 Marta Palomo1,2
Marta Palomo1,2 Ana Belén Moreno-Castaño1María Queralt Salas3
Ana Belén Moreno-Castaño1María Queralt Salas3 Silvia Escribano-Serrat1
Silvia Escribano-Serrat1 Montserrat Rovira3
Montserrat Rovira3 Elena Guillen-Olmos4
Elena Guillen-Olmos4 Sara Fernandez5
Sara Fernandez5 Helena Ventosa-Capell5Lina Youssef6,7Fatima Crispi6,8Meritxell Nomdedeu9
Helena Ventosa-Capell5Lina Youssef6,7Fatima Crispi6,8Meritxell Nomdedeu9 Julia Martinez-Sanchez1
Julia Martinez-Sanchez1 Blanca De Moner1,7
Blanca De Moner1,7 Maribel Diaz-Ricart1*
Maribel Diaz-Ricart1*- 1Hemostasis and Erythropathology Laboratory, Centre de Diagnòstic Biomèdic, Hospital Clínic de Barcelona, Institut de Recerca August Pi Sunyer, University of Barcelona, Barcelona, Spain
- 2Hematology External Quality Assessment Laboratory, Centre de Diagnòstic Biomèdic, Hospital Clínic de Barcelona, Barcelona, Spain
- 3Hematopoietic Stem Cell Transplantation Unit, Hematology Department, Institute of Cancer and Blood Diseases, Hospital Clínic de Barcelona, Institut de Recerca August Pi Sunyer, Barcelona, Spain
- 4Department of Nephrology and Kidney Transplantation, Hospital Clínic de Barcelona, Centro de Referencia en Enfermedad Glomerular Compleja del Sistema Nacional de Salud (CSUR), University of Barcelona, Barcelona, Spain
- 5Medical Intensive Care Unit, Hospital Clínic de Barcelona, Barcelona, Spain
- 6BCNatal – Barcelona Center for Maternal Fetal and Neonatal Medicine, Hospital Clínic de Barcelona and Hospital Sant Joan de Déu, Institut de Recerca August Pi Sunyer, University of Barcelona, Barcelona, Spain
- 7Josep Carreras Leukaemia Research Institute, Hospital Clinic, University of Barcelona, Barcelona, Spain
- 8Centre for Biomedical Research on Rare Diseases (CIBER-ER), Madrid, Spain
- 9Hemostasis and Hemotherapy Department, Institute of Cancer and Blood Diseases, Hospital Clínic de Barcelona, Barcelona, Spain
The endothelium is a biologically active interface with multiple functions, some of them common throughout the vascular tree, and others that depend on its anatomical location. Endothelial cells are continually exposed to cellular and humoral factors, and to all those elements (biological, chemical, or hemodynamic) that circulate in blood at a certain time. It can adapt to different stimuli but this capability may be lost if the stimuli are strong enough and/or persistent in time. If the endothelium loses its adaptability it may become dysfunctional, becoming a potential real danger to the host. Endothelial dysfunction is present in multiple clinical conditions, such as chronic kidney disease, obesity, major depression, pregnancy-related complications, septic syndromes, COVID-19, and thrombotic microangiopathies, among other pathologies, but also in association with cell therapies, such as hematopoietic stem cell transplantation and treatment with chimeric antigen receptor T cells. In these diverse conditions, evidence suggests that the presence and severity of endothelial dysfunction correlate with the severity of the associated disease. More importantly, endothelial dysfunction has a strong diagnostic and prognostic value for the development of critical complications that, although may differ according to the underlying disease, have a vascular background in common. Our multidisciplinary team of women has devoted many years to exploring the role of the endothelium in association with the mentioned diseases and conditions. Our research group has characterized some of the mechanisms and also proposed biomarkers of endothelial damage. A better knowledge would provide therapeutic strategies either to prevent or to treat endothelial dysfunction.
1. Introduction
At the conference on “Women and Science” organized by the European Parliament and Commission in April 1998 in Brussels, both institutions agreed in a formal statement on the “need to identify efforts to increase the presence of women in research in Europe.” Actually, as the scientific career advances, the proportion of women decreases (1). In our case, we are proud to constitute a group of multidisciplinary scientific women who address the same exciting topic: endothelial activation and damage in association with different pathologies and as a consequence of certain therapies.
For years, the vascular endothelium was considered an inert barrier, but, nowadays, it plays a fundamental role in human health and disease. This biologically active interface is constituted by 1 to 6 × 1013 endothelial cells (ECs) in an adult human being and covers a large surface area (between 4,000 and 7,000 m2) (2–4). The endothelium has a huge range of functions (5), including the maintenance of the vascular homeostatic balance, modulation of the vascular tone, participation in the recruitment of immune cells, and the generation of new blood vessels, among others.
All these functions are differentially regulated in space and time, showing the heterogeneity of the ECs in different organs in terms of morphology, structure, and barrier function (6, 7). The pulmonary endothelium is localized at a crucial interface and it is formed by an heterogenous cell monolayer that acts as a selective barrier between blood, airways, and lung parenchyma (8). Blood vessel endothelium crosses every tissue, exhibiting unique structural and functional properties in each vascular bed. As a result of organ-specific requirements, the vascular system varies in its organization and specifically in cell-to-cell junctions, which are crucial in the integrity of blood vessels, depending on the anatomical site. While tight junctions are well organized in arteries and brain microvessels, they are more unstructured in veins, capillaries, and organs where a higher rate of exchange is needed (9). Due to their location, the endothelium is directly exposed to all physiological and pathological stimuli (10, 11). These cells are able to adapt to a wide range of environmental conditions, however, noxious stimulus induce local or systemic endothelial activation (12).
Endothelial cell activation and damage imply a range of phenotypic changes in the endothelium and differ according to many physiological variables. If the activation stimuli are persistent in time and/or intense enough, the endothelium may become dysfunctional causing abnormal functional and structural changes, being the main consequence of the loss of vascular integrity with the detachment of ECs exposing a more thrombogenic extracellular matrix (ECM) (13). The relevance of endotheliopathy in the progression of several diseases has been increasingly accepted in the scientific community. The present review aims to summarize the complexity of this process in a range of pathological situations that share endothelial damage (ED) as a common feature.
Historically, our team initiated the research on endothelial activation and damage in the context of end-stage chronic kidney disease (CKD). Further collaboration with other teams in the Hospital Clinic and Institut de Recerca August Pi Sunyer helped us to expand the investigation into other pathologies with associated cardiovascular risk, such as obesity and major depression. A joint venture with obstetricians followed to explore the role of the endothelium in pregnancy-related pathologies, such as preeclampsia (PE), in which the dysregulation of the complement system plays a crucial role. Moreover, a tight partnership with the intensive care unit (ICU) prompted us to explore the ED in septic syndromes and how the severity of the disease could have a gradual impact on the endothelium. Then, with the global outbreak of the coronavirus SARS-CoV-2 that caused the COVID-19 pandemic starting in 2020, our efforts were focused on characterizing the associated endotheliopathy. Furthermore, in collaboration with the hematopoietic stem cell transplantation (HCT) unit at the Hospital Clínic, we were one of the first groups to demonstrate the role of the ED as a pathological substrate for the complications appearing in early post-transplantation. We are now progressing in the research of the role of the endothelium in the development of severe complications that may compromise the promising curative potential of new therapies, such as the use of chimeric antigen receptor T (CAR-T) cells. The investigation performed during these years to explore the mechanisms involved has already provided us with diagnostic and prognostic biomarkers and potential therapeutic targets. Future research should still generate additional tools focused on protecting the endothelium (Figures 1, 2).
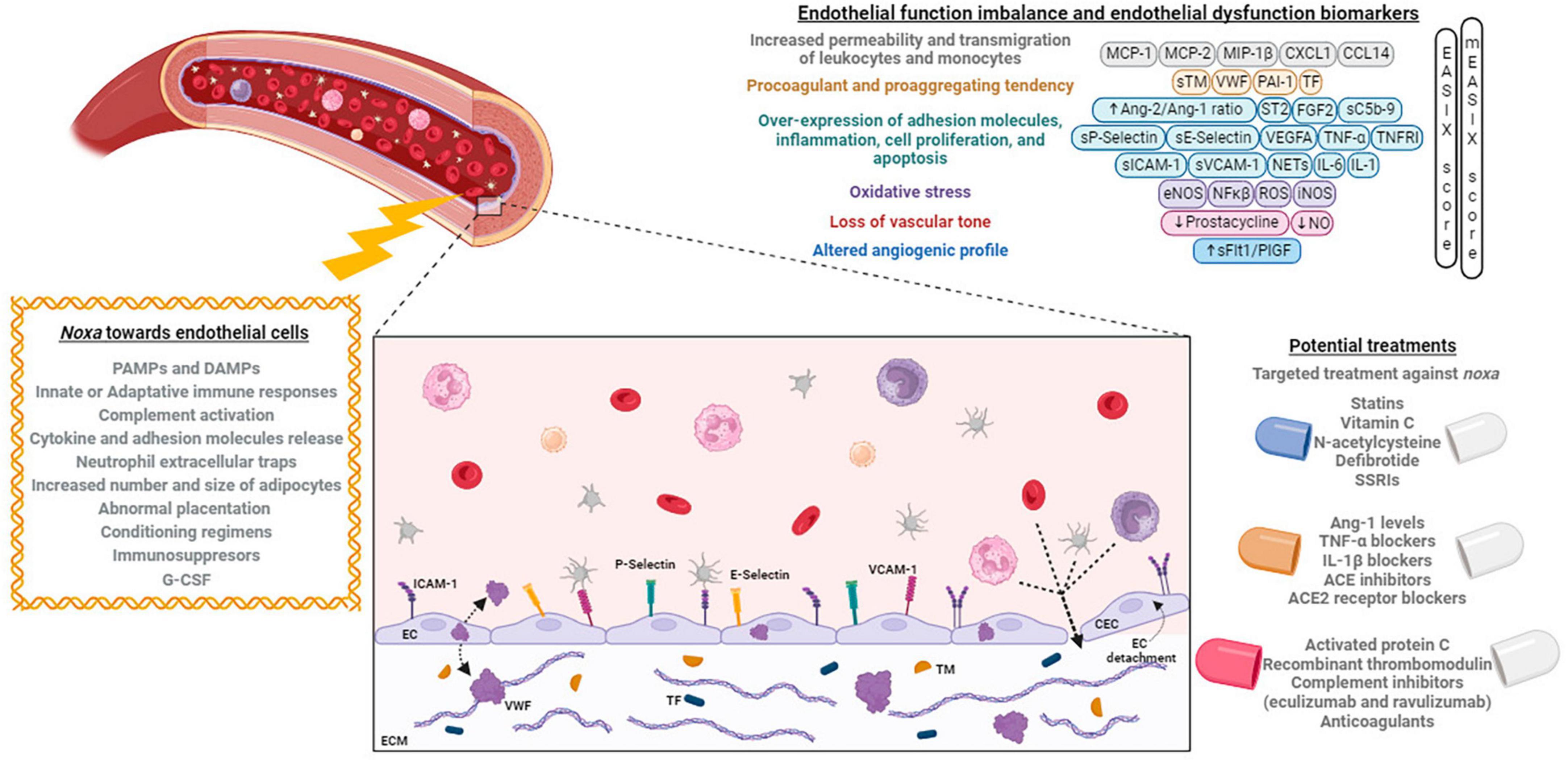
Figure 1. Noxa, mechanisms, and biomarkers involved in endothelial dysfunction, and its potential treatment. Different factors alter the endothelial cells phenotype causing an imbalance that can be identified by the expression of different biomarkers. At right, the principal developed treatments targeting the endothelium are exposed. Created with BioRender (available from https://www.biorender.com).
