 Juyi Li1†
Juyi Li1† Haichun Ni2†Peng Cheng1†Yujia Peng1†Lei Liu3Xiangyang Wang3Wei Cheng3Hengfei Li4Xiufang Wang5
Haichun Ni2†Peng Cheng1†Yujia Peng1†Lei Liu3Xiangyang Wang3Wei Cheng3Hengfei Li4Xiufang Wang5 Hongfeng Zhang2*Jifa Hu6*
Hongfeng Zhang2*Jifa Hu6* Aiping Deng1*Wei Cai3,7*
Aiping Deng1*Wei Cai3,7*- 1Department of Pharmacy, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Pathology, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Gastrointestinal Surgery, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 4Department of Infectious Diseases, Hubei Provincial Hospital of Traditional Chinese Medicine, Wuhan, China
- 5Department of Pain, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 6Department of Scientific Research, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 7Hubei Provincial Engineering Research Center of Intestinal Microecological Diagnostics, Therapeutics, and Clinical Translation, Wuhan, China
Purpose: This study aimed to examine pathogenic variations in three families clinically diagnosed with suspected Lynch syndrome (LS).
Methods: Three probands clinically diagnosed suspected LS were subjected to immunohistochemical analysis of DNA mismatch repair (MMR) protein. Whole-exome sequencing and Sanger sequencing were performed to screen pathogenic variations. I-TASSER and PyMOL were used to analyze changes in the functional domains of mutant proteins.
Results: A known missense variation (GRCh37 chr2:g.47702367G>A, MSH2:NM_000251:c.1963G>A:p.V655I), a known stop-gain variant (GRCh37 chr2:g.47709984G>T, MSH2:NM_000251:c.2701G>T:p.E901X), and a known frameshift insertion variation (GRCh37 chr2:g.48032124 dupA, MSH6:NM_000179:c.3514dupA:p.R1172Kfs*5) in Family 1, Family 2, and Family 3, respectively, were observed. The c.1963G>A variation caused the 655th amino acid of MSH2 to change from valine to isoleucine, and there were no significant changes in both the overall and local protein models in MSH2. Further, the c.2701G>T variation caused the 901st amino acid of MSH2 to change from glutamic acid to a premature stop codon in exon 16, and the deletion of amino-acids 901–934 caused changes in the Domain 5 of MSH2 protein. Furthermore, the c.3514dupA variation caused the 1172nd amino acid of MSH6 to change from arginine to lysine, followed by frameshift, which caused changes in the Domain 5 of MSH6 protein.
Conclusion: The missense variation (MSH2:NM_000251:c.1963G>A:p.V655I) and the stop-gain variation (MSH2:NM_000251:c.2701G>T:p.E901X) were considered uncertain significance for LS, and another pathogenic variation (MSH6:NM_000179:c.3514dupA:p.R1172Kfs*5) has been further confirmed.
Introduction
Colorectal cancer (CRC) is the third most common type of cancer worldwide (1). Lynch syndrome (LS) accounts for 3% patients with CRC and 2% of those with endometrial cancer (EC), and 10–15% of those with DNA mismatch repair (MMR)-deficient tumors (2, 3). LS patients have a high risk of developing CRC (52–82%), EC (40–60%), and several other types of cancer (4).
The LS is caused by germline variants in MMR genes, including MLH1, MSH2 (EPCAM), MSH6, and PMS2 accounting for 40–60%, 40–50%, 10–20%, and 2% of LS cases, respectively (5–7). Mutations in the above genes disrupt mismatch repair, which can accelerate the accumulation of somatic mutations and thus the occurrence of tumors (8).
Therefore, it is important to understand the mutation characteristics related to LS, and further conduct genetic counseling based on the results of gene testing, in the Chinese population. In our work, we performed gene sequencing on three families clinically diagnosed with suspected LS and identified three candidate pathogenic variants: a known missense variation (MSH2:NM_000251:c.1963G>A:p.V655I), a known stop-gain variant (MSH2:NM_000251:c.2701G>T:p.E901X), and a known frameshift insertion variant (MSH6:NM_000179:c.3514dupA:p.R1172Kfs*5). Next, we evaluate the spatial impact of candidate pathogenic variants on proteins, and finally, we provide personalized medication guidance to the carriers of these pathogenic variants.
Methods and materials
Patients
We obtained written informed consent from the study participants. The Ethics Committee of the Central Hospital of Wuhan approved this study (No. 2020-192). Three probands were clinically diagnosed with suspected LS and underwent partial colectomy or hysterectomy.
Immunohistochemistry
Tissue samples fixed in formalin and embedded in paraffin were used for pathological detection (hematoxylin-eosin, H&E). Slides were stained with mouse monoclonal antibodies for MLH1, PMS2, MSH2, and MSH6 (9, 10).
Next generation sequencing
Collect 2 milliliters of peripheral blood from each subject and extract genomic DNA. The Hybridization Capture procedure was performed using the Agilent SureSelect Human All Exon V7 enrichment kit. DNA fragments were sequenced using the NovaSeq™ 6000 Sequencing System (Illumina HiSeq 2500 Analyzer, United States) (11, 12). Annotate the genomic variations in this study based on the reference genome UCSC hg19 (9, 13, 14).
Sanger sequencing
DNA sequencing was performed using ABI 3500 (Thermos, United States). In Family 1, the forward and reverse primers were 5′-CAGGCTATGTAGAACCAATG-3′, 5′-GAGGACTGGCTCAAAGGTAA-3′, respectively; In Family 2, the forward and reverse primers were 5′-GGCAACATAGTGAGACCCTCGT-3′, 5′-TTGATAGCCCATGGGCACTGAC-3′, respectively; In Family 3, the forward and reverse primers were 5′-ATTCTAGGCATCTCAGTAGT-3′, 5′-AAAAGAGAGAGAGACTATGC-3′, respectively.
