 Alyona Sorokina
Alyona Sorokina Elizaveta Anchakova
Elizaveta Anchakova Erdem Dashinimaev
Erdem Dashinimaev- 1Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Pirogov Russian National Research Medical University, Moscow, Russia
- 2Moscow Institute of Physics and Technology (State University), Dolgoprudny, Russia
Human immunodeficiency virus type 1 (HIV-1) remains a significant challenge for global public health as limited therapeutic options are available for HIV-infected individuals receiving combination antiretroviral therapy. Additionally, individuals with HIV-1/acquired immunodeficiency syndrome (AIDS) complications have a reduced life expectancy. In recent decades, gene and cell-based strategies have shown promise in achieving a functional cure for HIV-1 infection. The outcomes of therapies with patients in Berlin and London have led to moderate optimism for a highly effective HIV-1 treatment. This review categorizes current strategies for HIV-1 treatment into RNA- and antibody-based therapies, cell and genome editing approaches, and methods for eradicating latent reservoirs. These findings demonstrate how the use of various anti-HIV-1 agents enhances our understanding of HIV-1 infection and may provide important insights for potential HIV-1 treatment.
1 Introduction
Human immunodeficiency virus type 1 belongs to the family Retroviridae of the genus Lentivirus. Two main types of the human immunodeficiency virus (HIV) exist, with HIV-1 being the first one discovered and the most common type found globally, whereas HIV type 2 is primarily found in West Africa (1). The viral genome (approximately 9.8 kb) consists of nine open reading frames that encode 15 proteins and govern all processes involved in the virus life cycle, including receptor binding, membrane fusion, reverse transcription, integration, protease processing, and virus assembly (2). The HIV-1 RNA genome contains structural genes (pol, gag, env), regulatory genes (tat, rev, vpr, nef), and accessory genes (vpu, vif) (3, 4).
Scientific advancements suggest that the threat of an HIV pandemic to public health may be eliminated by 2030, but currently, HIV-1 remains one of the most significant problems for global public health (5). HIV-1 causes acquired immunodeficiency syndrome (AIDS), and HIV-infected patients often suffer from opportunistic infections, cardiovascular and neurological diseases, as well as AIDS-related illnesses such as Hodgkin’s disease, non-Hodgkin’s lymphoma, lymphocytic leukemia, and other malignant and non-malignant complications that shorten the lifespan (4, 6, 7).
Approximately 79.3 million people have been infected with HIV-1 since the beginning of the pandemic, and approximately 50% of them have died from AIDS, according to statistics from the World Health Organization. In 2020, an estimated 1.5 million people were newly infected, and around 690,000 people died from illnesses related to AIDS, according to the Joint United Nations Programme on HIV/AIDS (UNAIDS) (8).
Existing methods for treating HIV-1 can be divided into two types: sterilizing and functional. The sterilizing method involves the complete elimination of replicative HIV-1 proviruses from the human body. The functional method, on the contrary, aims to control the replication of HIV-1 in the long term and maintain a normal level of CD4 + T cells, despite the presence of hidden HIV-1 reservoirs (hidden integrated provirus) and the absence of antiretroviral therapy (9–11).
Ideally, preventive HIV vaccines should prevent HIV infection by ensuring sterilizing immunity via stimulation of high titers of broadly neutralizing antibodies (12). Although such sterilizing agent is still far from available, several efforts have been made toward achieving functional cure of HIV-1 infection.
Currently, the standard treatment for HIV infection, known as combination antiretroviral therapy (cART), involves regular administration of a combination of antiretroviral drugs that block various stages of the virus replication cycle (13). Standard cART can greatly reduce the amount of virus in the blood of HIV-infected patients (viremia) to levels that cannot be detected and turn a fatal infection into a chronic disease that can be medically controlled (4, 14).
Despite the remarkable efficacy of cART, there are still several issues: (a) cART is unable to eradicate the hidden reservoir; (b) patients rely on daily and strict adherence to the treatment regimen; (c) potential side effects after negative drug interactions may occur; (d) cART can lead to drug resistance and limited therapeutic options in multi-class resistant HIV infection; (e) in some countries, this therapy is economically inefficient, and some individuals have limited access to antiretroviral drugs; and (f) there are limitations due to social issues, such as “social stigma.” Also, this therapy has side effects, such as chronic inflammation, CVD, frailty, etc. (15–17).
Therefore, in addition to cART, other promising approaches to achieving functional cure of HIV-1 have been studied in recent decades. Initially, gene therapy methods used RNA interference (RNAi) to suppress the expression of viral mRNA or host mRNA required for HIV-1 infection. This approach allowed for the development of sequence-specific agents to compensate for viral mutations, significantly expanding the number of therapeutic options beyond cART (18). With the passage of time, the technologies underlying the initial clinical trials of gene therapy for HIV-1 have been complemented by alternative gene therapy options such as the utilization of programmable nucleases. Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) systems, when compared to other programmable nucleases, provide enhanced efficacy and simplicity, and the capacity to execute multiplex genome engineering to target different stages of the virus’s life cycle (19, 20). The combination of CRISPR-Cas9 therapy and cell therapy based on CCR5-depleted hematopoietic stem cells (HSCs), in an attempt to recreate the CCR5Δ32 mutation, could be a valuable addition to the existing spectrum of cART (21).
The purpose of this review is to investigate certain important techniques for treating HIV-1 using approaches in cell and gene therapy. We will provide a brief overview of the molecular mechanisms utilized in therapeutic approaches to combat HIV-1, commonly used anti-HIV drugs, viral and cellular targets for HIV therapy, and the current challenges in the use and delivery of anti-HIV agents. HIV-1 treatment strategies have been categorized into RNA therapy, antibody-based therapy, cell therapy, genome editing strategies, and methods used to eradicate hidden reservoirs (Figure 1). Taken together, these findings demonstrate how the implementation of various agents advances our comprehension of HIV-1 infection and can offer significant insights for future HIV-1 treatments.
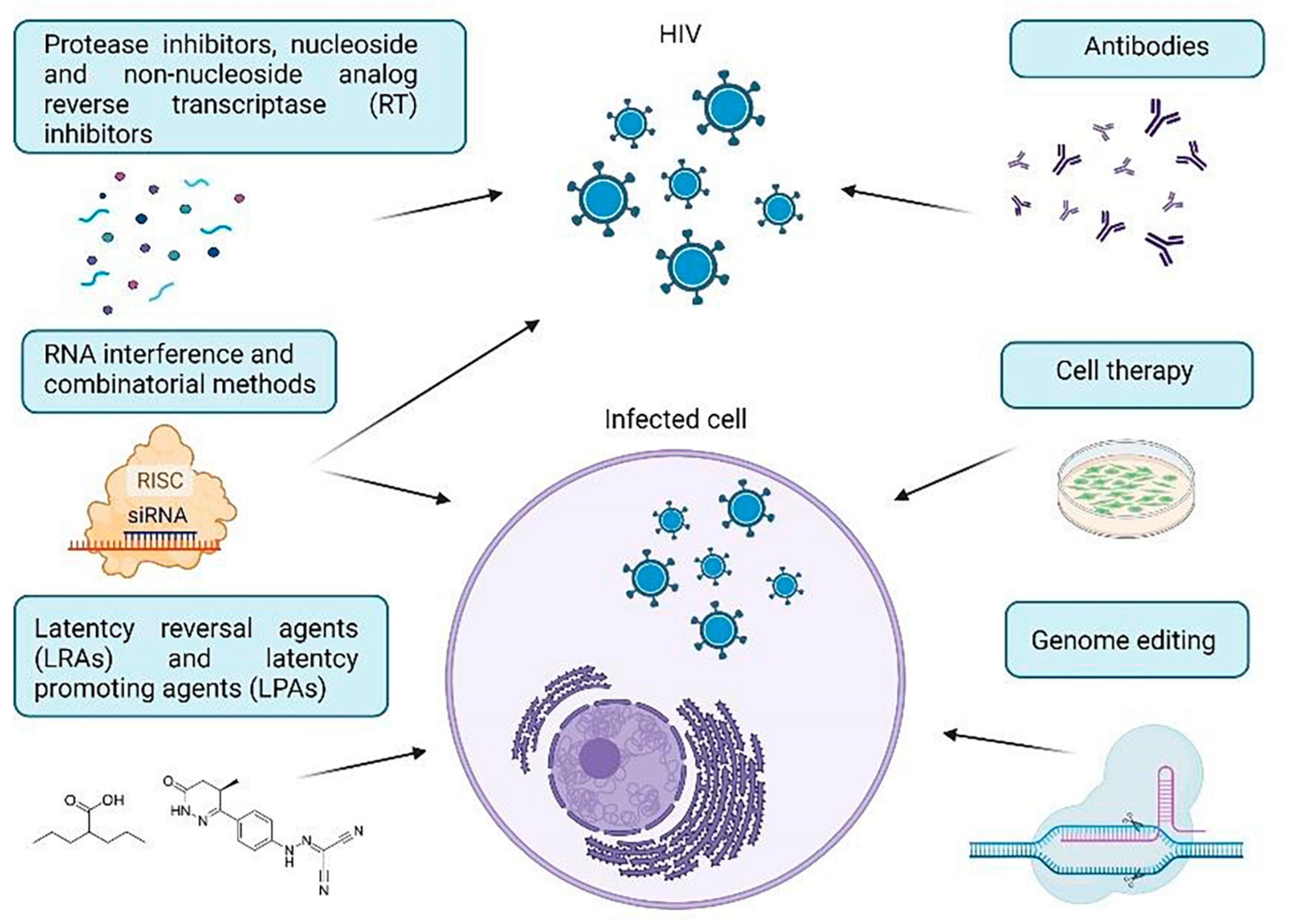
Figure 1. Current strategies for HIV-1 treatment.
2 RNA therapeutic strategies
2.1 RNA interference
The canonical pathway of RNA interference is initiated by the recognition and cleavage of intracellular long double-stranded RNA (dsRNA) intermediates or endogenous microRNAs (miRNA) into small interfering RNAs (siRNAs) of 21-23 nucleotides by the endoribonuclease Dicer (22, 23). The Argonaute protein, specifically Ago2 in mammals, plays a crucial role in the RNA-induced silencing complex (RISC) as an important component. One of the siRNAs becomes loaded onto Ago2, and RISC then utilizes the guide strand of the loaded siRNA to identify and cleave the mRNA that is complementary to it in the middle of the siRNA:mRNA duplex. Meanwhile, the non-targeted (passenger) strand of the loaded siRNA is eliminated (24, 25). Formation of the miRNA:mRNA duplex leads to the degradation of the mRNA fragment by cellular exonucleases (26). Besides, the canonical RNAi pathway functions in the cytoplasm, RNAi can also cause suppression of gene-specific transcription in the nucleus (18).
Anti-HIV-1 RNAs can be separated into two main groups according to their molecular targets: one group uses antisense mechanisms to target viral RNA or host factors, while the other group consists of RNA decoys and aptamers targeting proteins and acting as steric or competitive inhibitors (27, 28). There are different strategies to combat HIV-1 RNA, which use various active agents. Some of these agents include antisense oligonucleotides, small nuclear ribonucleoproteins U1, ribozymes, endoribonucleases, RNA aptamers, small RNA duplexes such as microRNAs, siRNAs, substrate RNA Dicer, short hairpin RNAs, and Ago/shRNAs (29–31).
Synthetic mature siRNA or short shRNA can be used to create artificial dcrRNA, which can be transfected into the cell, or synthetic miRNA can be synthesized, which can be expressed intracellularly from a transgenic construct (32). It is important to note that there are no miRNAs that are currently known to have anti-HIV properties, although some research groups have explored potential molecular candidates for this purpose (33). The therapeutic strategy known as “block and lock” also involves a more in-depth discussion of long non-coding RNAs (lncRNAs).
The most frequently used engineered miRNAs consist of two 21-nucleotide RNA strands with a two-nucleotide overhang at the 3′ end of every single chain (27). Previous studies indicate that dsRNA designs that require Dicer cleavage are more efficient than conventional siRNAs that have been developed based on sequence (34, 35). For the effective functioning of siRNA RISC, it is necessary to choose the right guide chain, as well as to ensure high thermodynamic stability of the siRNA ends, optimal G/C content and the absence of immunostimulating sequences in order to lower the chance of side effects (occurrence) (36–38). short hairpin RNAs (shRNAs) are synthetic oligonucleotides that contain a siRNA sequence followed by a 9-nucleotide loop and a sequence complementary to the siRNA sequence (39, 40). After being exported from the nucleus to the cytoplasm by Exportin-5, shRNA triggers the mechanism of RNAi. Additionally, it has been observed that shRNAs are 10 times more active than siRNA (41). A database named HIVsirDB is available for free and can predict the effectiveness of miRNAs against HIV-1. It encompasses 26 different strains of HIV and 28 different types of cells (42). Hammerhead or hairpin ribozymes can be constructed with ease, and unlike RNAi molecules, they do not depend on the presence of cellular factors such as Exportin-5, Drosha, or Dicer for processing or cleaving complementary RNA target sequences. They are also similar to aptamers in this regard (43). MazF endoribonuclease, derived from Escherichia coli, cleaves single-stranded RNA at 5′-ACA positions, providing an attractive tool for targeting HIV-1 RNA (31).
Single-stranded aptamers inhibit the activity of HIV-1 by directly interacting with the key proteins required for the virus replication cycle. They are generated from random sequence RNA libraries by an iterative selection and amplification procedure known as SELEX (Systematic Evolution of Ligands by EXponential enrichment) (44). In addition, there are ASOs, which are also single-stranded synthetic nucleic acids that bind to mRNA through base pairing and either induce degradation of their targets through an RNase-dependent mechanism or they can interrupt mRNA splicing or translation through a mechanism of steric blocking (45).
U1 snRNP, an important element of the splicing mechanism, is considered as a possible agent for the cure of the HIV-1 gene due to its composition of seven main proteins (SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE, SNRPF, and SNRPG), which are assembled into a heptamer ring to prevent polyadenylation (4).
2.1.1 Cellular and viral targets
Potential targets for therapy include structural genes (Pol, Gag, Env), regulatory genes (Tat, Rev, Vpr, Nef), and accessory genes (Vpu, Vif). These sites, including non-translated long terminal repeats (LTRs), may be targeted at spliced and non-spliced transcripts (18). Most strategies rely on manipulating RNAi to target viral transcripts in the cytoplasm, but the HIV-1 promoter can also be targeted to suppress gene transcription, leading to epigenetic silencing of the integrated provirus (26, 46).
Experiments have proved that siRNAs and shRNAs can be employed to target virtually all HIV-1 RNAs either upon viral uncoating or upon transcription from the proviral DNA (Figure 2B) (47, 48). The use of shRNAs or siRNAs that are complementary to the target gene or LTR can lead to significant reduction in the expression of viral proteins, thus ensuring protection of HIV-1 sensitive cells, including CD4 + T-cells, macrophages, monocytes and dendritic cells, through post-transcriptional gene silencing (49–52). For instance, inhibition of anti-HIV-1 miRNA transcription, constructed against gag and env in the viral genome (Figure 2A), has been detected in CD4-positive cells (3, 53). Moreover, hammerhead ribozymes targeting the HIV U5 and pol regions have been developed for the protection of Jurkat T-cell lines and PBMCs against both laboratory strains and clinical isolates of HIV (48). An RNA hammerhead ribozyme (Rz2) targeting overlapping vpr and tat ORFs of HIV-1 has also been developed and tested in macrophages, T-cell lines, and primary T-cells (54). The HIV-1 encapsidation signal (Ψ) is also an attractive target for gene therapy based on antisense RNA (55). Moreover, HIV genes encoding structural proteins (Gag, Pol, Nef, Tat) have often been considered as targets for RNAi-based gene therapy in clinical trials (26). Several early trials modified the viral regulatory proteins Rev and Tat to act as transdominant negative factors, in other words, these proteins blocked the export of viral RNA from the nucleus of infected cells (56). Zhu et al. conjugated an anti-CD4 aptamer with an siRNA molecule to target HIV-1 protease mRNA (Figure 2B) (57).
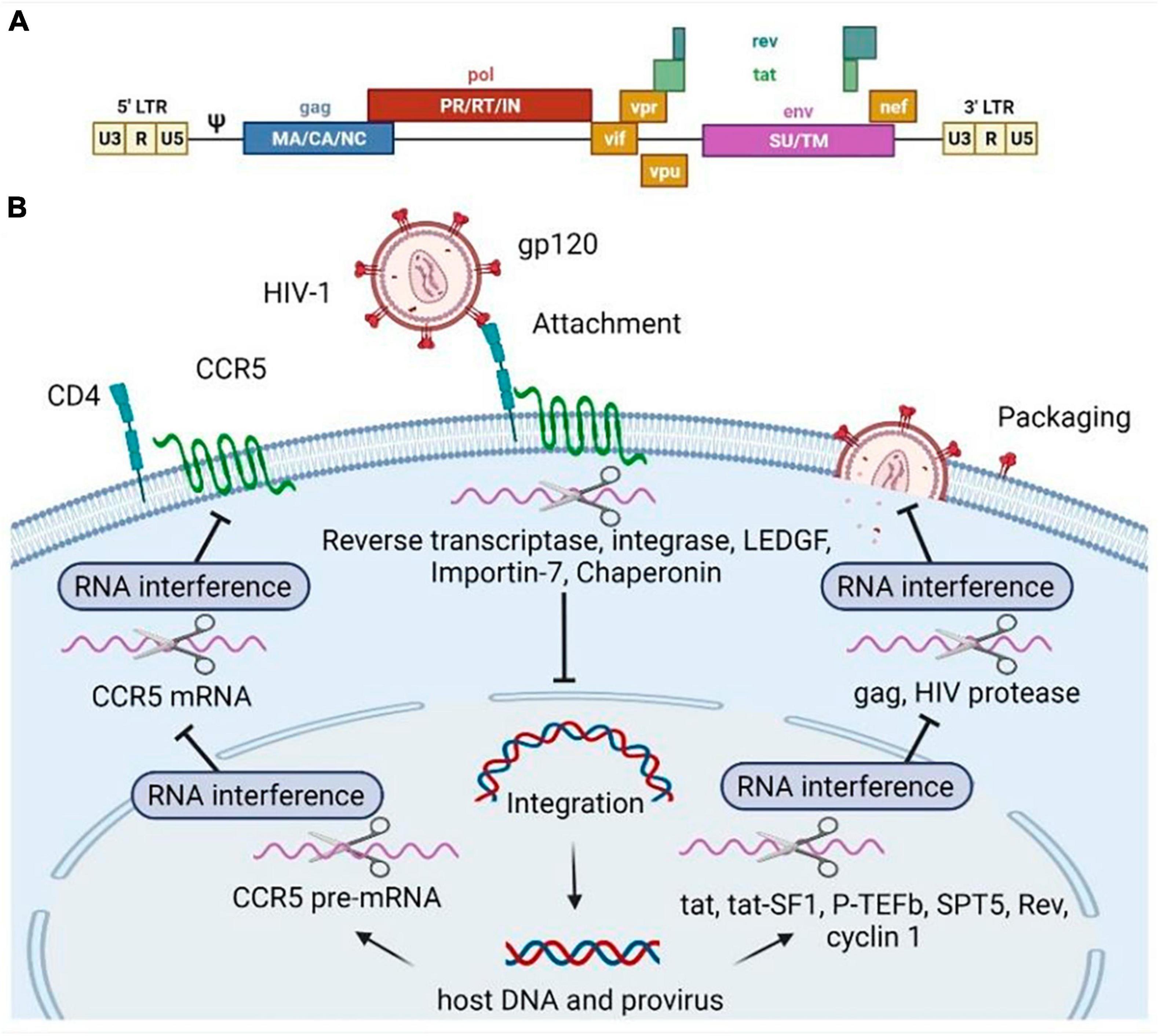
Figure 2. Human immunodeficiency virus type 1 (HIV-1) genome (A) and RNA-based inhibitors targeting various stages of the HIV-1 replicative cycle (B). Scissors illustrate RNA molecules. The RNAs can specifically bind to the mRNA encoding the host (pTEFb, tat-SF1, SPT5, cyclin T1, CCR5, LEDGF, Importin-7, Chaperonin) factors or HIV genes and regulatory regions (gag, rev, pol, tat), then the target mRNA degradation is initiated. Pol, polymerase enzyme; Gag, group-specific antigens; Env, envelope surface glycoprotein gp160, precursor; Tat, trans-activator of transcription; Rev, regulator of expression of virion proteins; Vpr, viral protein R; Nef, negative regulatory factor; Vpu, viral protein U; Vif, viral infectivity factor; PR, pol includes protease; RT, reverse transcriptase; IN, integrase. Gag consists of MA, matrix; CA, capsid, NC, nucleocapsid; p6 domains (not shown). The Env spike protein has a surface part (SU) and transmembrane (TM) part. CCR5 is known as C-C chemokine motif receptor type 5.
