 Chenyu Zhao1,2
Chenyu Zhao1,2 Hui Huang2*
Hui Huang2*- 1Department of Gastroenterology, Henan Provincial People’s Hospital, Zhengzhou University People’s Hospital, Zhengzhou, China
- 2Department of Medical Genetics, The Second Xiangya Hospital, Central South University, Changsha, China
Background: Rotor syndrome is a rare genetic disease inherited in an autosomal digenic recessive manner. It is caused by pathogenic mutations in both SLCO1B1 and SLCO1B3 genes, and characterized by predominantly conjugated hyperbilirubinemia.
Methods: Three Chinese patients clinically diagnosed with Rotor syndrome were included. Mutations in SLCO1B1/3 genes were identified using whole-exome sequencing.
Results: They all carried the same homozygous c.1738C>T mutation in SLCO1B1 and the c.481+22insLINE variant in SLCO1B3.
Conclusion: This study established a genetic diagnosis for the three patients and contributed to finding hotspot mutations in Rotor syndrome.
1 Introduction
Rotor syndrome (RS, OMIM*237450) is a rare and benign genetic disease characterized by low-grade, chronic or fluctuating, predominantly conjugated hyperbilirubinemia. It has no other features of hepatobiliary disorder (1, 2). The prevalence of RS is unknown but is very low (<1:1,000,000) (3). First described by Rotor and Florentin (4) in 1948, it is inherited in an autosomal recessive digenic manner. Biallelic pathogenic mutations in solute carrier organic anion transporter family member 1B1 (SLCO1B1) and SLCO1B3 genes cause RS. Organic anion transporting polypeptide 1B1 (OATP1B1) and OATP1B3 are encoded by SLCO1B1 and SLCO1B3 genes, respectively. They serve as transporters for hepatic uptake of conjugated bilirubin. Inactivation of both proteins together leads to RS, which does not affect life expectancy and usually requires no treatment (1). To date, 51 mutations in SLCO1B1 and 30 variants in SLCO1B3 have been described in the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php). In the present study, we reported three patients with RS and tested two disease-causing mutations: SLCO1B1 (NM_006446.5): c.1738C>T (p.R580*) and SLCO1B3 (NM_019844.4): c.481+22insLINE.
2 Materials and methods
2.1 Patients and ethics
Three unrelated Chinese patients (numbered 1–3) were enrolled in this study. Patients 1, 2, and 3 were aged 14, 16, and 20 years, respectively. They were clinically diagnosed as RS without other clinical comorbidities. They intermittently took S-adenosylmethionine or diammonium glycyrrhizinate for treatment. All patients or their guardians signed the written informed consent forms. This study was approved by the ethics committee of the Second Xiangya Hospital of Central South University.
2.2 Variant analysis
Genomic DNA was isolated from peripheral blood by the Blood gDNA Miniprep Kit (Hangzhou Beiwo Meditech Co., Ltd., Hangzhou, China). Whole-exome sequencing (WES) was performed on the three probands using the MGISEQ-2000 platform (MGI Tech Co., Ltd., Shenzhen, China). WES and basic bioinformatics analyses, including read mapping and variant detection, were performed by AmCare Genomics Lab Limited (Guangzhou, China).
The methods for filtering WES data are as follows: (1) Variants from the databases (1000G, ExAC, esp6500, gnomAD) with a minor allele frequency of >5% were excluded. (2) Variants in untranslated regions and synonymous mutations were excluded. (3) The candidate pathogenetic variants in bilirubin metabolism-related genes were retained. (4) The interpretation of mutation pathogenicity was guided by the American College of Medical Genetics and Genomics (ACMG) guideline (5). The potential variant from WES was validated by Sanger sequencing.
3 Results
This study included two male and one female patient, aged from 14 to 20 years. They were born to nonconsanguineous parents (Figures 1A–C) and presented with mild intermittent jaundice. The liver function test only showed predominantly conjugated hyperbilirubinemia. No abnormalities were observed in viral serologies (HBV and HCV), hemolysis test, coagulation function, autoimmune liver disease-associated antibodies, immunoglobulin G, serum ceruloplasmin testing, or abdominal ultrasound examination. The major clinical manifestations of the three patients with Rotor syndrome are summarized in Table 1. The WES indicated that all patients harbored the same homozygous c.1738C>T mutation in SLCO1B1 (Figure 1D) and c.481+22insLINE variant in SLCO1B3.
