







- 1 Department of Limnology, University of Vienna, Vienna, Austria
- 2 Natural Resources Ecology Laboratory, United States Geological Survey, Colorado State University, Fort Collins, CO, USA
- 3 Department of Chemical Ecology and Ecosystem Research, University of Vienna, Vienna, Austria
- 4 WasserCluster Lunz GmbH, Lunz am See, Austria
- 5 Department of Microbiology, Institute of Plant Biology, University of Zurich, Zurich, Switzerland
Fluvial ecosystems process large quantities of dissolved organic matter as it moves from the headwater streams to the sea. In particular, hyporheic sediments are centers of high biogeochemical reactivity due to their elevated residence time and high microbial biomass and activity. However, the interaction between organic matter and microbial dynamics in the hyporheic zone remains poorly understood. We evaluated how variance in resource chemistry affected the microbial community and its associated activity in experimentally grown hyporheic biofilms. To do this we fed beech leaf leachates that differed in chemical composition to a series of bioreactors filled with sediment from a sub-alpine stream. Differences in resource chemistry resulted in differences in diversity and phylogenetic origin of microbial proteins, enzyme activity, and microbial biomass stoichiometry. Specifically, increased lignin, phenolics, and manganese in a single leachate resulted in increased phenoloxidase and peroxidase activity, elevated microbial biomass carbon:nitrogen ratio, and a greater proportion of proteins of Betaproteobacteria origin. We used this model system to attempt to link microbial form (community composition and metaproteome) with function (enzyme activity) in order to better understand the mechanisms that link resource heterogeneity to ecosystem function in stream ecosystems.
Introduction
Headwater streams are increasingly being recognized as biogeochemical hotspots and key players in the processing of organic matter as it proceeds toward the sea (Battin et al., 2008, 2009; Mulholland et al., 2008; Beaulieu et al., 2010). Intimately connected with the landscape, headwaters receive significant amounts of terrestrial organic matter, which they retain, transform, and transport to larger downstream ecosystems (e.g., Wallace et al., 1997; Bernhardt et al., 2003; Fisher et al., 2004; Battin et al., 2008; Mulholland et al., 2008). It is generally recognized that most of the processing in headwater streams occurs in the hyporheic zone, the transition within the streambed where surface water mixes with groundwater (GW; Valett et al., 1996; Boulton et al., 1998). The sediments of the hyporheic zone also offer a very large surface area for colonization by microbial biofilms, the major contributors to biogeochemical transformations in streams (Battin et al., 2003, 2008). Therefore, a mechanistic understanding of the controls on these microbial communities is of central importance in developing a broader understanding of what constrains biogeochemistry in stream ecosystems.
Inputs in allochthonous organic matter in the headwaters of lotic ecosystem can vary in chemical composition due to regional differences in flora, driven by differences in geology, topography, and climate. This in turn influences the form and function of stream microbial communities. In a large spatial survey, differences in stream microbial community composition among biomes were linked to chemical differences of the organic matter entering these streams (Findlay and Sinsabaugh, 2006). In a different study, amendments with leaf leachates affected microbial community respiration, enzymatic activity, and composition in stream sediments (Findlay et al., 2003). Furthermore, experimentally amending organic matter and nutrients to benthic stream biofilms has shown that resource chemistry may influence bacterial communities at the phyla level (Olapade and Leff, 2005). But few studies have explicitly related changes in complex resource chemistry to community structure and function using the novel molecular techniques now available to microbial ecologists.
In this study, we assess the response of hyporheic biofilm community structure and function to variations in the chemical composition of the microbial resource pool. While there are generally large differences in chemical composition (both organic and inorganic) between plant species (Osborne et al., 2007), there can also be significant differences in elemental composition within a given species (Wanek et al., 2010). In order to better understand how differences in resource chemistry affect structure and function of hyporheic biofilms we constrained the resource landscape by collecting a single plant species (European Beech, Fagus sylvatica) from various regions in Austria. This allowed us to limit the diversity of chemistry present in complex plant communities and explore how more subtle variations in chemical composition affected the structure and function of microbial biofilms.
Materials and Methods
We operated 20 Plexiglass© bioreactors (∼660 mL; Battin et al., 1999) with hyporheic sediment from a third order sub-alpine stream (Steinbach, 47°47′42.62″N, 14°57′46.03″E, 552 m above sea level). Bioreactors were packed equally with hyporheic sediment that was sieved (2–4 mm) and maintained in the dark to avoid growth of phototrophs. Steinbach drains a pristine catchment dominated by Norway Spruce (Picea abies) and European Beech (Fagus Sylvatica). We fed bioreactors with carbonate-rich groundwater (GW) mixed with different beech leaf leachates at a dilution of ∼200:1 to simulate upwelling of shallow GW mixed with terrigenous resource supply. As is typical for high-gradient and karst streams, Steinbach has pronounced hydrodynamic exchange between sub-surface water and stream water. Bioreactors were operated for 94 days in a once-through flow mode at a rate of ∼110 mL h−1 using peristaltic pumps (Ecoline VC MS/CA2, ISMATEC). Average hydrologic residence times were approximately 4 h for all bioreactors and did not differ significantly between treatments as determined from conservative tracer injections (Uranin AP, 45350, Merck).
To test the effect of resource chemistry on the sediment microbial community composition and function, we amended GW with leachates from beech leaves that differed in their organic and inorganic chemistry. Substantial loads of terrigenous dissolved organic carbon (DOC) entering the stream via shallow subsurface flow paths have been shown to subsidize hyporheic microbial metabolism in our study catchment (Battin et al., 1999). To simulate this, leachates were made from beech leaves collected from multiple locations within Austria [Schottenwald (SW): 48°14′N, 16°15′E, 370 m above sea level, Klausen-Leopoldsdorf (KL): 48°07′N, 16°03′E, 510 m above sea level, and Ossiach (OS): 46°40′N, 14°40′E, 889 m above sea level]. Dried leaves (see Wanek et al., 2010 for collection and leaf processing details) were ground to a similar particle size and then extracted in MilliQ-water for ∼ 24 h at 4°C. After centrifugation and filtration (Whatman GF/F and 0.22 μm Millipore stericup filter cups), leaf extracts were diluted with MilliQ-water to a concentration of 500 mg C L−1. Leachate concentrate was further diluted online with untreated GW in 1 L glass bottles (Schott©), continuously stirred with a magnetic mixing bar to a final input concentration of ∼2 mg C L−1. Although we did not measure the pH of the leachate directly, the substantial dilution with carbonate-rich GW ensured that the influence of leachate pH on media fed to the bioreactors was negligible. Five replicate bioreactors were assigned for each of three treatments (OS, SW, and KL) and five additional bioreactors served as a control treatment and were fed untreated GW. Input DOC concentration in the GW treatment averaged 0.7 mg C L−1. At the end of the experiment, bioreactors were sacrificed and sediment from each bioreactor was equally partitioned into an inflow, middle, and outflow section (each ∼1/3 of the total bioreactor volume) and preserved (−80°C) for further processing and analyses.