Targeting host cell cofactors required for HIV-1 infection is a promising strategy for RNAi therapy because these endogenous genes are less susceptible to mutational escape compared to viral genes, which are prone to error-prone reverse transcription (18). However, cellular targets must be carefully evaluated as these molecules affect both cell growth and function, and also because they may be significant for transmitting cell signals that are not obvious in vitro (26).
The binding of the viral protein gp120 to the cellular receptor CD4 and its co-receptors CCR5 and CXCR4 initiates membrane fusion and subsequent cell entry by HIV-1 (58). Therefore, the CD4 receptor, and the entry coreceptors CCR5 and CXCR4 have been considered attractive targets for shRNA therapy to prevent the initiation of HIV-1 infection and inhibit the fusion of the host cell membrane with HIV-1 (see Figure 2B) (28, 39). For example, one of the effective shRNAs targeting the R region of HIV-1 LTR was validated in a humanized mouse model in combination with shRNA targeting CCR5 mRNA (54). Previously, Eekels et al. used multiple shRNAs targeting 30 human genes involved in HIV-1 replication and identified TRBP, ALIX, and AGT6 as the most suitable genes for long-term inhibition of HIV-1 replication with minimal toxicity in shRNA-transduced T lymphocyte cells (59). Several human proteins (cyclin T1, SOCS1, and RNA helicase DDX3), including co-factors of viral integrase (LEDGF/p75, importin-7, and chaperonin), elongation factors (P-TEFb, Tat-SF1, and SPT5), were also mentioned as promising candidates for RNA-based therapy (Figure 2B) (18).
2.1.2 Current status
Studies have shown that effective gene suppression is achieved only with a small number of RNA agents (13). RNAi may exhibit imperfection in RNA-RNA duplex formation, leading to off-target effects on unrelated mRNA (60). Additionally, if the passenger strand of the RNA molecule is loaded into RISC instead of the guide strand, it can cause RNAi side effects (37, 61).
Currently, the main problem is the delivery of RNA substances (as well as DNA plasmids, mini-circles and therapeutic transgenes) to infected cells or HIV-sensitive ones. Delivery systems play a fundamental role in facilitating the cellular uptake of RNA molecules and protecting them from nucleases degradation, thus minimizing the need for any chemical modifications that may alter the specificity and functionality of RNA (62, 63). Despite the successful application of anti-HIV-1 RNAi in vitro, current delivery methods have not been able to translate these achievements to in vivo conditions. Therefore, non-specific delivery methods or ex vivo methods such as electroporation, viral vectors, and nanoparticles are often used (18). Other methods of delivering target RNA include tissue-specific adeno-associated viruses (AAVs), nucleic acid aptamers, antibodies, and nanoparticles composed of cationic polymers (such as poly-L-lysine, polyethylenimine, polyamidoamine chitosan) and lipids. However, each of these approaches has its own challenges that require consideration in order to achieve optimal delivery solutions (18, 64).
2.2 RNA-vaccines
An mRNA vaccine is a synthetic vaccine that uses a DNA template to transcribe mRNA, which in turn triggers an immune response to the target pathogen. mRNA vaccines can be designed to express almost any antigen sequence, and the innate immune system’s ability to recognize viral RNA sequences enables the effective elicitation of an innate response along with the generation of cytokines and chemokines, that are crucial for a successful adaptive immune response (65, 66). It has also been shown that COVID-19 vaccines based on the mRNA platform have excellent immunogenicity and are able to stimulate B-cell and T-cell responses (67).
mRNA vaccines can be divided into several types, including self-amplifying mRNA vaccines (SAM), DC-mRNA vaccines, non-replicating mRNA vaccines, and mRNA vaccines against cancer. SAM vaccines create auxiliary equipment for the formation of double-stranded RNA and intermediates for replication and other products, and do not require mechanisms for replicating their RNA after introduction into cells (68, 69). They require a less dose to elicit a better immune response compared to non-amplifying RNA vaccines. SAM vaccines can be engineered by single-stranded positive-sense alphaviruses and delivered as virus replicon particles (VRP). mRNA is delivered through different strategies such as electroporation, cationic liposomes, or cationic Nano emulsion, and it carries the code for an RNA-dependent RNA polymerase together with the immunogen, which collectively contribute to a lasting immune response. The immune response can be measured by the number of Th1-type T cells generated after vaccination (70). All mRNA vaccines share common structural elements: cap, 5′UTR, 3′UTR, ORF, and poly(A) tail. They are generated through enzymatic transcription of a DNA template, and to create a vaccine, it is just required to change the sequence encoding the target antigen (71, 72).
2.2.1 Delivery to cells
Delivery of mRNA vaccine into antigen-presenting cells (APCs) is a limiting factor for vaccine efficacy (71). mRNA vaccine against HIV, containing free mRNA, faces challenges in stability preservation and penetration into the cytoplasm of APCs during transportation. Various strategies have been employed to enhance the delivery of HIV mRNA vaccine into APCs (73). The method of vaccine administration influences the immune response and mRNA uptake. Utilizing different strategies, such as mRNA delivery into dendritic cells (DCs) or through nanoparticle carriers, can improve mRNA penetration into APC cytoplasm and induce a high level of immune response, holding promise for the development of effective HIV vaccines (74–76).
2.2.2 Current status
Recent advancements in mRNA technology have led to increased utilization of vaccination experiments against HIV using improved delivery methods. However, clinical trials conducted on therapeutic vaccines against HIV-1 did not demonstrate significant clinical impact, despite achieving safety and efficacy in eliciting an immune response (77, 78).
2.3 CRISPR/Cas9
A variety of CRISPR-Cas systems have been identified, though only a select few have been leveraged as instruments for scientific research (20). Class 1 systems deploy a complex of multiple Cas proteins, while Class 2 systems utilize a single effector Cas protein (79, 80). The CRISPR-Cas9 system is most commonly utilized within human cells. The two-component RNA system, made up of CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA), has been streamlined into a single guide RNA (sgRNA) that aligns with a specific target sequence (81). For Cas9 to bind and cleave DNA, a protospacer adjacent motif (PAM) sequence is necessary; it lies downstream from the target sequence and results in either blunt or staggered double-strand breaks (DSBs) (20). Either homology-directed repair (HDR) or non-homologous end joining (NHEJ) can be employed for gene modification within mammalian cells (82).
A significant objective in combatting HIV-1 using the CRISPR/Cas9 strategy is to reduce or disable intact proviral sequences of HIV-1 (4). The fusion protein deficient in Cas9 (dCas9), in combination with sgRNAs targeting specific effector domains of DNA sequences, has been employed for gene activation or suppression of transcription (Figure 3) (83). Genome engineering supports the advancement of cell therapy, including a universal approach to introduce both CAR transgenes and CRISPR-Cas9 ribonucleoproteins (RNPs) into primary human T cells using engineered lentiviral particles (84). Additionally, a dual gene therapy strategy has been developed, involving a conditional suicide gene and CCR5 knockout, to overcome limitations associated with CCR5 knockout alone and receptor switching (85).
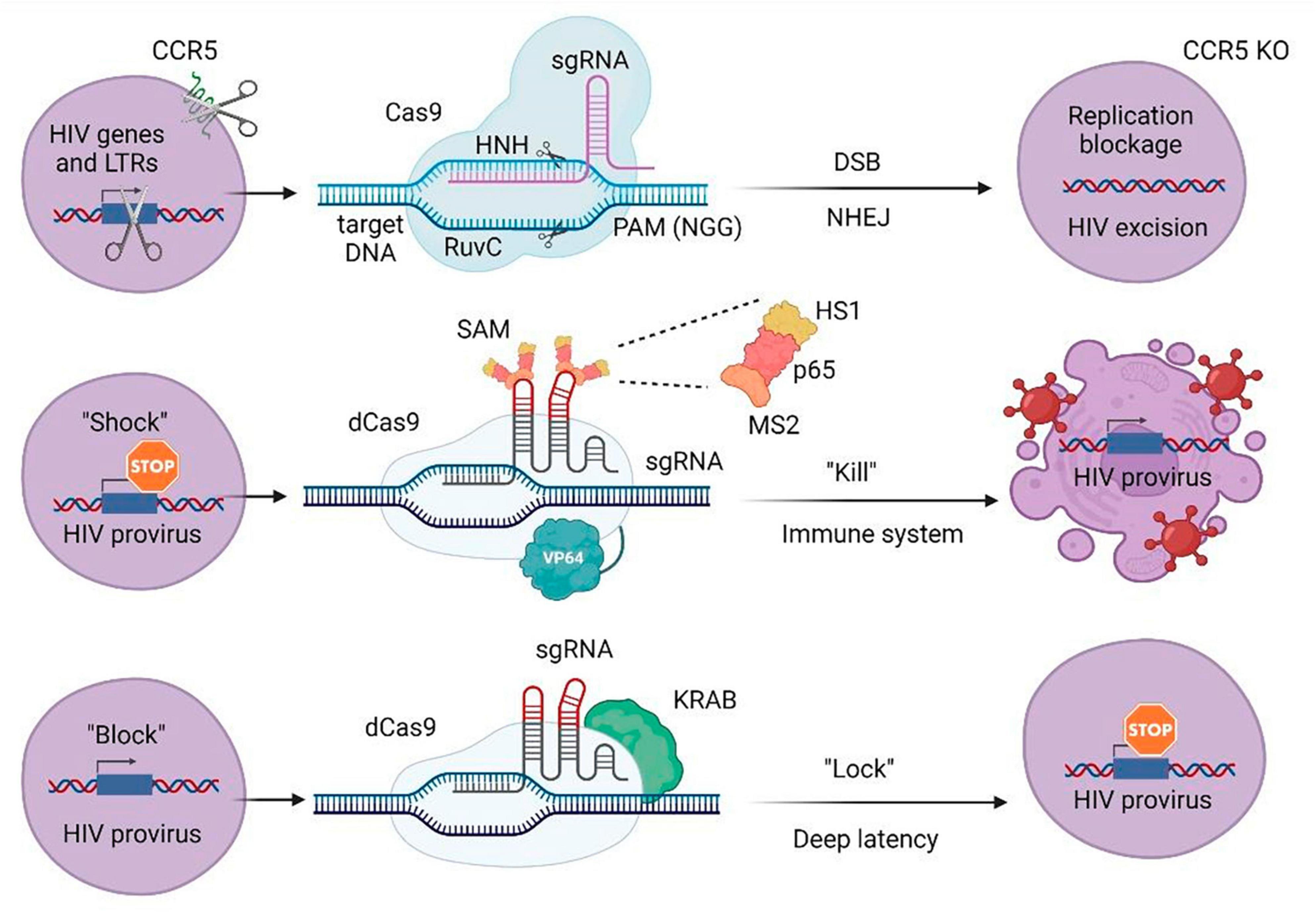
Figure 3. CRISPR/Cas approaches to attack HIV-1 or host-specific genes participating in the life cycle of the virus. LTRs, long terminal repeats; sgRNA, single guide RNA; PAM, protospacer adjacent motif; DSB, double stranded break; NHEJ, non-homologous end joining; KO, knockout; SAM, synergistic activation mediator; KRAB, Krüppel-associated box repressor; dCas9, nuclease-deactivated Cas9. NHN and RuvC are Cas9 catalytic domains that cut single-stranded DNA at the target site. MS2 protein via designed aptamers binds to the dCas9/VP64 fusion protein and involves additional activation domains (HS1 and p65).
2.3.1 Cellular and viral targets
The CRISPR/Cas9 strategy targets LTR as one of the most commonly used sites for removing HIV-1 proviral DNA (Figure 3) (86). Moreover, the CRISPR/Cas9 mechanism can also be used to remove regulatory genes of HIV-1, including tat and rev (87). For example, Herskovitz et al. developed a collection of gRNAs targeting the consensus sequence of the HIV-1 viral transcription regulator, tat (88). The integrated HIV-1 provirus is similarly controlled by an internal antisense lncRNA expressed from the nef gene located at the 3′ end of the viral genome, which makes the promoter region in nef a useful target for targeted CRISPR/Cas9 deletion or gene suppression (89). The CRISPR/Cas9 system can inhibit virus replication by targeting host factors and coreceptors, including CCR5 and CXCR4 (Figure 3) (90). In one of these approaches, HSCs are extracted from patients and transfused after CRISPR/Cas9 treatment through deletion in receptor genes. Bogerd et al. used a Cas9-based approach to induce the expression of restriction factors APOBEC3G (A3G) and APOBEC3B (A3B) in human cells (91). The CRISPR knockout screening approach can be used to identify new host genes involved in the virus replication cycle (92).
For the “shock and kill approach,” it was shown that the use of dCas9 aimed at LTR, together with the synergistic activator mediator (SAM) system, allowed to increase the activity of LTR-controlled gene expression. After that, the research team analyzed possible non-targeted effects leading to changes in transcription profiles associated with the use of dCas9-SAM. The authors demonstrated that of the tested genes, only two were significantly activated. Despite the fact that this study was limited by the small size of replication and the fact that further trials in other cell models (and possibly in vivo) would be required, this was the first evidence indicating the safety of this strategy for possible future therapeutic applications (Figure 3) (93).
dCas9 fused with the KRAB transcriptional repression domain was adopted to inhibit provirus activation. It was demonstrated that when LRA cells were stimulated, HIV-1 expression decreased by up to 60% compared to the control after delivery of specific gRNAs designed to direct dCas9 to the LTR promoter regions. This effect was associated with the presence of repressive epigenetic modifications, which indicates the possibility of developing a CRISPR system for the “block and block” approach (Figure 3) (94).
2.3.2 Current status
Although off-target effects are detected in all genome editing systems, the high occurrence of unpredictable off-target effects in the CRISPR/Cas9 technology is a serious drawback (95). Other limitations of this strategy include the requirement for a short PAM near the target locus, the complexity of packaging into AAV vectors due to the large size of the most popular, Streptococcus pyogenes, Cas9 (SpCas9), and the fact that CRISPR-induced DSBs often provoke apoptosis, leading to DNA damage and cellular toxicity (96–98). A sterilizing cure approach would require delivery of Cas9 and gRNA to all HIV-1 reservoir cells in vivo, but unfortunately, the effectiveness of Cas9 and gRNA delivery appears suboptimal (99). Additionally, immune reactions against the non-human Cas9 protein can complicate this strategy for in vivo HIV inactivation (91). Gene therapy and stem cell transplantation are high-cost and need advanced technologies. Nevertheless, recent studies show that the target gene may be gag. Thus, a study by Trisha H. Burda et al. found that SIV-induced rhesus monkeys receiving AAV9 CRISPR-Cas9 and two gRNAs were detected on SIV regions known as EBT-001. It was shown that there are signs of editing of SIV proviral DNA in all major viral reservoirs. EBT-001 was well tolerated in various dosages without obvious toxicity (when taking higher doses, a short-term increase in the level of liver enzymes was observed, which later returned to normal). Overall, this study confirms the potential of AAV9 delivered by CRISPR-Cas9 with dual gRNA, similar to EBT-001, in strategies aimed at eradicating HIV (100).
2.3.3 Supplement on genome editing methods
In recent years, the three main gene editing tools that use nucleases–transcription activator-like effector nucleases (TALENs), zinc finger nucleases (ZFNs), and CRISPR/Cas9–have been widely used in studies on the treatment of HIV/AIDS (83). These methods include inhibiting host factors, weakening transcription and replication of HIV-1, inactivating HIV LTR, suppressing proviral HIV expression, and eliminating latent HIV-1 provirus (4). A previous clinical trial involving 12 HIV-infected patients showed that introducing ZFN-modified autologous CD4 + T cells containing mutated CCR5 is a safe approach to combatting HIV (101). TALENs, in turn, are also site-specific nuclease-based tools used for first-generation genome editing approaches with greater safety than ZFNs (4). According to previous results, CRISPR/Cas9 is a more effective genome editing method for HIV treatment compared to ZFN and TALEN approaches (4, 102). Most CRISPR-based tools have undergone intensive review (103–106). A comparison of different nuclease-based genome engineering platforms has also been conducted (107).
3 Antibody based therapies and vaccines
T-cells, including CD4 + and CD8 + cell populations, and B-cell lymphocytes are very important members of the adaptive immune system. During the infectious response, CD8 + T cells are necessary for the direct destruction of infected cells, while CD4 + T cells promote the induction of CD8 + T cells and support the maturation of highly specific antibodies produced from B-lymphocytes (108, 109). B-cells undergo iterative cycles of proliferation, immunoglobulin mutation, and antigen selection for the generation of highly specific antibodies in specialized immune cells in the secondary lymphoid organs (germinal centers or GCs) (70, 108). After an antigen challenge, GCs activate the B-cells by antigen-specific B-cell surface receptors (BCRs) (110). Each GC generally focuses on one specific antigen and can produce a limited amount of antigen-specific B cells (108). This process contrasts with the early B-cell responses to antigens in the extrafollicular spaces that result in short-lived antibody-producing cells (plasmablasts) secreting non-mutated antibodies (70, 111). Eventually, the B-cells depart the GC and might turn out to be either plasmablasts, or memory B-cells (112).