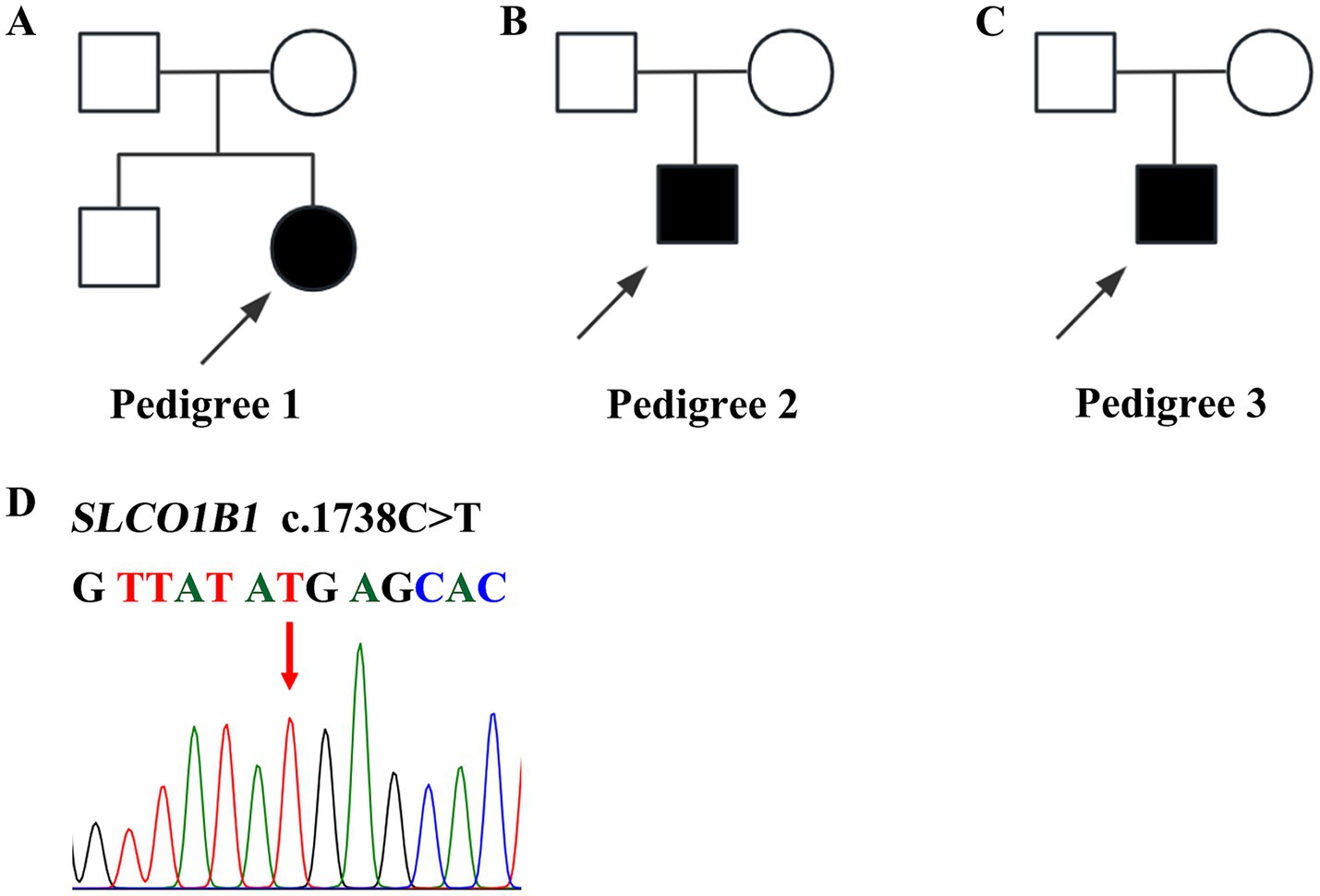
Figure 1. Pedigrees and variants of SLCO1B1 in patients with Rotor syndrome. (A–C) Three pedigrees affected with Rotor syndrome. (D) Homozygous c.1738 C>T SLCO1B1 mutation identified in three probands.