Three-dimensional structure
Three-dimensional (3D) structures of MSH2/MSH6 were analyzed and displayed using I-TASSER1 and PyMOL2 (15, 16).
Results
Clinical phenotypes
In family 1, the proband (II-1, 51 year old female, Figure 1A) underwent right colon surgery after being diagnosed with CRC (51 year old) because of changes in her bowel habits, and her father (I-1) died of cerebral hemorrhage and suspected gastrointestinal cancer. In family 2, the proband (II-1, 51 year old male, Figure 1B) underwent left colon surgery after being diagnosed with colon cancer (51 year old) owing to discomfort in her upper abdomen, and his mother (I-1) was also diagnosed with CRC. In family 3, the proband (II-1, 56 year old female, Figure 1C) underwent a total hysterectomy after being diagnosed with EC (51 year old) following vaginal bleeding. Her father (I-2) had died, but the cause of death was unknown.
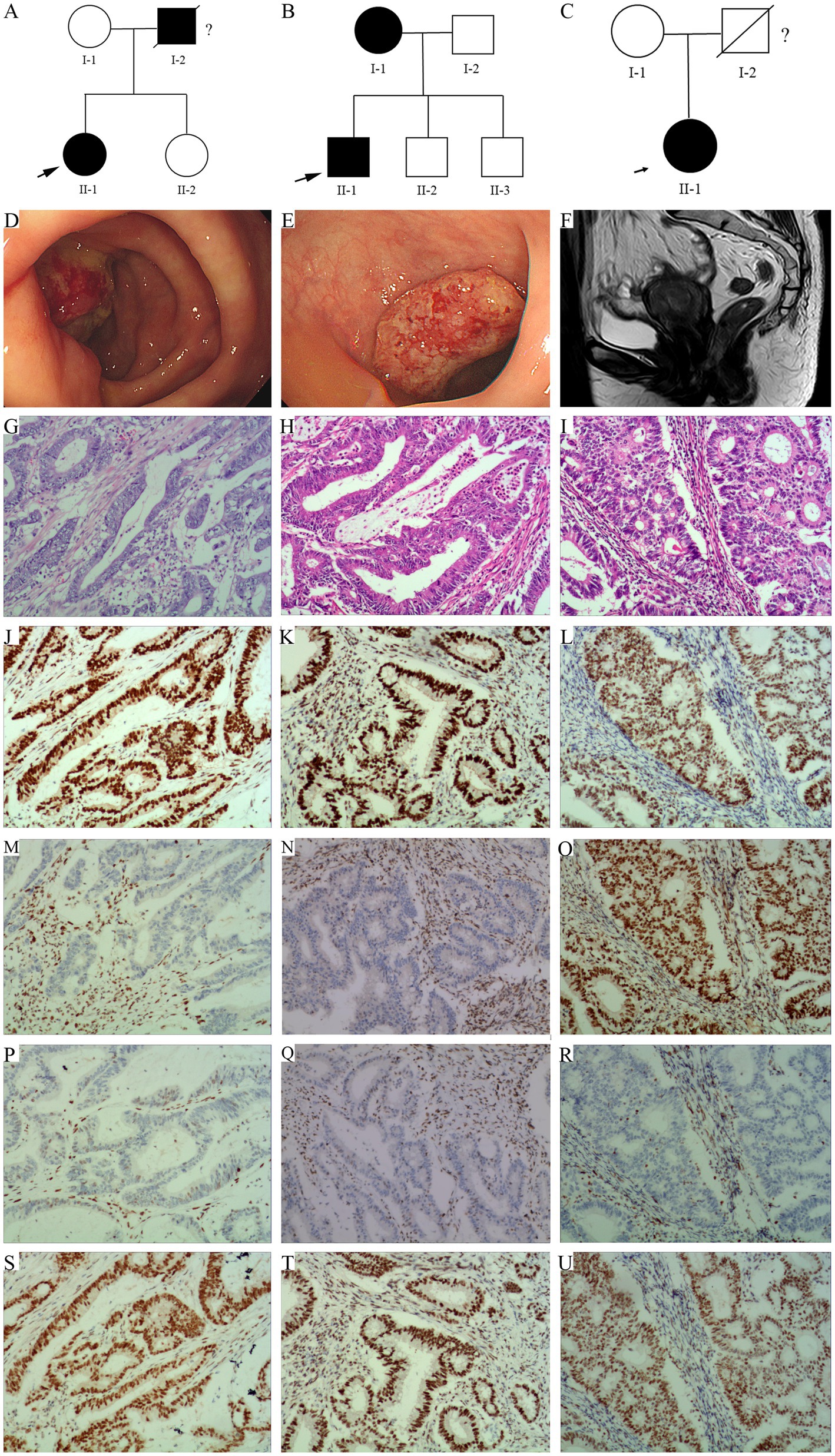
Figure 1. Pedigree structure of family 1, 2, and 3. (A) Family 1. (B) Family 2. (C) Family 3. The arrows indicate the probands. The proband is shown as II-1 in families 1, 2, and 3. Squares are males, circles are females, and crosses indicate deceased individuals. The dark shading represents individuals with LS associated cancer. (D) A new tumor in the cecum of the proband in family 1. (E) A huge new tumor in the sigmoid colon of the proband in family 2. (F) Irregular thickening of the endometrium at the bottom of the uterus, with local clusters protruding toward the uterine cavity in MRI scan in Family 3. (G–I) HE staining of the proband’s tumor tissue, G/H/I represents Family 1, Family 2, and Family 3, respectively. (J–U) Immunohistochemistry. From left to right, the staining of the proband’s tumor tissue from family 1, 2, and 3. From up to down, the antibodies in each line were specific for MLH1, MSH2, MSH6, and PMS2.