The antibody response to the viral Env, Gag, and Pol proteins, along with detection of p24 protein and viral RNA, can be used for tracking the early stages of HIV-1 progression (113). B-cells respond to HIV-1 infection for the first time within ∼1 week after viral RNA can be detected in the plasma and the immune response is initially observed in the form of virion-antibody immune complexes; subsequently free IgM antibodies to gp41 have been detected (114, 115). Protective neutralizing antibodies (NAbs) develop slowly and do not appear until 8–12 weeks after HIV-1 infection (114, 116). NAbs are produced against viral Env to neutralize the particular viral strain infecting the patient (autologous virus) (117). Only years after HIV-1 infection, could cross-reactive antibodies, able to neutralize heterologous viral isolates, frequently be found (114, 116). HIV-1-specific antibodies can also interact with the Fc gamma receptors (FcγRs) that have the potential to inhibit HIV-1 spread via antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated virus inhibition (ADCVI) (116, 118).
In fact, the development of a safe and effective protective vaccine against HIV-1 remains one of the highest priorities for global public health and the best long-term tool for controlling HIV-1 transmission (119). Preclinical and clinical trials have evaluated various approaches to creating HIV-1 vaccines, but the results have generally been unsatisfactory (120). Since vaccines were invented, five basic approaches have been used in the development of viral vaccines; however, the two most effective approaches (attenuated and inactivated organisms) have not been proven optimal for HIV vaccine development (12, 121, 122). Further, HIV vaccine development has shifted direction toward cellular immunity to induce HIV-specific cytotoxic T-lymphocyte (CTL) production. These cells recognize HIV epitopes on the cell surfaces and arrest the proliferation of HIV infection through apoptosis or the secretion of chemokines and cytokines, subsequently interfering with the next rounds of viral replication (12, 70). For instance, one of the approaches to T-cell-based vaccine development involves the induction of non-classical Major histocompatibility complex E (MHC-E) restricted CD8 + T-cell responses by a modified cytomegalovirus (CMV) vector (123, 124). Other approaches to vaccine development have used DNA plasmids and other viruses as vectors to deliver viral genes (lentiviral vectors, integrase-defective lentiviral vectors, recombinant adenovirus type 5 vectors) (12, 125, 126). Although DNA vaccines are safe because DNA plasmids stay episomal and act as expression vectors produced by peptides that can induce cellular immunity, they are not able to induce reliable T-cell levels or antibody responses (12). To boost the immune responses during DNA vaccination, the use of molecular adjuvants is also being explored (127, 128). Some approaches have used mRNA as a vector, also resulting in the induction of polyfunctional antibody responses (70, 127). A vaccine platform of mRNA incorporated into lipid nanoparticles (mRNA-LNPs) has lately been characterized for infectious diseases, notably for SARS-CoV-2 and for HIV (129). The strength of this approach, in addition to the positive aspects of using mRNA (e.g., Env–Gag mRNA designed by Zhang et al. in vaccine development, lies in the induction of both follicular helper cells (Tfh) and B-cells in the GC response (67, 120). Currently, tests are being conducted on various animal models, and the data obtained are hopeful for future use in humans (68, 70). However, it may be years before the most important scientific contribution made by the response to the SARS-CoV-2 epidemic can be applied to HIV vaccine research (130).
Vaccine development efforts are focused on the induction of neutralizing antibodies (NAbs), in particular broadly neutralizing antibodies (bNAbs) that cover 50–90% of transmitted viruses (131, 132). The serum of a small percentage of individuals (10–30%) living with HIV contains bNAbs, which has provided evidence that a bNAb-inducing vaccine is possible (118, 133). Broad neutralizing antibodies (bnAbs) neutralize multiple HIV-1 strains by targeting conserved epitopes of the virus (Figure 4) (134). The classification of HIV-1 reactive bnAbs includes naturally occurring and engineered antibodies (137). Moreover, there are twenty types of broadly neutralizing antibodies (bnAbs) that have been divided into six categories according to the specific Env residues they interact with. There’s also another class of bnAbs that targets the gp41 membrane proximal external region (MPER) (135, 136). bsAb molecules can also be separated into a class of IgG-like molecules and a class of non-IgG-like molecules (137). Additionally, first-generation antibodies (b12, 2G12, 4E10, and 2F5) and second-generation bnAbs (PG9, PG16, CH01, PGT145, PGT121, PGDM1400, 10-1074, 10E8, VRC01, 3BNC117, and CH103) with improved neutralizing ability and flexibility in technological manipulation have been classified (138, 139).
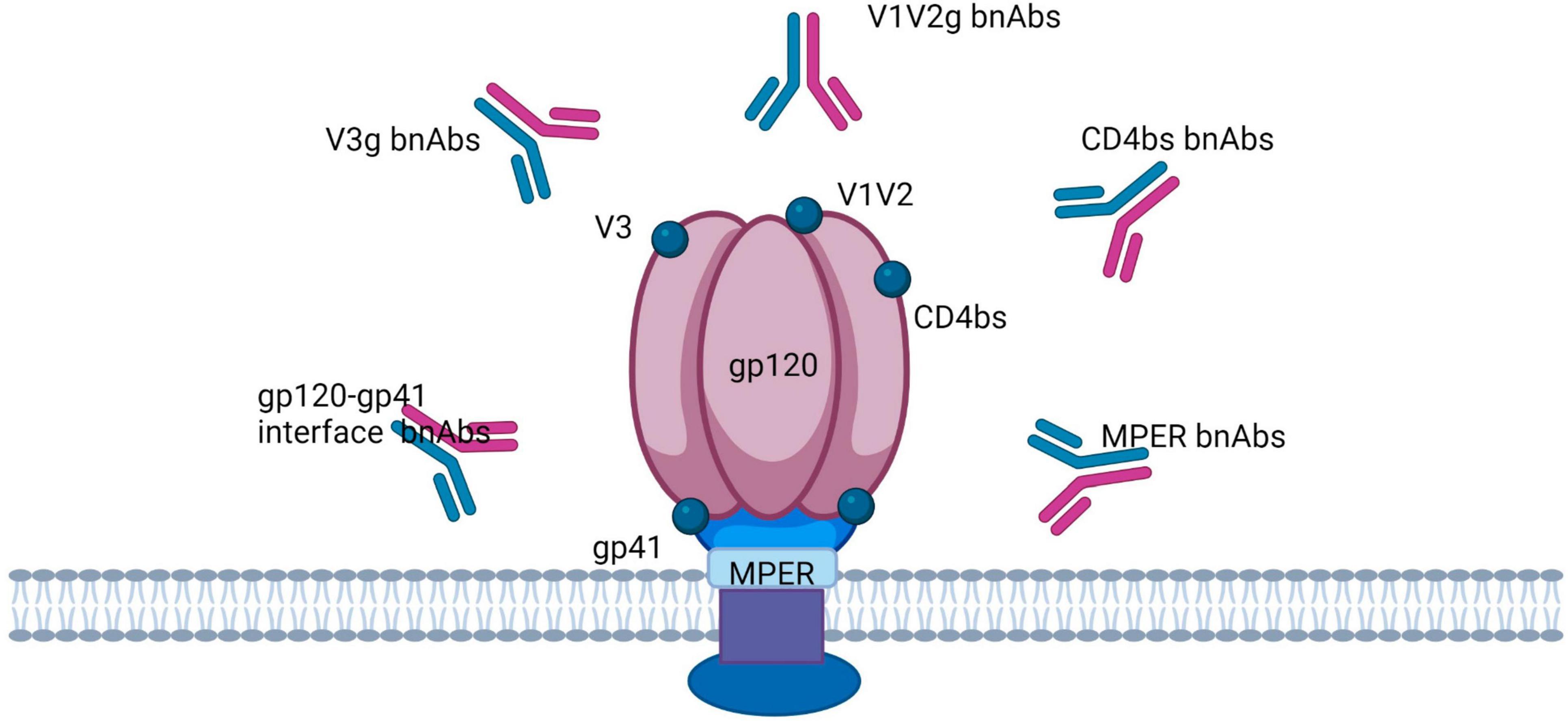
Figure 4. Broadly neutralizing antibodies target the HIV trimer Env in five regions: the CD4 binding site (CD4bs), V1/V2-binding sites, V3-glycan, glycoprotein (gp)41/gp120 interface, and the membrane proximal external region (MPER).
The first bnAb (b12) was discovered in 1991, and in recent years, there has been an increase in the number (more than 100) of available bnAbs through the introduction of new technologies like Env-specific sorting of individual B-cells, proteomic deconvolution, antibody cloning and high-throughput neutralization assays (118, 138, 140, 141). New technologies enable the construction of antibodies with two, three or four different binding sites on a single molecule (142). Thus, bsAbs are designed either to recognize two different HIV-1 env epitopes through the single-stranded variable fragment (scFv) of two independent bNAbs or to interact with cellular receptors with one scFv and one HIV-1 env epitope with another scFv. Bispecific (bsAbs) and trispecific bNAbs (tsAbs) represent a promising alternative to bNAb combination therapy because they recognize multiple targets on the viral Env protein (137). Some information about bnAbs is freely accessible on a website known as the Broadly Neutralizing Antibody Electronic Resource (bNAber), which is aimed at motivating researchers to design a reliable HIV vaccine (143). On the other hand, clinical and preclinical studies, for instance, the RV144 vaccine efficacy study, highlighted non-neutralizing antibodies (nNAb) that have no neutralizing activity in vitro, but may play an important role in protecting against viral infection in vivo (123, 124). Several human monoclonal antibodies with broadly neutralizing activities have been observed (F105, b12, 2F5, and 4E10) that are specific to the CD4+ T-cell binding site on gp120 or gp41 and demonstrate antiviral protection against different HIV clades (A, B, C, and D) in vitro or in experiments with neonatal macaques (144, 145). BG505 SOSIP is a well-studied, almost native recombinant HIV envelope trimer (Env) that holds promise as part of a successive anti-HIV immunogenic scheme for the induction of bnAbs (123, 146).
3.1 Viral and cellular targets
Effective HIV-1 vaccines depend upon T-cell-mediated immunity (CMI) focused on rather conserved viral proteins, which include Gag and Pol and/or non-neutralizing antibodies targeting the virus envelope, to prevent cell-to-cell transmission of the virus (147, 148). HIV-1 virions contain about 10–14 trimeric envelope glycoproteins (Env) on its surface, which mediate the penetration of the virus into host cells (149). Every HIV envelope spike protein (Env) comprises three external gp120 subunits that are non-covalently bound to three gp41 subunits attached to the membrane (Figure 4) (128). The Env glycoprotein is strongly glycosylated, and a dense shell of host-derived N-glycans protects the epitopes of the viral protein from antibody interactions (150). The new antibody group has been helpful in identifying a highly structured epitope present only on the trimeric envelope and including the conserved V2 and V3 regions that are symbolically represented in Figure 4 (116). Xu and group designed highly potent trispecific antibodies by combining the specificity of PGDM1400, VRC01, and 10E8v4 to interact with the membrane-proximal external region (MPER)-, CD4-, and V1/V2-binding sites (Figure 4) (151). bnAbs are thought to break through the glycan protection of the HIV trimer env in five regions, each of which is probably involved in the env function: the CD4 binding site (CD4bs), the variable loop 2 (V2)-apex, the V3-glycan, the glycoprotein (gp)41/gp120 interface, and the membrane proximal external region (MPER) (Figure 4) (152–154). It is believed that the apical region V2 is participating in maintenance of the metastability of the spike Env protein (155). The V3 glycan site is formed partially by the co-receptor site CCR5 and partially by the surrounding masking glycans (156). The interface region for glycoproteins gp120 and gp41 includes the fusion peptide (FP) and the cleavage site of gp160 into gp120 and gp41 (152, 157). In turn, MPER is part of the fusion machinery (152, 158).
3.2 Restrictions
Firstly, it is improbably that the HIV vaccine will be sufficient to stop a persistent HIV-1 infection (12). Secondly, HIV infection gradually disrupts the body’s immune response, which is necessary for the effectiveness of the vaccine (159). The ever-changing antigenic variations of HIV represent the third major problem for vaccine development (139). For, instance, based on whole-genome sequences, HIV is classified into three major groups: major (M), outlier (O), and non-M/non-O (N), and their prevalence varies according to geographical regions (12). Nine of the HIV subtypes belonging to group M form HIV subtypes or “clades” (A–D, F–H, J–K), which differ by ∼25–35% in their env sequences and ∼15% in their gag sequences (12, 123, 160). Moreover, the evolution of HIV-1 in infected individuals begins shortly after infection (123). New approaches to creating multivariate vaccines will probably be needed. The development of antibody production techniques is also strongly linked to the cost (138, 161). Other key difficulties are the delivery and stability of antibodies for HIV therapies.
4 Cell therapies
T-lymphocytes in the immune system can identify invading pathogens when they’re exposed to the pathogen’s specific immunogenic component, known as an antigen (162, 163). T-cell activation during infection happens through the T-cell receptor (TCR), which includes the CD3 receptor and either the CD4 or CD8 co-receptors. The CD4 receptor is utilized by helper T-cells to activate their ability to release cytokines, while cytotoxic T-cells use the CD8 receptor to enhance their capacity to kill cells (164). Once activated, T-cells start to multiply quickly, but after the infection is resolved, the population of activated T-cells, which was formed by clonal expansion, reduces and ultimately undergoes apoptosis (165). After this phase, a small group of memory cells remains. These cells can recognize the same antigen, quickly expand clonally, and differentiate to trigger a powerful and specific adaptive immune response (143, 151, 166). During ripening in the thymus, T cells that recognize their own peptides are destroyed to prevent autoimmune reactions (167). Additionally, the cytotoxic T-lymphocyte (CTL) response is a crucial aspect of host immunity against HIV infection (168, 169). It is generally believed that in elite controllers (a rare set of individuals who can control HIV replication for extended periods without anti-HIV treatment), the control of the virus is mostly mediated by the CD8 + T-cell response (170–172). CTLs also have the ability to direct the lysis of infected cells via major histocompatibility complex class I (MHC-I) molecules. However, HIV can potentially decrease the surface expression of MHC-I on infected cells to avoid this immune response (173, 174).
Chimeric antigen receptor (CAR) is an engineered TCR known to bind with a specific antigen, and after introducing CARs into T-lymphocytes, CAR T-cells can be obtained. The CAR transgene is a synthetic chimeric receptor comprising an scFv of the antibody and T-cell signaling domain(s), whereas another approach includes using a TCR transgene derived from a native TCR that has been partially modified to increase its affinity for the target antigen (Figure 5) (175). For most CAR T-cell therapies, cells are extracted from the peripheral blood mononuclear cells (PBMCs) of the patients by apheresis or leukapheresis (164). This sample represents a diversity of immune cells, including B-cells, macrophages, monocytes, natural killer (NK) cells, and T-cells. The first stage of preparation requires the identification of a subset of T-cells from the PBMCs, and this can be achieved by magnetic bead selection or selective expansion (176, 177). Anti-CD3 or a combination of anti-CD4 and anti-CD8 antibodies can be used for T-cell selection (178). Activated T-cells are then grown in culture to achieve the required number of cells and transduced with the CAR cassette (179). Gene delivery can be accomplished by stable integration of the cassette into the genomic DNA of the host T-cell assisted by viral vectors (e.g., lentiviral vectors) and by non-viral transfer or non-integrating transient delivery (180). Afterward, cell material is collected and cryopreserved at -120°C, and then the sample is returned to the clinic for intravenous infusion into the patient (Figure 5) (164). As an example, CD8 + T cells are harvested from HIV patients and transduced with CAR genes; after testing specificity and efficacy against HIV in vitro, functional HIV-specific CAR T cells are re-injected into patients to destroy HIV-infected cells (17).
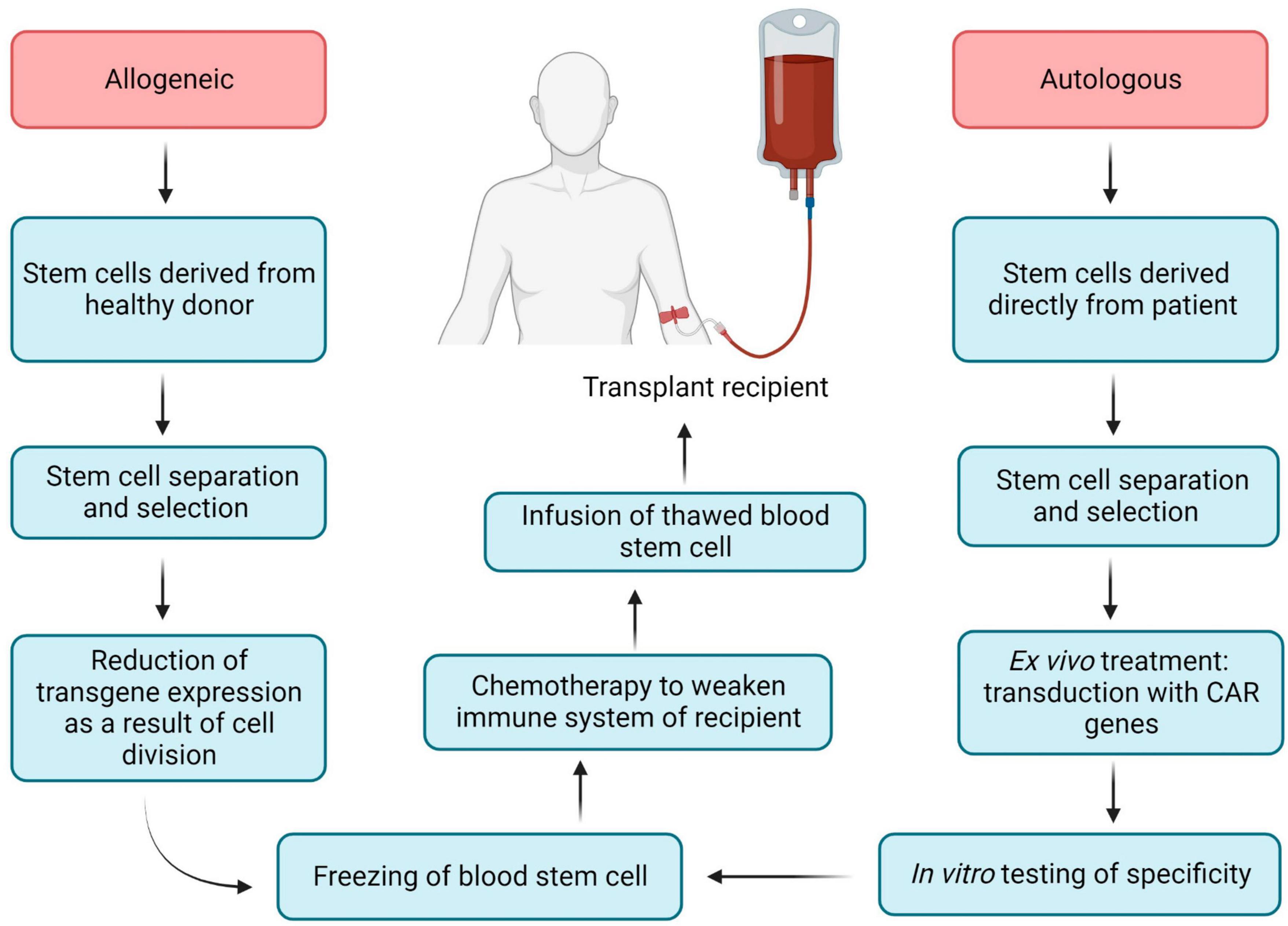
Figure 5. Allogeneic and autologous blood stem cell transplantation.