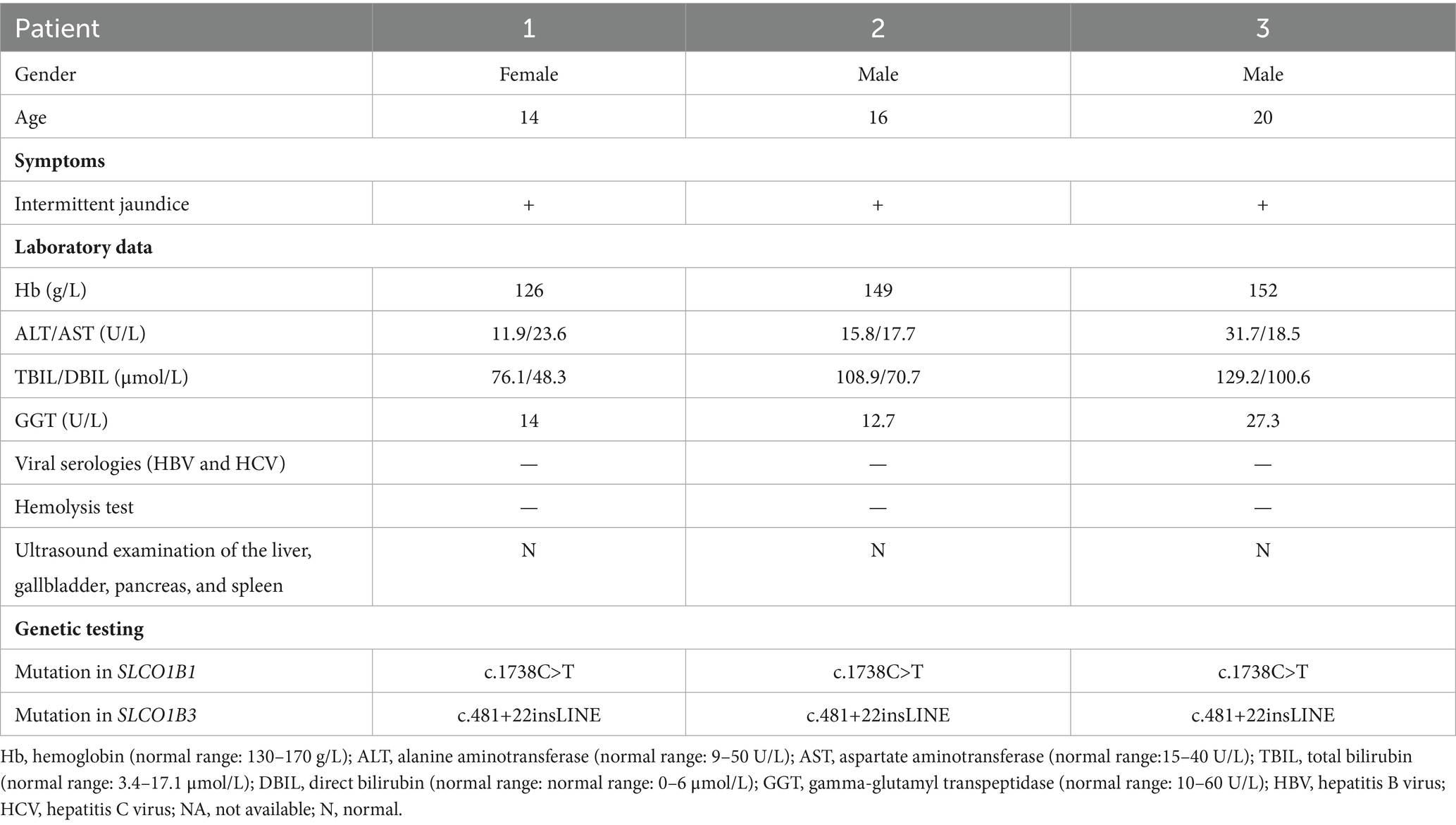
Table 1. Clinical characteristics and mutations in SLCO1B1/3.