Endoscopic examination revealed a huge new tumor in the cecum of the proband of family 1 (Figure 1D), and a huge new tumor in the sigmoid colon of the proband of family 2 (Figure 1E), all with surface ulceration. Magnetic resonance imaging (MRI) of the pelvic cavity revealed irregular thickening of the endometrium at the bottom of the uterus, with local clusters protruding toward the uterine cavity. Diffusion weighted imaging showed diffusion limitation, whereas an enhanced scan showed significant enhancement, involving the muscle layer at the bottom of the uterus of the proband of family 3 (Figure 1F).
Histological analysis of the tumor tissue
In family 1 (Figure 1G) and 2 (Figure 1H), the hematoxylin–eosin (HE) staining results showed that the tissue section locally presented an image of mucinous adenocarcinoma, with the cancer penetrating the intrinsic muscle layer and infiltrating into the subserosal fibrous adipose tissue. The proband of family 1 (Figure 1G) manifested with moderately differentiated adenocarcinoma in the right colon, the proband of family 2 (Figure 1H) manifested with moderately to well differentiated adenocarcinoma in the left colon. In family 3, HE staining indicated highly differentiated endometrioid in the uterus of the proband, with cancer cells invading the uterine muscle layer and penetrating to half of its thickness (Figure 1I).
Immunohistochemical staining of the proband’s tumor cells in family 1 and family 2 demonstrated strong positivity for MLH1 (Figures 1J,K) and PMS2 (Figures 1S,T), but no positivity for MSH2 (Figures 1M,N) and MSH6 (Figures 1P,Q) proteins. In family 3, strong positivity for MLH1 (Figure 1L), MSH2 (Figure 1O), and PMS2 (Figure 1U), not for MSH6 (Figure 1R) proteins, was observed.
Exome and sanger sequencing
We sequenced the exomes of the probands in the three families (Table 1), and average sequencing depth on the target of the probands exceeded 120. A known variant (GRCh37 chr2:g.47702367G>A, MSH2:NM_000251:c.1963G>A:p.V655I) was identified in MSH2 in family 1, namely rs549467183, and the allele frequency of this variant was 0.0000386 (GnomAD_exomes), 0.0002 (1000G_30X) and 0.000 (East Asian). Multiple statistical methods predicted that the variant will have harmful effects on genes or gene products. The MutationTaster score was 0.987549 and FATHMM score was −1.94, which were defined as deleterious. A known stop-gain variant (GRCh37 chr2:g.47709984G>T, MSH2:NM_000251:c.2701G>T:p.E901X) was identified in MSH2 in family 2. This variant frequency was not recorded in any database. Multiple statistical methods predicted that the variant will have harmful effects on genes or gene products. The LRT score was 0.000754 and MutationTaster score was 1, which were defined as deleterious. In family 3, a known frameshift insertion variant (GRCh37 chr2:g.48032124 dupA, MSH6:NM_000179:c.3514dupA: p.R1172Kfs*5) was identified in MSH6, namely rs63751327. The allele frequency is this variant was 0.0000100 (GnomAD_exomes), 0.00008 (GO-ESP) and 0.00 (East Asian), and the Clinical significance of this variant was defined as pathogenic. The results of Sanger sequencing confirmed the variants (rs549467183, MSH2:NM_000251:c.2701G>T:p.E901X and rs63751327) discovered by whole-exome sequencing in the above mentioned three families (Figure 2).
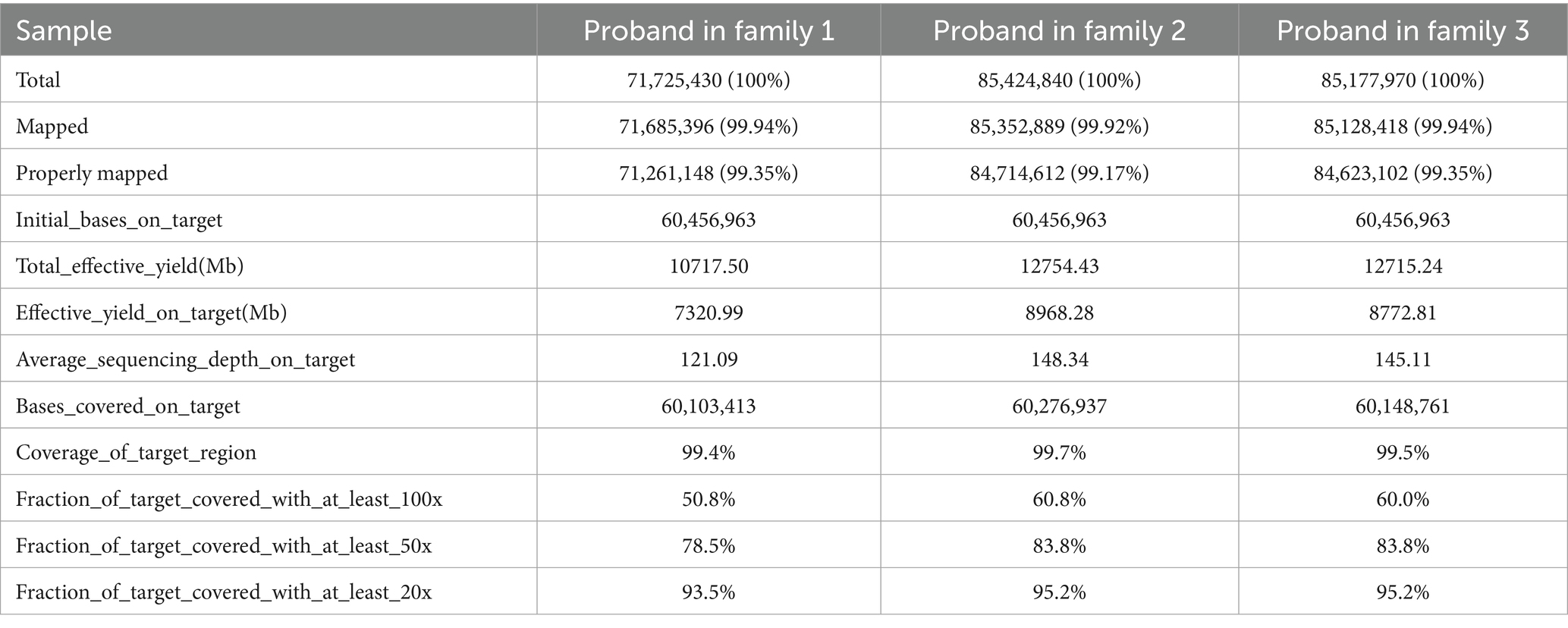
Table 1. Whole-exome sequencing detail of the proband in family 1, 2 and 3.