Hematopoietic stem cells (HSCs) perform two important functions: their ability to self-renew and the ability to differentiate into various hematopoietic lines, including lines that have the ability to kill HIV-infected cells (T-cells and NK-cells) (181). Hematopoietic stem cell transplantation (HSCT) has become a promising candidate for achieving a functional cure for HIV, mainly due to the clinical experience of the Berlin patient and the London patient (182–184). Both patients received HSPCs from donors with a naturally occurring 32 base pair deletion in the CCR5 gene corresponding to the second extracellular loop of the receptor (CCR5Δ32), resulting in a non-functional gene product that is not expressed on the cell surface due to frameshift and early termination (185, 186). The stable viral remission in these patients after cART interruption is presumably explained by a combination of a conditioning regimen that provided donor chimerism, destruction of host latent reservoirs during donor cell engraftment and almost full substitution of the host immune system by homozygous CCR5Δ32 donor cells (187–189). While the positive outcomes of these two patients’ therapies constitute a significant landmark in the effort to cure HIV, this method includes several limitations (risk of morbidity and mortality, limited prevalence of CCR5Δ32 donors, HIV mutation via a CCR5- to CXCR4 tropism shift) (16, 190). These limitations make allogeneic HSCT unfeasible for the vast majority of people living with HIV (187). HIV-specific CAR expression from genetically modified autologous HSPCs has the potential to bypass the limitations of the allogeneic HSCTs. Previously CD4ζ based CAR-modified HSPCs were differentiated into functional T-cells as well as NK cells in vivo in humanized mice, moreover, these cells are HIV-resistant (191). In addition, NK-cells can identify and eliminate HIV-infected signals using the mechanism of antibody-dependent cell-mediated cytotoxicity (ADCC) (191, 192). Novel genetically modifying HSPCs to express CD4 CAR are long-lived and proliferate in multiple tissues relevant to HIV infection and cancer (lymphoid germinal centers, brain, and gastrointestinal tract) for almost 2 years and have demonstrated multiphasic engraftment in macaques (187). All commercially accessible adoptive T-cell therapies have been autologous (cells derived directly from the patient), as the allogeneic approach has been shown to be complex to design through early HLA typing and stem cell technology (193).
In early clinical trials the first generation of CAR constructs had a single intracellular signaling domain from the CD3ζ of the TCR, fused to either the extracellular CD4 region (CD4ζ-CAR), or to the scFv of isolated monoclonal antibodies (scFv-CAR) (6, 181, 194). The second generation also has a CD28 signal, which promotes cell proliferation and cytokine secretion (195). The third generation of CARs supplemented the 4-1BB and OX40 regions to favor cell survival and to extend the persistence of CAR T-cells in vivo (Figure 6) (196). The latest generation, recognized as T-cells redirected for Universal cytokine-mediated destruction (TRUCKs), has been developed lately and carries specific cytokine signals that make CAR T cells resistant to the immunosuppressive effects of the tumor microenvironment (TME) (196, 197). However, when employing a panel of HIV-specific (CD4-based) CARs expressing distinct intracellular domains (ICDs) it was shown that only HIV-resistant, 4-1BB-stimulated CAR4 T-cells restrict HIV infection in vivo (198).
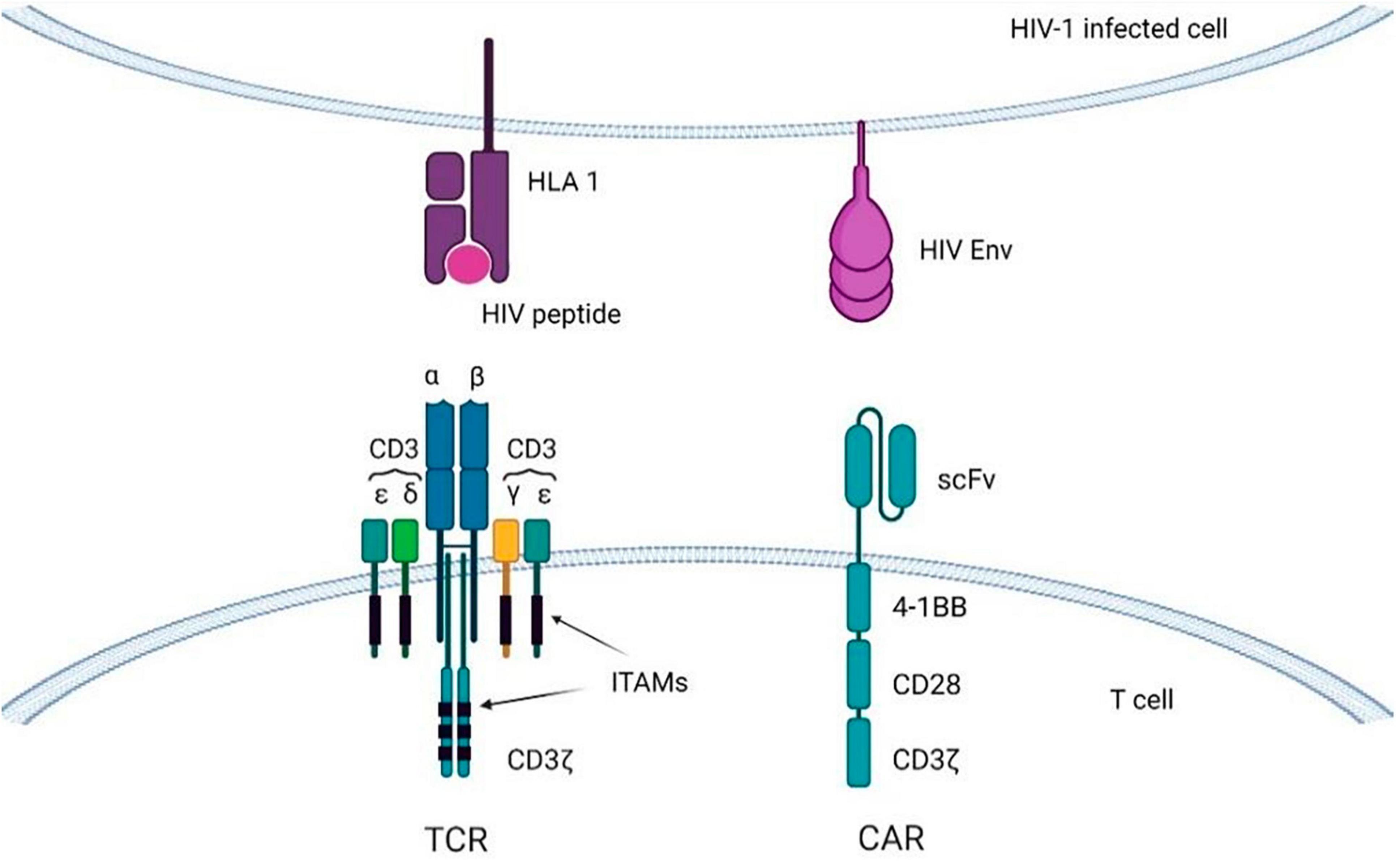
Figure 6. T-cells with CARs or transgenic TCRs targeting viral antigens displayed on HIV-infected cells. Immunoreceptor tyrosine-based activation motifs (ITAMs) are signaling motifs for adaptive immune responses regulation. CAR, chimeric antigen receptor; TCR, T-cell receptor; HLA, human leukocyte antigen.
The 2016 study included 28,992 patients. The average number of CD4-T cells at the beginning of treatment was 249 cells/ml. The average observed CD4 count after 6, 9, and 12 months was 382, 402, and 420 cells/μl. The two main factors explaining the change in CD4 count after 6 months were the stage of AIDS and the CD4 count at the beginning of cART. The median observed CD4 count at 6, 9, and 12 months was 382, 402, and 420 cells/μl. The two main factors explaining the change in the number of CD4 after 6 months were the stage of AIDS and the number of CD4 at the beginning of cART (199).
Additionally, with the discovery in the last few years of many potent bNAbs against HIV, CAR T therapy relying on bNAbs is thought to be a promising strategy for the treatment of HIV infection (200). Direct comparisons between bNAb- and CD4ζ-based CARs, or between bNAbs, have illustrated certain distinctions in breadth and potency and indicate that at least some antibody-derived scFVs may be more suitable than others for CAR T-cell administration (194).
4.1 Viral and cellular targets
Compared to CAR-T cells targeting tumor antigens such as CD19 and CD20, both of those are similarly expressed in healthy tissues, HIV-1-specific CAR-T cells target HIV-1 Env, that is expressed only on the surface of virus-producing cells (200). There are also two shared strategies for targeting HIV, one of which involves CD4 to target cells expressing HIV Env, while the second strategy involves aspects to prevent HIV infection of CD4-based CAR-T cells (6). Scholler et al. stated that, in general, CD4 receptor-based CAR T-cells exhibit a half-life of longer than 16 years with a stable engraftment level and safety (201). A recent study has demonstrated that primary T-cells transduced with a multispecific CAR (targeting the CD4-binding site gp120 and the co-receptor gp120 binding site) can potently inhibit cellular HIV infection by up to 99% in vitro and >97% in vivo (17, 202). To avoid identification of CD4-CARs HIV-1 as target cells, CD4-CARs are engineered to co-express HIV-1 fusion inhibitors, such as membrane-bound peptide C or peptides from the heptad repeat-2 domain of gp41 (203, 204). Alternative constructs have been developed for co-expression of shRNA targeting CCR5 and the LTR sequence to inhibit HIV-1 entry and to facilitate viral RNA degradation (205). The other strategy for avoiding infection of CD4ζ CAR T-cells is to modify the antigen-recognition part of the CAR molecule (181). Also, in vitro data showed that V1/V2-specific cars outperform CAR targeting CD4bs glycan and V3 on Env SIV. The transfer of anti-SIV CAR T cells to SIV-infected animals gave neither protection nor viral control. Unlike CAR-T-cell cancer therapy, CAR therapy against HIV requires a greater improvement in viral control.
Despite the recent FDA (Food and Drug Administration) approval of CAR-T cell therapy for B-cell leukemia and extensive research efforts in the field of cancer immunotherapy, about seven clinical trials of CAR-T cells against HIV have been initiated, five of which are currently in active status (NCT03240328, NCT04648046, NCT03617198, NCT01013415, NCT05077527), one completed (NCT03980691) and one in status unknown (NCT04863066) while clinical trials have previously confirmed the safety and efficacy of CAR-T cells against HIV validity of carcinotherapy.1
4.2 Restrictions
T-cells with CARs against the HIV envelope can increase the adaptive immune response, but are ineffective in controlling viremia (194). Another possible obstacle to CAR-therapy of HIV infection is an extremely low level of expression of the viral envelope on the surface of HIV-infected cells, moreover, the development of CAR-T-cell therapy is associated with a number of problems, including weak cell expansion, short vitality in vivo and significant side effects in patients (cytokine storms and neurological toxicity) (206). Strategies for improving the function and persistence of CART cells have been thoroughly researched for CARS based on CD19 and other tumor-specific antigens. In addition, research supporting the concept has shown the HSPCs are capable of lifelong engraftment and ensure the proper development of CAR-T cells in vivo (191). Although stem cell-based CAR-T-cell therapy against HIV has proven to be possible and effective in a humanized mouse model, there are limitations in the use of this model, which include a deficiency of the lymphoid structure and a graft vs. host reaction (207). Difficulties with the application of the CAR antibody are related to possible immunogenicity and the design of anti-idiotypic antibodies that can inhibit their activity (208). There is a risk that CAR may likewise attack healthy cells expressing the same or identical target antigen, that is recognized as an “off-target” effect (17).
5 Eradication of latent reservoirs
5.1 Establishment of HIV-1 reservoirs
Latent HIV reservoirs are the central barrier preventing a HIV-1 cure. A large number of cell types, including CD4 + T-cells, macrophages, and dendritic cells, are susceptible to HIV-1 infection, but CD4 + memory T-cells (primarily memory T-cells or TCMs and transient memory T-cells or TTMs) are widely considered as typical latent reservoirs for HIV-1, because of their active and resting physiological state, and the dynamic process of transforming effector cells into memory cells (209–211). HIV-1 reservoirs share a broad spectrum of anatomical localizations, including the lymph nodes, gut-associated lymphoid tissue (GALT), liver, genital tract, and brain (212). For instance, astrocytes and microglia are considered to be macrophage-like cells in the central nervous system (CNS) that are possible reservoirs of viruses in the brain (192). Viral DNA has also been found in HSCs from patients undergoing cART, which may indicate that these cells are involved in the persistence of HIV-1 (213). Some latent regions are shielded from cART penetration (the brain, testicles, and lymph node B-cell germinal centers) and present additional challenges to HIV treatment (214, 215). There is a lot of data from in vitro, in vivo, and animal models, but there is still no gold standard for defining the size of the latent reservoir (216). However, along with other techniques, digital droplet PCR assays can now detect intact, cell-associated, full-length genomic HIV DNA with increased sensitivity (217–219).
Human immunodeficiency virus type 1 reservoirs are a highly heterogeneous pool of infected cells that can be in different states of viral activation: (a) deep latency, in a state where no viral RNAs are expressed, (b) low transcriptional activation, when small amounts of viral RNAs are produced but not translated, or (c) dynamic viral activation, in this case there is a high level of expression of HIV-1 RNAs and a proportion of these RNAs are later translated into protein (193). The maintenance of the latent reservoir exists via clonal expansion of HIV-infected cells or through infection of long-term reservoir cells with both intact and defective proviruses (212, 220). It is suggested that the expansion of latently infected cells could be propelled by survival advantage and homeostatic cytokines such as IL-7. Moreover, latently infected CD4 + T-cells with antigen-specific TCR can divide in response to recurring exposure to antigens (220). Two primary models are proposed to clarify the latent infection of memory T-cells: the pre-activation and post-activation latency models (216). The pre-activation latency model posits that resting CD4 memory T-cells are infected with HIV-1 before being reactivated by environmental stimuli. However, this model might be inefficient due to the instability of the pre-integration complex, characterized by non-integrated linear and cytoplasmic forms of the viral genome with a half-life of about 1–6 days (221–223). The post-activation model suggests that activated CD4 T-cells are infected by HIV-1 while returning to a resting state, which results in the integration of the proviral genome into the host cell. This avoids creating favorable conditions for optimal viral gene expression, preventing the quick eradication of the infected T-cell (223, 224). Nevertheless, recent research implies that the relationship between T-cell activation and HIV-1 latency might not be as strongly correlated as previously assumed (216). This perspective proposes an additional model, suggesting that HIV-1 latency established in activated CD4 T-cells shortly after infection ensures greater survival capacity and possibly a return to a resting memory state. This process may facilitate the creation of long-lived latent reservoirs (224).
The selection of the HIV-1 integration site may contribute to the establishment of HIV-1 latency mechanisms in activated CD4 T-cells. HIV enters the host and replicates locally at the site of entry, then HIV rapidly circulates to the lymph nodes (within a few days) and further (within a few weeks) into the bloodstream (225). HIV-1 integration appears to be random, but prefers the introns of transcriptionally active genes found in gene-dense regions of the nuclear outer envelope near the nuclear pores (213). For example, pyrosequencing was used to identify 40,569 integration sites in Jurkat cells and another study revealed 6,719 integration sites in CD4 T-cells in a research study involving 13 individuals (226–228). The latency of HIV-1 is controlled by various related mechanisms acting at the transcriptional and post-transcriptional levels, and depends on the transcription program of the host cell (229, 230). In the latent period, the HIV-1 promoter is largely controlled by epigenetic mechanisms, including DNA methylation and post-translational histone modifications such as acetylation, methylation and crotonylation (213). Additionally, there are cellular cofactors of HIV-1 integration such as lens epithelium-derived growth factor (LEDGF/p75) (231).
The other possible interpretation of latency can be derived from the theory of stochastic gene expression. According to this theory, random mutations in a critically important HIV-1 Tat region can inhibit active HIV-1 transcription independently of the target cell activation (232, 233). The majority of investigations on this subject have been performed in vitro, which leaves a lot of questions about the level to which the different triggers contribute to the establishment of latency in vivo (216).
Strategies for the functional treatment of HIV, based on the “block and lock” approach, aim to induce transcriptional gene silencing (TGS) using latency-promoting agents (LPAs). This approach intends to block viral replication and lock the viral promoter into a dormant state through repressive epigenetic modifications (212). Epigenetic silencing can be initiated through various RNAs molecules (such as siRNAs, shRNAs, and lncRNAs) and small molecule inhibitors (including LEDGIN, epigenetic reader bromodomain and the extraterminal (BET) family proteins BRD4, Torin1, and pp242) (234–237). RNA-directed TGS results in the attraction of additional proteins in the nucleus, forming the RNA-induced transcriptional silencing (RITS) complex. This complex leads to the increase of repressive epigenetic markers such as histone and CpG methylation and the reduction of histone acetylation at the promoter (212).
The other strategy is the “shock and kill” approach when the latent HIV is reactivated by latency reversal agents (LRAs) followed by eradication of the cells with reactivated virus, achieved by enhancing the cytotoxic effect, immune clearance and additional procedures (210, 242). In these studies, the first generation of LRAs successfully induced viral RNA production, but only certain agents were able to cause the protein and viral particle generation (220). The next generations of LRAs in preclinical data demonstrate that small molecule antagonists of apoptosis (second mitochondria-derived activator of caspase or SMAC mimetic compounds) trigger the reversal of the latency (243). A number of LRAs have been developed based on in vitro and ex vivo systems, including HDAC inhibitors (HDACis), histone methyltransferases inhibitors (HMTis), and DNA methyltransferase inhibitors (DNMTis) (210). Immunostimulatory approaches based on Toll-like receptor (TLR)-7 agonists have displayed direct latency reversal activity in non-human primates, but their efficacy has not been confirmed in subsequent studies (244, 245). A further class of LRAs includes compounds that modulate protein kinases in signaling pathways upstream of the transcription factors that bind the LTR (246). Cytokines and LRA compounds can also be used in combination with recombinant macromolecules (CRISPR/nuclease deficient Cas9 (dCas9) and zinc finger proteins) to reverse HIV latency (247). The main LPAs and LRAs are summarized in Table 1.