Analysis of Resource Chemistry
To evaluate the resource pool, we determined the elemental and organic chemistry of each leachate before dilution with GW. DOC was analyzed using a Sievers TOC analyzer (GE, Boulder, CO, USA). Inorganic nutrients (P–PO4, N–NO3, N–NO2, N–NH4) were determined using a flow-through autoanalyzer (SYSTEA Analytical Technologies, Anagni, Italy) and metal concentrations were determined using ICP-MS. Differences in organic chemistry between leachates were determined by Curie-point-pyrolysis–GC/MS using a GSG Pyromat (GSG Mess und Analysegeräte, Bruchsal, Germany) coupled to a Thermo Trace GC and a DSQ MS detector (Thermo Scientific). Organic material was pyrolyzed at 590°C and pyrolysates separated on a carbowax column (Supelcowax 10, Sigma-Aldrich) with a linear temperature gradient from 50 to 260°C in 30 min. Peaks were assigned based on NiSt 05 MS library and comparison with reference material measured. Pyrolysis products were assigned to their substances of origin by comparison to reference material and structural similarity in accordance with Schellekens et al. (2009). In total 39 peaks were selected for high peak area and diagnostic value. Peak areas are reported as a fraction of total area of all integrated peaks of a sample (Table 1).
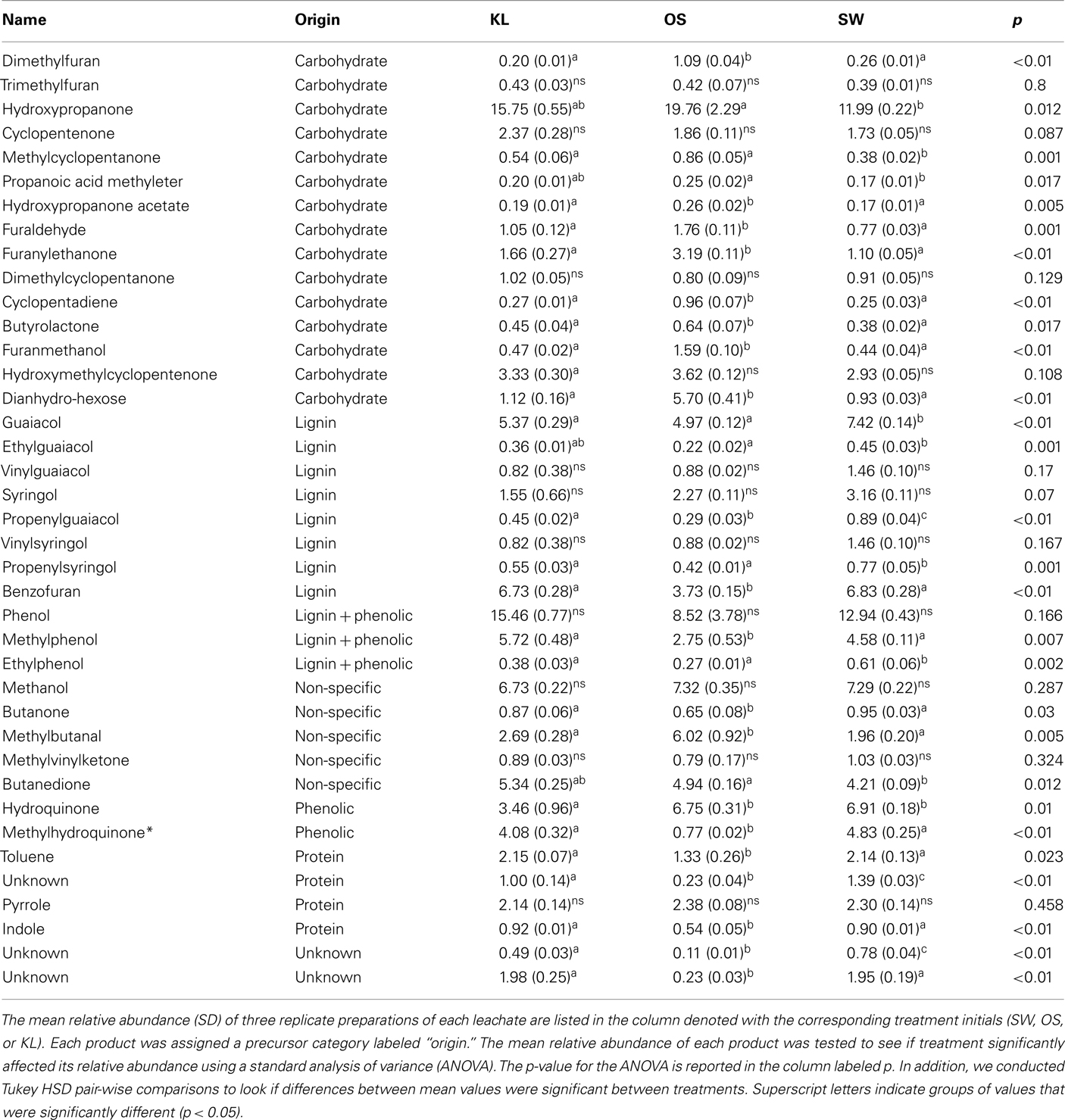
Table 1. Lists the 39 pyrolysis products analyzed using Curie-point-pyrolysis–GC/MS.
Biofilm Analyses
Enzyme Assays
For all assays, after deconstruction of the bioreactors, samples of alliquoted sediment were sonicated gently (1 min, Branson Digital Sonifier 250, 13% amplitude, 1 s pulse) to detach biofilms from the sediments as described below. The slurry was placed into triplicate microtiter well containing the respective substrate and the incubation was carried out at room temperature over 48 h with repeated measurements (Sinsabaugh et al., 1999). Fluorometrically labeled [4-methylumbelliferone (MUF) and 7-amino-4-methyl coumarin (AMC)] substrates were used for the measurement of cellulase (MUF-cellobioside), chitinase (MUF-triacetylchitotrioside), phosphatase (MUF-phosphate), and protease (leucine-AMC) at 1, 0.5, 2, and 0.5 mM concentrations, respectively. Peroxidase and phenoloxidase activity were also measured fluorometrically by adding sample and substrate [20 mM L-3,4-dihydroxyphenylalanin (DOPA) in Na-acetate buffer] in a 1:1 ratio (200 μl sample + 200 μl substrate) for phenoloxidase activity. Peroxidase activity was measured in the same manner as phenoloxidase, but with additional 10 μl of 0.3% H2O2 added 10 min prior to addition of DOPA as a substrate (Sinsabaugh et al., 1999).