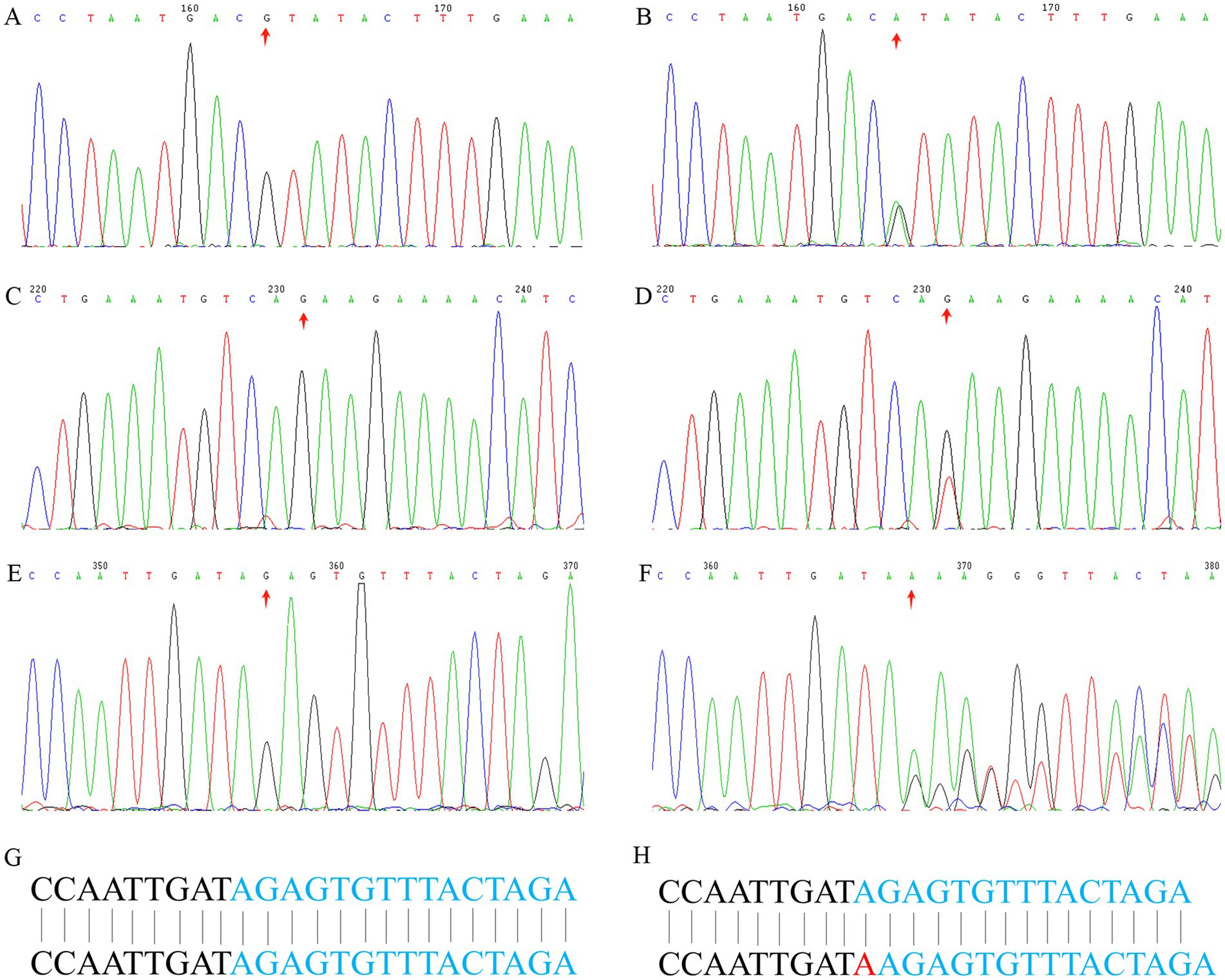
Figure 2. Sanger sequencing analysis. Sanger sequencing of MSH2 gene (c.1963G>A) of the proband in family 1: (A) wild type, (B) mutant type. A stop-gain variant (MSH2: c.2701G>T) in Family 2: (C) wild type, (D) mutant type. A frameshift insertion variant (MSH6: c.3514dupA) in Family 3: (E) wild type, (F) mutant type, (G) wild type base sequence, (H) mutant type base sequence.