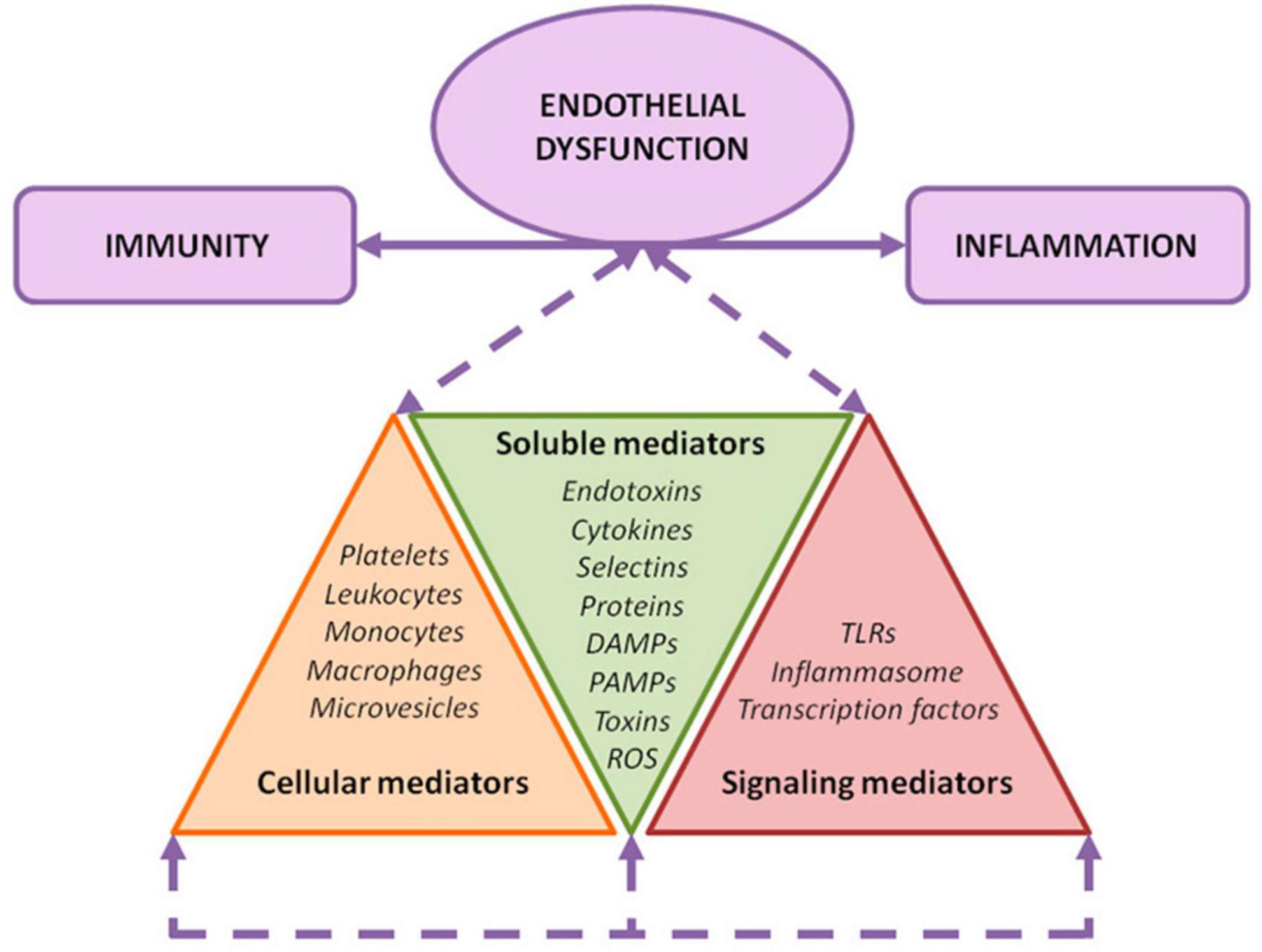
Figure 2. Endothelial damage mechanisms. Exposure of the endothelium to different noxa leads to a convergence among inflammation, immunity, and endothelial activation. As depicted, several soluble, cellular, and signaling mediators are involved in this orchestrated response.
2. Endothelial activation and damage in end-stage chronic kidney disease
Chronic kidney disease is a major public health issue with an increasing prevalence. It is associated with poorer quality of life, reduced life expectancy, and, therefore, high rates of morbidity and mortality, and increased hospitalization costs (14, 15). An increment in cardiovascular diseases has been highlighted as the main cause of the increased morbidity and mortality in this population; however, it cannot be fully explained by the presence of traditional cardiovascular risk factors. Endothelial activation and damage in CKD patients have been described as related to sustained toxic, oxidative stress, and inflammatory conditions. Endothelial dysfunction has been proposed as a pathophysiological substrate for accelerated atherothrombosis, hemostasis alterations, inflammatory activity, and impaired immune response in these patients (16, 17).
Endothelial activation in CKD may be attributed to different factors: the pulsatile blood flow and disturbed shear stress (18), the presence of uremic toxins, such as indoxyl sulfate, and the production and release of oxidative stress and inflammation related products to the circulation (19). All these elements constitute the uremic environment, and could be classified into three major categories of mediators: (i) soluble, (ii) cellular, and (iii) signal transduction mediators (19).
Innate immune system alterations have been also reported in association with end-stage renal disease, aggravated by dialysis procedures. In addition, CKD is related to gut dysbiosis, with a significant loss of the gut microbiota diversity due to the uremic condition, dietary restrictions, administered drugs (antibiotics, phosphate binders, and oral iron supplementation), and hypervolemia, leading to intestinal wall congestion and edema (20, 21). In addition to the impaired renal function, there is also a reduced function of the intestinal barrier with increased permeability to different size molecules. All together enrich the toxic milieu in uremia
There is in vivo and in vitro evidence demonstrates endothelial activation and damage in association with CKD, causing impaired endothelium-dependent vasodilatation and increased plasma levels of circulating cell adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin (22–24). Also, other ED-derived proteins, such as monocyte chemoattractant protein-1 (MCP-1) (25), angiopoietin-2 (Ang-2) (26), tissue factor (TF) (27–29), and total von Willebrand factor (VWF) (30, 31) are elevated in circulation. Endothelial activation is considered an early trigger for atherosclerosis and, therefore, in the setting of CKD may explain the increased cardiovascular risk in this population, beyond traditional cardiovascular risk factors (32).
Our research group has carefully studied the ED in uremia by exposing ECs to culture media supplemented with sera from patients on renal replacement therapy. ECs exhibited alterations in their morphology, with accelerated proliferation (33). They also showed signs of inflammation, expressing VCAM-1, ICAM-1 on their surface, with activation of the intracellular protein p38MAPK (34). The ECM produced by these ECs was characterized by a less intricate network of fibrils (27) and an increased thrombogenicity, mainly due to a higher expression of TF (27), VWF (34), and thrombomodulin (TM). No changes in ADAMTS-13 activity, the VWF metalloprotease, were detected in patients’ plasma (35).
The analysis of the proteome of ECs grown under uremic conditions versus control (36) provided information on the differentially expressed proteins. Also, antioxidant enzymes, such as glutathione peroxidase, superoxide dismutase, and peroxiredoxin, were detected to be increased, suggesting an adaptive response to the oxidative stress induced by uremic media. Most of the proteins found to be unregulated in the uremic ECs are related to nuclear factor kappa B (NF-κB) (35). This protein is key in mediating inflammatory and immunological responses and oxidative stress. In addition, elements participating in innate immunity, such as Toll-like receptor 4 and the inflammasome nucleotide-binding oligomerization domain-like receptor prying domain-containing-3 (NLRP3, also known as NALP3) were also upregulated in ECs exposed to the uremic milieu (37). ED can also develop into apoptotic changes (38, 39).
Therefore, endothelial activation is associated with inflammation, oxidative stress, and alterations of immune mechanisms in CKD patients. Therapies focused on protecting the endothelium at different levels could diminish the accelerated cardiovascular risk in this population.
3. The endothelium in obesity
Obesity is a chronic systemic metainflammation. Is associated with oxidative stress, endothelial dysfunction, and vasculopathy, and, therefore, constitutes an important risk factor for atherothrombotic cardiovascular morbidity and mortality (40, 41). In addition, obesity is related to dyslipidemia, hypertension, insulin resistance, and diabetes mellitus, which are conditions that also have a deleterious impact on the endothelium.
The increase in the number and size of adipocytes appears to be the initial event of adipose tissue dysfunction, resulting in hypoxia and defects in the lipids accumulation, together with infiltration of inflammatory macrophages, the switch of adipose tissue-resident macrophages to a proinflammatory phenotype, and conversion of preadipocytes to macrophages. Activation of non-adipocyte stromal cells and secretion of factors from the adipose tissue lead to an increased presence of chemokines and cytokines in plasma, which may participate in the development of chronic inflammation, angiogenesis, and atherothrombotic changes (42–46). There is evidence of the secretion of cytoadipokines from different adipose tissue depots (42, 47–49).
In studies performed by our group, cultured ECs were exposed to the secretome of adipose tissue from visceral and subcutaneous locations of obese and non-obese individuals (50). The cytokines secreted by the adipose tissue of obese subjects caused an adverse effect on the cultured ECs (50), characterized by increased proliferation, morphology alteration, higher expression of VCAM-1, ICAM-1, and VWF, and production of a more thrombogenic ECM. The visceral secretomes induced the strongest expression of these markers, which occurred through NF-κB activation in ECs, together with an increased presence of proinflammatory cytokines (interleukin-6, IL-6), and neutrophil and monocyte chemo-attractants (MCP-1, MCP-2, MIP-1β, CXCL1, and CCL14), in the secretomes from obese adipose tissue (42–47). All these alterations could be involved in vascular and systemic aging (51, 52). Our findings are in agreement with observations by several groups of a better cardiovascular risk profile for those obese individuals with low levels of visceral adipose tissue, known as “healthy obesity.”
We believe that excessive fat accumulation causes activation of the stromal cell fraction, altering the adipose tissue secretion pattern. The synergic proinflammatory and prothrombotic profiles of the obese secretome are responsible for the systemic macrovascular endothelial activation observed in obesity.
4. Major depression and cardiovascular risk: role of the endothelium
Major depression and cardiovascular disease are two comorbid conditions highly prevalent that constitute an important health concern in developed countries Signs of ED have been demonstrated in major depression (53, 54). In this regard, inflammation and ED are mechanisms potentially connecting depression to cardiovascular disease (55–57).
Our group was able to demonstrate significant elevation in circulation of different biomarkers of ED, such as circulating endothelial cell (CEC), VWF, and soluble VCAM-1, in patients with the diagnose of major depression (58). Moreover, treatment with the selective serotonin reuptake inhibitor (SSRI) escitalopram exhibited a protective role since biomarker levels decreased substantially in a gradual manner. In addition, when cultured ECs were exposed to the sera from these patients, these findings were reproducible. ECs exhibited signs of inflammation, oxidative stress, and increased thrombogenicity of the ECM generated, which were inhibited significantly by the presence of escitalopram in vitro (58).
There is strong evidence on the role of serotonin in the cardiovascular system. Platelets, the main carriers of serotonin (59), play a key role in the development of cardiovascular events. In experimental studies performed by our group, exogenous addition of serotonin to blood samples potentiated platelet functions, increasing their procoagulant behavior, and enhancing thrombus formation on damaged vascular surfaces, effects that were inhibited by the presence of SSRIs (60, 61). In addition, in experiments performed with blood samples from patients with major depression, a pronounced procoagulant profile, with increased platelet thrombus and fibrin formation, was observed at the moment of diagnosis and normalized after 24 weeks of treatment with escitalopram (62). Altogether, our results suggest that both platelets and endothelium are two key hemostatic components, whose responses may be altered and may be acting synergistically in major depression (63).
5. Endothelial damage in pregnancy-associated complications
Preeclampsia is a life-threatening pregnancy-associated disorder that affects 2–8% of pregnancies (64). It is defined as new-onset hypertension with other signs of endothelial systemic damage, accompanied by signs of end-organ damage, such as proteinuria, acute kidney failure, liver dysfunction, hemolysis or thrombocytopenia, occurring after 20 weeks of gestation (65). PE is a heterogenous disorder, with a large variability in its associated risk factors and clinical presentation (66). The exact pathophysiology remains unclear despite exhaustive investigation (67). However, the most accepted hypothesis is known as the two-stage model claiming that complications originate during abnormal placentation at the beginning of pregnancy, followed by generalized inflammation and progressive maternal ED (68). Resulting placental insufficiency and overt clinical signs of PE do not manifest usually until the last pregnancy trimester (69) with a possibly rapid and unexpected progression from mild to severe PE (70). Unfortunately, there is no effective treatment and delivery if the only available intervention (71).
Endothelial dysfunction has been accepted as one of the key mechanisms in PE development (72). In normal pregnancy, the uterine vasculature undergoes significant adaptations to ensure proper blood supply to the developing fetus (73). These adaptations mainly involve increased vasodilation and decreased vasoconstriction, allowing for higher blood flow to the uterus (73). However, these adaptations are disrupted in PE, leading to vasoconstriction and inadequate blood supply to the placenta and fetus (74).
The dysfunctional endothelium promotes the production and release of pro-inflammatory cytokines (75), such as tumor necrosis factor-alpha (TNF-α), interleukin 6 and 1b (IL-6 and IL-1b), and adhesion molecules (VCAM-1 and ICAM-1) (76). The cytokines activate immune cells, causing excessive inflammatory responses (77), and the adhesion molecules promote the breakdown of EC-cell contacts (78). Prolonged ECs activation results in a cycle of inflammation and oxidative stress. This inflammatory environment contributes to the extensive ED and organ dysfunction seen in PE, including kidney, liver, and brain involvement (79). Furthermore, this ED is associated with a dysregulation of the complement system (80). The maternal innate immune system is crucial throughout pregnancy, providing protection against pathogens while inducing tolerance to semi-allogeneic fetal and placental development (81). Dysregulation of the maternal immune system during PE leads to overstimulation of the complement system as a compensatory mechanism (82), with recruitment of phagocytic cells and neutrophils to the site of stimulation (83). This phenomenon manifests with elevated plasma C5b9 in PE mothers and C5b9 deposits on ECs (76, 83).
In addition, there is imbalance in the coagulation system (80). Damaged endothelium does not produce sufficient anticoagulant factors, such as tissue factor pathway inhibitor (TFPI), and TM (84), in association with increased VWF (85). This prothrombotic state increases the risk of thrombosis and microvascular fibrin deposition, further impairing placental blood flow (86) and contributing to the development of maternal organ dysfunction (87).
Angiogenic factors have emerged as the most specific biomarkers of PE ever described and have recently been incorporated as essential components in the prediction, diagnosis and prognosis of PE (88). A proper angiogenic balance during pregnancy is critical for adequate development of fetus and placenta together with appropriate maternal cardiovascular adaptation to pregnancy (89). Indeed, angiogenic factors are essential not only for new vessel formation but also to keep the maternal endothelium healthy by promoting vasorelaxation, adequate permeability and cell survival (90). In PE, placental inflammation and increased oxidative stress cause release of larger amounts of sFlt1 over PIGF (91), with an antiangiogenic profile reflecting placental malfunctioning and maternal endothelial dysfunction (92).