4 Discussion
OATP1B1/3 are expressed in the hepatocyte basolateral membrane, which are also called SLCO1B1/3. They uptake endogenous substances, such as conjugated bilirubin, bile acids (BAs), eicosanoids, prostaglandins, and hormones. Unconjugated bilirubin (UCB) enters hepatocytes through passive diffusion and/or transporters, which may include OATP1B1/3. Uridine-diphospho glucuronosyl transferase 1A1 (UGT1A1) catalyzes the conversion of UCB to bilirubin glucuronides (BG) in the endoplasmic reticulum. BG is secreted into bile by ABCC2 and ABCG2. A substantial fraction of BG is rerouted by ABCC3 to the blood. It can be taken up by downstream hepatocytes via OATP1B1/3 transporters (2). In RS, the absence or dysfunction of the OATP1B1/3 may disrupt the uptake of BG (Figure 2), which causes predominantly conjugated hyperbilirubinemia.

Figure 2. Schematic view of bilirubin transport by the hepatocyte in Rotor syndrome. In hepatic metabolism, UGT1A1 catalyzes the conversion of UCB to water-soluble BG within the endoplasmic reticulum. The generated BG is secreted into bile by ABCC2 and ABCG2 transporters, while a substantial fraction is rerouted to the bloodstream via ABCC3. Downstream hepatocytes can uptake BG from the circulation through OATP1B1/3 transporters. In RS, the absence or dysfunction of OATP1B1/3 transporters disrupts hepatic uptake of BG. UGT1A1, uridine diphosphate glucuronosyltransferase 1A1; UCB, unconjugated bilirubin; BG, bilirubin glucuronides; RS, Rotor syndrome.
The accurate diagnosis of RS is of paramount clinical importance, as it directly influences pharmaceutical safety by preventing unwarranted exposure to drugs that rely on functional OATP1B1/3 transporters for hepatic uptake and systemic clearance. The dysfunction of OATP1B1/3 transporters can profoundly impact numerous drug metabolisms, particularly statins, ezetimibe, methotrexate, irinotecan, cabazitaxel, sunitinib, and sartans (3, 6–8). They could exhibit significantly increased systemic exposure in RS patients. After a confirmed diagnosis of RS, clinicians can proactively select alternative drugs with minimal OATP dependency and implement therapeutic drug monitoring (TDM) for high-risk agents.
The c.1738C>T is a nonsense variant in SLCO1B1, which was very strong evidence of pathogenicity (PVS1). The mutation is located in a mutational hotspot (PM1). It has been reported in multiple clinical cases of RS (6, 9–12). Prediction software, specifically MutationTaster (13), predicted a deleterious effect of the variant (PP3). Therefore, according to ACMG guidelines, the variant is classified as a pathogenic mutation.
The c.481+22insLINE mutation in SLCO1B3 is a long-interspersed element (LINE) insertion variant about 6.1 kb in size. It could affect normal editing of mRNA and cause abnormal skipping of exons (PS3) (10). It is a common mutation of RS in Asian populations (PM1) (10). The variant is not found in either the 1000G or EXAC databases (PM2). The patients carried both the homozygous c.1738C>T variant in SLCO1B1 and the c.481+22insLINE mutation in SLCO1B3. The findings were consistent with the digenic recessive pattern of RS (PM3). Therefore, the variants were also classified as a pathogenic mutation.
This study provided evidence that the detected genetic mutations in SLCO1B1 and SLCO1B3 are common in Rotor syndrome, which is consistent with previous research (6, 9–12, 14). However, precise information on the frequencies of c.1738C>T (SLCO1B1) and c.481+22insLINE (SLCO1B3) mutations is limited. There is insufficient data to assess its distribution in RS and the general population. In resource-limited settings across East Asia, targeted PCR assays for the two mutations might be considered as a screening method for RS; however, validation in larger sample sizes is required.