Protein structure prediction
In family 1, the c.1963G>A variant caused the 655th amino acid of MSH2 to change from valine (Figure 3A) to isoleucine (Figure 3C). Protein model predictions showed that this variant was located in the β-fold region of the protein, and two hydrogen bonds with L634 were observed at a distance of 2.9 Å before (Figure 3B) and after (Figure 3D) the variant. There were no significant changes in both the overall and local protein models. In family 2, c.2701G>T variant caused the 901st amino acid of MSH2 to change from glutamic acid to a premature stop codon. The deletion of amino acids 901–934 caused changes in the Domain 5 region sequence of MSH2 protein (Figure 3E). In family 3, c.3514dupA variant caused the 1172nd amino acid of MSH6 to change from arginine to lysine, followed by frameshift, causing changes in the Domain 5 region of the MSH6 protein (Figure 3G, wild type of MSH6 shown in Figure 3F).
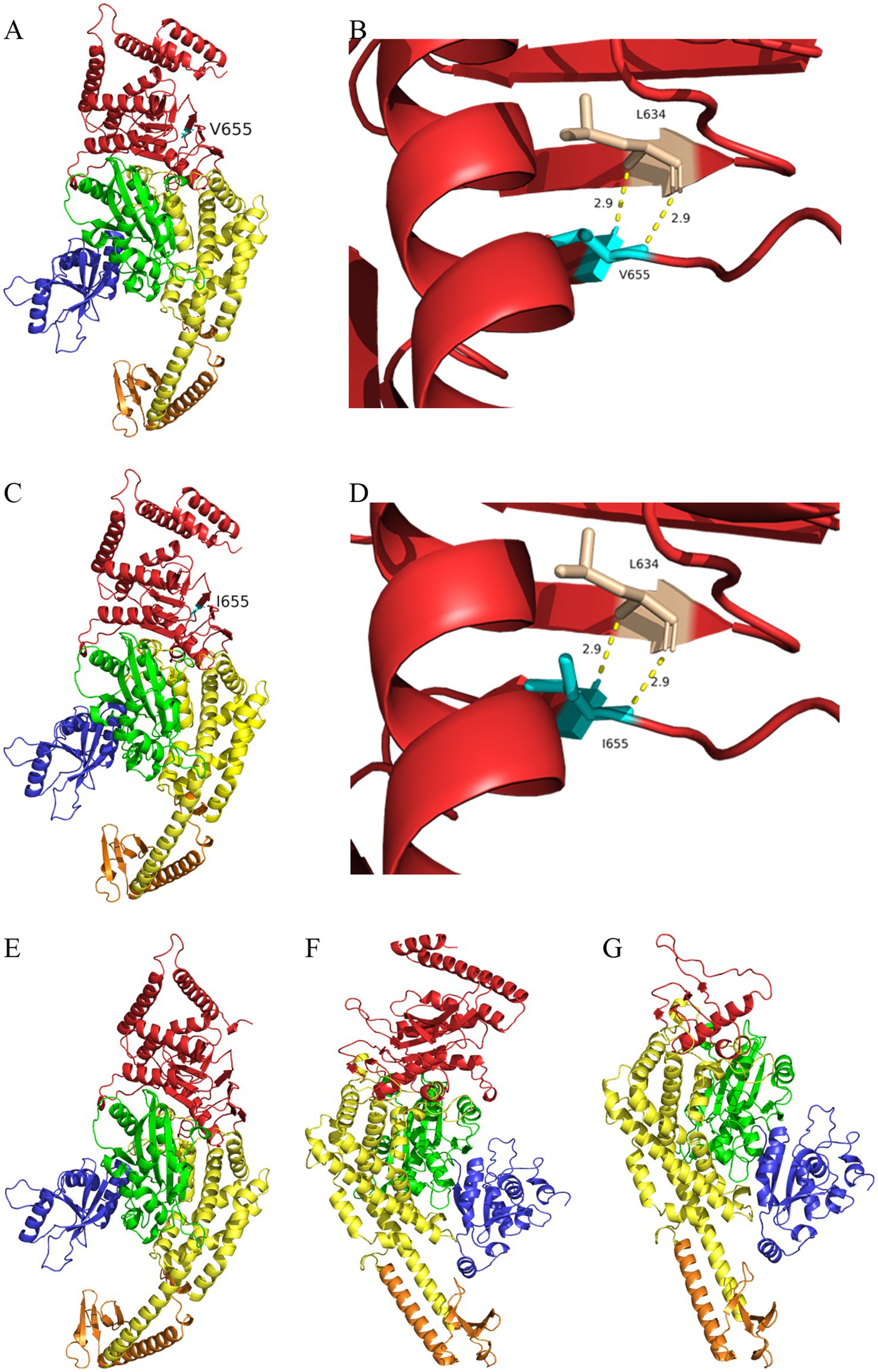
Figure 3. I-TASSER predicts the protein structure of the wild type and mutant of MSH2/MSH6 proteins. (A) Three dimensional structure of MSH2 wild-type protein (V655). (B) Partial three-dimensional structure of MSH2 wild-type protein (V655). (C) Three dimensional structure of MSH2 mutant protein (I655). (D) Partial three-dimensional structure of MSH2 mutant protein (I655). (E) The deletion of amino acids 901–934 in the mutant MSH2 protein model causing changes in the Domain 5 (red region) region sequence of MSH2 protein. (F) Three-dimensional structure of MSH6 wild-type protein. (G) The c.3514dupA variant leads to amino acid frameshift, which in turn causes changes in Domain 5 region sequence of MSH6 protein (red region).
Discussion
In this study, the missense variation (MSH2:NM_000251:c.1963G>A:p.V655I) was considered uncertain significance (PP3 + PP4), the stop-gain variant (MSH2:NM_000251:c.2701G>T:p.E901X) in MSH2 was also defined as uncertain significance (PM2 + PP3 + PP4). In addition, the known frameshift insertion variant (MSH6:NM_000179:c.3514dupA:p.R1172Kfs*5) in MSH6 was confirmed as pathogenic.
