacogene dezaparvovec gene therapy in the treatment of hemophilia B. Curr Med Res Opin. (2023) 39:227–37. doi: 10.1080/03007995.2022.2133492
28. Lillicrap, D. Evaluating the potential benefits of the extravascular pool of factor IX. Blood Coagul Fibrinolysis. (2021) 32:68–9. doi: 10.1097/MBC.0000000000000969
29. Carter, B. Adeno-associated virus and the development of adeno-associated virus vectors: a historical perspective. Mol Ther. (2004) 10:981–9. doi: 10.1016/j/mythe.2004.09.011
30. Nault, J, Datta, S, Imbeaud, S, Franconi, A, Mallet, M, Couchy, G, et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet. (2015) 47:1187–93. doi: 10.1038/ng.3389
31. Russell, D, and Grompe, M. Adeno-associated virus finds its disease. Nat Genet. (2015) 47:1104–5. doi: 10.1038/ng.3407
32. Buening, H, and Schmidt, M. Adeno-associated vector toxicity-to be or not to be? Mol Ther. (2015) 23:1673–5. doi: 10.1038/mt.2015.182
33. Dave, U, and Cornetta, K. AAV joins the rank of genotoxic vectors. Mol Ther. (2021) 29:418–9. doi: 10.1016/j.mthe.2021.01.007
34. Chandler, R, Sands, M, and Venditti, C. Recombinant adeno-associated viral integration and genotoxicity: insights from animal models. Hum Gene Ther. (2017) 28:314–22. doi: 10.1089/hum.2017.009
35. Monahan, P, Negrier, C, Tarantino, M, Valentino, L, and Mingozzi, F. Emerging immunogenicity and genotoxicity of adeno-associated virus vector gene therapy for Hemophilia. J Clin Med. (2021) 10:2471. doi: 10.3390/jcm10112471
36. Dalwadi, D, Torrens, L, Abril-Fornaguera, J, Pinyol, R, Willoughby, C, Posey, J, et al. Liver injury increases the incidence of HCC following AAV gene therapy in mice. Mol Ther. (2021) 29:680–90. doi: 10.1016/j.jmthe.2020.10.018
37. Nguyen, G, Everett, J, Kafle, S, Roche, A, Raymond, H, Leiby, J, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. (2021) 39:47–55. doi: 10.1038/s41587-020-0741-7
38. Batty, P, Mo, A, Hurlbut, D, Ishida, B, Yates, B, Brown, C, et al. Long-term follow-up of liver-directed, adeno-associated vector-mediated gene therapy in the canine model of hemophilia A. Blood. (2022) 140:2672–83. doi: 10.1182/blood.2021014735
39. Sabatino, D, Bushman, F, Chandler, R, Crystal, R, Davidson, B, Dolmetsch, R, et al. Evaluating the state of the science for adeno-associated virus integration: an integrated perspective. Mol Ther. (2022) 30:2646–63. doi: 10.1016/j.jmthe.2022.06.004
40. Niemeyer, G, Herzog, R, Mount, J, Arruda, V, Tillson, D, Hathcock, J, et al. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood. (2009) 113:797–806. doi: 10.1182/blood-2008-10-181479
41. Fong, S, Yates, B, Sihn, C, Mattis, A, Mitchell, N, Liu, S, et al. Interindividual variability in transgene mRNA and protein production following adeno-associated virus gene therapy for hemophilia A. Nat Med. (2022) 28:789–97. doi: 10.1038/s41591-022-01751-0
42. Dalwadi, D, Calabria, A, Tiyaboonchai, A, Posey, J, Naugler, W, Montini, E, et al. AAV integration in human hepatocytes. Mol Ther. (2021) 29:2898–909. doi: 10.1016/j.ymthe.2021.08.031
43. Konkle, B, Walsh, C, Escobar, M, Josephson, N, Young, G, von Drygalski, A, et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. (2021) 137:763–74. doi: 10.1182/blood.2019004625
44. Gardner, J. BiopharmaDIVE. Biomarin reports cancer case in hemophilia gene therapy trial. (2023) Available at: https://www.biopharmadive.com/news/biomarin-reports-cancer-case-in-hemophilia-gene-therapy-trial/631648/ (Accessed 30 June 2023)
45. Gorder, L, Doshi, B, Willis, E, Lanza, M, Arruda, V, Callan, M, et al. Vector analysis of multicentric lymphoma in a severe Hemophilia A dog after AAV gene therapy. Blood. (2022) 140:4919–20. doi: 10.1182/blood-2022-164440
46. Fahs, S, Hille, M, Shi, Q, Weiler, H, and Montgomery, R. A conditional knockout mouse model reveals endothelial cells as the principal and possible exclusive source of plasma factor VIII. Blood. (2014) 123:3706–13. doi: 10.1182/blood-2014-02-555151
47. Poothong, J, Pottekat, A, Siirin, M, Campos, A, Paton, A, Paton, J, et al. Factor VIII exhibits chaperone-dependent and glucose-regulated reversible amyloid formation in the endoplasmic reticulum. Blood. (2020) 135:1899–911. doi: 10.1182/blood.2019002867