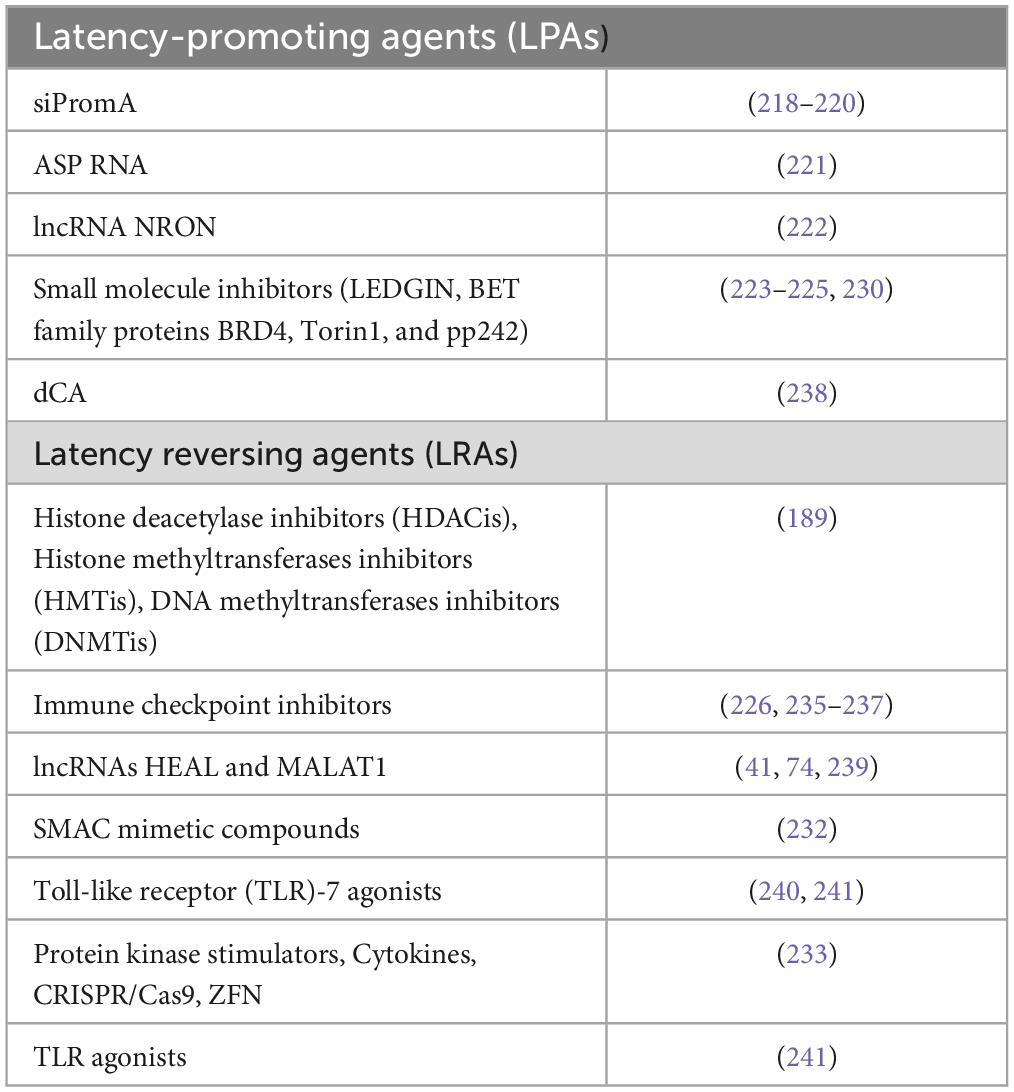
Table 1. The main latency reversing agents (LRAs) able to eradicate the latent HIV-1 reservoirs and latency promoting agents (LPAs) to make the proviral state of HIV-1 deeper.
In persistent viral infections, the large volume of antigens continually stimulates T-cells, leading to a gradual loss of functionality known as T-cell exhaustion (248). During this phase, there is an increased expression of immune checkpoint molecules (ICs) on T-cells. These molecules include programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG-3), T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT), T-cell immunoglobulin and mucin domain 3 (TIM-3), CD160, and 2B4 (CD244) (19). IC expression leads to suppression of the immune response and serves as a marker for HIV latently infected cells with a higher tendency to viral transcription (249). Immune checkpoint blockade in HIV has been intensively studied (250, 251). For example, PD-1 or Interleukin-10 (IL-10) blockade have been shown to reactivate CD4 + T-cell function in vitro and to restore NK-cell support (252).
5.2 Viral and cellular targets
Latency-promoting agents target the NF-κB sites, the interaction complex of LEDGF/p75 and HIV integrase, Tat, and the mammalian target of rapamycin (mTOR) signaling pathway as an important modulator of HIV-1 latency, triggering recruitment of chromatin-remodeling complexes, including DNA methyltransferase 3 alpha (DNMT3a), histone-lysine N-methyltransferase enzyme (EZH2) and histone deacetylase 1 (HDAC-1) (212). For instance, SiPromA was identified in 2005 as the first anti-HIV agent to induce TGS, because it targeted NF-κB sites in the HIV-1 promoter (253–255). Further, HIV-1 encoding antisense protein (ASP) was identified. ASP RNA recruits the repressive Polycomb group 2 complex (PRC2) to the 5′ LTR HIV-1 promoter, resulting in repressive epigenetic modifications (an increase in H3K27me3 marks and a decrease in RNA polymerase II occupancy) (212, 256). Moreover, an lncRNA named NRON was found in resting CD4 + T-cells suppressing viral transcription by causing degradation of Tat (248). However, the virus promoter can be activated by lncRNA HEAL and lncRNA MALAT1 (257–259). McBrien et al. used a provocative method to activate HIV-1 provirus. They used the drug N-803 to induce IL-15, a protein that promotes viral transcription, and an antibody to deplete the CD8 + T-cells that appear to play a role in stabilizing viral latency (260).
5.3 Restrictions
A major barrier to HIV-1 eradication is the multiplex mechanism of establishing HIV latency, and the way the latent reservoir recovers and produces infectious HIV virions when ART is terminated (228). Targeting and reactivating latent cells is problematic because of the highly heterogeneous nature of the viral reservoirs. In addition, some studies indicate controversial effects of LRA on NK cell function and on cytotoxic T-cell lymphocytes (CTLs) (213). Escape mutations in dominant CTL epitopes prevent the targeting of induced cells and certain LRAs suppress CTL function (261). The eradication strategy requires better LRA penetration into the tissues by improving the drug delivery system and, most importantly, enhancing the killing of LRA-activated cells by stimulating the CD8 + T response (194). In addition to low efficacy in the clinic, other disadvantages of many LRAs are their adverse effects and toxicity (237).
6 Conclusion
According to the latest update (March 2022) from the U.S. National Institutes of Health (see text footnote 1), 630 clinical trials associated with HIV-1 therapy have been initiated. Although some clinical trials have shown that gene-based therapeutic approaches in combination with conventional therapies can eliminate the HIV-1 virus, most gene-based clinical tests are still in the early stages (4). All mentioned advantages and limitations of HIV-1 cure strategies have been included in Table 2.
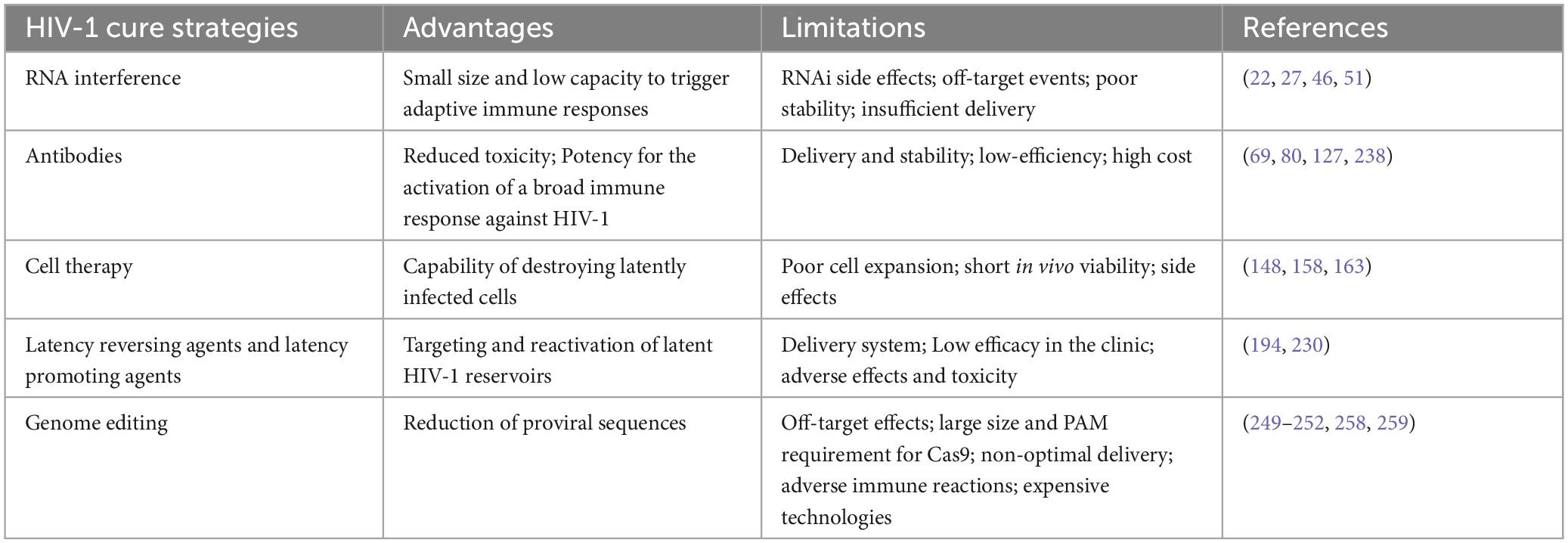
Table 2. The main advantages and limitations of HIV-1 cure strategies.
As with cART, strategies combining multiple antiviral approaches should be considered to avoid the escape of HIV-1 mutants (Table 3). Although, there is evidence that viruses have developed mechanisms to escape the RNAi defensive mechanism, RNAi-based therapeutics can be enhanced by using a combination of different siRNAs or by coupling the siRNAs with ribozymes, aptamers and antiviral proteins (such as RevM10) (28). Strand selection can be biased by constructing asymmetric siRNAs or by chemical modifications at one or both ends of the siRNA (4). Dicer-independent Ago/shRNAs have the potential to demonstrate an improved safety profile and to reduce off-target effects compared to conventional shRNAs, but identifying their overall benefit requires additional laboratory testing (60, 262, 263).
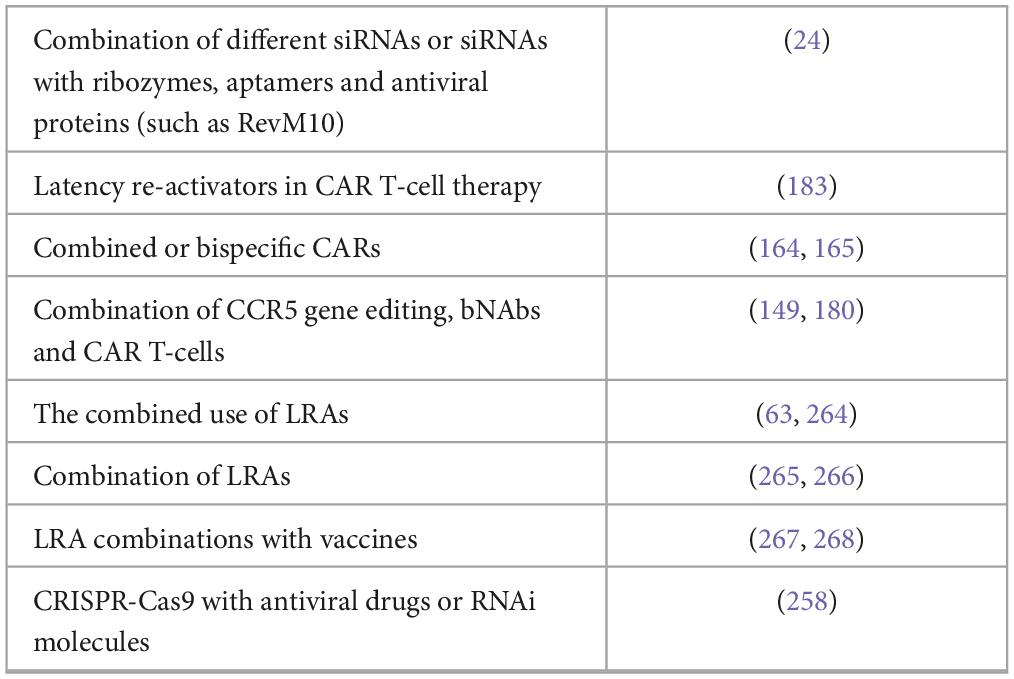
Table 3. Combinatorial strategies for functional treatment of HIV-1.
To date, the accumulated data indicate that no single factor will define the ultimate achievement of a bNAb-inducing HIV-1 vaccine, that probably requires a combination of effective priming of B-cell precursors, optimization of Env design and presentation, as well as sustained enhancement of heterologous Env (120). To resolve the problems of antibody delivery and stability, antibodies may be conjugated with cell-penetrating peptides (CPPs) derived from various sources to penetrate the cell cytoplasm (269). Smaller antibodies such as Fabs, scFvs and single-domain antibodies or sdAbs (with Fc removed) have also been created for this goal (138, 270, 271). Modifications that increase the half-life, potency, Fc-receptor (FcR) binding, and polyfunctionality are thought to bypass the several disadvantages of bnAbs (272).
Combined or bispecific CARs may be necessary to overcome the well-documented capacity of HIV-1 to mutate and escape treatment or host immune responses, for example, bispecific CARs have displayed improved efficacy against several primary HIV-1 isolates compared to single CD4ζ CARs and this approach deserves additional in vivo studies (273, 274). CAR approaches for people with HIV may be improved with a combination of therapies, such as CCR5 gene editing, the use of individual bNAbs targeting various regions of the viral envelope and the development of next-generation CAR T-cells capable of acting on multiple antigens (181, 191). The incorporation of co-stimulatory domains, including CD28, 4-1BB, CD28 + 4-1BB, OX40, ICOS, and CD27, or the engineering of CD4-ζ CARs in second- and third-generations could increase the proliferation and killing efficiency of these cells (181, 275, 276). In addition, the use of latent re-activators in CAR-T cell therapy could potentially allow CAR-T cells to act on latent reservoirs, since these CAR-T cells are able to move into various types of tissue reservoirs, including the central nervous system, which is a potentially significant refuge for latent HIV (276).
The combined use of LRAs with synergistic effects is currently an actively studied area of research (9, 172). Combined approaches, which include LRAs with several different types of mechanisms, are being studied to obtain more effective shocks (277, 278). HIV reservoirs are often hidden in sites, such as lymphatic, gut or brain tissues, but the development of nanoparticle-packed cART drugs or CRISPR-Cas9 system have the ability to directly target the provirus and to destroy the HIV reservoirs (265). LRA combinations with vaccines targeting conservative HIV-1 epitopes have also generated interesting results (266, 279).
CRISPR-Cas9 can be combined with other anti-HIV therapies (antiviral drugs or RNAi molecules) to reduce viral replication, but these combinations also increase the genetic threshold at which viral escape can occur (99). Current efforts are focusing on reducing the number of CRISPR off-targets by creating alternative Cas9 variants (SaCas9, Cas12a, Cas13a, Cas13d, base editors and prime editing systems) and improving the architecture of gRNAs (239, 280, 281). Moreover, non-viral delivery systems also have been extensively investigated and reviewed (63, 240, 264, 267, 268).
Eventually, the high initial cost of cell and gene therapies will become more cost-effective than conventional cART, if a single treatment can be sufficient (31). Combined cell and gene therapies have come a long way, and their great potential will open up new opportunities for the development of HIV cures.
Author contributions
AS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review and editing. EA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review and editing. ED: Conceptualization, Funding acquisition, Project administration, Validation, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This publication was supported by a grant No. 075-15-2019-1789 from the Ministry of Science and Higher Education of the Russian Federation allocated to the Center for Precision Genome Editing and Genetic Technologies for Biomedicine.
Acknowledgments
Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Nyamweya S, Hegedus A, Jaye A, Rowland-Jones S, Flanagan K, Macallan D. Comparing HIV-1 and HIV-2 infection: lessons for viral immunopathogenesis. Rev Med Virol. (2013) 23:221–40. doi: 10.1002/rmv.1739
2. Watts J, Dang K, Gorelick R, Leonard C, Bess J, Swanstrom R, et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. (2009) 460:711–6. doi: 10.1038/nature08237
3. Reza S, Mim F, Quader M, Khan M, Hossain M, Uddin K, et al. The possibility of nucleic acids to act as anti-viral therapeutic agents-A review. Open J Med Microbiol. (2021) 11:198–248.