MMRs play a critical role in DNA replication, genome stability, and mutation avoidance (17–19). Under normal circumstances, strong nuclear staining is a characteristic of MMR proteins, but their loss or decrease in expression indicates a defect in the MMR system (17, 20). LS-related cancers typically exhibit characteristic loss of MMR protein expression, mainly involving MLH1 and PMS2. This loss is usually attributed to germline variations in the MLH1 gene or high methylation of the MLH1 gene promoter. This disrupts its function as a heterodimer of PMS2, resulting in the loss of immunohistochemical expression of MLH1 and PMS2. In contrast, MSH2 gene variants disrupt its function as a heterodimer of MSH6, accompanied by immunohistochemistry deletions of both MSH2 and MSH6 (17, 21, 22).
In family 1, immunohistochemical analysis of MMR proteins suggested double loss of MSH2 and MSH6 expression, and the known variant (MSH2:NM_000251:c.1963G>A:p.V655I) was identified in the proband. The variant was very rare, and the clinical significance of this variant was defined as uncertain significance in ClinVar. This variant was located in the β-fold region of the protein, and showed no significant changes in both the overall and local protein models. However, multiple statistical methods predict that the variant can have harmful effects on genes or gene products. The variant had a FATHMM score of −1.94 and MutationTaster score of 0.987549, and was defined as deleterious. MSH2 mutations account for 36% of patients with the MMR variations (23, 24). The vast majority of variants are nonsense or frameshift mutations, which lead to loss of protein function. However, 18% of MSH2 variants are single base variants, which may cause changes in one amino acid, and the ultimate impact on protein function is often uncertain, therefore, which posing a challenge for doctors and genetic counselors who must manage the disease and determine cancer risk. Therefore, the specific pathogenic mechanism of the missense variant (MSH2:NM_000251:c.1963G>A:p.V655I) needs further research.
The DNA repair system is crucial for repairing errors causing DNA replication. The MSH2-MSH6 protein complex plays an important role in maintaining the mismatch repair mechanism. An interface mutation between the two proteins can impair their function during the repair process (25, 26). In family 1, the missense variation in MSH2 could result in a large number of small fragment deletions or insertions, leading to DNA instability, however, MSH2 forms a dimer with MSH6 genes, ultimately causing double loss of MSH2 and MSH6 expression in the result of immunohistochemical staining. In family 2, the stop-gain variant of MSH2 (NM_000251:c.2701G>T:p.E901X) caused the deletion of amino acids 901–934, leading to changes in the Domain 5 region sequence of MSH2 protein. The nonsense variant in MSH2 gene leaded to the truncation of MSH2 protein or mRNA, resulting in nonsense mediated attenuation, however, MSH2 forms a dimer with MSH6 genes, ultimately causing double loss of MSH2 and MSH6 expression in the result of immunohistochemical staining. In family 3, the known frameshift insertion variant MSH6:NM_000179:c.3514dupA:p.R1172Kfs*5 caused amino acid frameshift, wherein the fifth amino acid encountered a stop codon after frameshift, ultimately resulting in partial deletion of the Domain 5 region sequence of the MSH6 protein. The frameshift variant in MSH6 resulted in the truncation of MSH6 protein or mRNA, leading to nonsense mediated attenuation and ultimately causing loss of MSH6 expression, however, MSH2 expression remained positive, which requires further exploration. Therefore, partial deletion of the structural domain of MSH2 or MSH6 might impair the MSH2-MSH6 complex, thereby impair the activity of the complex and ultimately impair the DNA mismatch repair function of the MSH2-MSH6 complex.
CRCs with deficient MMRs have sustained responses to immune checkpoint inhibitor such as monoclonal antibody against program death 1 (PD-1) (27–29). In 2017, the FDA approved pembrolizumab for the treatment of unresectable or metastatic solid tumors with microsatellite instability-high or MMR defects that have progressed after previous treatment and for adult and pediatric patients without satisfactory alternative treatment options, as well as for the treatment of CRCs with inoperable or metastatic microsatellite instability-high or MMR defects that have progressed well after treatment with fluoropyrimidine, oxaliplatin, and irinotecan (27, 28, 30). Microsatellite instability and MMR defects are interchangeably used as the first pan-cancer biomarkers for the prediction of response to anti-PD-1/PD-L1-therapy (31). Pembrolizumab is the first FDA approved cancer treatment indication based on common biomarkers rather than primary sources (28). In addition, patients with colon cancer who demonstrate microsatellite instability-high or MMR deficiencies have shown improved survival (32). A large amount of preclinical and clinical evidence suggests a possible resistance to 5-FU in these tumors with microsatellite instability-high (33, 34). Therefore, patients with LS will not benefit from fluorouracil treatment, but may be sensitive to PD-1/PD-L1 inhibitors.
At present, less than 10% of individuals undergo genetic testing for CRC in the United States, and the incidence rate of LS is severely underestimated. Furthermore, the proportion of individuals undergoing genetic testing in China is also estimated to be very low. It is recommended that family members with LS undergo genetic counseling and that LS patients or carriers of pathogenic mutations undergo gastroscopy/colonoscopy every 1–2 years (35).