Elemental analysis of microbial biomass
To analyze elemental content of the microbial communities, a known volume of sediment was mixed with MilliQ-water (millipore; 15 mL) in sterile centrifuge tubes (BD Falcon). Samples were thoroughly vortexed (10 s) and sonicated (1 min, Branson Digital Sonifier 250, 13% amplitude, 1 s pulse, e.g., Battin et al., 2003). After sonication the supernatant was decanted, the sediment rinsed twice with MilliQ-water (total 10 mL), and decanted again. The supernatant containing the microbial biomass was frozen (−80°C) and lyophilized. Between 1.5 and 5 mg of lyophilized biomass (including mineral particles) was weighed into Eppendorf tubes and treated with HCl (10% w/v, p.a.) to remove inorganic carbon. Samples were dried (60°C, 48 h), re-suspended in MilliQ-water, transferred into tin capsules (Lüdi AG, cat. no. 184.9927.26) and dried again (60°C, 48 h). Carbon and nitrogen content were determined using an elemental analyzer (EA1110, CE Instruments ThermoFisher), and phosphorus content was determined using the persulfate digestion method (Eaton et al., 2005).
Microbial community analysis
To assess relative differences between the most abundant taxa among treatments terminal-restriction fragment length polymorphism (T-RFLP) fingerprinting was conducted on all three sections of each bioreactor. In addition, in order to evaluate differences in total microbial diversity between treatments, for a subset of the samples (inflow section from three replicates of each of the four treatments), we analyzed the community 16S rRNA genes with 454 pyrosequencing. For both analyses DNA was extracted from sediments using a Power Soil DNA extraction Kit (MoBIO, cat no. 12888-100; Besemer et al., 2009). For T-RFLP we used the hexachlorofluorescein-labeled bacteria-specific primer 27F (5′-AGR GTT TGA TCM TGG CTC AG-3′) and the universal primer 519R (5′-GWA TTA CCG CGG CKG CTG-3′; Eiler and Bertilsson, 2007). The 16S rRNA gene was amplified using an initial denaturation step of 94°C for 3 min, followed by 30 cycles of denaturation at 94°C (40 s), annealing at 55°C (40 s), and extension at 72°C (1 min). After PCR, amplicons were cleaned using the PCR Purification Kit (Qiagen cat no. #28106; Moeseneder et al., 1999) and were digested with the restriction enzyme Hha1 (Besemer et al., 2009). T-RFLP profiles were produced by running the samples on a 3130 XL sequencer (Applied Biosystems) using the default settings for fragment analysis with 50 cm capillary length and POP7 gel matrix (Applied Biosystems). Size standards (MM-1000-ROX, BioVentures Inc.) and operational taxonomic units (OTUs) were assigned using a freeware designed for T-RFLP analysis in the computing environment R (Jones and McMahon, 2009).
For 454 pyrosequencing, we amplified the V4 and V5 regions of bacterial 16S rRNA genes using the fusion primers adaptor A-MID-515F (GTG NCA GCM GCC GCG GTA A) and adaptor B-926R (CCG YCA ATT YMT TTR AGT TT; Quince et al., 2011) containing the GS FLX Titanium sequencing adapters and in case of the forward primer a sample specific 8 bp multiplex identifier (Hamady et al., 2008). PCR was performed using Phusion High-Fidelity DNA Polymerase (Finnzymes) according to the manufacturers recommendations. Samples were amplified using a 25-cycle touchdown PCR protocol with an initial annealing temperature of 62°C decreasing 0.5°C with every cycle. Each sample was amplified using two different multiplex identifiers and each PCR was performed in duplicate reactions to account for PCR drift. PCR-products were cleaned with the QIAquick Gel Extraction Kit (Qiagen) and quantified using agarose gel electrophoresis and the Low DNA Mass Ladder (Invitrogen). Equal amounts of the barcoded PCR-products were mixed and submitted to the Centre for Genomic Research (Liverpool, UK) for pyrosequencing on a 454 GS20 FLX Titanium platform. Pyrosequencing data were cleaned using the software package AmpliconNoiseV1.21 following the procedure published by Quince et al. (2011). The cleaned reads were clustered to OTUs with a complete linkage algorithm on a 97% sequence identity level, yielding a clean dataset of 47,267 reads constituting 1,509 OTUs. The microbial diversity of the samples was calculated from Bayesian fitting of Sichel distribution curves to the abundance distributions and as the Chao estimator of richness using the Bayesian Diversity Estimation software (Quince et al., 2008). The sequence data have been submitted to the NCBI Sequence Read Archive under accession number SRX099855.
Metaproteome analysis
Total proteins from the inflow section of three replicate bioreactors were extracted from ca. 40 g of sediment in 20 mL of extraction buffer [50 mM Tris, pH 7.5 (NaOH), 1% SDS]. Samples were vortexed vigorously (2 min), sonicated (5 min) on ice, boiled (15 min), and gently shaken (4°C, 1 h). Samples were then centrifuged (1000 g, 5 min, 4°C) and the supernatant containing the extracted proteins was transferred into a sterile tube. This procedure was repeated on the sediment, and the supernatants pooled. Proteins in the supernatant were concentrated and purified by acetone precipitation (acetone: sample 5:1) overnight (−20°C). The resulting pellets were re-suspended in 1 mL 50 mM Tris, pH 7.5 (NaOH), 1% SDS. Equal amounts (30 μl) of re-suspended protein pellet were separated by SDS-PAGE (Laemmli, 1970) using 12% polyacrylamide gels. Protein lanes were cut into four slices and gel slices were immediately subjected to in-gel tryptic digestion (Shevchenko et al., 1996). The resulting peptide mixtures were analyzed on a hybrid LTQ-Orbitrap mass spectrometer (ThermoFischer Scientific, Bremen, Germany) interfaced with a nanoelectrospray ion source as described earlier (Schneider et al., 2010)
We conducted protein database searches using the Mascot Search Engine (version no. 2.2.04). The reference database contained all proteins from UniRef100 (9808438 entries, downloaded from the European Bioinformatics Institute webpage http://www.ebi.ac.uk/uniref/on January, 26th 2010) and protein sequence information from a metagenome of microbial community of a Minnesota farm silage soil (Tringe et al., 2005; 184,374 entries, downloaded from http://img.jgi.doe.gov on October 15th, 2009) as well as common contaminants like keratin and trypsin (total number of entries 9,993,117). All MS/MS ion searches were performed as described in Schneider et al. (2010).
To validate and quantify MS/MS-based peptide and protein identifications we used Scaffold (version Scaffold 2.02.01, Proteome Software Inc., Portland, OR, USA). Peptide identifications were accepted if they could be established at greater than 95% probability as specified by the Peptide Prophet algorithm (Keller et al., 2002). Protein identifications were accepted if they could be established at greater than 90% probability and contained at least one identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003).