In conclusion, endothelial dysfunction is key in the pathogenesis of PE. Impaired NO production, increased vasoconstriction, inflammation, innate immunity dysregulation, and coagulation and angiogenic imbalance contribute to the hypertension, poor placental perfusion, and multiple organ damage occurring in this condition. Other pathways, like altered lipid metabolism (93), mitochondrial dysfunction (94), and maternal response to circulating trophoblast-derived extracellular vesicles (95) may be also involved. Furthermore, endothelial dysfunction seems to act as a cardiometabolic stressor that may culminate in long-term cardiovascular complications in women who developed PE during pregnancy (96). Further elucidation of the molecular mechanisms involved is critical for the development of potential therapeutic strategies aim at preventing or reducing the adverse consequences associated with this syndrome.
6. Endothelial alterations in septic syndromes
Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection (97). The endothelium orchestrates a beneficial local host response to infection by regulating the vasomotor tone, leukocyte trafficking, vascular permeability and hemostasis. However, when the response is overproduced, a systemic and untargeted dysregulated inflammatory response leads to endothelial hyperactivation, resulting in tissue hypoperfusion and subsequent multi-organ failure and death. In the lung, this translates as a localized injury to the alveolar-capillary membrane, fostering the onset of acute respiratory distress syndrome (ARDS). ARDS is characterized by an acute onset of respiratory failure typically requiring mechanical ventilation and radiographic bilateral pulmonary opacities of non-cardiogenic origin. Direct ARDS occurs after a direct insult to the lung tissue, leading to an increase in the capillary hydrostatic pressure and interstitial and alveolar flooding, impaired gas exchange, and decreased lung compliance. Indirect ARDS is triggered by a systemic insult and the release of inflammatory mediators that eventually damage the pulmonary endothelium. Its most common cause is sepsis (98).
Activation of the endothelium in sepsis occurs directly by recognition of pathogen-associated molecular patterns (PAMPs) through patron recognition receptors, such as Toll-like receptors (TLRs) expressed in ECs, and, indirectly, by released proinflammatory cytokines, such as TNF-α, IL-6, and IL-1, complement components or neutrophil extracellular traps (NETs) (99). Damage-associated molecular patterns (DAMPs) can also be recognized by ECs, contributing to the amplification of the inflammatory cascade. This endothelial activation leads to a proadhesive, proinflammatory, prothrombotic, and proapoptotic phenotype.
The endothelial barrier integrity is altered during sepsis due to the action of inflammatory mechanisms, such as metalloproteinases and heparanase, causing glycocalyx deconstruction and disruption of ECs junctions, and increasing endothelial permeability, albumin extravasation, and capillary leak (100). Glycocalyx degradation may be potentially compounded by fluid resuscitation practices (101). In addition, there is dysregulation of the NO pathway with a decreased activity of the endothelial nitric oxide synthase (eNOS) and an increased NO production by inducible nitric oxide synthase (iNOS), altering the vascular vasomotor tone toward vasodilatation and producing reactive nitrogen species (102). Moreover, infection triggers ECs to produce ROS, leading to EC apoptosis and production of proinflammatory cytokines, acting not only as a victim but also as an active participant and amplifier of the inflammation. This proinflammatory environment increases expression of adhesion molecules (VCAM-1, ICAM-1, and selectins) on the ECs surface, promoting subsequent leukocyte rolling, adhesion and transmigration, contributing to inflammation and progression of endothelial dysfunction (103). The activated endothelium also interacts with platelets, which are activated directly by PAMPs or indirectly by innate immune cells, promoting a prothrombotic phenotype in which an increased production of VWF seems to play a major role. ADAMTS-13 activity has been found to be decreased in patients with sepsis, which may increase platelet-vessel wall interaction by an increased presence of ultra-large VWF multimers (104, 105). Furthermore, activated platelets also contribute to inflammation by releasing proinflammatory proteins that help establish a “cross-talk” with the endothelium (106). Indeed, thrombocytopenia is frequently seen in sepsis, probably due to excessive peripheral consumption, and it is associated with increased disease severity and mortality (107).
Upon sepsis-induced endothelial activation, there is also an increased expression of TF with subsequent extrinsic coagulation pathway activation. This procoagulant state is favored by a dysregulation of the endothelial anticoagulant and fibrinolytic properties, with decreased protein C activation, TM and TFPI and increased PAI-1 release. This procoagulant state favors thrombosis in the microvasculature, potentially causing disseminated intravascular coagulation (DIC), which is associated with poor prognosis in patients with sepsis (108, 109).
Neutrophil extracellular traps have also an important role in sepsis-induced endothelial dysfunction, causing the expression of adhesion molecules, participating in the prothrombotic state by increasing platelet adhesion on the ECs surface, and contributing to thrombin-mediated fibrin generation (110, 111).
This ED significantly alters the microcirculation, with decreased vascular flow and subsequent organ hypoperfusion, with mitochondrial dysfunction leading to organ failure and death. Experimental and clinical studies have demonstrated a correlation between endothelial dysfunction and sepsis severity, highlighting the crucial role of the endothelium in the pathophysiology of sepsis-induced organ dysfunction, and arising as an attractive therapeutic target (112–114). Thus, in the last decades, experimental and clinical studies have strived to find effective treatments targeting sepsis-induced endothelial dysfunction (111), however, none of them have shown survival improvement in large randomized clinical trials. Our group has demonstrated the utility of an in vitro model of ED in sepsis, able to show a gradual effect depending on the severity of the disease, which may constitute a useful tool to explore different treatments.
7. The endotheliopathy developed in COVID-19
The COVID-19 pandemic, caused by the emergence and worldwide spread of the SARS-CoV-2 virus, exerted profound and far-reaching impacts on global healthcare and the economy. It is now widely acknowledged that the endothelium plays a pivotal role in its pathogenesis and manifestations (115, 116). Intracellular penetration of SARS-CoV-2 into human cells occurs via union of its spike protein to the angiotensin converting enzyme 2 (ACE2) (117). While ACE2 receptors are ubiquitous, they are particularly overexpressed on the alveoli and the ECs (118), leading to an immediate interaction with the endothelium from the very initial stage of infection. As viral replication progresses, more severe symptoms may appear accompanied by a hyperinflammatory activation of host immunity. Key features at this stage are elevated acute phase reactants, such as C-reactive protein, D-dimer, and ferritin, as well as increased circulating cytokine levels. As discussed below, the endothelium is a primary contributor in sustaining and intensifying this maladaptive immune response (119, 120).
Critically ill COVID-19 patients develop pulmonary infiltrates leading to acute respiratory distress syndrome with an eventual need for respiratory support. The development of ARDS in COVID-19 involves a complex interplay of immune responses and inflammatory processes. Both direct (lung tissue-specific) and indirect (systemically triggered) mechanisms are involved in the pathogenesis of ARDS, but there is no evidence of a specific phenotype related to COVID-19 (121). The clinical diagnosis of ARDS is based on the Berlin definition (122) of sudden refractory hypoxemia and bilateral shadows in the lung fields. The endothelium plays a pivotal role in the spreading of inflammation and damage to the lung and the alveolar-capillary membrane, which culminates in fluid accumulation, compromised gas exchange, and pulmonary vasculature hypertension. The initial virus mediated pneumocyte injury triggers local cytokine production and, via paracrine cell communication, the alveolar capillary ECs sense the signaling distress (123). This activates the endothelium, which responds with the induction of a proinflammatory cell recruitment state, mediated by the expression of cell adhesion molecules (124). As a result, oxidative stress, endotheliitis and EC dysfunction ensue. Multiple mechanisms are involved in the switching of the endothelium into a hypercoagulative state, such as TF coagulation activation, platelet pro-aggregation due to increased release of VWF, NET-mediated thrombin generation and fibrinolytic shutdown. This is supported by the fact that COVID-19 patients present with increased circulating endothelial stress products (proinflammatory cytokine levels, VWF, soluble VCAM-1 and heparan sulfate) in correlation with disease severity (105). Also, biomarkers of complement activation, fibrinolysis inhibition, proangiogenic factors, and NET formation have shown to be significantly higher in the serum of COVID-19 patients (125–127). Several postmortem histological lung studies (128, 129) have widely evidenced the aforementioned mechanisms involved in the cascade of endothelial modifications, showing increased levels of inflammatory cytokines, such as IL-6 and TNF-α (130), and upregulation of ICAM-1 and VWF (131).
Other organs are often affected in the spectrum of COVID-19 manifestations, notably the cardiovascular system (132, 133). Thrombosis, coronary infarction and stroke are more frequently observed in the setting of critical illness (134). Further evidence pointing toward endotheliopathy as a predictor of COVID-19 morbidity and mortality emerges from the strong association between pre-existing cardiovascular risk factors (i.e., advanced age, hypertension, and obesity) and the likelihood of disease progression (135). In the brain, neurologic symptoms such as anosmia, ageusia or even encephalopathy were widely reported during the pandemics. In the skin, cutaneous lesions have also demonstrated endothelial activation. Histological studies evidenced complement-mediated vascular injury (136), particularly in severe COVID-19.
Current strategies primarily focus on high-efficacy antivirals and immunosuppressants. The combination of therapies targeting inflammation, coagulopathy, and endotheliopathy is a promising strategy to address disease complications. Many publications have explored other approaches to reduce ED, such as defibrotide (137), ACE inhibitors and ACE2 receptor blockers, statins, heparins, and direct oral anticoagulants. These therapies appear to be promising due to their pleiotropic mechanisms of action and their ability to regulate the endothelium (138).
Lastly, evidence of persistent endothelial dysfunction has been found in the subset of patients who develop long-term sequelae after acute infection. Elevated inflammatory markers, cytokine levels, and cytotoxic T cells subsist in convalescent patients (139), and vascular barrier injury is considered to be responsible for the disruption of normal organ physiology (140), leading to severely impairing systemic symptoms. While days of high infection incidence and overwhelming ICU admissions may be behind us, the implications of endothelial dysregulation remain relevant in the current era of persisting COVID-19. Therefore, endothelial protection remains a valid target for preventing both acute critical illness and long-term COVID-19 complications.
Our group of researchers has contributed to improving the knowledge of the endotheliopathy associated with COVID-19. We have provided evidence on biomarkers that may be useful for the stratification of disease severity and also to guide specific therapeutic strategies to prevent endotheliopathy progression. Some of these biomarkers help to differentiate COVID-19 endotheliopathy from the one that occurs in septic syndromes, in which ED is also a pathological substrate (120). Similarly, we have demonstrated that preeclampsia and severe COVID-19, which may be clinically similar, exhibit distinctive biomarker profiles related to ED, coagulopathy, and angiogenic imbalance. Therefore, differential diagnosis of these entities could be done based on these results.
8. The endothelium as a central player in thrombotic microangiopathies
Thrombotic microangiopathies (TMAs) are a group of disorders characterized by microangiopathic hemolytic anemia and ischemic organ dysfunction, resulting in a wide spectrum of symptoms. The most commonly affected organs in TMAs are the brain, kidneys, and gastrointestinal system (141). Although uncommon, these are life-threatening conditions that require urgent management (142). ED is the common underlying mechanism among different forms of TMAs, leading to the microvasculature thrombosis observed histologically (143).
Classically, primary TMAs have been classified according to the identification of the following pathogenic mechanisms: thrombotic thrombocytopenic purpura (TTP), mediated by a deficiency in the activity of ADAMTS-13 enzyme; typical hemolytic uremic syndrome, caused by a Shiga-toxin–producing Escherichia coli (STEC-HUS); and primary atypical HUS (aHUS), due to the dysregulation of the alternative complement pathway (ACP) (142).
Thrombotic thrombocytopenic purpura is characterized by ADAMTS-13 deficiency, resulting in a deficient excision of the ultra-large VWF multimers presented in the VWF molecule on ECs, causing platelet adhesion and aggregation with rapid generation of disseminated microthrombi. However, evidence generated from clinical, in vitro, and in vivo studies suggests that ADAMTS-13 deficiency may be a necessary but not sufficient condition to induce TTP. Weibel–Palade bodies (WPBs) are endothelial-specific organelles that contain molecules involved in the regulatory functions of the endothelium, such as ultra-large VWF multimers (proaggregating), P-selectin (proinflammatory) or Endothelin-1 (vasoconstricting). The “second hit” model suggests that in TTP, besides ADAMTS-13 deficiency, endogenous (antibodies or cytokines) and/or exogenous (virus or drugs) factors induce endothelial activation leading to an uncontrolled WPBs degranulation and, finally, to endothelial dysfunction (144).
In STEC-HUS, Shiga-toxin (Stx) is thought to be the key element in the pathogenesis of ED through several mechanisms. Stx induces the production of adhesive molecules (E-selectin, ICAM-1, and VCAM-1) and chemokines (MCP-1, IL-8, and fractalkine), leading to the adhesion of leukocytes to cultured human ECs. Moreover, Stx induces rapid release of ultra-large VWF multimers inhibiting its cleavage by ADAMTS-13, therefore enhancing platelet adhesion and clot formation in the microvasculature. In addition, Stx modifies gene expression, with mRNA production, and release of chemokines and cytokines that may aggravate ED (145, 146).
As mentioned before, dysregulation of the ACP occurs as the primary event in aHUS, prompting the activation of the terminal complement phase and the deposit of the lytic complex C5b-9 on the EC surface (146). Eculizumab, a humanized monoclonal antibody against C5, and ravulizumab, a long-acting C5 inhibitor, are first-line treatments for aHUS (147). In this regard, it has been observed that the measurement of C5b-9 deposits on ECs constitutes a reliable tool to explore the complement system dysregulation in aHUS, as well as to monitor the response efficiency to eculizumab treatment in these patients (83).