In summary, in this study, the missense variation (MSH2:NM_000251:c.1963G>A:p.V655I) and the stop-gain variation (MSH2:NM_000251:c.2701G>T:p. E901X) were considered uncertain significance for LS, and the pathogenic variation (MSH6:NM_000179:c.3514dupA:p.R1172Kfs*5) was further confirmed. Genetic testing is crucial for the diagnosis and treatment of LS. Finally, patients with LS should not be treated with fluorouracil drugs, and anti-PD1/PD-L1 may be preferred.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI SRA, PRJNA1292369.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Central Hospital of Wuhan. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JL: Funding acquisition, Writing – review & editing, Writing – original draft, Data curation. HN: Writing – review & editing, Methodology, Investigation, Resources. PC: Writing – review & editing, Data curation, Investigation, Validation. YP: Writing – review & editing, Visualization, Supervision. LL: Validation, Writing – review & editing, Data curation, Supervision. XiaW: Writing – review & editing, Resources, Supervision, Data curation. WCh: Writing – review & editing, Resources, Data curation, Validation. HL: Writing – review & editing, Validation, Investigation, Visualization. XiuW: Investigation, Visualization, Formal analysis, Supervision, Writing – review & editing. HZ: Resources, Validation, Data curation, Investigation, Writing – review & editing. JH: Software, Writing – review & editing, Conceptualization, Supervision. AD: Writing – review & editing, Funding acquisition, Project administration, Resources. WCa: Project administration, Supervision, Writing – review & editing, Data curation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Major Projects of Wuhan Municipal Health Commission (No. WX18M02 and No. WX23J03), Wuhan Central Hospital Horizontal Research Project (No. 2023-024 and No. 2024-066).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Wang, T, Tao, Y, Gan, G, Chen, L, Xu, Y, Sun, F, et al. Application of high-dose-rate endorectal brachytherapy in the treatment of locally advanced rectal cancer. Precis Radiat Oncol. (2025) 9:133–42. doi: 10.1002/pro6.70004
2. Peltomaki, P, Nystrom, M, Mecklin, JP, and Seppala, TT. Lynch syndrome genetics and clinical implications. Gastroenterology. (2023) 164:783–99. doi: 10.1053/j.gastro.2022.08.058
3. Pellat, A, Netter, J, Perkins, G, Cohen, R, Coulet, F, Parc, Y, et al. Lynch syndrome: what is new? Bull Cancer. (2019) 106:647–55. doi: 10.1016/j.bulcan.2018.10.009
4. Llach, J, Pellise, M, and Monahan, K. Lynch syndrome; towards more personalized management? Best Pract Res Clin Gastroenterol. (2022) 58–59:101790. doi: 10.1016/j.bpg.2022.101790
5. Sehgal, R, Sheahan, K, O’Connell, P, Hanly, A, Martin, S, and Winter, D. Lynch syndrome: an updated review. Genes. (2014) 5:497–507. doi: 10.3390/genes5030497
6. Chen, W, Swanson, BJ, and Frankel, WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. (2017) 12:24. doi: 10.1186/s13000-017-0613-8
7. Dominguez-Valentin, M, Haupt, S, Seppälä, TT, Sampson, JR, Sunde, L, Bernstein, I, et al. Mortality by age, gene and gender in carriers of pathogenic mismatch repair gene variants receiving surveillance for early cancer diagnosis and treatment: a report from the prospective Lynch syndrome database. EClinicalMedicine. (2023) 58:101909. doi: 10.1016/j.eclinm.2023.101909
8. Cohen, SA, and Leininger, A. The genetic basis of Lynch syndrome and its implications for clinical practice and risk management. Appl Clin Genet. (2014) 7:147–58. doi: 10.2147/TACG.S51483
9. Liu, Y, Wang, M, Chen, Q, Zheng, Q, Li, G, Cheng, Q, et al. A novel heterozygous large deletion of MSH6 gene in a Chinese family with Lynch syndrome. Gene. (2019) 704:103–12. doi: 10.1016/j.gene.2019.04.011
10. Tian, T, Li, J, Xue, T, Yu, B, Li, X, Zhou, X, et al. Microsatellite instability and its associations with the clinicopathologic characteristics of diffuse large B-cell lymphoma. Cancer Med. (2020) 9:2330–2342. doi: 10.1002/cam4.2870
11. Fu, F, Tao, X, Jiang, Z, Gao, Z, Zhao, Y, Li, Y, et al. Identification of germline mutations in East-Asian young never-smokers with lung adenocarcinoma by whole-exome sequencing. Phenomics. (2023) 3:182–9. doi: 10.1007/s43657-022-00062-1
12. Cheng, WZ, Wang, WH, Deng, AP, Dang, X, Liu, C, Wang, XC, et al. Identification of an LDLR variant in a Chinese familial hypercholesterolemia and its relation to ROS/NLRP3-mediated pyroptosis in hepatic cells. J Geriatr Cardiol. (2023) 20:341–9. doi: 10.26599/1671-5411.2023.05.003
13. Bonnycastle, LL, Chines, PS, Hara, T, Huyghe, JR, Swift, AJ, Heikinheimo, P, et al. Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes. (2013) 62:3943–50. doi: 10.2337/db13-0571
14. Morazán-Fernández, D, Mora, J, and Molina-Mora, JA. In silico pipeline to identify tumor-specific antigens for cancer immunotherapy using exome sequencing data. Phenomics. (2022) 3:130–7. doi: 10.1007/s43657-022-00084-9
15. Warren, JJ, Pohlhaus, TJ, Changela, A, Iyer, RR, Modrich, PL, and Beese, LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. (2007) 26:579–92. doi: 10.1016/j.molcel.2007.04.018