Semi-quantitative analyses of protein abundances were performed on the basis of the number of unique spectra that were assigned to a protein by the Scaffold software. Finally, proteins were assigned to functional and phylogenetic groups followed by a validation of this process and quantification of protein abundances based on normalized spectral abundance factors (Florens et al., 2006) using the proteomics result pruning and homology group annotation engine (ProPHANE, Schneider et al., 2011).
Statistical Analyses
All statistical analyses were performed with the analytical software JMP or using the software and statistical computing environment R. Direct comparisons of values between treatments were analyzed with an ANOVA and using a post hoc Tukey HSD for direct pair-wise comparisons as specified in the text.
Results
Resource Chemistry
Although all leachates were made from the same plant species we found marked differences in the chemistry (both organic and inorganic) of the individual treatments. Most elements increased in elemental content from KL to SW to OS, respectively (Figure 1). However, SW was significantly (Tukey HSD, p < 0.05) depleted in Na and enriched in Mn relative to the other treatments. Notably the Mn concentration in SW was at least twice as high as in the other two leachates (Figure 1). GW, which was the base of the resource supply, contained insignificant amounts of all elements with the exception of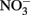 , which was three orders of magnitude higher than that of the leachates and thus negated any difference in dissolved nitrogen between treatments (Figure 1). In addition, the leachates differed in organic compounds as determined by Curie-point-pyrolysis–GC/MS (Table 1). Notably, the SW treatment was significantly enriched for three of eight lignin products, and one of three products attributed to phenolics and lignin, relative to the other two treatments (Table 1). When pyrolysis products were grouped by category, SW was significantly enriched in both lignin and phenolics categories, while OS was significantly enriched in carbohydrate biomarkers and depleted in protein biomarkers relative to the other treatments (Table 2).
, which was three orders of magnitude higher than that of the leachates and thus negated any difference in dissolved nitrogen between treatments (Figure 1). In addition, the leachates differed in organic compounds as determined by Curie-point-pyrolysis–GC/MS (Table 1). Notably, the SW treatment was significantly enriched for three of eight lignin products, and one of three products attributed to phenolics and lignin, relative to the other two treatments (Table 1). When pyrolysis products were grouped by category, SW was significantly enriched in both lignin and phenolics categories, while OS was significantly enriched in carbohydrate biomarkers and depleted in protein biomarkers relative to the other treatments (Table 2).
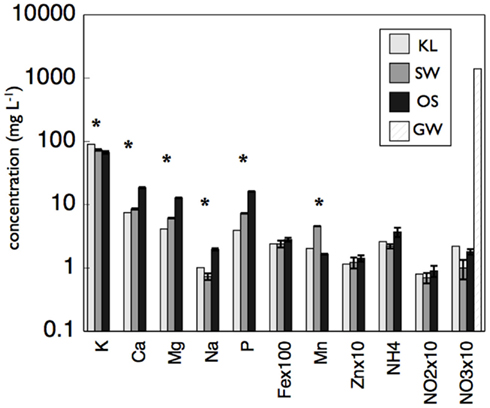
Figure 1. Concentration of metals, NH4, NOx, and phosphorus (P) in concentrated beech leaf extract at a dissolved organic carbon concentration of 500 mg L−1. We report average concentration from concentrate made on three separate dates. Error bars represent ±1 SD. An asterisk indicates a significant difference (p < 0.05) between all three leachates as determined by Tukey HSD post hoc analysis. The leachate treatments were not significantly different in 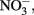 but the GW was significantly higher than all leachate treatments.
but the GW was significantly higher than all leachate treatments.
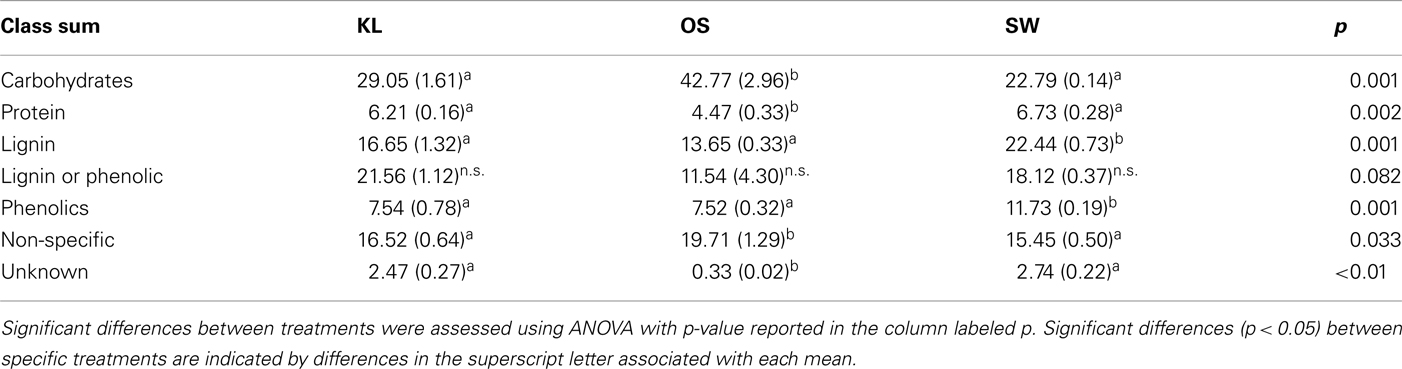
Table 2. Reports the relative mean value (SD) of curie-point-pyrolysis–GC/MS product classes.
Microbial Community Composition
The biofilm associated microbial communities differed in their composition between treatments as assessed by T-RFLP (Figure 2A). A multi-dimensional scaling (MDS) of the Jacquard similarity index indicated that the GW, KL, and OS treatments clustered separately from each other, with some overlap, and were spread along the same plane in MDS space. A portion of the within-cluster variance could be attributed to the section of the bioreactor from which the community was derived (Figure 2A). The SW treatment also showed differences in the community composition between sections of the bioreactor. However, the SW treatment primarily varied along the primary MDS axis while the other three treatments were spread equally along both axes (Figure 2A). A MDS plot of a Bray–Curtis dissimilarity index generated from 454 pyrosequencing data of the inflow section of each treatment showed that the communities separated clearly by treatment with the GW treatment being the most dissimilar from the other treatments (Figure 2B). However, there were no pronounced differences in the most abundant phylogenetic groups between treatments (Figure 3). Generally, the Proteobacteria (alpha-, beta-, gamma-, and delta-proteobacteria) constituted a higher percentage in KL, SW, and OS compared to the GW treatment, with the highest contribution to the SW community. Microbial diversity calculated as the Chao estimator and the Bayesian estimate of richness (Quince et al., 2008) was similar between treatments (Figure 4). Thus, while there were treatment level differences in the community composition as assessed by T-RFLP and, for the inflow section communities, as assessed by pyrosequencing, the microbial diversity did not differ significantly between treatments that received different leachates (Figure 4).