Although aHUS is the prototype of complement-mediated TMAs, the contribution of dysregulated complement activation to ED has been widely demonstrated in other TMA forms. Increased levels of soluble C5b-9 (sC5b-9) have been detected in patients with acute TTP, probably due to the activation of the classical complement pathway by immunocomplexes of ADAMTS-13 and anti-ADAMTS-13 antibodies (146). In STEC-HUS, the ED is caused by different mechanisms driven by Stx, including ACP activation, resulting in increased levels of C3a, Bb, and sC5b-9 during the active phase of the disease. In this regard, eculizumab has been employed for the treatment of children and adults with STEC-HUS, with no systematic assessment of its efficacy or safety (145).
Secondary forms of TMAs may occur in multiple clinical settings: autoimmune diseases, cancer, pregnancy, solid organ and HCT, certain medications, and infections (141). The underlying pathogenic mechanism has been attributed to a direct ED conferring a more procoagulant and proinflammatory phenotype in ECs, inducing a “second-hit” in which complement activation and its perpetuation occurs, aggravating TMA (148). Among them, TMAs after kidney transplantation should be highlighted due to the well-known involvement of ED in its pathogenesis, which can occur due to the combination of multiple triggers: ischemia-reperfusion injury, immunosuppressive drugs (calcineurin and m-TOR inhibitors), viral infections (mainly cytomegalovirus), and acute or chronic humoral rejection (148).
Thrombotic microangiopathies differential diagnosis is a major challenge due to their variable clinical presentation and the absence of pathognomonic histological findings or specific biomarkers (141). Moreover, differentiation between primary or secondary forms may be difficult in clinical practice because triggering factors, such as infections or drugs, are often identified in patients with primary aHUS (142). Therefore, there is an urgent need for new diagnostic tools, based on functional and genetic studies, to assess the involvement of complement dysregulation in the pathogenesis of the different forms of TMAs (146). In this regard, the use of complement-targeted therapies in patients with secondary refractory TMAs to traditional therapy could be useful. The duration of the treatment should be tailored based on the presence of complement abnormalities and response to therapy (149).
9. Hematopoietic stem cell transplantation and endothelial damage
Hematopoietic stem cell transplantation is the best-known form of cellular therapy, widely used for the treatment of malignant and non-malignant hematologic, metabolic, or autoimmune disorders (150). During the last decades, HCT has experienced significant improvements in terms of donor selection, in allogeneic HCT (allo-HCT), and treatment refinement in both, allo- and autologous HCT (auto-HCT) (151, 152), allowing the expansion of HCT to older adults or patients with comorbidities. More recently, the scope of cellular therapy has expanded through the emergence of immunotherapies based on the cytotoxic effect of autologous cells, as CAR-T cells and tumor-infiltrating lymphocytes (TIL), for refractory/relapsed hematological malignancies and solid tumors, respectively. Despite the curative potential of cell based therapies, there are associated complications that may compromise the success of the treatment. In these complications, the endothelium seems to play a main role.
Sinusoidal obstruction syndrome (SOS), formerly known as veno-occlusive disease, was the first post-HCT complication where endotheliopathy was proven as its pathophysiological substrate and targeted for its treatment (153, 154). Consecutively, growing evidence points to endothelial dysfunction underlying other highly incident HCT-related complications, such as acute graft-versus-host disease (aGVHD) (155, 156), and the main CAR-T cell-related toxicities (157, 158).
Endotheliopathy has not only shown to be involved in the pathogenesis of the complications of cellular therapies but also to be the result of different harms toward ECs before and during the treatment. Mainly due to drugs used during the induction or consolidation chemotherapeutic schemes (159, 160) or the ones used for the conditioning treatment before and after the infusion of the autologous or allogeneic cells (161–165). Consequently, it is essential to understand the involvement of the endothelium and other associated pathways in the pathogenesis of cellular therapies-complications in order to better-stratify risk patients and develop targeted treatments and preventive strategies.
The HCT treatment itself induces endothelial dysregulation, leading to a hypercoagulable state. Studies demonstrate that while procoagulant molecules increase, levels of the main natural-anticoagulant molecules decrease in the context of HCT (166, 167). Furthermore, the innate and adaptative immune reactions, the PAMPs resulting from infections occurring during the HCT process, together with the toxic agents included in the preparative regimens, have been identified as noxa toward the endothelium.
Endothelial dysfunction after HCT has multiple origins and varies according to the time after HCT and anatomical location. In most cases, it implies increased leukocyte adhesion and transmigration, molecule extravasations, platelet activation, and cytokine liberation (168). The ED occurring after HCT and derived from the mentioned stressors would consist of the following: (1) increased synthesis of Ang-2, which is involved in endothelial inflammation increasing its permeability (169), (2) overexpression of adhesion molecules (such as ICAM-1, VCAM-1, E-selectin, and P-selectin), responsible for leukocyte recruitment and transmigration through the endothelium (170), (3) dysregulation of the vascular tone, since the endothelial synthesis of NO and prostacyclin is reduced, and (4) elevation of angiogenic molecules, such as vascular endothelial growth factor A (VEGFA), fibroblast growth factor 2 (FGF2), and Ang-2, molecules that have their respective receptors (VEGFR1 and VEGFR2, FGR1, and TIE-2) (171).
The complex link existing between endothelial activation and progression to endothelial dysfunction occurring during the HCT process has been investigated in different in vitro and pre-clinical studies. Results obtained indicate that ECs are activated and damaged by different factors, including drugs used in the conditioning regimen, radiotherapy, cytokines released by the injured tissues, endogenous microbial products that translocated through damaged mucosal barriers, immunosuppressants in allo-HCT, the engraftment process itself, and allo-reactivity (163, 168, 172, 173). Studies by our group and others, using an in vitro model of ED, allowed the investigation of the specific responses of the endothelium to controlled and well-known stimuli at different stages of the HCT, and also to molecules associated with HCT, such as lipopolysaccharide or TNF-α (174).
In the HCT setting, the ED occurring since the start of the conditioning regimen and during the post-transplant process is involved in a group of early and potentially life-threatening post-HCT endothelial complications (175, 176). These events generally appear during the first 100 days after the stem cell infusion, their diagnosis is mainly based on medical signs and symptoms, and all of them seem to begin at the capillary level and result from an endothelial dysfunction occasioned by the administration of chemotherapy, calcineurin inhibitors, G-CSF, infections, and allogeneic-derived reactivity (12, 168, 177).
Historically, SOS was the first complication in which the role of ED was proposed. Nevertheless, there is increasing evidence of the implication of the endothelium in the pathophysiology of other post-HCT vascular endothelial complications, such as engraftment syndrome, capillary leak syndrome, transplant-associated thrombotic microangiopathy (TA-TMA), GVHD, and the vascular idiopathic pneumonia syndrome.
Several groups, in which ours is included, have investigated the presence of circulating biomarkers for diagnosis and prognostication of post-HCT vascular endothelial complications (177–180). In general, the majority of the studies show increased levels of ED biomarkers, especially in patients with HCT complications (179, 181–183). The complexity of the diagnosis and clinical management required to treat post-HCT vascular endothelial complications enhances the need to increase the knowledge of predictors and clinical manifestations for the early detection of these syndromes to decrease mortality after HCT (168). Furthermore, the increasing knowledge of the physiopathology of these complications opens up potential pharmacologic interventions to prevent and treat ED and, therefore, to improve the outcome of patients receiving HCT.
10. Endotheliopathy in CAR-T cell immunotherapy
Treatment with CAR-T cells has risen as a viable and safe procedure for the treatment of relapsed/refractory hematological malignancies. CAR-T cell technology is based on the cytotoxic effect of T lymphocytes of the patient modified in vitro to target antigens present in tumoral cells. This immunotherapy has proven to be effective for the treatment of acute lymphoblastic leukemia and lymphomas expressing CD19 antigen and for myeloma patients when targeting B-cell maturation antigen. To date, only a few therapeutic schemes and constructs have been approved by Food and Drug Administration and European Medicines Agency (184, 185) whereas many others are still being under assessment in clinical assays (186). Despite the promising remission rates of these novel therapies, cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), are highly incident toxicities, and potentially life-threatening (187–189). Growing evidence sustains that endotheliopathy underlies and promotes the onset of these toxicities. However, not all endothelial dysfunction can be attributed to the administration of the construct but to different noxa before and during immunotherapy.
The use of conditioning, or lymphodepletion, before CAR-T cell infusion has proven to enhance the in vivo expansion of the construct, and both its engraftment and anti-tumoral function (190–192). The most commonly used drugs are cyclophosphamide (Cy) and fludarabine (Flu) in combination. Both, Cy and Flu, have shown to cause deleterious effects on ECs (173, 193) in in vitro assays. In fact, Flu has been observed to increase the incidence of CAR-T related toxicities (194, 195). Nevertheless, the individual impact of these drugs, in vivo, and the incidence of endothelial-related complications has to be elucidated.
Up to 80% of patients treated with CAR-T cell immunotherapy present CRS of any grade, clinically ranging from fever ± hypoxemia, hypotension, capillary leak and/or signs of specific-organ toxicity, depending on the severity of the case (196). Less incident, ICANS can be suspected by a wide range of symptoms and signs, such as headache, cognitive or motor impairment, delirium and seizures. Since ICANS is predisposed in the vast majority of cases by severe/early onset CRS (157, 197), the pathways involved in their development seem to be common. For this reason, the pathophysiology of CRS and ICANS will be reviewed altogether except when otherwise specified.
Clinically, different risk factors have been associated with an increased risk of developing CRS and ICANS: lymphodepletion schemes containing Flu, high burden/bone marrow involvement of the basal disease, and infusion of high doses of the CAR-T cell construct leading to high peaks of in vivo proliferation (188, 194). Biologically, elevations of proinflammatory cytokines (IL-6, interferon-γ, and TNF-α) after the construct infusion were firstly reported as the potential cause of CRS/ICANS in correlation with the clinical severity (187, 198).
Endothelial dysfunction appearing as a consequence of the mentioned cytokine storm, among other causes (199), has been hypothesized as a relevant pathway in the development of CAR-T cell-related toxicities (200). In the specific context of ICANS, the increased permeability within the endothelium of the blood-brain barrier was proven after observing the presence of CAR-T cells on the cerebrospinal fluid (157, 201–203). Recently, scores based on indirect biomarkers of endotheliopathy, such as EASIX or modified EASIX, have demonstrated to be reliable tools to predict the incidence or severe CRS and/or ICANS and their related decrease of the progression-free survival (204–207). Similar to other diseases with ED (112, 208, 209), innate immune activation and hemostasis imbalance are linked pathways also altered in CAR-T cell patients developing toxicities (210–213).
Coagulopathy is an underestimated adverse effect of CAR T-cell therapies (214) that usually derives from severe CRS or ICANS (157, 211). While isolated changes in coagulation parameters, such as elevation in D-dimer, increase in fibrinogen degradation product, decrease of fibrinogen levels and prolongation of activated partial thromboplastin time (aPTT), can be observed in a high proportion of patients (215, 216), the analytical and clinical phenotype of DIC occurs only in cases of high-grade toxicities and is related to an increase of the non-relapse mortality (215). More specifically, a recent study has described increased prothrombin time (PT) and aPTT, fibrinogen, D-dimer, factor VIII (FVIII) and VWF antigen levels in ≥grade 2 CRS. The manifestation of ICANS was associated with elevated PT, D-dimer, FVIII and VWF antigen levels and decreased fibrinogen and platelet count (207). Moreover, patients with high-grade ICANS were found to present higher levels of Ang-2 and VWF, lower levels of ADAMTS-13 metalloprotease and loss of VWF high molecular weight multimers than patients with lower severity grades (157, 198, 200). Although the consumptive mechanism seems to be the predominant one for the development of DIC, impairment of the liver function has also been described as an early indicator (217).
Recently, we have demonstrated that different circulating biomarkers of endotheliopathy, innate-immunity activation, hemostasis alterations and fibrinolytic imbalance may be good early-predictors of severe CRS/ICANS (ST2, Ang-2, and NETs). Also, the use of some of these biomarkers could be a feasible discriminating tool for the differential diagnosis between CAR T-cell-related severe toxicities and sepsis (Ang-2, NETs, sC5b-9, VWF antigen, and PAI-1 antigen) (213).
11. Pharmacological armamentarium
Targeted treatments against the noxa responsible for endothelial activation and dysfunction is the ideal first line therapy, though, in most cases we do not have that kind of treatments available. In terms of pharmacological armamentarium targeting the endothelium, different options are under assessment. For instance, statins, vitamin C, direct oral anticoagulants, heparins, and N-acetylcysteine are cheap and safe drugs that have demonstrated a protective effect toward the endothelium by reducing the oxidative stress (218–224). The use of drugs able to restore the anti-inflammatory and anticoagulant properties of the endothelium, such as defibrotide, activated protein C, recombinant TM, and Ang-1 levels (an endothelium stabilizer molecule as opposed to Ang-2) have shown promising effects in in vitro and in murine models of endothelial dysfunction (225–231). However, disappointing results were found in clinical trials using activated protein C for severe sepsis, highlighting the complexity of the mechanisms involved in ED. In addition, the use of complement inhibitors, such as eculizumab and ravulizumab, in diseases in which terminal complement activation prevails like aHUS, has shown to play a prominent role (83, 147). Other compounds, like TNF-α, IL-1β, and ACE2 receptor blockers, ACE inhibitors or SSRIs, used in synergy with other treatments (i.e., those for CAR T-related toxicities, COVID-19 or major depression) could be worth exploring in clinical trials as potential useful novel therapies in the specific context in which ED is implied (232).
12. Conclusion and future perspectives
The endothelium is an endocrine organ that plays essential functions in maintaining homeostasis. It regulates the vascular tone, hemostasis and fibrinolysis; it shows anti-inflammatory and anticoagulant actions; and it participates in angiogenesis, among other functions. The failure of the endothelial adaptability to the different circulating stimuli, independently of their nature, may cause in the loss of endothelial integrity and function, which is critical for the development of cardiovascular disease.