4. Sheykhhasan M, Foroutan A, Manoochehri H, Khoei S, Poondla N, Saidijam M. Could gene therapy cure HIV? Life Sci. (2021) 277:119451. doi: 10.1016/j.lfs.2021.119451
5. Cabrera-Rodríguez R, Pérez-Yanes S, Estévez-Herrera J, Márquez-Arce D, Cabrera C, Espert L, et al. The interplay of HIV and autophagy in early infection. Front Microbiol. (2021) 12:661446. doi: 10.3389/fmicb.2021.661446
6. Wagner T. Quarter century of anti-HIV CAR T cells. Curr HIV/AIDS Rep. (2018) 15:147–54. doi: 10.1007/s11904-018-0388-x
7. Nchioua R, Bosso M, Kmiec D, Kirchhoff F. Cellular factors targeting HIV-1 transcription and viral RNA transcripts. Viruses. (2020) 12:495. doi: 10.3390/v12050495
9. Moranguinho I, Valente S. Block-and-lock: new horizons for a cure for HIV-1. Viruses. (2020) 12:1443. doi: 10.3390/v12121443
10. Woldemeskel B, Kwaa A, Blankson J. Viral reservoirs in elite controllers of HIV-1 infection: implications for HIV cure strategies. Ebiomedicine. (2020) 62:103118. doi: 10.1016/j.ebiom.2020.103118
11. Ward A, Mota T, Jones R. Immunological approaches to HIV cure. Semin Immunol. (2021) 51:101412. doi: 10.1016/j.smim.2020.101412
12. Duerr A, Wasserheit J, Corey L. HIV vaccines: new frontiers in vaccine development. Clin Infect Dis. (2006) 43:500–11. doi: 10.1086/505979
13. Course M, Gudsnuk K, Desai N, Chamberlain J, Valdmanis P. Endogenous MicroRNA competition as a mechanism of shRNA-induced cardiotoxicity. Mol Ther Nucleic Acids. (2020) 19:572–80. doi: 10.1016/j.omtn.2019.12.007
14. Martinez-Navio J, Fuchs S, Pantry S, Lauer W, Duggan N, Keele B, et al. Adeno-associated virus delivery of anti-HIV monoclonal antibodies can drive long-term virologic suppression. Immunity. (2019) 50:567–75.e5. doi: 10.1016/j.immuni.2019.02.005
15. Kang W, Sun Y. Moving toward a functional cure for HIV-1. Infect Dis Immun. (2021) 1:161–8. doi: 10.1097/ID9.0000000000000025
16. Kalidasan V, Theva Das K. Lessons learned from failures and success stories of HIV breakthroughs: are we getting closer to an HIV cure? Front Microbiol. (2020) 11:46. doi: 10.3389/fmicb.2020.00046
17. Qi J, Ding C, Jiang X, Gao Y. Advances in developing CAR T-cell therapy for HIV cure. Front Immunol. (2020) 11:361. doi: 10.3389/fimmu.2020.00361
18. Bobbin M, Burnett J, Rossi J. RNA interference approaches for treatment of HIV-1 infection. Genome Med. (2015) 7:50. doi: 10.1186/s13073-015-0174-y
19. Darcis G, Berkhout B, Pasternak A. The quest for cellular markers of HIV reservoirs: any color you like. Front Immunol. (2019) 10:2251. doi: 10.3389/fimmu.2019.02251
20. Hendriks D, Clevers H, Artegiani B. CRISPR-Cas tools and their application in genetic engineering of human stem cells and organoids. Cell Stem Cell. (2020) 27:705–31. doi: 10.1016/j.stem.2020.10.014
21. Atkins A, Allen A, Dampier W, Haddad E, Nonnemacher M, Wigdahl B. HIV-1 cure strategies: why CRISPR? Expert Opin Biol Ther. (2021) 21:781–93. doi: 10.1080/14712598.2021.1865302
22. Berkhout B. RNAi-mediated antiviral immunity in mammals. Curr Opin Virol. (2018) 32:9–14. doi: 10.1016/j.coviro.2018.07.008
23. Ding S, Voinnet O. Antiviral immunity directed by small RNAs. Cell. (2007) 130:413–26. doi: 10.1016/j.cell.2007.07.039
24. Svoboda P. Key mechanistic principles and considerations concerning RNA interference. Front Plant Sci. (2020) 11:1237. doi: 10.3389/fpls.2020.01237
25. Hossbach M, Gruber J, Osborn M, Weber K, Tuschl T. Gene silencing with siRNA duplexes composed of target-mRNA-complementary and partially palindromic or partially complementary single-stranded siRNAs. RNA Biol. (2006) 3:82–9. doi: 10.4161/rna.3.2.3110
26. Delgado R, Regueiro B. The future of HIV infection: gene therapy and RNA interference. Enferm Infecc Microbiol Clin. (2005) 23 Suppl. 2::68–83.
27. Scarborough R, Gatignol A. RNA interference therapies for an HIV-1 functional cure. Viruses. (2017) 10:8. doi: 10.3390/v10010008
28. Zhou J, Rossi J. Current progress in the development of RNAi-based therapeutics for HIV-1. Gene Ther. (2011) 18:1134–8. doi: 10.1038/gt.2011.149
29. Rettig G, Behlke M. Progress toward in vivo use of siRNAs-II. Mol Ther. (2012) 20:483–512. doi: 10.1038/mt.2011.263
30. Dorman N, Lever A. RNA-based gene therapy for HIV infection. HIV Med. (2001) 2:114–22. doi: 10.1046/j.1468-1293.2001.00052.x
31. Cornu T, Mussolino C, Müller M, Wehr C, Kern W, Cathomen T. HIV gene therapy: an update. Hum Gene Ther. (2021) 32:52–65. doi: 10.1089/hum.2020.159
32. Zeng Y, Wagner E, Cullen B. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. (2002) 9:1327–33. doi: 10.1016/s1097-2765(02)00541-5
33. Dey R, Soni K, Saravanan S, Balakrishnan P, Kumar V, Boobalan J, et al. Anti-HIV microRNA expression in a novel Indian cohort. Sci Rep. (2016) 6:28279. doi: 10.1038/srep28279
34. Jacque J, Triques K, Stevenson M. Modulation of HIV-1 replication by RNA interference. Nature. (2002) 418:435–8. doi: 10.1038/nature00896
35. Kim D, Behlke M, Rose S, Chang M, Choi S, Rossi J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. (2005) 23:222–6. doi: 10.1038/nbt1051
36. Petri S, Meister G. siRNA design principles and off-target effects. Methods Mol Biol. (2013) 986:59–71. doi: 10.1007/978-1-62703-311-4_4
37. Khvorova A, Reynolds A, Jayasena S. Functional siRNAs and miRNAs exhibit strand bias. Cell. (2003) 115:209–16. doi: 10.1016/s0092-8674(03)00801-8
38. Schwarz D, Hutvágner G, Du T, Xu Z, Aronin N, Zamore P. Asymmetry in the assembly of the RNAi enzyme complex. Cell. (2003) 115:199–208. doi: 10.1016/s0092-8674(03)00759-1
39. Subramanya S, Kim S, Manjunath N, Shankar P. RNA interference-based therapeutics for human immunodeficiency virus HIV-1 treatment: synthetic siRNA or vector-based shRNA? Expert Opin Biol Ther. (2010) 10:201–13. doi: 10.1517/14712590903448158
40. Yi R, Qin Y, Macara I, Cullen B. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. (2003) 17:3011–6. doi: 10.1101/gad.1158803
41. Xu W, Jiang X, Huang L. RNA interference technology. Comprehens Biotechnol. (2019) 5:560–75. doi: 10.1016/B978-0-444-64046-8.00282-2
42. Tyagi A, Ahmed F, Thakur N, Sharma A, Raghava G, Kumar M. HIVsirDB: a database of HIV inhibiting siRNAs. PLoS One. (2011) 6:e25917. doi: 10.1371/journal.pone.0025917
43. Zeng W, Chen Y, Bai Y, Trang P, Vu G, Lu S, et al. Effective inhibition of human immunodeficiency virus 1 replication by engineered RNase P ribozyme. PLoS One. (2012) 7:e51855. doi: 10.1371/journal.pone.0051855
44. Held D, Kissel J, Patterson J, Nickens D, Burke D. HIV-1 inactivation by nucleic acid aptamers. Front Biosci. (2006) 11:89–12. doi: 10.2741/1782
45. Takahashi M, Li H, Zhou J, Chomchan P, Aishwarya V, Damha M, et al. Dual mechanisms of action of self-delivering, anti-HIV-1 FANA oligonucleotides as a potential new approach to HIV therapy. Mol Ther Nucleic Acids. (2019) 17:615–25. doi: 10.1016/j.omtn.2019.07.001
46. Ahlenstiel C, Suzuki K, Marks K, Symonds G, Kelleher A. Controlling HIV-1: non-coding RNA gene therapy approaches to a functional cure. Front Immunol. (2015) 6:474. doi: 10.3389/fimmu.2015.00474
47. Rossi J. RNAi as a treatment for HIV-1 infection. Biotechniques. (2006) Suppl:25–9. doi: 10.2144/000112167
48. Zeller S, Kumar P. RNA-based gene therapy for the treatment and prevention of HIV: from bench to bedside. Yale J Biol Med. (2011) 84:301–9.
49. Lee N, Dohjima T, Bauer G, Li H, Li M, Ehsani A, et al. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol. (2002) 20:500–5. doi: 10.1038/nbt0502-500
50. Chery J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. (2016) 4:35–50. doi: 10.14304/surya.jpr.v4n7.5
51. Kole R, Krainer A, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. (2012) 11:125–40. doi: 10.1038/nrd3625
52. von Eije K, Berkhout B. RNA-interference-based gene therapy approaches to HIV type-1 treatment: tackling the hurdles from bench to bedside. Antivir Chem Chemother. (2009) 19:221–33. doi: 10.1177/095632020901900602
53. Park W, Miyano-Kurosaki N, Hayafune M, Nakajima E, Matsuzaki T, Shimada F, et al. Prevention of HIV-1 infection in human peripheral blood mononuclear cells by specific RNA interference. Nucleic Acids Res. (2002) 30:4830–5. doi: 10.1093/nar/gkf627
54. Kim N, Baek J, Choi H, Chung I, Shin S, Lee J, et al. Short-hairpin RNA-mediated gene expression interference in Trichoplusia ni cells. J Microbiol Biotechnol. (2012) 22:190–8. doi: 10.4014/jmb.1108.08045
55. Chadwick D, Lever A. Antisense RNA sequences targeting the 5’ leader packaging signal region of human immunodeficiency virus type-1 inhibits viral replication at post-transcriptional stages of the life cycle. Gene Ther. (2000) 7:1362–8. doi: 10.1038/sj.gt.3301254
56. Bahner I, Zhou C, Yu X, Hao Q, Guatelli J, Kohn D. Comparison of trans-dominant inhibitory mutant human immunodeficiency virus type 1 genes expressed by retroviral vectors in human T lymphocytes. J Virol. (1993) 67:3199–207. doi: 10.1128/JVI.67.6.3199-3207.1993
57. Zhu Q, Shibata T, Kabashima T, Kai M. Inhibition of HIV-1 protease expression in T cells owing to DNA aptamer-mediated specific delivery of siRNA. Eur J Med Chem. (2012) 56:396–9. doi: 10.1016/j.ejmech.2012.07.045
58. Didigu C, Doms R. Novel approaches to inhibit HIV entry. Viruses. (2012) 4:309–24. doi: 10.3390/v4020309
59. Eekels J, Geerts D, Jeeninga R, Berkhout B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antiviral Res. (2011) 89:43–53. doi: 10.1016/j.antiviral.2010.11.005
60. Herrera-Carrillo E, Gao Z, Berkhout B. CRISPR therapy towards an HIV cure. Brief Funct Genomics. (2020) 19:201–8. doi: 10.1093/bfgp/elz021
61. Gu S, Jin L, Zhang F, Huang Y, Grimm D, Rossi J, et al. Thermodynamic stability of small hairpin RNAs highly influences the loading process of different mammalian Argonautes. Proc Natl Acad Sci U S A. (2011) 108:9208–13. doi: 10.1073/pnas.1018023108
62. Lam J, Chow M, Zhang Y, Leung S. siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucleic Acids. (2015) 4:e252. doi: 10.1038/mtna.2015.23
63. Yin H, Kanasty R, Eltoukhy A, Vegas A, Dorkin J, Anderson D. Non-viral vectors for gene-based therapy. Nat Rev Genet. (2014) 15:541–55. doi: 10.1038/nrg3763
64. Levanova A, Poranen M. RNA interference as a prospective tool for the control of human viral infections. Front Microbiol. (2018) 9:2151. doi: 10.3389/fmicb.2018.02151
65. Maruggi G, Zhang C, Li J, Ulmer J, Yu D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther. (2019) 27:757–72. doi: 10.1016/j.ymthe.2019.01.020
66. Liang F, Lindgren G, Lin A, Thompson E, Ols S, Röhss J, et al. Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol Ther. (2017) 25:2635–47. doi: 10.1016/j.ymthe.2017.08.006
67. Morris L. mRNA vaccines offer hope for HIV. Nat Med. (2021) 27:2082–4. doi: 10.1038/s41591-021-01602-4
68. Pardi N, Hogan M, Porter F, Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. (2018) 17:261–79. doi: 10.1038/nrd.2017.243
69. Khalid K, Padda J, Khedr A, Ismail D, Zubair U, Al-Ewaidat O, et al. HIV and messenger RNA (mRNA) vaccine. Cureus. (2021) 13:e16197. doi: 10.7759/cureus.16197
70. Mu Z, Haynes B, Cain D. HIV mRNA vaccines-progress and future paths. Vaccines. (2021) 9:134. doi: 10.3390/vaccines9020134
71. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. (2015) 16:343–53. doi: 10.1038/ni.3123
72. Pardi N, Muramatsu H, Weissman D, Karikó K. In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol. (2013) 969:29–42. doi: 10.1007/978-1-62703-260-5_2
73. Martinon F, Krishnan S, Lenzen G, Magné R, Gomard E, Guillet J, et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. (1993) 23:1719–22. doi: 10.1002/eji.1830230749
74. Romani B, Kavyanifard A, Allahbakhshi E. Antibody production by in vivo RNA transfection. Sci Rep. (2017) 7:10863. doi: 10.1038/s41598-017-11399-3
75. Reichmuth A, Oberli M, Jaklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv. (2016) 7:319–34. doi: 10.4155/tde-2016-0006
76. Moyo N, Vogel A, Buus S, Erbar S, Wee E, Sahin U, et al. Efficient induction of T cells against conserved HIV-1 regions by mosaic vaccines delivered as self-amplifying mRNA. Mol Ther Methods Clin Dev. (2018) 12:32–46. doi: 10.1016/j.omtm.2018.10.010
77. Gay C, DeBenedette M, Tcherepanova I, Gamble A, Lewis W, Cope A, et al. Immunogenicity of AGS-004 dendritic cell therapy in patients treated during acute HIV infection. AIDS Res Hum Retroviruses. (2018) 34:111–22. doi: 10.1089/aid.2017.0071
78. de Jong W, Aerts J, Allard S, Brander C, Buyze J, Florence E, et al. iHIVARNA phase IIa, a randomized, placebo-controlled, double-blinded trial to evaluate the safety and immunogenicity of iHIVARNA-01 in chronically HIV-infected patients under stable combined antiretroviral therapy. Trials. (2019) 20:361. doi: 10.1186/s13063-019-3409-1
79. Koonin E, Makarova K. Origins and evolution of CRISPR-Cas systems. Philos Trans R Soc Lond B Biol Sci. (2019) 374:20180087. doi: 10.1098/rstb.2018.0087
80. Makarova K, Wolf Y, Iranzo J, Shmakov S, Alkhnbashi O, Brouns S, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. (2020) 18:67–83. doi: 10.1038/s41579-019-0299-x
81. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna J, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. (2012) 337:816–21. doi: 10.1126/science.1225829
82. Cho S, Kim S, Kim J, Kim J. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. (2013) 31:230–2. doi: 10.1038/nbt.2507
83. Xiao Q, Guo D, Chen S. Application of CRISPR/Cas9-based gene editing in HIV-1/AIDS therapy. Front Cell Infect Microbiol. (2019) 9:69. doi: 10.3389/fcimb.2019.00069
84. Hamilton J, Tsuchida C, Nguyen D, Shy B, McGarrigle E, Sandoval Espinoza C, et al. Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep. (2021) 35:109207. doi: 10.1016/j.celrep.2021.109207
85. Mehmetoglu-Gurbuz T, Yeh R, Garg H, Joshi A. Combination gene therapy for HIV using a conditional suicidal gene with CCR5 knockout. Virol J. (2021) 18:31. doi: 10.1186/s12985-021-01501-7
86. Tang X. CRISPR/Cas9-based genome engineering in HIV gene therapy. E3S Web Conf. (2021) 233:02004.
87. Ophinni Y, Inoue M, Kotaki T, Kameoka M. CRISPR/Cas9 system targeting regulatory genes of HIV-1 inhibits viral replication in infected T-cell cultures. Sci Rep. (2018) 8:7784. doi: 10.1038/s41598-018-26190-1
88. Herskovitz J, Hasan M, Patel M, Blomberg W, Cohen J, Machhi J, et al. CRISPR-Cas9 mediated exonic disruption for HIV-1 elimination. Ebiomedicine. (2021) 73:103678. doi: 10.1016/j.ebiom.2021.103678
89. Saayman S, Ali S, Morris K, Weinberg M. The therapeutic application of CRISPR/Cas9 technologies for HIV. Expert Opin Biol Ther. (2015) 15:819–30. doi: 10.1517/14712598.2015.1036736
90. Vergara-Mendoza M, Gomez-Quiroz L, Miranda-Labra R, Fuentes-Romero L, Romero-Rodríguez D, González-Ruiz J, et al. Regulation of Cas9 by viral proteins Tat and Rev for HIV-1 inactivation. Antiviral Res. (2020) 180:104856. doi: 10.1016/j.antiviral.2020.104856
91. Bogerd H, Kornepati A, Marshall J, Kennedy E, Cullen B. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc Natl Acad Sci U S A. (2015) 112:E7249–56. doi: 10.1073/pnas.1516305112
92. Magro G, Calistri A, Parolin C. Targeting and understanding HIV latency: the CRISPR system against the provirus. Pathogens. (2021) 10:1257. doi: 10.3390/pathogens10101257
93. Zhang Y, Arango G, Li F, Xiao X, Putatunda R, Yu J, et al. Comprehensive off-target analysis of dCas9-SAM-mediated HIV reactivation via long noncoding RNA and mRNA profiling. BMC Med Genom. (2018) 11:78. doi: 10.1186/s12920-018-0394-2
94. Olson A, Basukala B, Lee S, Gagne M, Wong W, Henderson A. Targeted chromatinization and repression of HIV-1 provirus transcription with repurposed CRISPR/Cas9. Viruses. (2020) 12:1154. doi: 10.3390/v12101154
95. Fernandez J, Vejnar C, Giraldez A, Rouet R, Moreno-Mateos M. Optimized CRISPR-Cpf1 system for genome editing in zebrafish. Methods. (2018) 150:11–8. doi: 10.1016/j.ymeth.2018.06.014
96. Bhardwaj A, Nain V. TALENs-an indispensable tool in the era of CRISPR: a mini review. J Genet Eng Biotechnol. (2021) 19:125. doi: 10.1186/s43141-021-00225-z
97. Robb G. Genome editing with CRISPR-Cas: an overview. Curr. Protoc. Essent. Lab. Tech. (2019) 19:e36. doi: 10.1002/cpet.36
98. Park J, Moon B, Park J, Thornton J, Park Y, Seo K. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci Rep. (2017) 7:44929. doi: 10.1038/srep44929
99. Das A, Binda C, Berkhout B. Elimination of infectious HIV DNA by CRISPR-Cas9. Curr Opin Virol. (2019) 38:81–8. doi: 10.1016/j.coviro.2019.07.001
100. Burdo T, Chen C, Kaminski R, Sariyer I, Mancuso P, Donadoni M, et al. Preclinical safety and biodistribution of CRISPR targeting SIV in non-human primates. Gene Ther. (2023): doi: 10.1038/s41434-023-00410-4 [Epub ahead of print].