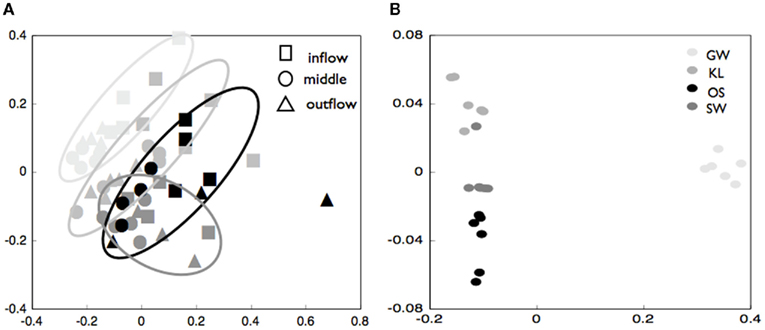
Figure 2. (A) Multi-dimensional scaling (MDS) plot of Jacquard similarity index of terminal-restriction fragment length polymorphism data for community biofilms from the inflow, middle, and outflow section of each reactor as noted in the figure legend. The GW treatment is plotted is in the lightest color, with KL light gray, SW intermediate gray, and OS black symbols. Symbols are grouped with a hand-drawn ellipses to better illustrate how symbols cluster in MDS space. (B) Multi-dimensional scaling (MDS) of Bray–Curtis similarity index for pyrosequencing data for the inflow slice of triplicate reactors from each of four treatments.
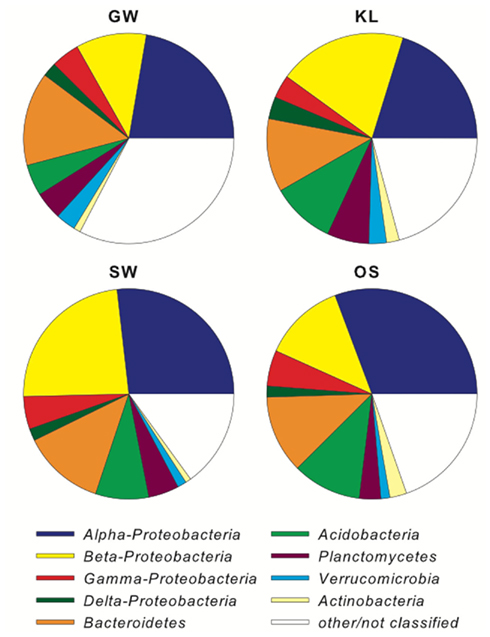
Figure 3. Shows the phylogenetic distribution of the major bacterial phyla between treatments as determined by pyrosequencing of the 16S rRNA gene amplicons.
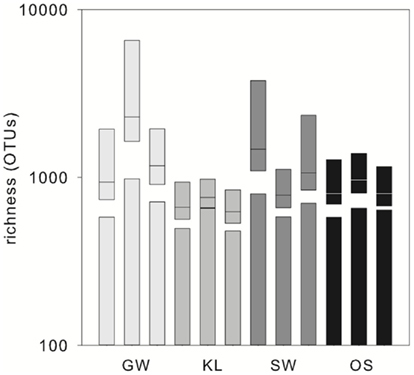
Figure 4. OTU richness in the inflow sections of the triplicate bioreactors. Bars represent the Chao estimator of richness, boxplots represent the richness estimates (medians with 95% confidence interval), calculated by fitting Sichel distribution curves to the abundance distributions obtained from the pyrosequencing data.
Biomass Stoichiometry and Extracellular Enzyme Activity
We also found significant differences in both biomass stoichiometry and enzyme activity between leachate treatments (Figures 5 and 6), but only when sections of the bioreactors were evaluated separately. Biomass stoichiometry differed significantly between treatments primarily in the inflow section of the bioreactor (Figure 5). SW had higher C:N but lower C:P and N:P biomass than either KL or OS, however this difference was only significant (ANOVA, p = 0.001) for C:N. In addition, while the mean values for elemental ratios were similar within each treatment between sections of the bioreactors, variance in biomass stoichiometry increased along the flowpath of the reactor (Figure 5). This was most pronounced for C:N of all treatments.
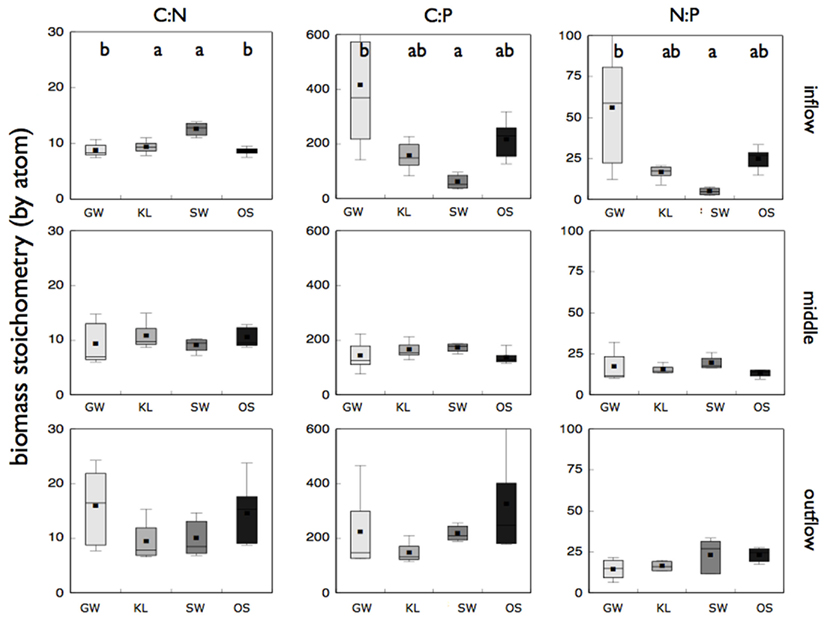
Figure 5. Reports the molar ratios of the microbial biofilm biomass from the inflow, middle, and outflow section of the bioreactors for each treatment as labeled. Treatments that are significantly different (p < 0.05) from one another are denoted with a different letter (a, b, or c) as determined by a Tukey HSD post hoc analysis. If no letters are present each treatment was statistically indistinguishable for that particular bioreactor section and molar ratio combination.