Endothelial damage, and alterations of several linked pathways, are increasingly being proposed as a pathophysiological substrate for different pathologies and cell therapy complications. The knowledge generated during the last years has promoted the development of panels composed of ED biomarkers for the early prediction of these complications, to stratify their risk, and to facilitate their follow-up. In this regard, new and old therapeutic and prophylactic strategies focused on endothelial protection are being proposed. However, their impact on the incidence of complications and non-relapse mortality should be further explored.
More basic research is needed to elucidate the whole bunch of mechanisms by which the endothelium becomes dysfunctional in a variety of pathological conditions, and more investment in clinical assays is necessary to demonstrate the effect of potentially useful drugs to prevent and treat the endothelium. Additionally, considering that the endothelium is the biggest organ in the body, probably followed by the gastrointestinal tract, we are convinced that the future investigations on the endothelium should consider the crosstalk between both organs, in which the microbiome could be cornerstone (233).
Author contributions
MP: Conceptualization, Formal analysis, Investigation, Supervision, Writing – original draft, Writing – review and editing. AM-C: Writing – original draft, Writing – review and editing. MS: Writing – original draft, Writing – review and editing. SE-S: Conceptualization, Writing – original draft, Writing – review and editing. JM-S: Writing – original draft, Writing – review and editing. BD: Writing – original draft, Writing – review and editing. MR: Writing – original draft, Writing – review and editing. EG-O: Writing – original draft, Writing – review and editing. SF: Writing – original draft, Writing – review and editing. HV-C: Writing – original draft, Writing – review and editing. LY: Writing – original draft, Writing – review and editing. FC: Writing – original draft, Writing – review and editing. MN: Writing – original draft, Writing – review and editing. MD-R: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Work partially supported by Fundació Marató de TV3 (202026-10), Instituto de Salud Carlos III (PI14/00226, INT21/00027, PI17/00675, PI18/00073, PI19/00888, PI20/00246, PI22/00684, and PI22/00240) integrados en el Plan Nacional de I+D+I y cofinanciados por el ISCIII-Subdirección General de Evaluación y el Fondo Europeo de Desarrollo Regional (FEDER) “Una manera de hacer Europa” (Spain), Departament de Recerca i Universitats de la Generalitat de Catalunya (2017-SGR-1531 and 2021-SGR-01118), Contrato Clínico de investigación “Emili Letang-Josep Font,” Centro de Investigación Biomédica en Red de Enfermedades Raras (ERPR04G719/2016) (Spain), Cerebra Foundation for the Brain Injured Child (Carmarthen, Wales, UK), and Fundación Mutua Madrileña (AP180722022). LY had received support from Juan de la Cierva grant FJC2021-048123-I, funded by MCIN/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR. FC had received support from Fundación para la Investigación Jesus Serra.
Acknowledgments
We would like to acknowledge other members of the Research Group, especially Drs. Ginés Escolar, Enric Carreras, Aleix Cases, Pedro Castro, Víctor Navarro, Manel Vera, and Miquel Blasco for their contribution to the research performed during the last 30 years in the field of the endothelial pathophysiology. We would also like to thank the technicians Marc Pino, Patricia Molina, Lidia Martín, Estefanía García, Pilar Gómez, and Paula de la Gala for their technical support at different stages of the research.
Conflict of interest
MD-R has received honoraria from Jazz and research funding from Zacros (Fujimori Kogyo Co., Ltd., Japan), Cellphire Therapeutics (USA), CSL Behring (Spain), and Sysmex Europe GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Científicas en cifras. Publicación incluida en el Programa Editorial 2023 del Ministerio de Ciencia e Innovación. (2023). Available online at: https://www.ciencia.gob.es/dam/jcr:f4f6bb28-cae5-4da2-85f4-067508c410eb/CientiificasCifras2023.pdf (accessed July 12, 2023).
2. Lemichez E, Lecuit M, Nassif X, Bourdoulous S. Breaking the wall: targeting of the endothelium by pathogenic bacteria. Nat Rev Microbiol. (2010) 8:93–104. doi: 10.1038/nrmicro2269
3. Augustin H, Kozian D, Johnson R. Differentiation of endothelial cells: analysis of the constitutive and activated endothelial cell phenotypes. Bioessays. (1994) 16:901–6.
4. Cines D, Pollak E, Buck C, Loscalzo J, Zimmerman G, McEver R, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. (1998) 91:3527–61.
6. Yano K, Gale D, Massberg S, Cheruvu P, Monahan-Earley R, Morgan E, et al. Phenotypic heterogeneity is an evolutionarily conserved feature of the endothelium. Blood. (2007) 109:613–5.
7. Reiterer M, Branco C. Endothelial cells and organ function: applications and implications of understanding unique and reciprocal remodelling. FEBS J. (2020) 287:1088–100. doi: 10.1111/febs.15143
8. Goldenberg N, Kuebler W. Endothelial cell regulation of pulmonary vascular tone, inflammation, and coagulation. 1st ed. In: Prakash Y editor. Comprehensive Physiology. New York, NY: Wiley (2015). p. 531–59.
9. Bazzoni G, Tonetti P, Manzi L, Cera M, Balconi G, Dejana E. Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J Cell Sci. (2005) 118(Pt 3):623–32. doi: 10.1242/jcs.01661
10. Aird W. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. (2007) 100:158–73.
11. Aird W. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. (2007) 100:174–90. doi: 10.1161/01.RES.0000255690.03436.ae
12. Carreras E, Diaz-Ricart M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transpl. (2011) 46:1495–502. doi: 10.1038/bmt.2011.65
14. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. (2013) 382:260–72.
15. Collaboration G. Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2020) 395:709–33. doi: 10.1016/S0140-6736(20)30045-3
16. Matsushita K, van der Velde M, Astor B, Woodward M, Levey A, de Jong P, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. (2010) 375:2073–81.
17. Weiner D, Tighiouart H, Elsayed E, Griffith J, Salem D, Levey A, et al. The Framingham predictive instrument in chronic kidney disease. J Am Coll Cardiol. (2007) 50:217–24.
18. Stenvinkel P, Carrero J, Axelsson J, Lindholm B, Heimbürger O, Massy Z. Emerging biomarkers for evaluating cardiovascular risk in the chronic kidney disease patient: how do new pieces fit into the uremic puzzle? Clin J Am Soc Nephrol. (2008) 3:505–21. doi: 10.2215/CJN.03670807
19. Diaz-Ricart M, Torramade-Moix S, Pascual G, Palomo M, Moreno-Castaño A, Martinez-Sanchez J, et al. Endothelial damage, inflammation and immunity in chronic kidney disease. Toxins. (2020) 12:361.
20. Cigarran Guldris S, González Parra E, Cases Amenós A. Gut microbiota in chronic kidney disease. Nefrologia. (2017) 37:9–19.
21. Vaziri N, Zhao Y, Pahl M. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: the nature, mechanisms, consequences and potential treatment. Nephrol Dial Transpl. (2016) 31:737–46. doi: 10.1093/ndt/gfv095
22. Yilmaz M, Stenvinkel P, Sonmez A, Saglam M, Yaman H, Kilic S, et al. Vascular health, systemic inflammation and progressive reduction in kidney function; clinical determinants and impact on cardiovascular outcomes. Nephrol Dial Transpl. (2011) 26:3537–43.
23. Recio-Mayoral A, Banerjee D, Streather C, Kaski J. Endothelial dysfunction, inflammation and atherosclerosis in chronic kidney disease–a cross-sectional study of predialysis, dialysis and kidney-transplantation patients. Atherosclerosis. (2011) 216:446–51. doi: 10.1016/j.atherosclerosis.2011.02.017
24. Chen J, Hamm L, Mohler E, Hudaihed A, Arora R, Chen C, et al. Interrelationship of multiple endothelial dysfunction biomarkers with chronic kidney disease. PLoS One. (2015) 10:e0132047. doi: 10.1371/journal.pone.0132047
25. Tumur Z, Shimizu H, Enomoto A, Miyazaki H, Niwa T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am J Nephrol. (2010) 31:435–41. doi: 10.1159/000299798
26. David S, Kümpers P, Lukasz A, Fliser D, Martens-Lobenhoffer J, Bode-Böger S, et al. Circulating angiopoietin-2 levels increase with progress of chronic kidney disease. Nephrol Dial Transpl. (2010) 25:2571–6.
27. Serradell M, Díaz-Ricart M, Cases A, Zurbano M, Aznar-Salatti J, López-Pedret J, et al. Uremic medium disturbs the hemostatic balance of cultured human endothelial cells. Thromb Haemost. (2001) 86:1099–105.
28. Addi T, Dou L, Burtey S. Tryptophan-derived uremic toxins and thrombosis in chronic kidney disease. Toxins Basel. (2018) 10:412. doi: 10.3390/toxins10100412
29. Gondouin B, Cerini C, Dou L, Sallée M, Duval-Sabatier A, Pletinck A, et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. (2013) 84: 733–44.
30. Thambyrajah J, Landray M, McGlynn F, Jones H, Wheeler D, Townend J. Abnormalities of endothelial function in patients with predialysis renal failure. Heart. (2000) 83:205–9.
31. van der Vorm L, Visser R, Huskens D, Veninga A, Adams D, Remijn J, et al. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-stage renal disease. Clin Kidney J. (2020) 13:72–4.
32. Vila Cuenca M, Hordijk P, Vervloet M. Most exposed: the endothelium in chronic kidney disease. Nephrol Dial Transpl. (2020) 35:1478–87.
33. Serradell M, Díaz-Ricart M, Cases A, Petriz J, Ordinas A, Escolar G. Uraemic medium accelerates proliferation but does not induce apoptosis of endothelial cells in culture. Nephrol Dial Transpl. (2003) 18:1079–85.
34. Serradell M, Díaz-Ricart M, Cases A, Zurbano M, López-Pedret J, Arranz O, et al. Uremic medium causes expression, redistribution and shedding of adhesion molecules in cultured endothelial cells. Haematologica. (2002) 87:1053–61.
35. Caballo C, Palomo M, Cases A, Galán A, Molina P, Vera M, et al. NFκB in the development of endothelial activation and damage in uremia: an in vitro approach. PLoS One. (2012) 7:e43374. doi: 10.1371/journal.pone.0043374
36. Carbó C, Arderiu G, Escolar G, Fusté B, Cases A, Carrascal M, et al. Differential expression of proteins from cultured endothelial cells exposed to uremic versus normal serum. Am J Kidney Dis. (2008) 51:603–12. doi: 10.1053/j.ajkd.2007.11.029
37. Martin-Rodriguez S, Caballo C, Gutierrez G, Vera M, Cruzado J, Cases A, et al. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur J Clin Invest. (2015) 45:160–9. doi: 10.1111/eci.12392
38. Escolar G, Díaz-Ricart M, Cases A. Uremic platelet dysfunction: past and present. Curr Hematol Rep. (2005) 4:359–67.
39. Kim K, Kwon S, Yun J, Jeong H, Kim H, Lee E, et al. STAT3 activation in endothelial cells is important for tumor metastasis via increased cell adhesion molecule expression. Oncogene. (2017) 36:5445–59.
40. Berg A, Scherer P. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. (2005) 96:939–49.
42. Fain J. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm. (2006) 74:443–77. doi: 10.1016/S0083-6729(06)74018-3
43. Hocking S, Wu L, Guilhaus M, Chisholm D, James D. Intrinsic depot-specific differences in the secretome of adipose tissue, preadipocytes, and adipose tissue-derived microvascular endothelial cells. Diabetes. (2010) 59:3008–16. doi: 10.2337/db10-0483
44. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. (2007) 56:901–11.
45. Lumeng C, Bodzin J, Saltiel A. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84.
46. Mack I, BelAiba R, Djordjevic T, Görlach A, Hauner H, Bader B. Functional analyses reveal the greater potency of preadipocytes compared with adipocytes as endothelial cell activator under normoxia, hypoxia, and TNFalpha exposure. Am J Physiol Endocrinol Metab. (2009) 297:E735–48. doi: 10.1152/ajpendo.90851.2008
47. Maury E, Ehala-Aleksejev K, Guiot Y, Detry R, Vandenhooft A, Brichard S. Adipokines oversecreted by omental adipose tissue in human obesity. Am J Physiol Endocrinol Metab. (2007) 293:E656–65.
48. Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arter Thromb Vasc Biol. (2007) 27:996–1003.
49. Faber D, de Groot P, Visseren F. Role of adipose tissue in haemostasis, coagulation and fibrinolysis. Obes Rev. (2009) 10:554–63.
50. Hanzu F, Palomo M, Kalko S, Parrizas M, Garaulet M, Escolar G, et al. Translational evidence of endothelial damage in obese individuals: inflammatory and prothrombotic responses. J Thromb Haemost. (2011) 9:1236–45. doi: 10.1111/j.1538-7836.2011.04285.x
51. Cunha M, Mattos S, Klein M, Neves M. Early vascular aging in obese individuals with low cardiovascular health. High Blood Press Cardiovasc Prev Off J Ital Soc Hypertens. (2023) 30:45–54.
52. Hwang H, Kim N, Herman A, Gorospe M, Lee J. Factors and pathways modulating endothelial cell senescence in vascular aging. Int J Mol Sci. (2022) 23:10135.
53. Sherwood A, Hinderliter A, Watkins L, Waugh R, Blumenthal J. Impaired endothelial function in coronary heart disease patients with depressive symptomatology. J Am Coll Cardiol. (2005) 46:656–9.
54. Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al. The vascular endothelium and human diseases. Int J Biol Sci. (2013) 9:1057–69.