16. Wang, X, Ni, H, Zhu, L, Huang, H, Deng, A, Hu, J, et al. Analyzing pathogenic variants in mismatch repair genes: personalized prevention strategies for Lynch syndrome in Chinese families. Front Med. (2025) 12:1527249. doi: 10.3389/fmed.2025.1527249
17. Miolo, G, Marus, W, Buonadonna, A, Da Ros, L, Della Puppa, L, and Corona, G. Null mismatch repair proteins expression reveals the temporal molecular events in Lynch syndrome-related cancers. Diagnostics. (2024) 14:888. doi: 10.3390/diagnostics14090888
18. Harfe, BD, and Jinks-Robertson, S. DNA mismatch repair and genetic instability. Annu Rev Genet. (2000) 34:359–99. doi: 10.1146/annurev.genet.34.1.359
19. Liu, D, Keijzers, G, and Rasmussen, LJ. DNA mismatch repair and its many roles in eukaryotic cells. Mutat Res Rev Mutat Res. (2017) 773:174–87. doi: 10.1016/j.mrrev.2017.07.001
20. Bateman, AC. DNA mismatch repair protein immunohistochemistry—an illustrated guide. Histopathology. (2021) 79:128–38. doi: 10.1111/his.14367
21. Hampel, H, Frankel, WL, Martin, E, Arnold, M, Khanduja, K, Kuebler, P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. (2005) 352:1851–60. doi: 10.1056/NEJMoa043146
22. Gomez, R, and Spampinato, CP. Mismatch recognition function of Arabidopsis thaliana MutSgamma. DNA Repair. (2013) 12:257–64. doi: 10.1016/j.dnarep.2013.01.002
23. Cyr, JL, Brown, GD, Stroop, J, and Heinen, CD. The predicted truncation from a cancer-associated variant of the MSH2 initiation codon alters activity of the MSH2-MSH6 mismatch repair complex. Mol Carcinog. (2012) 51:647–58. doi: 10.1002/mc.20838
24. Peltomaki, P, and Vasen, H. Mutations associated with HNPCC predisposition—update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. (2004) 20:269–76. doi: 10.1155/2004/305058
25. Thirumal Kumar, D, Susmita, B, Judith, E, Priyadharshini Christy, J, George Priya Doss, C, and Zayed, H. Elucidating the role of interacting residues of the MSH2-MSH6 complex in DNA repair mechanism: a computational approach. Adv Protein Chem Struct Biol. (2019) 115:325–50. doi: 10.1016/bs.apcsb.2018.11.005
26. Drotschmann, K, Hall, MC, Shcherbakova, PV, Wang, H, Erie, DA, Brownewell, FR, et al. DNA binding properties of the yeast Msh2-Msh6 and Mlh1-Pms1 heterodimers. Biol Chem. (2002) 383:969–75. doi: 10.1515/BC.2002.103
27. Le, DT, Durham, JN, Smith, KN, Wang, H, Bartlett, BR, Aulakh, LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733
28. Marcus, L, Lemery, SJ, Keegan, P, and Pazdur, R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. (2019) 25:3753–8. doi: 10.1158/1078-0432.CCR-18-4070
29. Lizardo, DY, Kuang, C, Hao, S, Yu, J, Huang, Y, and Zhang, L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: from bench to bedside. Biochim Biophys Acta Rev Cancer. (2020) 1874:188447. doi: 10.1016/j.bbcan.2020.188447
30. Le, DT, Uram, JN, Wang, H, Bartlett, BR, Kemberling, H, Eyring, AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
31. Rasmussen, M, Lim, K, Rambech, E, Andersen, MH, Svane, IM, Andersen, O, et al. Lynch syndrome-associated epithelial ovarian cancer and its immunological profile. Gynecol Oncol. (2021) 162:686–93. doi: 10.1016/j.ygyno.2021.07.001
32. Li, Z, Su, Y, Cui, Y, Yin, Y, and Li, Z. Multi-sequence MRI-based clinical-radiomics models for the preoperative prediction of microsatellite instability-high status in endometrial cancer. Precis Radiat Oncol. (2025) 9:43–53. doi: 10.1002/pro6.70000
33. Gelsomino, F, Barbolini, M, Spallanzani, A, Pugliese, G, and Cascinu, S. The evolving role of microsatellite instability in colorectal cancer: a review. Cancer Treat Rev. (2016) 51:19–26. doi: 10.1016/j.ctrv.2016.10.005
34. Sargent, DJ, Marsoni, S, Monges, G, Thibodeau, SN, Labianca, R, Hamilton, SR, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. (2010) 28:3219–26. doi: 10.1200/JCO.2009.27.1825
35. Patel, SG, Ahnen, DJ, Kinney, AY, Horick, N, Finkelstein, DM, Hill, DA, et al. Knowledge and uptake of genetic counseling and colonoscopic screening among individuals at increased risk for Lynch syndrome and their endoscopists from the family health promotion project. Am J Gastroenterol. (2016) 111:285–93. doi: 10.1038/ajg.2015.397
Keywords: whole exome sequencing, Lynch syndrome, mismatch repair gene, genetic counseling, three-dimensional structure
Citation: Li J, Ni H, Cheng P, Peng Y, Liu L, Wang X, Cheng W, Li H, Wang X, Zhang H, Hu J, Deng A and Cai W (2025) Molecular analysis of three DNA mismatch repair protein variants in Chinese families with suspected Lynch syndrome. Front. Med. 12:1635964. doi: 10.3389/fmed.2025.1635964
Edited by:
Hao Zhang, The Affiliated Hospital of Qingdao University, ChinaReviewed by:
Zheng Jin Tu, Cleveland Clinic, United StatesJoseph Christopher, University of Cambridge, United Kingdom
Anthony Bayega, McGill University, Canada
Copyright © 2025 Li, Ni, Cheng, Peng, Liu, Wang, Cheng, Li, Wang, Zhang, Hu, Deng and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongfeng Zhang, emhmMTUyQDE2My5jb20=; Jifa Hu, amlmYWh1QHNpbmEuY29t; Aiping Deng, ZGFweXhiQDE2My5jb20=; Wei Cai, Y2Fpd2VpOTk5OUBzb2h1LmNvbQ==
†These authors have contributed equally to this work