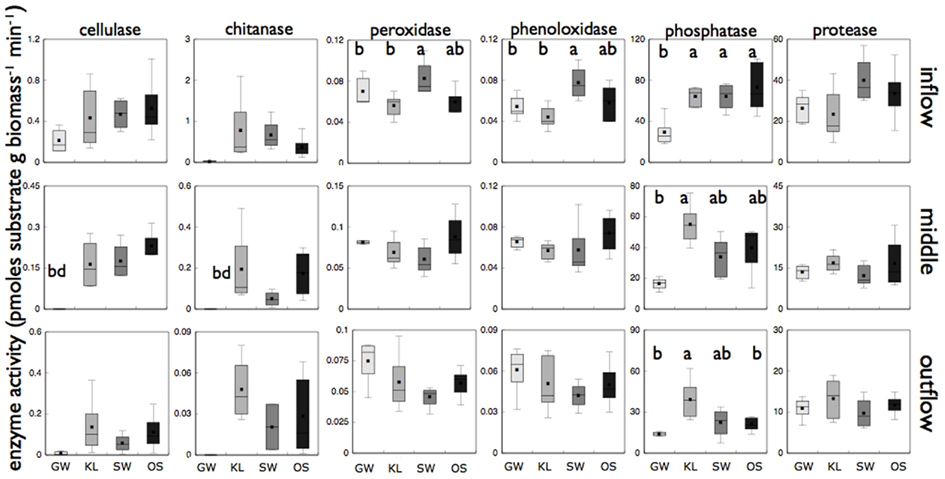
Figure 6. Reports biomass specific enzyme activity for each of six different enzyme assays for microbial biofilms sampled from the inflow, middle, and outflow section of each reactor. “bd” Indicates enzyme activity was below detection for that treatment. Treatments that are significantly different from one another are denoted with a different letter (a, b, or c) as determined by Tukey HSD post hoc analysis. If no letters are present each treatment was statistically indistinguishable.
The SW treatment also had elevated levels of oxidative enzymes as compared to the other treatments (Figure 6). Both phenoloxidase activity and peroxidase activity were significantly higher (Tukey HSD, p < 0.05) for SW than for KL and GW while OS was intermediate between each in the inflow slice. In the outflow slice, only phosphatase activity was significantly higher in the KL (the treatment with the lowest phosphorus concentration) than SW and GW treatment while OS was intermediate. Similar to community composition there were differences in enzyme activity between sections of the bioreactor, with enzyme activity decreasing along the flowpath of the bioreactor. However, with the exception for low phosphatase activity in the GW treatment relative to the other treatments we found no significant differences in enzyme activity between treatments in the middle section or for any of the other enzymes we assayed (Figure 6).
Metaproteome
Differences in enzyme activity between treatments led us to evaluate the origin of the proteins in our samples. To do so we analyzed the metaproteome for the inflow section for three bioreactors from each of the four treatments. While we did not directly identify any proteins in the metaproteome that were related to oxidative enzyme activity, we were able to gain insight into differences of the active microbial communities between treatments. Specifically, between 60 and 95% of all spectra to which a phylogenetic origin could be assigned were contributed by bacteria with additional spectra associated with protists or metazoans, and virtually none from fungi or plants (Figure 7A). Further analysis of the metaproteome revealed differences in the total number of assigned spectra in each treatment. While OS had the highest total spectra (891), SW had the most unique spectra (464, Figure 7B). In addition, while 13% of all assigned spectra were cosmopolitan (i.e., shared by all treatments), SW shared only 12–13% additional spectra with KL or OS while KL and OS shared ca. 40% of spectra between each other (Figure 7B). Differences in spectra abundance and percent of shared spectra between treatments were supported by differences in phylogenetic assignment of spectra. While virtually all identifiable bacterial spectra were assigned to Proteobacteria (Figure 7C). SW treatment had significantly more alpha and betaproteobacteria spectra, and fewer gammaproteobacteria spectra than either OS or KL (Figure 8). Comparison of the metaproteome with the pyrosequencing data of the Proteobacteria alone showed a clearly higher proportion of Gammaproteobacteria and a clearly lower proportion of Alphaproteobacteria in the metaproteome (Figure 8).
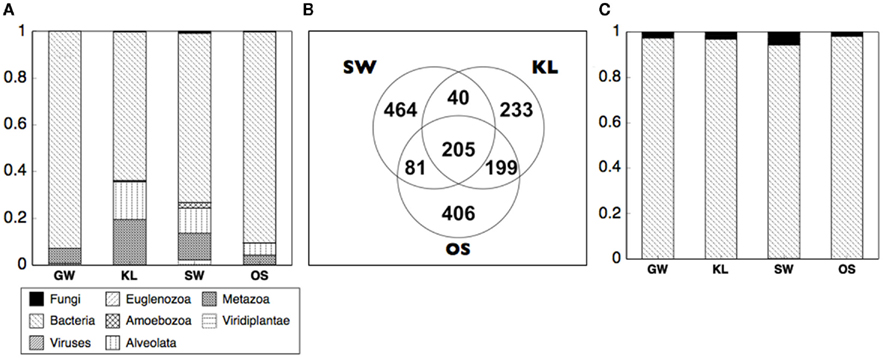
Figure 7. Summarizes the analysis of the metaproteome from the inflow section of the bioreactors for each treatment. (A) The proportional abundance of spectra that could be designated to a phylogenetic origin are dominated by spectra of bacterial origin. (B) SW treatment had the most endemic spectra and shared the fewest spectra with the other two leachate treatments. Within the bacteria (C) spectra were almost entirely from the Proteobacteria.
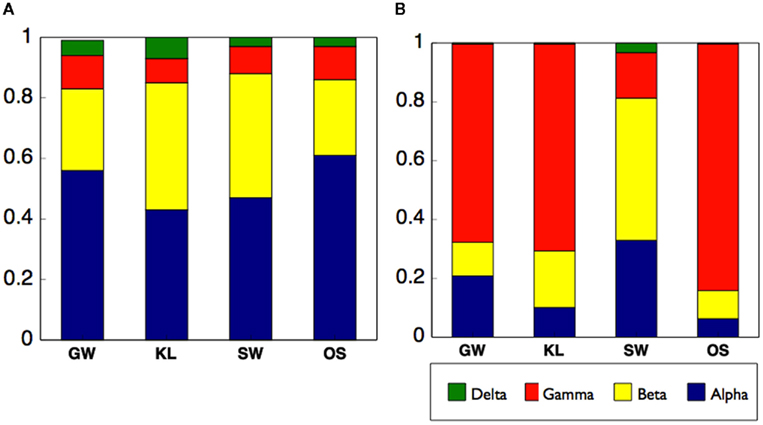
Figure 8. Compares the distribution of the Proteobacteria among treatments using (A) 16S rRNA sequences as a metric of the total Proteobacteria community, and (B) phylogenetic assignment of the metaproteome as a metric of the active Proteobacteria community.