In conclusion, RS is a rare inherited disorder that causes predominantly conjugated hyperbilirubinemia. Genetic testing is a useful tool for efficient diagnosis. This study supported that c.1738C>T in SLCO1B1 and c.481+22insLINE in SLCO1B3 are common pathogenic mutations in the East Asian RS patients.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Second Xiangya Hospital of Central South University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
CZ: Software, Funding acquisition, Writing – original draft, Conceptualization, Project administration, Visualization, Methodology, Formal analysis. HH: Supervision, Data curation, Writing – review & editing, Resources, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Department of Science and Technology of Henan Province, China (Grant No. 252300421609).
Acknowledgments
The authors would like to thank the patients for their participation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Memon, N, Weinberger, BI, Hegyi, T, and Aleksunes, LM. Inherited disorders of bilirubin clearance. Pediatr Res. (2016) 79:378–86. doi: 10.1038/pr.2015.247
2. Erlinger, S, Arias, IM, and Dhumeaux, D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. (2014) 146:1625–38. doi: 10.1053/j.gastro.2014.03.047
3. Jirsa, M, Knisely, AS, Schinkel, A, and Kmoch, S. Rotor syndrome In: MP Adam, J Feldman, GM Mirzaa, RA Pagon, SE Wallace, and A Amemiya, editors. Genereviews®. Seattle, WA: University of Washington (1993)
4. Rotor, AB, and Florentin, A. Familial nonhemolytic jaundice with direct van den Bergh reaction. Acta Med Phil. (1948) 5:37–49.
5. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
6. Zhou, D, Qi, S, Zhang, W, Wu, L, Xu, A, Li, X, et al. Insertion of Line-1 retrotransposon inducing exon inversion causes a rotor syndrome phenotype. Front Genet. (2019) 10:1399. doi: 10.3389/fgene.2019.01399
7. Niemi, M, Pasanen, MK, and Neuvonen, PJ. Organic anion transporting polypeptide 1b1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. (2011) 63:157–81. doi: 10.1124/pr.110.002857
8. Shitara, Y. Clinical importance of OATP1B1 and OATP1B3 in drug–drug interactions. Drug Metab Pharmacokinet. (2011) 26:220–7. doi: 10.2133/dmpk.DMPK-10-RV-094
9. Kim, SR, Saito, Y, Sai, K, Kurose, K, Maekawa, K, Kaniwa, N, et al. Genetic variations and frequencies of major haplotypes in SLCO1B1 encoding the transporter OATP1B1 in Japanese subjects: SLCO1B1*17 is more prevalent than *15. Drug Metab Pharmacokinet. (2007) 22:456–61. doi: 10.2133/dmpk.22.456
10. Kagawa, T, Oka, A, Kobayashi, Y, Hiasa, Y, Kitamura, T, Sakugawa, H, et al. Recessive inheritance of population-specific intronic line-1 insertion causes a Rotor syndrome phenotype. Hum Mutat. (2015) 36:327–32. doi: 10.1002/humu.22745
11. van de Steeg, E, Stránecký, V, Hartmannová, H, Nosková, L, Hřebíček, M, Wagenaar, E, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. (2012) 122:519–28. doi: 10.1172/jci59526
12. Kimura, A, Kagawa, T, Takei, H, Maruo, Y, Sakugawa, H, Sasaki, T, et al. Rotor syndrome: glucuronidated bile acidemia from defective reuptake by hepatocytes. Hepatol Commun. (2021) 5:629–33. doi: 10.1002/hep4.1660
13. Schwarz, JM, Cooper, DN, Schuelke, M, and Seelow, D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
Keywords: Rotor syndrome, SLCO1B1, SLCO1B3, mutation, hyperbilirubinemia
Citation: Zhao C and Huang H (2025) Recurrent SLCO1B1 and SLCO1B3 mutations identified in three patients with Rotor syndrome. Front. Med. 12:1630360. doi: 10.3389/fmed.2025.1630360
Edited by:
Yulei Li, Hubei University of Arts and Science, ChinaReviewed by:
Mohammad A. Alshabeeb, King Abdullah International Medical Research Center (KAIMRC), Saudi ArabiaMariana B. Morais, Universidade de Lisboa, Portugal
Copyright © 2025 Zhao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Huang, aHVpaHVhbmcwOTE2QGNzdS5lZHUuY24=