55. Shimbo D, Chaplin W, Crossman D, Haas D, Davidson K. Role of depression and inflammation in incident coronary heart disease events. Am J Cardiol. (2005) 96:1016–21.
56. Steyers C, Miller F. Endothelial dysfunction in chronic inflammatory diseases. Int J Mol Sci. (2014) 15:11324–49.
57. Grzywocz P, Mizia-Stec K, Wybraniec M, Chudek J. Adipokines and endothelial dysfunction in acute myocardial infarction and the risk of recurrent cardiovascular events. J Cardiovasc Med Hagerstown. (2015) 16:37–44. doi: 10.2459/JCM.0000000000000042
58. Lopez-Vilchez I, Diaz-Ricart M, Navarro V, Torramade S, Zamorano-Leon J, Lopez-Farre A, et al. Endothelial damage in major depression patients is modulated by SSRI treatment, as demonstrated by circulating biomarkers and an in vitro cell model. Transl Psychiatry. (2016) 6:e886. doi: 10.1038/tp.2016.156
59. McNicol A, Israels S. Platelet dense granules: structure, function and implications for haemostasis. Thromb Res. (1999) 95:1–18.
60. Galan A, Lopez-Vilchez I, Diaz-Ricart M, Navalon F, Gomez E, Gasto C, et al. Serotonergic mechanisms enhance platelet-mediated thrombogenicity. Thromb Haemost. (2009) 102:511–9. doi: 10.1160/TH08-12-0810
61. Lopez-Vilchez I, Diaz-Ricart M, White J, Escolar G, Galan A. Serotonin enhances platelet procoagulant properties and their activation induced during platelet tissue factor uptake. Cardiovasc Res. (2009) 84:309–16. doi: 10.1093/cvr/cvp205
62. Lopez-Vilchez I, Serra-Millas M, Navarro V, Rosa Hernandez M, Villalta J, Diaz-Ricart M, et al. Prothrombotic platelet phenotype in major depression: downregulation by antidepressant treatment. J Affect Disord. (2014) 159:39–45. doi: 10.1016/j.jad.2014.02.022
64. Dimitriadis E, Rolnik D, Zhou W, Estrada-Gutierrez G, Koga K, Francisco R, et al. Pre-eclampsia. Nat Rev Primer. (2023) 9:8.
65. Brown M, Magee L, Kenny L, Karumanchi S, McCarthy F, Saito S, et al. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension. (2018) 72:24–43.
66. Mol B, Roberts C, Thangaratinam S, Magee L, de Groot C, Hofmeyr G. Pre-eclampsia. Lancet. (2016) 387:999–1011.
67. Ives C, Sinkey R, Rajapreyar I, Tita A, Oparil S. Preeclampsia-pathophysiology and clinical presentations: JACC state-of-the-art review. J Am Coll Cardiol. (2020) 76:1690–702. doi: 10.1016/j.jacc.2020.08.014
68. Redman C, Sargent I, Staff A. IFPA Senior Award Lecture: making sense of pre-eclampsia - two placental causes of preeclampsia? Placenta. (2014) 35(Suppl):S20–5. doi: 10.1016/j.placenta.2013.12.008
69. Rana S, Lemoine E, Granger J, Karumanchi S. Preeclampsia: pathophysiology, challenges, and perspectives. Circ Res. (2019) 124:1094–112.
70. Burton G, Redman C, Roberts J, Moffett A. Pre-eclampsia: pathophysiology and clinical implications. BMJ. (2019) 366:l2381.
71. ACOG. ACOG Practice Bulletin No. 202: gestational hypertension and preeclampsia. Obstet Gynecol. (2019) 133:1.
72. Mannaerts D, Faes E, Gielis J, Van Craenenbroeck E, Cos P, Spaanderman M, et al. Oxidative stress and endothelial function in normal pregnancy versus pre-eclampsia, a combined longitudinal and case control study. BMC Pregnancy Childbirth. (2018) 18:60. doi: 10.1186/s12884-018-1685-5
73. Ridder A, Giorgione V, Khalil A, Thilaganathan B. Preeclampsia: the relationship between uterine artery blood flow and trophoblast function. Int J Mol Sci. (2019) 20:3263.
74. Tian Y, Yang X. A review of roles of uterine artery doppler in pregnancy complications. Front Med Lausanne. (2022) 9:813343. doi: 10.3389/fmed.2022.813343
75. Aggarwal R, Jain A, Mittal P, Kohli M, Jawanjal P, Rath G. Association of pro- and anti-inflammatory cytokines in preeclampsia. J Clin Lab Anal. (2019) 33:e22834.
76. Palomo M, Youssef L, Ramos A, Torramade-Moix S, Moreno-Castaño A, Martinez-Sanchez J, et al. Differences and similarities in endothelial and angiogenic profiles of preeclampsia and COVID-19 in pregnancy. Am J Obstet Gynecol. (2022) 227:277.e1–277.e16. doi: 10.1016/j.ajog.2022.03.048
77. Pinheiro M, Martins-Filho O, Mota A, Alpoim P, Godoi L, Silveira A, et al. Severe preeclampsia goes along with a cytokine network disturbance towards a systemic inflammatory state. Cytokine. (2013) 62:165–73. doi: 10.1016/j.cyto.2013.02.027
78. Krieglstein C, Granger D. Adhesion molecules and their role in vascular disease. Am J Hypertens. (2001) 14(6 Pt 2):44S–54S.
79. Wang Y, Li B, Zhao Y. Inflammation in preeclampsia: genetic biomarkers, mechanisms, and therapeutic strategies. Front Immunol. (2022) 13:883404. doi: 10.3389/fimmu.2022.883404
80. Youssef L, Miranda J, Blasco M, Paules C, Crovetto F, Palomo M, et al. Complement and coagulation cascades activation is the main pathophysiological pathway in early-onset severe preeclampsia revealed by maternal proteomics. Sci Rep. (2021) 11:3048. doi: 10.1038/s41598-021-82733-z
81. Bouças A, de Souza B, Bauer A, Crispim D. Role of innate immunity in preeclampsia: a systematic review. Reprod Sci. (2017) 24:1362–70.
82. Aneman I, Pienaar D, Suvakov S, Simic T, Garovic V, McClements L. Mechanisms of key innate immune cells in early- and late-onset preeclampsia. Front Immunol. (2020) 11:1864. doi: 10.3389/fimmu.2020.01864
83. Palomo M, Blasco M, Molina P, Lozano M, Praga M, Torramade-Moix S, et al. Complement activation and thrombotic microangiopathies. Clin J Am Soc Nephrol. (2019) 14:1719–32.
84. Turner R, Bloemenkamp K, Bruijn J, Baelde H. Loss of thrombomodulin in placental dysfunction in preeclampsia. Arter Thromb Vasc Biol. (2016) 36:728–35.
85. Stepanian A, Cohen-Moatti M, Sanglier T, Legendre P, Ameziane N, Tsatsaris V, et al. Von Willebrand factor and ADAMTS13: a candidate couple for preeclampsia pathophysiology. Arter Thromb Vasc Biol. (2011) 31:1703–9. doi: 10.1161/ATVBAHA.111.223610
86. Paules C, Youssef L, Rovira C, Crovetto F, Nadal A, Peguero A, et al. Distinctive patterns of placental lesions in pre-eclampsia vs small-for-gestational age and their association with fetoplacental Doppler. Ultrasound Obstet Gynecol. (2019) 54:609–16. doi: 10.1002/uog.20350
87. Raia-Barjat T, Edebiri O, Ni Ainle F. Preeclampsia and venous thromboembolism: pathophysiology and potential therapy. Front Cardiovasc Med. (2022) 9:856923. doi: 10.3389/fcvm.2022.856923
88. Karumanchi S. Angiogenic factors in preeclampsia: from diagnosis to therapy. Hypertension. (2016) 67:1072–9.
90. Wang W, Lin D, Jarman E, Polacheck W, Baker B. Functional angiogenesis requires microenvironmental cues balancing endothelial cell migration and proliferation. Lab Chip. (2020) 20:1153–66. doi: 10.1039/c9lc01170f
91. Rana S, Burke S, Karumanchi S. Imbalances in circulating angiogenic factors in the pathophysiology of preeclampsia and related disorders. Am J Obstet Gynecol. (2022) 226:S1019–34.
92. Zeisler H, Llurba E, Chantraine F, Vatish M, Staff A, Sennström M, et al. Predictive Value of the sFlt-1:PlGF ratio in women with suspected preeclampsia. N Engl J Med. (2016) 374:13–22.
93. Youssef L, Simões R, Miranda J, García-Martín M, Paules C, Crovetto F, et al. Paired maternal and fetal metabolomics reveal a differential fingerprint in preeclampsia versus fetal growth restriction. Sci Rep. (2021) 11:14422. doi: 10.1038/s41598-021-93936-9
94. Jahan F, Vasam G, Green A, Bainbridge S, Menzies K. Placental mitochondrial function and dysfunction in preeclampsia. Int J Mol Sci. (2023) 24:4177.
95. Liu X, Fei H, Yang C, Wang J, Zhu X, Yang A, et al. Trophoblast-derived extracellular vesicles promote preeclampsia by regulating macrophage polarization. Hypertension. (2022) 79:2274–87. doi: 10.1161/HYPERTENSIONAHA.122.19244
96. Hauspurg A, Ying W, Hubel C, Michos E, Ouyang P. Adverse pregnancy outcomes and future maternal cardiovascular disease. Clin Cardiol. (2018) 41:239–46.
97. Singer M, Deutschman C, Seymour C, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10.
98. Cusack R, Bos L, Povoa P, Martin-Loeches I. Endothelial dysfunction triggers acute respiratory distress syndrome in patients with sepsis: a narrative review. Front Med. (2023) 10:1203827. doi: 10.3389/fmed.2023.1203827
99. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun. (2015) 21:827–46.
100. Yang X, Meegan J, Jannaway M, Coleman D, Yuan S. A disintegrin and metalloproteinase 15-mediated glycocalyx shedding contributes to vascular leakage during inflammation. Cardiovasc Res. (2018) 114:1752–63. doi: 10.1093/cvr/cvy167
101. Chappell D, Jacob M. Role of the glycocalyx in fluid management: small things matter. Best Pr Res Clin Anaesthesiol. (2014) 28:227–34. doi: 10.1016/j.bpa.2014.06.003
102. Ait-Oufella H, Maury E, Lehoux S, Guidet B, Offenstadt G. The endothelium: physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. (2010) 36:1286–98.
103. Dolmatova E, Wang K, Mandavilli R, Griendling K. The effects of sepsis on endothelium and clinical implications. Cardiovasc Res. (2021) 117:60–73.
104. Aibar J, Castro P, Espinosa G, Fernández S, Hernández C, Rinaudo M, et al. ADAMTS-13 in Critically Ill patients with septic syndromes and noninfectious systemic inflammatory response syndrome. Shock. (2015) 43:556–62.
105. Fernández S, Moreno-Castaño A, Palomo M, Martinez-Sanchez J, Torramadé-Moix S, Téllez A, et al. Distinctive biomarker features in the endotheliopathy of COVID-19 and septic syndromes. Shock. (2022) 57:95–105. doi: 10.1097/SHK.0000000000001823
106. Schouten M, Wiersinga W, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. (2008) 83:536–45.
107. McDonald B, Dunbar M. Platelets and intravascular immunity: guardians of the vascular space during bloodstream infections and sepsis. Front Immunol. (2019) 10:2400. doi: 10.3389/fimmu.2019.02400
108. Levi M, de Jonge E, van der Poll T. Sepsis and disseminated intravascular coagulation. J Thromb Thrombolysis. (2003) 16:43–7.
109. Ikezoe T. Thrombomodulin/activated protein C system in septic disseminated intravascular coagulation. J Intensive Care. (2015) 3:1. doi: 10.1186/s40560-014-0050-7
110. Czaikoski P, Mota J, Nascimento D, Sônego F, Castanheira F, Melo P, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One. (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
111. Maneta E, Aivalioti E, Tual-Chalot S, Emini Veseli B, Gatsiou A, Stamatelopoulos K, et al. Endothelial dysfunction and immunothrombosis in sepsis. Front Immunol. (2023) 14:1144229. doi: 10.3389/fimmu.2023.1144229
112. Fernández S, Palomo M, Molina P, Díaz-Ricart M, Escolar G, Téllez A, et al. Progressive endothelial cell damage in correlation with sepsis severity. Defibrotide as a contender. J Thromb Haemost. (2021) 19:1948–58. doi: 10.1111/jth.15343
113. Uchimido R, Schmidt E, Shapiro N. The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit Care. (2019) 23:16.
114. Shapiro N, Schuetz P, Yano K, Sorasaki M, Parikh S, Jones A, et al. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care. (2010) 14:R182. doi: 10.1186/cc9290
115. Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. (2020) 41:3038–44.
116. Evans P, Rainger G, Mason J, Guzik T, Osto E, Stamataki Z, et al. Endothelial dysfunction in COVID-19: a position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc Res. (2020) 116:2177–84. doi: 10.1093/cvr/cvaa230
117. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8.
118. Aleksova A, Gagno G, Sinagra G, Beltrami A, Janjusevic M, Ippolito G, et al. Effects of SARS-CoV-2 on cardiovascular system: the dual role of angiotensin-converting enzyme 2 (ACE2) as the virus receptor and homeostasis regulator-review. Int J Mol Sci. (2021) 22:4526. doi: 10.3390/ijms22094526
119. Passi R, Brittan M, Baker A. The role of the endothelium in severe acute respiratory syndrome coronavirus 2 infection and pathogenesis. Curr Opin Physiol. (2023) 34:100670.