Discussion
The exceptional chemical and biological diversity inherent in aquatic ecosystems makes it challenging to understand the mechanisms that underlie the relationship between the two. We attempted to limit the chemical diversity by focusing on the extracted DOC from leaves of a single tree species. While the system we chose to address remains reasonably complicated, we were able to arrive at some insight into the drivers of microbial community structure and function. In all of the treatments we noticed shifts in community composition (T-RFLP), variance in biomass stoichiometry, and enzyme activity along the flow path of the reactor indicative of well described patterns of resource gradients that occur in hyporheic sediments (Boulton et al., 1998). However, we focused on the pronounced differences between the resource treatments, and in particular in SW relative to the other treatments, to identify how resource chemistry affected microbial community composition and function.
Specifically, we found that differences between the beech leaf leachates led to differences in composition and activity of the microbial community in SW relative to the other leachate treatments. Notably the SW treatment significantly differed from the other two treatments in biomass stoichiometry, enzyme activity, and metaproteome composition. Analysis of the resource pool for each treatment suggested that the form and function of the microbial community of the SW treatment was affected by significant differences in organic chemistry (i.e., elevated levels of compounds assigned to the lignin and phenolics class) and Mn content of the SW leachate. Furthermore, differences in community composition of the most abundant phyla (as determined by T-RFLP) were also most pronounced in SW relative to the other treatments (Figure 2A), although there were no clear differences in total microbial diversity (as indicated by the 454 pyrosequencing data) between treatments that received leachate (Figure 4). Thus, multiple results indicated that differences in the composition of the SW leachate drove differences in community composition and function of the microbial communities in the SW treatment. This suggests a relationship between microbial form and function that was detectable at a relatively coarse level of phylogenetic resolution. The idea that composition and function of microbial communities may be evident at higher taxonomic levels has been suggested previously (Phillippot et al., 2010). In the current study, we show that these broad phylogenetic shifts in the active community (metaproteome) may be attributed to relatively few phyla and thus suggests one approach to dealing with the exceptional diversity of environmental microbial communities.
The SW community, compared to the other two leachate treatments, had elevated peroxidase and phenoloxidase activity on the same order of magnitude as those reported for soil ecosystems (Figure 6; Sinsabaugh, 2010). Enzymes that express peroxidase and phenoloxidase activity are involved in the breakdown of complex organic substrates, such as lignin. Activity of both of these families of enzymes is known to increase with increasing levels of lignin and complex organic compounds (Sinsabaugh, 2010). Differences in the organic composition among our leachates should have been greatly reduced by using leachate extracted from a single tree species. However, we found SW to be enriched in both lignin and phenolic derivatives relative to the other two leachates (Tables 1 and 2). These differences in organic chemistry between treatments suggested that an increased abundance of complex organics (i.e., organic carbon molecules enriched in aromatic moieties) in the SW treatment may have stimulated phenoloxidase and peroxidase activity of the microbial community.
Previous work on stream biofilms have shown explicit links between organic matter composition and differences in bacterial community composition. For example, a study on biofilms grown on substrate-amended clay pots found that low molecular weight labile carbon enriched for Gammaproteobacteria relative to Alpha and Betaproteobacteria (Olapade and Leff, 2005). A more recent marine study similarly showed that production of high levels of labile dissolved organic matter also enriched for Gammaproteobacteria (Condon et al., 2011). In the current study the SW treatment only showed slightly different total community composition in the T-RFLP analysis, and was not more dissimilar than the other two leachate treatments when the more rare phyla were considered using pyrosequencing data (Figure 2).
However, SW did stand apart from the other treatments in our analysis of the active community as indicated by the metaproteome. The phylogenetic assignment of the spectra from the metaproteome revealed shifts in the relative abundance of phyla between treatments with SW being most distinct from the other two treatments that received leachate (Figure 7B). Specifically, we saw a decrease in spectra assigned to Gammaproteobacteria and an increase in the Alpha- and Beta-proteobacteria in the SW treatment (Figure 8B). While an enrichment of spectra of Gammaproteobacteria origin in the metaproteome is consistent with an increased supply of labile carbon in the KL and OS treatments, we did not see a pronounced relative increase in the Gammaproteobacteria sequences as determined by sequencing of the 16S amplicon (Figure 8A). This may be due to the dynamic nature of the flow-through bioreactor system with differences in enzyme activity being reflected in the structure of the active community (metaproteome) but not the total community (16S rRNA gene).
It is also possible that differences in phenoloxidase and peroxidase activity were stimulated by the elevated level of Mn which was approximately two times greater in SW than the other two treatments (Figure 1). There are a set of phenoloxidases and peroxidases whose activity is associated with Mn (Sinsabaugh, 2010). Mn is oxidized by both phenoloxidase and peroxidase enzymes and then reacts with the bonds in complex carbon molecules such as lignin. In previous studies the breakdown of terrestrial organic matter has been shown to be stimulated by Mn concentration in leaf litter. A study of pine species from a wide range of habitats found Mn to be the single most important factor in determining the decomposition rates of pine litter (Berg et al., 2010). A separate study of a more diverse litter types (i.e., pine, spruce, alder, birch) also found that late stage decomposition was strongly correlated with a threshold Mn concentration near 2 mg Mn g−1 dry mass litter (Berg et al., 2007). Mn concentrations of the dry litter used to produce leachate in this study were 0.74, 1.37, and 2.15 mg Mn g−1 litter dry mass for OS, KL, and SW, respectively (unpublished). Thus, at least in the dry leaf litter, Mn concentration of SW was in the range that has been shown to have a significant effect on carbon processing in terrestrial decomposition studies. These differences in Mn between treatments were maintained in the dissolved phase with Mn concentrations in the SW being at least twice as high as in OS or KL (Figure 1).
While the above studies used inherent variation in litter Mn concentration as we did, other studies have more directly evaluated the effect of Mn concentrations on litter decomposition and microbial activity by experimentally manipulating Mn levels. Additions of Mn to beech leaf litter stimulated microbial respiration, leading to increased CO2 production and decreased DOC release from the remaining litter over the course of a year-long incubation (Trum et al., 2010). In addition, Mn has also been shown to stimulate peroxidase activity at the organismal level. A study using a pure culture of a forest soil Basidiomycete showed that increased Mn concentrations increased the expression of Mn peroxidase resulting in increased decomposition of humic acids in the culture (Steffen et al., 2002). This last study provides direct mechanistic support for Mn stimulation of microbially derived enzyme activity involved in breakdown on complex organic substrates (Steffen et al., 2002).
While in the current study fungi played a minor role, if any, there are several bacteria known to be directly involved in the oxidation of Mn including Leptothrix sp. and members of the Hyphomicrobium (Nealson, 2006). Our 16S pyrosequence data revealed four hits for Leptothrix sp. and 21 hits for Hyphomicrobium, however, these were distributed across the treatments and were not overrepresented in SW compared to the other treatments. Other known Mn oxidizers in the Aurantimonas family and Metallogenium group were absent from the 454 pyrosequence data and the metaproteome.