48. Lange, A, Altynova, E, Nguyen, G, and Sabatino, D. Overexpression of factor VIII after AAV delivery is transiently associated with cellular stress in hemophilia A mice. Mol Ther. (2016) 3:16064. doi: 10.1038/mtm.2016.64
49. Samelson-Jones, B, and George, L. Adeno-associated virus gene therapy for hemophilia. Annu Rev Med. (2023) 74:231–47. doi: 10.1146/annurev-med-043021-033013
50. Butterfield, J, Yamada, K, Bertolini, T, Syed, F, Kumar, S, Li, X, et al. Il-15 blockade, and rapamycin rescue multifactorial loss of factor VIII from AAV-transduced hepatocytes in hemophilia A mice. Mol Ther. 12:3552–69. doi: 10.1016/j.ymthe.2022.07.005
51. Kapelanski-Lamoureaux, A, Chen, Z, Gao, Z, Deng, R, Lasaris, A, Lebeaupin, C, et al. Ectopic expression and misfolding in hepatocytes as a cause for hepatocellular carcinoma. Mol Ther. (2022) 30:3542–51. doi: 10.1016/j.ymthe.2022.10.004
52. Mendell, J, Al-Zaidy, S, Shell, R, Arnold, W, Rodino-Klapac, L, Prior, T, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) 377:1713–22. doi: 10.1056/NEJMoa1706198
53. Day, J, Mendell, J, Mercuri, E, Finkel, R, Strauss, K, Kleyn, A, et al. Clinical trial and post marketing safety of Onasemnogene Abeparvovec therapy. Drug Saf. (2021) 44:1109–19. doi: 10.1007/s40264-021-01107-6
54. Ertl, H. Immunogenicity and toxicity of AAV gene therapy. Front Immunol. (2022) 13:975803. doi: 10.3389/fimmu.2022.975803
55. United States Food and Drug Administration Adverse Events Reporting System (FAERS) Public Dashboard. Search Zolgensma. (2022) Available at: https://fis.fda.gov. (Accessed 22 December 2022).
56. Pagliarulo, N. Novartis reports deaths of two patients treated with Zolgensma gene therapy. (2023) Available at: https://www.biopharmadive.com/news/novartis-zolgensma-patient-death-liver-injury/629542/ (Accessed 30 June 2023).
57. Coratti, G, Cutrona, C, Pera, M, Bovis, F, Ponzano, M, Chieppa, F, et al. Motor function in type 2 and 3 SMA patients treated with Nusinersen: a critical review and meta-analysis. Orphanet J Rare Dis. (2021) 16:430. doi: 10.1186/s13023-021-02065-z
58. Genentech. New data for Genentech’s Evrysdi (risdiplam) demonstrates long-term efficacy and safety in a broad population of people with spinal muscular atrophy. (2023) Available at: https://www.gene.com/media/press-releases/14945/2022-03-15/new-data-for-genentechs-evrysdi-risdipla. (Accessed 30 June 2023).
59. Ribero, V, Daigl, M, Marti, Y, Gorni, K, Evans, R, Scott, A, et al. How does risdiplam compare with other treatments for types 1-3 spinal muscular atrophy: a systematic literature review and indirect treatment comparison. J Comp Eff Res. (2022) 11:347–70. doi: 10.2217/cer-2021-0216
60. Hordeaux, J, Buza, E, Dyer, C, Goode, T, Mitchell, T, Richman, L, et al. Adeno-associated virus induced dorsal root ganglion pathology. Hum Gene Ther. (2021) 31:808–19. doi: 10.1089/hum.2020.167
61. Buss, N, Lanigan, L, Zeller, J, Cissell, D, Metea, M, and Adams, E. Characterization of AAV-mediated dorsal root ganglionopathy. Mol Ther Methods Clin Dev. (2022) 24:342–54. doi: 10.1016/j.omtm.2022.01.013
62. Sidonio, R, Pipe, S, Callaghan, M, Valentino, L, Monahan, P, and Croteau, S. Discussing investigational AAV gene therapy with hemophilia patients: a guide. Blood Rev. (2021) 47:100759. doi: 10.1016/j.blre.2020.100759
63. Mannucci, P. Hemophilia treatment innovations: 50 years of progress and more to come. J Thromb Haemost. (2023) 21:403–12. doi: 10.1016/j.jtha.2022.12.029
64. Murphy, L, Schwartz, T, Helmick, C, Renner, J, Tudor, G, Koch, G, et al. Lifetime risk of symptomatic knee osteoarthritis. Arthritis Rheum. (2008) 59:1207–13. doi: 10.1002/art.24021
Keywords: hemophilia, gene therapy, adeno-associated virus, factor VIII, factor IX, hepatocellular carcinoma, thrombotic microangiopathy
Citation: Puetz J (2023) Emergent data influences the risk/benefit assessment of hemophilia gene therapy using recombinant adeno-associated virus. Front. Med. 10:1256919. doi: 10.3389/fmed.2023.1256919
Edited by:
Majed Refaai, University of Rochester, United StatesReviewed by:
Tim Nichols, University of North Carolina at Chapel Hill, United StatesBagirath Gangadharan, Takeda, United States
Copyright © 2023 Puetz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John Puetz, am9obi5wdWV0ekBoZWFsdGguc2x1LmVkdQ==