101. Tebas P, Stein D, Tang W, Frank I, Wang S, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. (2014) 370:901–10. doi: 10.1056/NEJMoa1300662
102. Kwarteng A, Ahuno S, Kwakye-Nuako G. The therapeutic landscape of HIV-1 via genome editing. AIDS Res Ther. (2017) 14:32. doi: 10.1186/s12981-017-0157-8
103. Doudna J. The promise and challenge of therapeutic genome editing. Nature. (2020) 578:229–36. doi: 10.1038/s41586-020-1978-5
104. Anzalone A, Koblan L, Liu D. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. (2020) 38:824–44. doi: 10.1038/s41587-020-0561-9
105. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. (2018) 9:1911. doi: 10.1038/s41467-018-04252-2
106. Nidhi S, Anand U, Oleksak P, Tripathi P, Lal J, Thomas G, et al. Novel CRISPR-cas systems: an updated review of the current achievements, applications, and future research perspectives. Int J Mol Sci. (2021) 22:3327. doi: 10.3390/ijms22073327
107. Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther. (2020) 5:1. doi: 10.1038/s41392-019-0089-y
108. Brenna E, McMichael A. The importance of cellular immune response to HIV: implications for antibody production and vaccine design. DNA Cell Biol. (2022) 41:38–42. doi: 10.1089/dna.2021.0520
109. Swain S, McKinstry K, Strutt T. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. (2012) 12:136–48. doi: 10.1038/nri3152
110. Nowosad C, Spillane K, Tolar P. Germinal center B cells recognize antigen through a specialized immune synapse architecture. Nat Immunol. (2016) 17:870–7. doi: 10.1038/ni.3458
111. Victora G, Nussenzweig M. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
112. Mesin L, Ersching J, Victora G. Germinal center B cell dynamics. Immunity. (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
113. Fiebig E, Wright D, Rawal B, Garrett P, Schumacher R, Peddada L, et al. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS. (2003) 17:1871–9. doi: 10.1097/00002030-200309050-00005
114. Munier C, Andersen C, Kelleher A. HIV vaccines: progress to date. Drugs. (2011) 71:387–414. doi: 10.2165/11585400-000000000-00000
115. Tomaras G, Yates N, Liu P, Qin L, Fouda G, Chavez L, et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol. (2008) 82:12449–63. doi: 10.1128/JVI.01708-08
116. Overbaugh J, Morris L. The antibody response against HIV-1. Cold Spring Harb Perspect Med. (2012) 2:a007039. doi: 10.1101/cshperspect.a007039
117. Wei X, Decker J, Wang S, Hui H, Kappes J, Wu X, et al. Antibody neutralization and escape by HIV-1. Nature. (2003) 422:307–12. doi: 10.1038/nature01470
118. Su B, Dispinseri S, Iannone V, Zhang T, Wu H, Carapito R, et al. Update on Fc-mediated antibody functions against HIV-1 beyond neutralization. Front Immunol. (2019) 10:2968. doi: 10.3389/fimmu.2019.02968
119. Kim J, Rerks-Ngarm S, Excler J, Michael N. HIV vaccines: lessons learned and the way forward. Curr Opin HIV AIDS. (2010) 5:428–34. doi: 10.1097/COH.0b013e32833d17ac
120. Zhang P, Narayanan E, Liu Q, Tsybovsky Y, Boswell K, Ding S, et al. A multiclade env-gag VLP mRNA vaccine elicits tier-2 HIV-1-neutralizing antibodies and reduces the risk of heterologous SHIV infection in macaques. Nat Med. (2021) 27:2234–45. doi: 10.1038/s41591-021-01574-5
121. Learmont J, Cook L, Dunckley H, Sullivan J. Update on long-term symptomless HIV type 1 infection in recipients of blood products from a single donor. AIDS Res Hum Retroviruses. (1995) 11:1. doi: 10.1089/aid.1995.11.1
122. Learmont J, Tindall B, Evans L, Cunningham A, Cunningham P, Wells J, et al. Long-term symptomless HIV-1 infection in recipients of blood products from a single donor. Lancet. (1992) 340:863–7. doi: 10.1016/0140-6736(92)93281-q
123. Stephenson K, Wagh K, Korber B, Barouch D. Vaccines and broadly neutralizing antibodies for HIV-1 prevention. Annu Rev Immunol. (2020) 38:673–703. doi: 10.1146/annurev-immunol-080219-023629
124. Hansen S, Ford J, Lewis M, Ventura A, Hughes C, Coyne-Johnson L, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. (2011) 473:523–7. doi: 10.1038/nature10003
125. Hokello J, Sharma A, Tyagi M. An update on the HIV DNA vaccine strategy. Vaccines. (2021) 9:605. doi: 10.3390/vaccines9060605
126. Ng’uni T, Chasara C, Ndhlovu Z. Major scientific hurdles in HIV vaccine development: historical perspective and future directions. Front Immunol. (2020) 11:590780. doi: 10.3389/fimmu.2020.590780
127. Sobia P, Archary D. Preventive HIV vaccines-leveraging on lessons from the past to pave the way forward. Vaccines. (2021) 9:1001. doi: 10.3390/vaccines9091001
128. Ratnapriya S, Perez-Greene E, Schifanella L, Herschhorn A. Adjuvant-mediated enhancement of the immune response to HIV vaccines. FEBS J. (2022) 289:3317–34. doi: 10.1111/febs.15814
129. Saunders K, Pardi N, Parks R, Santra S, Mu Z, Sutherland L, et al. Lipid nanoparticle encapsulated nucleoside-modified mRNA vaccines elicit polyfunctional HIV-1 antibodies comparable to proteins in nonhuman primates. NPJ Vaccines. (2021) 6:50. doi: 10.1038/s41541-021-00307-6
130. Kublin J. HIV vaccines beyond COVID-19: merits of trust. J Int AIDS Soc. (2021) 24:e25742. doi: 10.1002/jia2.25742
131. Koff W, Russell N, Walport M, Feinberg M, Shiver J, Karim S, et al. Accelerating the development of a safe and effective HIV vaccine: HIV vaccine case study for the decade of vaccines. Vaccine. (2013) 31 Suppl. 2:B204–8. doi: 10.1016/j.vaccine.2012.10.115
132. Burton D, Desrosiers R, Doms R, Feinberg M, Gallo R, Hahn B, et al. Public health. A sound rationale needed for phase III HIV-1 vaccine trials. Science. (2004) 303:316. doi: 10.1126/science.1094620
133. Johnston M, Scarlatti G, Pitisutthithum P, Bekker L. HIV vaccines: progress and promise. J Int AIDS Soc. (2021) 24 Suppl. 7(Suppl. 7):e25828. doi: 10.1002/jia2.25828
134. Burton D, Hangartner L. Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu Rev Immunol. (2016) 34:635–59. doi: 10.1146/annurev-immunol-041015-055515
135. Williams W, Wiehe K, Saunders K, Haynes B. Strategies for induction of HIV-1 envelope-reactive broadly neutralizing antibodies. J Int AIDS Soc. (2021) 24 Suppl. 7(Suppl. 7):e25831. doi: 10.1002/jia2.25831
136. Chuang G, Zhou J, Acharya P, Rawi R, Shen C, Sheng Z, et al. Structural survey of broadly neutralizing antibodies targeting the HIV-1 env trimer delineates epitope categories and characteristics of recognition. Structure. (2019) 27:196–206.e6. doi: 10.1016/j.str.2018.10.007
137. Tuyishime M, Ferrari G. Engineering antibody-based molecules for HIV treatment and cure. Curr Opin HIV AIDS. (2020) 15:290–9. doi: 10.1097/COH.0000000000000640
138. Awi N, Teow S. Antibody-mediated therapy against HIV/AIDS: where are we standing now? J Pathog. (2018) 2018:8724549. doi: 10.1155/2018/8724549
139. McCoy L, Weiss R. Neutralizing antibodies to HIV-1 induced by immunization. J Exp Med. (2013) 210:209–23. doi: 10.1084/jem.20121827
140. Sajadi M, Dashti A, Rikhtegaran Tehrani Z, Tolbert W, Seaman M, Ouyang X, et al. Identification of near-pan-neutralizing antibodies against HIV-1 by deconvolution of plasma humoral responses. Cell. (2018) 173:1783–95.e14. doi: 10.1016/j.cell.2018.03.061
141. Rujas E, Cui H, Burnie J, Aschner C, Zhao T, Insausti S, et al. Engineering pan-HIV-1 neutralization potency through multispecific antibody avidity. Proc Natl Acad Sci U S A. (2022) 119:e2112887119. doi: 10.1073/pnas.2112887119
142. Hsu D, Mellors J, Vasan S. Can broadly neutralizing HIV-1 antibodies help achieve an ART-free remission? Front Immunol. (2021) 12:710044. doi: 10.3389/fimmu.2021.710044
143. Eroshkin A, LeBlanc A, Weekes D, Post K, Li Z, Rajput A, et al. bNAber: database of broadly neutralizing HIV antibodies. Nucleic Acids Res. (2014) 42:D1133–9. doi: 10.1093/nar/gkt1083
144. Ferrantelli F, Ruprecht R. Neutralizing antibodies against HIV – back in the major leagues? Curr Opin Immunol. (2002) 14:495–502. doi: 10.1016/s0952-7915(02)00362-x
145. Ferrantelli F, Rasmussen R, Buckley K, Li P, Wang T, Montefiori D, et al. Complete protection of neonatal rhesus macaques against oral exposure to pathogenic simian-human immunodeficiency virus by human anti-HIV monoclonal antibodies. J Infect Dis. (2004) 189:2167–73. doi: 10.1086/420833
146. Zhao F, Joyce C, Burns A, Nogal B, Cottrell C, Ramos A, et al. Mapping neutralizing antibody epitope specificities to an HIV env trimer in immunized and in infected rhesus macaques. Cell Rep. (2020) 32:108122. doi: 10.1016/j.celrep.2020.108122
147. Rolland M, Nickle D, Mullins J. HIV-1 group M conserved elements vaccine. PLoS Pathog. (2007) 3:e157. doi: 10.1371/journal.ppat.0030157
148. Mekonnen Z, Grubor-Bauk B, Masavuli M, Shrestha A, Ranasinghe C, Bull R, et al. Toward DNA-based T-cell mediated vaccines to target HIV-1 and hepatitis C virus: approaches to elicit localized immunity for protection. Front Cell Infect Microbiol. (2019) 9:91. doi: 10.3389/fcimb.2019.00091
149. Herschhorn A, Sodroski J. An entry-competent intermediate state of the HIV-1 envelope glycoproteins. Receptors Clin Investig. (2017) 4:e1544. doi: 10.14800/rci.1544
150. Derking R, Sanders R. Structure-guided envelope trimer design in HIV-1 vaccine development: a narrative review. J Int AIDS Soc. (2021) 24 Suppl. 7(Suppl. 7):e25797. doi: 10.1002/jia2.25797
151. Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. (2017) 358:85–90. doi: 10.1126/science.aan8630
152. Sok D, Burton D. Recent progress in broadly neutralizing antibodies to HIV. Nat Immunol. (2018) 19:1179–88. doi: 10.1038/s41590-018-0235-7
153. Miner M, Corey L, Montefiori D. Broadly neutralizing monoclonal antibodies for HIV prevention. J Int AIDS Soc. (2021) 24 Suppl. 7(Suppl. 7):e25829. doi: 10.1002/jia2.25829
154. Caillat C, Guilligay D, Sulbaran G, Weissenhorn W. Neutralizing antibodies targeting HIV-1 gp41. Viruses. (2020) 12:1210. doi: 10.3390/v12111210
155. Lee J, Andrabi R, Su C, Yasmeen A, Julien J, Kong L, et al. A broadly neutralizing antibody targets the dynamic HIV envelope trimer apex via a long, rigidified, and anionic β-hairpin structure. Immunity. (2017) 46:690–702. doi: 10.1016/j.immuni.2017.03.017
156. Sok D, Pauthner M, Briney B, Lee J, Saye-Francisco K, Hsueh J, et al. A prominent site of antibody vulnerability on HIV envelope incorporates a motif associated with CCR5 binding and its camouflaging glycans. Immunity. (2016) 45:31–45. doi: 10.1016/j.immuni.2016.06.026
157. Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science (1998) 280:1884–8.
158. Molinos-Albert L, Clotet B, Blanco J, Carrillo J. Immunologic insights on the membrane proximal external region: a major human immunodeficiency virus type-1 vaccine target. Front Immunol. (2017) 8:1154. doi: 10.3389/fimmu.2017.01154
159. Lu G, Shan S, Zainab B, Ayaz Z, He J, Xie Z, et al. Novel vaccine design based on genomics data analysis: a review. Scand J Immunol. (2021) 93:e12986. doi: 10.1111/sji.12986
160. Kandathil A, Ramalingam S, Kannangai R, David S, Sridharan G. Molecular epidemiology of HIV. Indian J Med Res. (2005) 121:333–44.
161. Salazar G, Zhang N, Fu T, An Z. Antibody therapies for the prevention and treatment of viral infections. NPJ Vaccines. (2017) 2:19. doi: 10.1038/s41541-017-0019-3
162. Newell E, Davis M. Beyond model antigens: high-dimensional methods for the analysis of antigen-specific T cells. Nat Biotechnol. (2014) 32:149–57. doi: 10.1038/nbt.2783
163. Zhang S, Ma K, Schonnesen A, Zhang M, He C, Sun E, et al.. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat Biotechnol. (2018). doi: 10.1038/nbt.4282 [Epub ahead of print].
164. Labbé R, Vessillier S, Rafiq Q. Lentiviral vectors for T cell engineering: clinical applications, bioprocessing and future perspectives. Viruses. (2021) 13:1528. doi: 10.3390/v13081528
165. Pennock N, White J, Cross E, Cheney E, Tamburini B, Kedl R. T cell responses: naive to memory and everything in between. Adv Physiol Educ. (2013) 37:273–83. doi: 10.1152/advan.00066.2013
166. Baliu-Piqué M, Verheij M, Drylewicz J, Ravesloot L, de Boer R, Koets A, et al. Short lifespans of memory T-cells in bone marrow, blood, and lymph nodes suggest that T-cell memory is maintained by continuous self-renewal of recirculating cells. Front Immunol. (2018) 9:2054. doi: 10.3389/fimmu.2018.02054
167. Germain R. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. (2002) 2:309–22. doi: 10.1038/nri798
168. Kalos M, Levine B, Porter D, Katz S, Grupp S, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
169. Schmitz J, Kuroda M, Santra S, Sasseville V, Simon M, Lifton M, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. (1999) 283:857–60. doi: 10.1126/science.283.5403.857
170. Okulicz J, Lambotte O. Epidemiology and clinical characteristics of elite controllers. Curr Opin HIV AIDS. (2011) 6:163–8. doi: 10.1097/COH.0b013e328344f35e
171. Loffredo J, Sidney J, Bean A, Beal D, Bardet W, Wahl A, et al. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B*08 and HLA-B*2705, bind peptides with sequence similarity. J Immunol. (2009) 182:7763–75. doi: 10.4049/jimmunol.0900111
172. Sáez-Cirión A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, et al. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci U S A. (2007) 104:6776–81. doi: 10.1073/pnas.0611244104
173. Collins K, Chen B, Kalams S, Walker B, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. (1998) 391:397–401. doi: 10.1038/34929
174. Schwartz O, Maréchal V, Le Gall S, Lemonnier F, Heard J. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. (1996) 2:338–42. doi: 10.1038/nm0396-338
175. Watanabe K, Nishikawa H. Engineering strategies for broad application of TCR-T- and CAR-T-cell therapies. Int Immunol. (2021) 33:551–62. doi: 10.1093/intimm/dxab052
176. Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolyt. (2016) 3:16015. doi: 10.1038/mto.2016.15
177. Tyagarajan S, Spencer T, Smith J. Optimizing CAR-T cell manufacturing processes during pivotal clinical trials. Mol Ther Methods Clin Dev. (2019) 16:136–44. doi: 10.1016/j.omtm.2019.11.018
178. Levine B, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. (2016) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
179. Neelapu S, Locke F, Bartlett N, Lekakis L, Miklos D, Jacobson C, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
180. Mohanty R, Chowdhury C, Arega S, Sen P, Ganguly P, Ganguly N. CAR T cell therapy: a new era for cancer treatment (Review). Oncol Rep. (2019) 42:2183–95. doi: 10.3892/or.2019.7335
181. Zhen A, Carrillo M, Kitchen S. Chimeric antigen receptor engineered stem cells: a novel HIV therapy. Immunotherapy. (2017) 9:401–10. doi: 10.2217/imt-2016-0121
182. Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. (2009) 360:692–8. doi: 10.1056/NEJMoa0802905
183. Gupta R, Abdul-Jawad S, McCoy L, Mok H, Peppa D, Salgado M, et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature. (2019) 568:244–8. doi: 10.1038/s41586-019-1027-4
184. Gupta R, Peppa D, Hill A, Gálvez C, Salgado M, Pace M, et al. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV. (2020) 7:e340–7. doi: 10.1016/S2352-3018(20)30069-2
185. Hütter G, Bodor J, Ledger S, Boyd M, Millington M, Tsie M, et al. CCR5 targeted cell therapy for HIV and prevention of viral escape. Viruses. (2015) 7:4186–203. doi: 10.3390/v7082816
186. Parmentier M. CCR5 and HIV infection, a view from Brussels. Front Immunol. (2015) 6:295. doi: 10.3389/fimmu.2015.00295
187. Barber-Axthelm I, Barber-Axthelm V, Sze K, Zhen A, Suryawanshi G, Chen I, et al. Stem cell-derived CAR T cells traffic to HIV reservoirs in macaques. JCI Insight. (2021) 6:e141502. doi: 10.1172/jci.insight.141502
188. Salgado M, Kwon M, Gálvez C, Badiola J, Nijhuis M, Bandera A, et al. Mechanisms that contribute to a profound reduction of the HIV-1 reservoir after allogeneic stem cell transplant. Ann Intern Med. (2018) 169:674–83. doi: 10.7326/M18-0759
189. Peterson C, Kiem H. Lessons from London and Berlin: designing a scalable gene therapy approach for HIV cure. Cell Stem Cell. (2019) 24:685–7. doi: 10.1016/j.stem.2019.04.010
190. Kordelas L, Verheyen J, Beelen D, Horn P, Heinold A, Kaiser R, et al. Shift of HIV tropism in stem-cell transplantation with CCR5 Delta32 mutation. N Engl J Med. (2014) 371:880–2. doi: 10.1056/NEJMc1405805
191. Mu W, Carrillo M, Kitchen S. Engineering CAR T cells to target the HIV reservoir. Front Cell Infect Microbiol. (2020) 10:410. doi: 10.3389/fcimb.2020.00410
192. Chung A, Navis M, Isitman G, Wren L, Silvers J, Amin J, et al. Activation of NK cells by ADCC antibodies and HIV disease progression. J Acquir Immune Defic Syndr. (2011) 58:127–31. doi: 10.1097/QAI.0b013e31822c62b9
193. Townsend M, Bennion K, Robison R, O’Neill K. Paving the way towards universal treatment with allogenic T cells. Immunol Res. (2020) 68:63–70. doi: 10.1007/s12026-020-09119-7
194. Kuhlmann A, Peterson C, Kiem H. Chimeric antigen receptor T-cell approaches to HIV cure. Curr Opin HIV AIDS. (2018) 13:446–53. doi: 10.1097/COH.0000000000000485
195. Blaeschke F, Stenger D, Kaeuferle T, Willier S, Lotfi R, Kaiser A, et al. Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia. Cancer Immunol Immunother. (2018) 67:1053–66. doi: 10.1007/s00262-018-2155-7
196. Brentjens R, Davila M, Riviere I, Park J, Wang X, Cowell L, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
197. Hurton L, Singh H, Najjar A, Switzer K, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. (2016) 113:E7788–97. doi: 10.1073/pnas.1610544113
198. Maldini C, Gayout K, Leibman R, Dopkin D, Mills J, Shan X, et al. HIV-resistant and HIV-specific CAR-modified CD4+ T cells mitigate HIV disease progression and confer CD4+ T cell help in vivo. Mol Ther. (2020) 28:1585–99. doi: 10.1016/j.ymthe.2020.05.012
199. Bouteloup V, Sabin C, Mocroft A, Gras L, Pantazis N, Le Moing V, et al. Reference curves for CD4 T-cell count response to combination antiretroviral therapy in HIV-1-infected treatment-naïve patients. HIV Med. (2017) 18:33–44. doi: 10.1111/hiv.12389
200. Liu B, Zhang W, Zhang H. Development of CAR-T cells for long-term eradication and surveillance of HIV-1 reservoir. Curr Opin Virol. (2019) 38:21–30. doi: 10.1016/j.coviro.2019.04.004
201. Scholler J, Brady T, Binder-Scholl G, Hwang W, Plesa G, Hege K, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. (2012) 4:132ra53. doi: 10.1126/scitranslmed.3003761
202. Anthony-Gonda K, Bardhi A, Ray A, Flerin N, Li M, Chen W, et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci Transl Med. (2019) 11:eaav5685. doi: 10.1126/scitranslmed.aav5685
203. MacLean A, Walker E, Sahu G, Skowron G, Marx P, von Laer D, et al. A novel real-time CTL assay to measure designer T-cell function against HIV Env(+) cells. J Med Primatol. (2014) 43:341–8. doi: 10.1111/jmp.12137
204. Leslie G, Wang J, Richardson M, Haggarty B, Hua K, Duong J, et al. Potent and broad inhibition of HIV-1 by a peptide from the gp41 heptad repeat-2 domain conjugated to the CXCR4 amino terminus. PLoS Pathog. (2016) 12:e1005983. doi: 10.1371/journal.ppat.1005983
205. Zhen A, Kamata M, Rezek V, Rick J, Levin B, Kasparian S, et al. HIV-specific immunity derived from chimeric antigen receptor-engineered stem cells. Mol Ther. (2015) 23:1358–67. doi: 10.1038/mt.2015.102
206. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall E. The other side of CAR T-cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. (2019) 39:433–44. doi: 10.1200/EDBK_238691
207. Akkina R, Allam A, Balazs A, Blankson J, Burnett J, Casares S, et al. Improvements and limitations of humanized mouse models for HIV research: NIH/NIAID meet the experts 2015 workshop summary. AIDS Res Hum Retroviruses. (2016) 32:109–19. doi: 10.1089/AID.2015.0258
208. Dotti G, Gottschalk S, Savoldo B, Brenner M. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. (2014) 257:107–26. doi: 10.1111/imr.12131
209. Shan L, Deng K, Gao H, Xing S, Capoferri A, Durand C, et al. Transcriptional reprogramming during effector-to-memory transition renders CD4+ T cells permissive for latent HIV-1 infection. Immunity. (2017) 47:766–75.e3. doi: 10.1016/j.immuni.2017.09.014
210. Zhao J, Deng K. Heterogeneity of HIV-1 latent reservoirs. Chin Med J. (2020) 133:2867–73. doi: 10.1097/CM9.0000000000001085
211. Murray A, Kwon K, Farber D, Siliciano R. The latent reservoir for HIV-1: how immunologic memory and clonal expansion contribute to HIV-1 persistence. J Immunol. (2016) 197:407–17. doi: 10.4049/jimmunol.1600343
212. Ahlenstiel C, Symonds G, Kent S, Kelleher A. Block and lock HIV cure strategies to control the latent reservoir. Front Cell Infect Microbiol. (2020) 10:424. doi: 10.3389/fcimb.2020.00424
213. Ait-Ammar A, Kula A, Darcis G, Verdikt R, De Wit S, Gautier V, et al. Current status of latency reversing agents facing the heterogeneity of HIV-1 cellular and tissue reservoirs. Front Microbiol. (2020) 10:3060. doi: 10.3389/fmicb.2019.03060
214. Eisele E, Siliciano R. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. (2012) 37:377–88. doi: 10.1016/j.immuni.2012.08.010
215. Fletcher C, Staskus K, Wietgrefe S, Rothenberger M, Reilly C, Chipman J, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A. (2014) 111:2307–12. doi: 10.1073/pnas.1318249111
216. Vanhamel J, Bruggemans A, Debyser Z. Establishment of latent HIV-1 reservoirs: what do we really know? J Virus Erad. (2019) 5:3–9.