120. Castro P, Palomo M, Moreno-Castaño A, Fernández S, Torramadé-Moix S, Pascual G, et al. Is the endothelium the missing link in the pathophysiology and treatment of COVID-19 complications? Cardiovasc Drugs Ther. (2022) 36:547–60. doi: 10.1007/s10557-021-07207-w
121. Reddy M, Subramaniam A, Chua C, Ling R, Anstey C, Ramanathan K, et al. Respiratory system mechanics, gas exchange, and outcomes in mechanically ventilated patients with COVID-19-related acute respiratory distress syndrome: a systematic review and meta-analysis. Lancet Respir Med. (2022) 10:1178–88. doi: 10.1016/S2213-2600(22)00393-9
122. Ards Definition Task Force, Ranieri V, Rubenfeld G, Thompson B, Ferguson N, Caldwell E, et al. Acute Respiratory Distress Syndrome: the berlin definition. JAMA. (2012) 307:2526–33.
123. Zhang J, Tecson K, McCullough P. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev Cardiovasc Med. (2020) 21:315–9.
124. Meyer K, Patra T, Vijayamahantesh, Ray R. SARS-CoV-2 spike protein induces paracrine senescence and leukocyte adhesion in endothelial cells. J Virol. (2021) 95:e0079421. doi: 10.1128/JVI.00794-21
125. Wright F, Vogler T, Moore E, Moore H, Wohlauer M, Urban S, et al. Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 Infection. J Am Coll Surg. (2020) 231:193–203.e1.
126. Mohebbi A, Haybar H, Nakhaei Moghaddam F, Rasti Z, Vahid M, Saki N. Biomarkers of endothelial dysfunction are associated with poor outcome in COVID-19 patients: a systematic review and meta-analysis. Rev Med Virol. (2023) 33:e2442.
127. Andrianto, Al-Farabi M, Nugraha R, Marsudi B, Azmi Y. Biomarkers of endothelial dysfunction and outcomes in coronavirus disease 2019 (COVID-19) patients: a systematic review and meta-analysis. Microvasc Res. (2021) 138:104224.
128. Varga Z, Flammer A, Steiger P, Haberecker M, Andermatt R, Zinkernagel A, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8.
129. Ackermann M, Verleden S, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8.
130. Nagashima S, Mendes M, Camargo Martins A, Borges N, Godoy T, Miggiolaro A, et al. Endothelial dysfunction and thrombosis in patients with COVID-19-brief report. Arter Thromb Vasc Biol. (2020) 40:2404–7. doi: 10.1161/ATVBAHA.120.314860
131. Birnhuber A, Fließer E, Gorkiewicz G, Zacharias M, Seeliger B, David S, et al. Between inflammation and thrombosis: endothelial cells in COVID-19. Eur Respir J. (2021) 58:2100377.
132. Greistorfer T, Jud P. Pathophysiological aspects of COVID-19-associated vasculopathic diseases. Thromb Haemost. (2023) 123:931–44. doi: 10.1055/s-0043-1768969
133. Yugar-Toledo J, Yugar L, Sedenho-Prado L, Schreiber R, Moreno H. Pathophysiological effects of SARS-CoV-2 infection on the cardiovascular system and its clinical manifestations-a mini review. Front Cardiovasc Med. (2023) 10:1162837. doi: 10.3389/fcvm.2023.1162837
134. Louis D, Saad M, Vijayakumar S, Ilyas S, Kokkirala A, Aronow H. The cardiovascular manifestations of COVID-19. Cardiol Clin. (2022) 40:277–85.
135. Matsushita K, Ding N, Kou M, Hu X, Chen M, Gao Y, et al. The relationship of COVID-19 severity with cardiovascular disease and its traditional risk factors: a systematic review and meta-analysis. Glob Heart. (2020) 15:64.
136. Magro C, Nuovo G, Mulvey J, Laurence J, Harp J, Crowson A. The skin as a critical window in unveiling the pathophysiologic principles of COVID-19. Clin Dermatol. (2021) 39:934–65. doi: 10.1016/j.clindermatol.2021.07.001
137. Calabretta E, Moraleda J, Iacobelli M, Jara R, Vlodavsky I, O’Gorman P, et al. COVID-19-induced endotheliitis: emerging evidence and possible therapeutic strategies. Br J Haematol. (2021) 193:43–51. doi: 10.1111/bjh.17240
138. García-Bernal D, Richardson E, Vlodavsky I, Carlo-Stella C, Iacobelli M, Jara R, et al. Endothelial dysfunction and its critical role in COVID-19-associated coagulopathy: defibrotide as an endothelium-protective, targeted therapy. EJHaem. (2021) 2:680–1. doi: 10.1002/jha2.198
139. Nicolai L, Kaiser R, Stark K. Thromboinflammation in long COVID-the elusive key to postinfection sequelae? J Thromb Haemost. (2023) 21:2020–31. doi: 10.1016/j.jtha.2023.04.039
140. Wu X, Xiang M, Jing H, Wang C, Novakovic V, Shi J. Damage to endothelial barriers and its contribution to long COVID. Angiogenesis. (2023) [Online ahead of print]. doi: 10.1007/s10456-023-09878-5
141. Blasco M, Guillén E, Quintana L, Garcia-Herrera A, Piñeiro G, Poch E, et al. Thrombotic microangiopathies assessment: mind the complement. Clin Kidney J. (2021) 14:1055–66. doi: 10.1093/ckj/sfaa195
142. Arnold D, Patriquin C, Nazy I. Thrombotic microangiopathies: a general approach to diagnosis and management. CMAJ. (2017) 189:E153–9.
143. Campistol J, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. (2015) 35:421–47.
144. Cauchois R, Muller R, Lagarde M, Dignat-George F, Tellier E, Kaplanski G. Is endothelial activation a critical event in thrombotic thrombocytopenic purpura? J Clin Med. (2023) 12:758.
145. Petruzziello-Pellegrini T, Moslemi-Naeini M, Marsden P. New insights into Shiga toxin-mediated endothelial dysfunction in hemolytic uremic syndrome. Virulence. (2013) 4:556–63. doi: 10.4161/viru.26143
146. Blasco M, Guillén-Olmos E, Diaz-Ricart M, Palomo M. Complement mediated endothelial damage in thrombotic microangiopathies. Front Med Lausanne. (2022) 9:811504. doi: 10.3389/fmed.2022.811504
147. Tomazos I, Hatswell A, Cataland S, Chen P, Freemantle N, Lommele Å, et al. Comparative efficacy of ravulizumab and eculizumab in the treatment of atypical hemolytic uremic syndrome: an indirect comparison using clinical trial data. Clin Nephrol. (2022) 97:261–72. doi: 10.5414/CN110516
148. Praga M, Rodríguez de Córdoba S. Secondary atypical hemolytic uremic syndromes in the era of complement blockade. Kidney Int. (2019) 95:1298–300.
149. Palma L, Sridharan M, Sethi S. Complement in secondary thrombotic microangiopathy. Kidney Int Rep. (2021) 6:11–23.
150. Snowden J, Sánchez-Ortega I, Corbacioglu S, Basak G, Chabannon C, de la Camara R, et al. Indications for haematopoietic cell transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2022. Bone Marrow Transpl. (2022) 57:1217–39. doi: 10.1038/s41409-022-01691-w
151. Kanate A, Majhail N, Savani B, Bredeson C, Champlin R, Crawford S, et al. Indications for hematopoietic cell transplantation and immune effector cell therapy: guidelines from the american society for transplantation and cellular therapy. Biol Blood Marrow Transpl. (2020) 26:1247–56.
152. Passweg J, Baldomero H, Chabannon C, Basak G, de la Cámara R, Corbacioglu S, et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: monitoring of activities and trends over 30 years. Bone Marrow Transpl. (2021) 56:1651–64. doi: 10.1038/s41409-021-01227-8
153. Shulman H, Hinterberger W. Hepatic veno-occlusive disease–liver toxicity syndrome after bone marrow transplantation. Bone Marrow Transpl. (1992) 10:197–214.
154. Richardson P, Carreras E, Iacobelli M, Nejadnik B. The use of defibrotide in blood and marrow transplantation. Blood Adv. (2018) 2:1495–509.
155. Tichelli A, Gratwohl A. Vascular endothelium as ‘novel’ target of graft-versus-host disease. Best Pr Res Clin Haematol. (2008) 21:139–48.
156. Ghimire S, Weber D, Mavin E, Wang X, Dickinson A, Holler E. Pathophysiology of GvHD and Other HSCT-related major complications. Front Immunol. (2017) 8:79. doi: 10.3389/fimmu.2017.00079
157. Gust J, Hay K, Hanafi L, Li D, Myerson D, Gonzalez-Cuyar L, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T Cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
158. Gavriilaki E, Sakellari I, Gavriilaki M, Anagnostopoulos A. A new era in endothelial injury syndromes: toxicity of CAR-T cells and the role of immunity. Int J Mol Sci. (2020) 21:3886. doi: 10.3390/ijms21113886
159. Kebriaei P, Cutler C, de Lima M, Giralt S, Lee S, Marks D, et al. Management of important adverse events associated with inotuzumab ozogamicin: expert panel review. Bone Marrow Transpl. (2018) 53:449–56. doi: 10.1038/s41409-017-0019-y
160. Martinez-Sanchez J, Palomo M, Torramade-Moix S, Moreno-Castaño A, Rovira M, Gutiérrez-García G, et al. The induction strategies administered in the treatment of multiple myeloma exhibit a deleterious effect on the endothelium. Bone Marrow Transpl. (2020) 55:2270–8. doi: 10.1038/s41409-020-0947-9
161. Palomo M, Diaz-Ricart M, Carbo C, Rovira M, Fernandez-Aviles F, Escolar G, et al. The release of soluble factors contributing to endothelial activation and damage after hematopoietic stem cell transplantation is not limited to the allogeneic setting and involves several pathogenic mechanisms. Biol Blood Marrow Transpl. (2009) 15:537–46. doi: 10.1016/j.bbmt.2009.01.013
162. Palomo M, Diaz-Ricart M, Carbo C, Rovira M, Fernandez-Aviles F, Martine C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transpl. (2010) 16:985–93. doi: 10.1016/j.bbmt.2010.02.008
163. Carmona A, Díaz-Ricart M, Palomo M, Molina P, Pino M, Rovira M, et al. Distinct deleterious effects of cyclosporine and tacrolimus and combined tacrolimus-sirolimus on endothelial cells: protective effect of defibrotide. Biol Blood Marrow Transpl. (2013) 19:1439–45. doi: 10.1016/j.bbmt.2013.07.001
164. Gutiérrez-García G, Rovira M, Magnano L, Rosiñol L, Bataller A, Suárez-Lledó M, et al. Innovative strategies minimize engraftment syndrome in multiple myeloma patients with novel induction therapy following autologous hematopoietic stem cell transplantation. Bone Marrow Transpl. (2018) 53:1541–7. doi: 10.1038/s41409-018-0189-2
165. Rodríguez-Lobato L, Martínez-Roca A, Castaño-Díez S, Palomino-Mosquera A, Gutiérrez-García G, Pedraza A, et al. The avoidance of G-CSF and the addition of prophylactic corticosteroids after autologous stem cell transplantation for multiple myeloma patients appeal for the at-home setting to reduce readmission for neutropenic fever. PLoS One. (2020) 15:e0241778. doi: 10.1371/journal.pone.0241778
166. Harper P, Jarvis J, Jennings I, Luddington R, Marcus R. Changes in the natural anticoagulants following bone marrow transplantation. Bone Marrow Transpl. (1990) 5:39–42.
167. Gordon B, Haire W, Kessinger A, Duggan M, Armitage J. High frequency of antithrombin 3 and protein C deficiency following autologous bone marrow transplantation for lymphoma. Bone Marrow Transpl. (1991) 8:497–502.
168. Palomo M, Diaz-Ricart M, Carreras E. Endothelial Dysfunction in Hematopoietic Cell Transplantation. Clin Hematol Int. (2019) 1:45–51.
169. Akwii R, Sajib M, Zahra F, Mikelis C. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells. (2019) 8:471.
170. Vestweber D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev. (2007) 218:178–96.
171. Pober J, Sessa W. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15.
172. Richard S, Seigneur M, Blann A, Adams R, Renard M, Puntous M, et al. Vascular endothelial lesion in patients undergoing bone marrow transplantation. Bone Marrow Transpl. (1996) 18:955–9.
173. Eissner G, Multhoff G, Gerbitz A, Kirchner S, Bauer S, Haffner S, et al. Fludarabine induces apoptosis, activation, and allogenicity in human endothelial and epithelial cells: protective effect of defibrotide. Blood. (2002) 100:334–40. doi: 10.1182/blood.v100.1.334
174. Eissner G, Kohlhuber F, Grell M, Ueffing M, Scheurich P, Hieke A, et al. Critical involvement of transmembrane tumor necrosis factor-alpha in endothelial programmed cell death mediated by ionizing radiation and bacterial endotoxin. Blood. (1995) 86:4184–93.
175. Catani L, Gugliotta L, Vianelli N, Nocentini F, Baravelli S, Bandini G, et al. Endothelium and bone marrow transplantation. Bone Marrow Transpl. (1996) 17:277–80.
176. Pagliuca S, Michonneau D, Sicre de Fontbrune F, Sutra Del Galy A, Xhaard A, Robin M, et al. Allogeneic reactivity-mediated endothelial cell complications after HSCT: a plea for consensual definitions. Blood Adv. (2019) 3:2424–35. doi: 10.1182/bloodadvances.2019000143
177. Hildebrandt G, Chao N. Endothelial cell function and endothelial-related disorders following haematopoietic cell transplantation. Br J Haematol. (2020) 190:508–19.