217. Rutsaert S, Bosman K, Trypsteen W, Nijhuis M, Vandekerckhove L. Digital PCR as a tool to measure HIV persistence. Retrovirology. (2018) 15:16. doi: 10.1186/s12977-018-0399-0
218. Strain M, Lada S, Luong T, Rought S, Gianella S, Terry V, et al. Highly precise measurement of HIV DNA by droplet digital PCR. PLoS One. (2013) 8:e55943. doi: 10.1371/journal.pone.0055943
219. Levy C, Hughes S, Roychoudhury P, Reeves D, Amstuz C, Zhu H, et al. A highly multiplexed droplet digital PCR assay to measure the intact HIV-1 proviral reservoir. Cell Rep Med. (2021) 2:100243. doi: 10.1016/j.xcrm.2021.100243
220. Cohn L, Chomont N, Deeks S. The biology of the HIV-1 latent reservoir and implications for cure strategies. Cell Host Microbe. (2020) 27:519–30. doi: 10.1016/j.chom.2020.03.014
221. Marsden M, Zack J. Establishment and maintenance of HIV latency: model systems and opportunities for intervention. Future Virol. (2010) 5:97–109. doi: 10.2217/fvl.09.70
222. Pierson T, McArthur J, Siliciano R. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol. (2000) 18:665–708. doi: 10.1146/annurev.immunol.18.1.665
223. Chun T, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano R. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med. (1995) 1:1284–90. doi: 10.1038/nm1295-1284
224. Finzi D, Hermankova M, Pierson T, Carruth L, Buck C, Chaisson R, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. (1997) 278:1295–300. doi: 10.1126/science.278.5341.1295
225. Cohen M, Shaw G, McMichael A, Haynes B. Acute HIV-1 infection. N Engl J Med. (2011) 364:1943–54. doi: 10.1056/NEJMra1011874
226. Wang G, Ciuffi A, Leipzig J, Berry C, Bushman F. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. (2007) 17:1186–94. doi: 10.1101/gr.6286907
227. Cohn L, Silva I, Oliveira T, Rosales R, Parrish E, Learn G, et al. HIV-1 integration landscape during latent and active infection. Cell. (2015) 160:420–32. doi: 10.1016/j.cell.2015.01.020
228. Khanal S, Schank M, El Gazzar M, Moorman J, Yao Z. HIV-1 latency and viral reservoirs: existing reversal approaches and potential technologies targets, and pathways involved in HIV latency studies. Cells. (2021) 10:475. doi: 10.3390/cells10020475
229. Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology. (2013) 10:67. doi: 10.1186/1742-4690-10-67
230. Bradley T, Ferrari G, Haynes B, Margolis D, Browne E. Single-cell analysis of quiescent HIV infection reveals host transcriptional profiles that regulate proviral latency. Cell Rep. (2018) 25:107–17.e3. doi: 10.1016/j.celrep.2018.09.020
231. Debyser Z, Christ F, De Rijck J, Gijsbers R. Host factors for retroviral integration site selection. Trends Biochem Sci. (2015) 40:108–16. doi: 10.1016/j.tibs.2014.12.001
232. Weinberger L, Burnett J, Toettcher J, Arkin A, Schaffer D. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. (2005) 122:169–82. doi: 10.1016/j.cell.2005.06.006
233. Weinberger A, Weinberger L. Stochastic fate selection in HIV-infected patients. Cell. (2013) 155:497–9. doi: 10.1016/j.cell.2013.09.039
234. Christ F, Voet A, Marchand A, Nicolet S, Desimmie B, Marchand D, et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol. (2010) 6:442–8. doi: 10.1038/nchembio.370
235. Bisgrove D, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A. (2007) 104:13690–5. doi: 10.1073/pnas.0705053104
236. Besnard E, Hakre S, Kampmann M, Lim H, Hosmane N, Martin A, et al. The mTOR complex controls HIV latency. Cell Host Microbe. (2016) 20:785–97. doi: 10.1016/j.chom.2016.11.001
237. Vansant G, Bruggemans A, Janssens J, Debyser Z. Block-and-lock strategies to cure HIV infection. Viruses. (2020) 12:84. doi: 10.3390/v12010084
238. Mousseau G, Aneja R, Clementz M, Mediouni S, Lima N, Haregot A, et al. Resistance to the tat inhibitor didehydro-cortistatin A is mediated by heightened basal HIV-1 transcription. mBio (2019) 10:e1750–1718. doi: 10.1128/mBio.01750-18
239. Jonlin E. Informed consent for human embryo genome editing. Stem Cell Rep. (2020) 14:530–7. doi: 10.1016/j.stemcr.2020.03.010
240. Gutierrez-Guerrero A, Abrey Recalde M, Mangeot P, Costa C, Bernadin O, Périan S, et al. Baboon envelope pseudotyped nanoblades carrying Cas9/gRNA complexes allow efficient genome editing in human T, B, and CD34+ cells and knock-in of AAV6-encoded donor DNA in CD34+ Cells. Front Genome Ed. (2021) 3:604371. doi: 10.3389/fgeed.2021.604371
241. Macedo A, Novis C, De Assis C, Sorensen E, Moszczynski P, Huang S, et al. Dual TLR2 and TLR7 agonists as HIV latency-reversing agents. JCI Insight. (2018) 3:e122673. doi: 10.1172/jci.insight.122673
242. Elsheikh M, Tang Y, Li D, Jiang G. Deep latency: a new insight into a functional HIV cure. Ebiomedicine. (2019) 45:624–9. doi: 10.1016/j.ebiom.2019.06.020
243. Pache L, Dutra M, Spivak A, Marlett J, Murry J, Hwang Y, et al. BIRC2/cIAP1 is a negative regulator of HIV-1 transcription and can be targeted by smac mimetics to promote reversal of viral latency. Cell Host Microbe. (2015) 18:345–53. doi: 10.1016/j.chom.2015.08.009
244. Del Prete G, Alvord W, Li Y, Deleage C, Nag M, Oswald K, et al. TLR7 agonist administration to SIV-infected macaques receiving early initiated cART does not induce plasma viremia. JCI Insight. (2019) 4:e127717. doi: 10.1172/jci.insight.127717
245. Lim S, Osuna C, Hraber P, Hesselgesser J, Gerold J, Barnes T, et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci Transl Med. (2018) 10:eaao4521. doi: 10.1126/scitranslmed.aao4521
246. Sadowski I, Hashemi F. Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs. Cell Mol Life Sci. (2019) 76:3583–600. doi: 10.1007/s00018-019-03156-8
247. Van der Sluis R, Kumar N, Pascoe R, Zerbato J, Evans V, Dantanarayana A, et al. Combination immune checkpoint blockade to reverse HIV latency. J Immunol. (2020) 204:1242–54. doi: 10.4049/jimmunol.1901191
249. Hurst J, Hoffmann M, Pace M, Williams J, Thornhill J, Hamlyn E, et al. Immunological biomarkers predict HIV-1 viral rebound after treatment interruption. Nat Commun. (2015) 6:8495. doi: 10.1038/ncomms9495
250. Castelli V, Lombardi A, Palomba E, Bozzi G, Ungaro R, Alagna L, et al. Immune checkpoint inhibitors in people living with HIV/AIDS: facts and controversies. Cells. (2021) 10:2227. doi: 10.3390/cells10092227
251. Gubser C, Chiu C, Lewin S, Rasmussen T. Immune checkpoint blockade in HIV. Ebiomedicine. (2022) 76:103840. doi: 10.1016/j.ebiom.2022.103840
252. Porichis F, Hart M, Massa A, Everett H, Morou A, Richard J, et al. Immune checkpoint blockade restores HIV-specific CD4 T cell help for NK cells. J Immunol. (2018) 201:971–81. doi: 10.4049/jimmunol.1701551
253. Suzuki K, Shijuuku T, Fukamachi T, Zaunders J, Guillemin G, Cooper D, et al. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J RNAi Gene Silencing. (2005) 1:66–78.
254. Suzuki K, Ahlenstiel C, Marks K, Kelleher A. Promoter targeting RNAs: unexpected contributors to the control of HIV-1 transcription. Mol Ther Nucleic Acids. (2015) 4:e222. doi: 10.1038/mtna.2014.67
255. Méndez C, Ledger S, Petoumenos K, Ahlenstiel C, Kelleher A. RNA-induced epigenetic silencing inhibits HIV-1 reactivation from latency. Retrovirology. (2018) 15:67. doi: 10.1186/s12977-018-0451-0
256. Zapata J, Campilongo F, Barclay R, DeMarino C, Iglesias-Ussel M, Kashanchi F, et al. The human immunodeficiency virus 1 ASP RNA promotes viral latency by recruiting the Polycomb Repressor Complex 2 and promoting nucleosome assembly. Virology. (2017) 506:34–44. doi: 10.1016/j.virol.2017.03.002
257. Chao T, Zhang Q, Li Z, Tiwari S, Qin Y, Yau E, et al. The long noncoding RNA HEAL regulates HIV-1 replication through epigenetic regulation of the HIV-1 promoter. mBio (2019) 10:e2016–9. doi: 10.1128/mBio.02016-19
258. Qu D, Sun W, Li L, Ma L, Sun L, Jin X, et al. Long noncoding RNA MALAT1 releases epigenetic silencing of HIV-1 replication by displacing the polycomb repressive complex 2 from binding to the LTR promoter. Nucleic Acids Res. (2019) 47:3013–27. doi: 10.1093/nar/gkz117
259. Boliar S, Russell D. Lnc(ing)RNAs to the shock and kill strategy for HIV-1 cure. Mol Ther Nucleic Acids. (2021) 23:1272–80. doi: 10.1016/j.omtn.2021.02.004
260. McBrien J, Mavigner M, Franchitti L, Smith S, White E, Tharp G, et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature. (2020) 578:154–9. doi: 10.1038/s41586-020-1946-0
261. Sengupta S, Siliciano R. Targeting the latent reservoir for HIV-1. Immunity. (2018) 48:872–95. doi: 10.1016/j.immuni.2018.04.030
262. Liu Y, Schopman N, Berkhout B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. (2013) 41:3723–33. doi: 10.1093/nar/gkt036
263. Grimm D, Wang L, Lee J, Schürmann N, Gu S, Börner K, et al. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J Clin Invest. (2010) 120:3106–19. doi: 10.1172/JCI43565
264. Indikova I, Indik S. Highly efficient ‘hit-and-run’ genome editing with unconcentrated lentivectors carrying Vpr.Prot.Cas9 protein produced from RRE-containing transcripts. Nucleic Acids Res. (2020) 48:8178–87. doi: 10.1093/nar/gkaa561
265. Dash P, Kaminski R, Bella R, Su H, Mathews S, Ahooyi T, et al. Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat Commun. (2019) 10:2753. doi: 10.1038/s41467-019-10366-y
266. Abner E, Jordan A. HIV “shock and kill” therapy: in need of revision. Antiviral Res. (2019) 166:19–34.
267. Horodecka K, Düchler M. CRISPR/Cas9: principle, applications, and delivery through extracellular vesicles. Int J Mol Sci. (2021) 22:6072. doi: 10.3390/ijms22116072
268. Sung Y, Kim S. Recent advances in the development of gene delivery systems. Biomater Res. (2019) 23:8. doi: 10.1186/s40824-019-0156-z
269. Dinca A, Chien W, Chin M. Intracellular delivery of proteins with cell-penetrating peptides for therapeutic uses in human disease. Int J Mol Sci. (2016) 17:263. doi: 10.3390/ijms17020263
270. Teow SY. Cell-Penetrating Antibodies for Targeting HIV-1 p24 Capsid Protein. Ph.D. Thesis. Penang: Universiti Sains Malaysia (2015). p. 281–329.
271. Muller S, Zhao Y, Brown T, Morgan A, Kohler H. TransMabs: cell-penetrating antibodies, the next generation. Expert Opin Biol Ther. (2005) 5:237–41. doi: 10.1517/14712598.5.2.237
272. Kumar R, Qureshi H, Deshpande S, Bhattacharya J. Broadly neutralizing antibodies in HIV-1 treatment and prevention. Ther Adv Vaccines Immunother. (2018) 6:61–8. doi: 10.1177/2515135518800689
273. Liu L, Patel B, Ghanem M, Bundoc V, Zheng Z, Morgan R, et al. Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J Virol. (2015) 89:6685–94. doi: 10.1128/JVI.00474-15
274. Ghanem M, Bolivar-Wagers S, Dey B, Hajduczki A, Vargas-Inchaustegui D, Danielson D, et al. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy. (2018) 20:407–19. doi: 10.1016/j.jcyt.2017.11.001
275. Zhen A, Carrillo M, Mu W, Rezek V, Martin H, Hamid P, et al. Robust CAR-T memory formation and function via hematopoietic stem cell delivery. PLoS Pathog. (2021) 17:e1009404. doi: 10.1371/journal.ppat.1009404
276. Wu C, Roybal K, Puchner E, Onuffer J, Lim W. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. (2015) 350:aab4077. doi: 10.1126/science.aab4077
277. Bouchat S, Delacourt N, Kula A, Darcis G, Van Driessche B, Corazza F, et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol Med. (2016) 8:117–38. doi: 10.15252/emmm.201505557
278. Spivak A, Planelles V. Novel latency reversal agents for HIV-1 cure. Annu Rev Med. (2018) 69:421–36. doi: 10.1146/annurev-med-052716-031710
279. Pollard R, Rockstroh J, Pantaleo G, Asmuth D, Peters B, Lazzarin A, et al. Safety and efficacy of the peptide-based therapeutic vaccine for HIV-1, Vacc-4x: a phase 2 randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. (2014) 14:291–300. doi: 10.1016/S1473-3099(13)70343-8
280. Zhang Z, Hou W, Chen S. Updates on CRISPR-based gene editing in HIV-1/AIDS therapy. Virol Sin. (2022) 37:1–10. doi: 10.1016/j.virs.2022.01.017
Keywords: HIV-1, AIDS, cART, CCR5, CRISPR/Cas9 findings number
Citation: Sorokina A, Anchakova E and Dashinimaev E (2023) Strategies for HIV-1 suppression through key genes and cell therapy. Front. Med. 10:1259995. doi: 10.3389/fmed.2023.1259995
Received: 18 July 2023; Accepted: 06 November 2023;
Published: 29 November 2023.
Edited by:
Ian James Martins, University of Western Australia, AustraliaReviewed by:
Claudio Mussolino, University of Freiburg Medical Center, GermanyGee Jun Tye, University of Science Malaysia (USM), Malaysia
Copyright © 2023 Sorokina, Anchakova and Dashinimaev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erdem Dashinimaev, ZGFzaGluaW1hZXZAZ21haWwuY29t