178. Dignat-George F, Sampol J. Circulating endothelial cells in vascular disorders: new insights into an old concept. Eur J Haematol. (2000) 65:215–20. doi: 10.1034/j.1600-0609.2000.065004215.x
179. Erdbruegger U, Dhaygude A, Haubitz M, Woywodt A. Circulating endothelial cells: markers and mediators of vascular damage. Curr Stem Cell Res Ther. (2010) 5:294–302.
180. Moreno-Castaño A, Salas M, Palomo M, Martinez-Sanchez J, Rovira M, Fernández-Avilés F, et al. Early vascular endothelial complications after hematopoietic cell transplantation: role of the endotheliopathy in biomarkers and target therapies development. Front Immunol. (2022) 13:1050994. doi: 10.3389/fimmu.2022.1050994
181. Nürnberger W, Michelmann I, Burdach S, Göbel U. Endothelial dysfunction after bone marrow transplantation: increase of soluble thrombomodulin and PAI-1 in patients with multiple transplant-related complications. Ann Hematol. (1998) 76:61–5. doi: 10.1007/s002770050364
182. Akil A, Zhang Q, Mumaw C, Raiker N, Yu J, Velez de Mendizabal N, et al. Biomarkers for diagnosis and prognosis of sinusoidal obstruction syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transpl. (2015) 21:1739–45.
183. Luft T, Benner A, Jodele S, Dandoy C, Storb R, Gooley T, et al. EASIX in patients with acute graft-versus-host disease: a retrospective cohort analysis. Lancet Haematol. (2017) 4:e414–23.
184. Castella M, Boronat A, Martín-Ibáñez R, Rodríguez V, Suñé G, Caballero M, et al. Development of a novel anti-CD19 chimeric antigen receptor: a paradigm for an affordable CAR T cell production at academic institutions. Mol Ther Methods Clin Dev. (2019) 12:134–44. doi: 10.1016/j.omtm.2018.11.010
185. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, He B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
186. Sengsayadeth S, Savani B, Oluwole O, Dholaria B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. EJHaem. (2022) 3(Suppl 1):6–10. doi: 10.1002/jha2.338
187. Brudno J, Kochenderfer J. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30.
188. Brudno J, Kochenderfer J. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
189. Azoulay É, Castro P, Maamar A, Metaxa V, de Moraes A, Voigt L, et al. Outcomes in patients treated with chimeric antigen receptor T-cell therapy who were admitted to intensive care (CARTTAS): an international, multicentre, observational cohort study. Lancet Haematol. (2021) 8:e355–64. doi: 10.1016/S2352-3026(21)00060-0
190. Gattinoni L, Finkelstein S, Klebanoff C, Antony P, Palmer D, Spiess P, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. (2005) 202:907–12.
191. Wrzesinski C, Paulos C, Kaiser A, Muranski P, Palmer D, Gattinoni L, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. (2010) 33:1–7.
192. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. (2015) 125:3905–16. doi: 10.1182/blood-2015-01-621474
193. Martinez-Sanchez J, Pascual-Diaz R, Palomo M, Moreno-Castaño A, Ventosa H, Salas M, et al. Mafosfamide, a cyclophosphamide analog, causes a proinflammatory response and increased permeability on endothelial cells in vitro. Bone Marrow Transpl. (2023) 58:407–13. doi: 10.1038/s41409-023-01912-w
194. Sievers S, Watson G, Johncy S, Adkins S. Recognizing and grading CAR T-cell toxicities: an advanced practitioner perspective. Front Oncol. (2020) 10:885. doi: 10.3389/fonc.2020.00885
195. Hong R, Zhao H, Wang Y, Chen Y, Cai H, Hu Y, et al. Clinical characterization and risk factors associated with cytokine release syndrome induced by COVID-19 and chimeric antigen receptor T-cell therapy. Bone Marrow Transpl. (2021) 56:570–80. doi: 10.1038/s41409-020-01060-5
196. Jain M, Smith M, Shah N. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141:2430–42. doi: 10.1182/blood.2022017414
197. Morris E, Neelapu S, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96.
198. Gust J, Ponce R, Liles W, Garden G, Turtle C. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
199. Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. (2008) 103:398–406.
200. Hay K, Hanafi L, Li D, Gust J, Liles W, Wurfel M, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. (2017) 130:2295–306. doi: 10.1182/blood-2017-06-793141
201. Mackall C, Miklos D. CNS endothelial cell activation emerges as a driver of CAR T cell-associated neurotoxicity. Cancer Discov. (2017) 7:1371–3.
202. Santomasso B, Park J, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
203. Rice J, Nagle S, Randall J, Hinson H. Chimeric antigen receptor T cell-related neurotoxicity: mechanisms, clinical presentation, and approach to treatment. Curr Treat Options Neurol. (2019) 21:40.
204. Greenbaum U, Strati P, Saliba R, Torres J, Rondon G, Nieto Y, et al. CRP and ferritin in addition to the EASIX score predict CAR-T-related toxicity. Blood Adv. (2021) 5:2799–806. doi: 10.1182/bloodadvances.2021004575
205. Pennisi M, Sanchez-Escamilla M, Flynn J, Shouval R, Alarcon Tomas A, Silverberg M, et al. Modified EASIX predicts severe cytokine release syndrome and neurotoxicity after chimeric antigen receptor T cells. Blood Adv. (2021) 5:3397–406. doi: 10.1182/bloodadvances.2020003885
206. Korell F, Penack O, Mattie M, Schreck N, Benner A, Krzykalla J, et al. EASIX and severe endothelial complications after CD19-directed CAR-T cell therapy-a cohort study. Front Immunol. (2022) 13:877477. doi: 10.3389/fimmu.2022.877477
207. Galli E, Sorà F, Hohaus S, Fresa A, Pansini I, Autore F, et al. Endothelial activation predicts disseminated intravascular coagulopathy, cytokine release syndrome and prognosis in patients treated with anti-CD19 CAR-T cells. Br J Haematol. (2023) 201:86–94. doi: 10.1111/bjh.18596
208. Kumar S, Gupta E, Kaushik S, Srivastava V, Saxena J, Mehta S, et al. Quantification of NETs formation in neutrophil and its correlation with the severity of sepsis and organ dysfunction. Clin Chim Acta. (2019) 495:606–10. doi: 10.1016/j.cca.2019.06.008
209. Iba T, Levi M, Thachil J, Levy J. Disseminated intravascular coagulation: the past, present, and future considerations. Semin Thromb Hemost. (2022) 48:978–87. doi: 10.1055/s-0042-1756300
210. Giavridis T, van der Stegen S, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
211. Jiang H, Liu L, Guo T, Wu Y, Ai L, Deng J, et al. Improving the safety of CAR-T cell therapy by controlling CRS-related coagulopathy. Ann Hematol. (2019) 98:1721–32. doi: 10.1007/s00277-019-03685-z
212. Jess J, Yates B, Dulau-Florea A, Parker K, Inglefield J, Lichtenstein D, et al. CD22 CAR T-cell associated hematologic toxicities, endothelial activation and relationship to neurotoxicity. J Immunother Cancer. (2023) 11:e005898. doi: 10.1136/jitc-2022-005898
213. Moreno-Castaño A, Fernández S, Ventosa H, Palomo M, Martinez-Sanchez J, Ramos A, et al. Characterization of the endotheliopathy, innate-immune activation and hemostatic imbalance underlying CAR-T cell toxicities: laboratory tools for an early and differential diagnosis. J Immunother Cancer. (2023) 11:e006365. doi: 10.1136/jitc-2022-006365
214. Song Z, Tu D, Tang G, Liu N, Tai Z, Yang J, et al. Hemophagocytic lymphohistiocytosis and disseminated intravascular coagulation are underestimated, but fatal adverse events in chimeric antigen receptor T-cell therapy. Haematologica. (2023) 108:2067–79. doi: 10.3324/haematol.2022.281455
215. Wang Y, Qi K, Cheng H, Cao J, Shi M, Qiao J, et al. Coagulation disorders after chimeric antigen receptor T Cell therapy: analysis of 100 patients with relapsed and refractory hematologic malignancies. Biol Blood Marrow Transpl. (2020) 26:865–75. doi: 10.1016/j.bbmt.2019.11.027
216. Buechner J, Grupp S, Hiramatsu H, Teachey D, Rives S, Laetsch T, et al. Practical guidelines for monitoring and management of coagulopathy following tisagenlecleucel CAR T-cell therapy. Blood Adv. (2021) 5:593–601. doi: 10.1182/bloodadvances.2020002757
217. Liu R, Lv Y, Hong F, Zhao W, Lei B, Liu J, et al. A comprehensive analysis of coagulopathy during anti-B cell maturation antigen chimeric antigen receptor-T therapy in multiple myeloma, a retrospective study based on LEGEND-2. Hematol Oncol. (2023) 41:704–17. doi: 10.1002/hon.3155
218. Matsumoto T, D’uscio L, Eguchi D, Akiyama M, Smith L, Katusic Z. Protective effect of chronic vitamin C treatment on endothelial function of apolipoprotein E-deficient mouse carotid artery. J Pharmacol Exp Ther. (2003) 306:103–8. doi: 10.1124/jpet.103.049163
219. Wolfrum S, Jensen K, Liao J. Endothelium-dependent effects of statins. Arter Thromb Vasc Biol. (2003) 23:729–36.
220. Bourraindeloup M, Adamy C, Candiani G, Cailleret M, Bourin M, Badoual T, et al. N-acetylcysteine treatment normalizes serum tumor necrosis factor-alpha level and hinders the progression of cardiac injury in hypertensive rats. Circulation. (2004) 110:2003–9. doi: 10.1161/01.CIR.0000143630.14515.7C
222. Rotta M, Storer B, Storb R, Martin P, Flowers M, Vernon M, et al. Impact of recipient statin treatment on graft-versus-host disease after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transpl. (2010) 16:1463–6.
223. Chen J, Reheman A, Gushiken F, Nolasco L, Fu X, Moake J, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. (2011) 121:593–603. doi: 10.1172/JCI41062
224. Zheng P, Wu Q, Li B, Chen P, Nie D, Zhang R, et al. Simvastatin ameliorates graft-vs-host disease by regulating angiopoietin-1 and angiopoietin-2 in a murine model. Leuk Res. (2017) 55:49–54. doi: 10.1016/j.leukres.2017.01.017
225. Papapetropoulos A, García-Cardeña G, Dengler T, Maisonpierre P, Yancopoulos G, Sessa W. Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab Invest. (1999) 79:213–23.
226. Gamble J, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas L, et al. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res. (2000) 87:603–7. doi: 10.1161/01.res.87.7.603
227. Novotny N, Lahm T, Markel T, Crisostomo P, Wang M, Wang Y, et al. Angiopoietin-1 in the treatment of ischemia and sepsis. Shock. (2009) 31:335–41.
228. Ikezoe T, Yang J, Nishioka C, Yokoyama A. Thrombomodulin alleviates murine GVHD in association with an increase in the proportion of regulatory T cells in the spleen. Bone Marrow Transpl. (2015) 50:113–20. doi: 10.1038/bmt.2014.208
229. Martinez-Sanchez J, Hamelmann H, Palomo M, Mir E, Moreno-Castaño A, Torramade S, et al. Acute graft-vs.-host disease-associated endothelial activation. Front Immunol. (2019) 10:2339. doi: 10.3389/fimmu.2019.02339
230. Sinha R, Flynn R, Zaiken M, Paz K, Gavin A, Nemazee D, et al. Activated protein C ameliorates chronic graft-versus-host disease by PAR1-dependent biased cell signaling on T cells. Blood. (2019) 134:776–81. doi: 10.1182/blood.2019001259
231. García-Bernal D, Palomo M, Martínez C, Millán-Rivero J, García-Guillén A, Blanquer M, et al. Defibrotide inhibits donor leucocyte-endothelial interactions and protects against acute graft-versus-host disease. J Cell Mol Med. (2020) 24:8031–44. doi: 10.1111/jcmm.15434
232. Chen Y, Li R, Shang S, Yang X, Li L, Wang W, et al. Therapeutic Potential of TNFα and IL1β Blockade for CRS/ICANS in CAR-T Therapy. Front Immunol. (2021) 12:623610. doi: 10.3389/fimmu.2021.623610
Keywords: endothelial damage, cardiovascular disease, chronic kidney disease, obesity, major depression, sepsis, COVID-19, hematopoietic stem cell transplantation
Citation: Palomo M, Moreno-Castaño AB, Salas MQ, Escribano-Serrat S, Rovira M, Guillen-Olmos E, Fernandez S, Ventosa-Capell H, Youssef L, Crispi F, Nomdedeu M, Martinez-Sanchez J, De Moner B and Diaz-Ricart M (2023) Endothelial activation and damage as a common pathological substrate in different pathologies and cell therapy complications. Front. Med. 10:1285898. doi: 10.3389/fmed.2023.1285898
Received: 30 August 2023; Accepted: 30 October 2023;
Published: 14 November 2023.
Edited by:
Eleni Gavriilaki, G. Papanikolaou General Hospital, GreeceReviewed by:
Nektarios Barabutis, University of Louisiana at Monroe, United StatesBenjamin Florian Koch, Goethe University Frankfurt, Germany
Styliani Kokoris, National and Kapodistrian University of Athens, Greece
Copyright © 2023 Palomo, Moreno-Castaño, Salas, Escribano-Serrat, Rovira, Guillen-Olmos, Fernandez, Ventosa-Capell, Youssef, Crispi, Nomdedeu, Martinez-Sanchez, De Moner and Diaz-Ricart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maribel Diaz-Ricart, bWRpYXpAY2xpbmljLmNhdA==