Further analysis of the metaproteome spectra that were enriched in SW relative to the other treatments did not reveal any spectra that were related to oxidases or associated with Mn (Table 3). However, the shifts in Proteobacteria classes observed in the metaproteome between SW and the other leachate treatments could be attributed to relatively few phyla within each class of Proteobacteria. The decrease in the SW treatment of the Gammaproteobacteria was primarily due to the absence of spectra attributed to members of the family Pseudomonadaceae and in particular Pseudomonas aeruginosa (Table 3). Similarly increases in spectra attributed to the Betaproteobacteria in SW relative to the other treatments were due to increases in spectra attributed to Dechloromonas aromatica and a handful of other species, while increases in Alphaproteobacteria classes could be attributed to increases in spectra from the family Sphingomonadaceae and Sphingopyxis alaskensis in particular (Table 3).
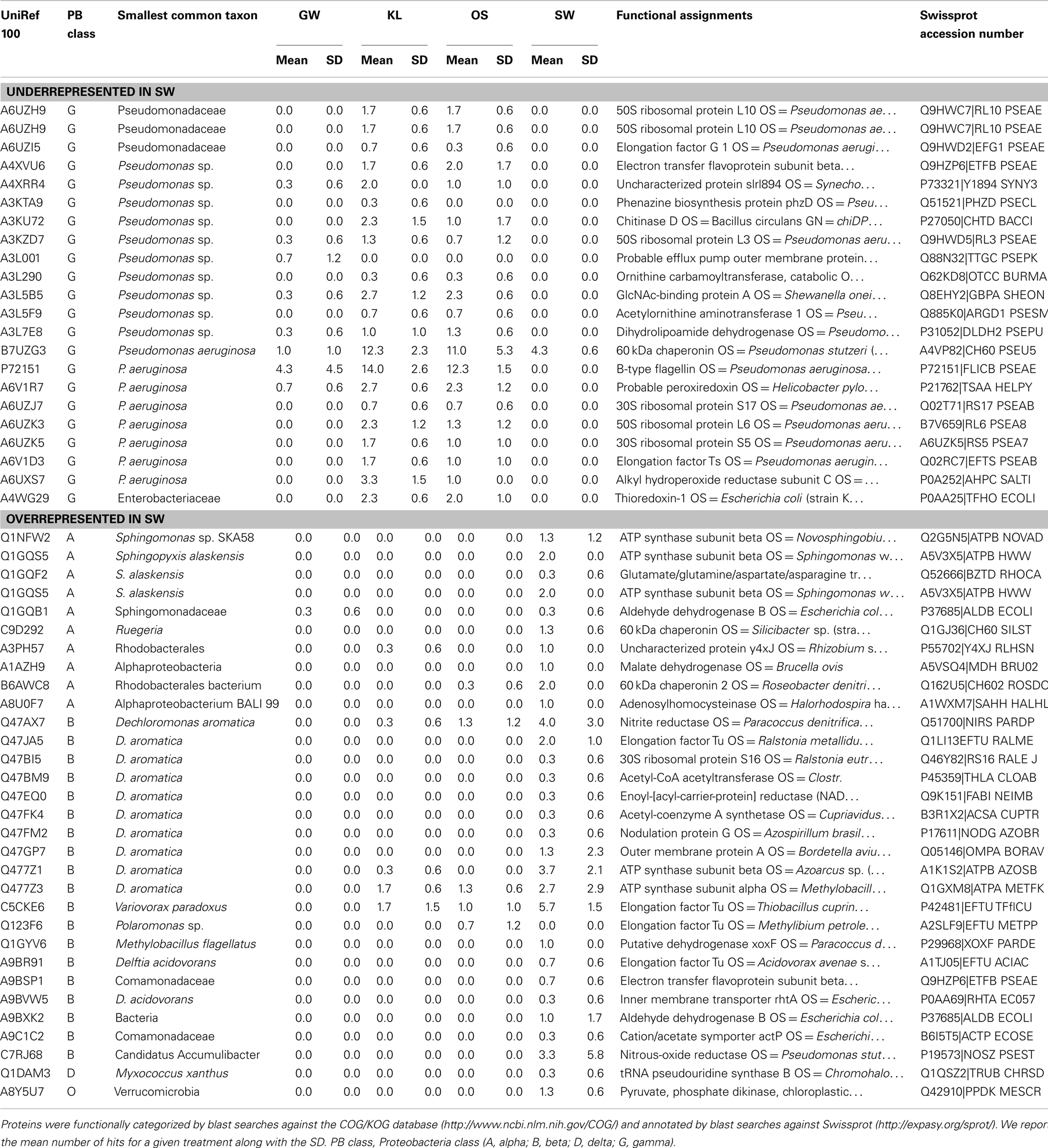
Table 3. Phylogenetic and functional assignment of identified proteins that were either overrepresented in the SW treatment or those that were underrepresented in the SW treatment.
While P. aeruginosa is a cosmopolitan species with diverse metabolism, D. aromatica and S. alexensis are a noted for unique metabolisms (Cavicchioli et al., 2003; Salinero et al., 2009), specifically their ability to degrade aromatic compounds and proliferate in oligotrophic environments, respectively. Enrichment of D. aromatica in the SW treatment is consistent with the higher level of phenol and lignin in the SW treatment. In addition, an overview of the annotated genes in the genome of each D. aromatica and S. alaskensis showed several genes that code for functions that required Mn and/or were involved in oxidative processes. These included a series of Mn or Mn/Zn transporters present in D. aromatica (e.g., ABC-type Mn/Zn transport systems, COG1121 and COG1108, and a zinc/manganese transport system substrate-binding protein) and superoxide dismutases with a Mn co-factor thought to be involved in oxidative stress present in the genome of both D. aromatica and S. alaskensis [e.g., superoxide dismutase, Fe–Mn family (EC:1.15.1.1)]. However, the ABC-type Mn/Zn transport systems were also present in the P. aeruginosa genome, while the superoxide dismutases present in both the D. aromatica and S. alaskensis were absent in P. aeruginosa. We interpret these results to suggest that either the increase in phenolics and lignin and/or the additional Mn in the SW leachate stimulated activity by D. aromatica and to a lesser extent S. alaskensis. The absence of spectra directly related to oxidative function may be due to the flow-through nature of our experimental system and/or the low abundance of functional proteins relative to total proteins associated with the complete proteome of any given phyla.
While it is possible that the intermediate levels among treatments of the other elements led to differences in activity and composition of the communities, we know of no mechanism that would explain those differences. Rather we suggest that increased amounts of lignin, phenolics, and Mn of the SW leachate led to differences in the community structure and function of the microbial biofilm communities. The elevated levels of phenoloxidase and peroxidase activity in the leachate with higher Mn, phenolic, and lignin contents, the evidence of stimulation of these enzymes systems by Mn in microbial culture, and enhanced decomposition with increasing Mn levels in a wide range of litter, including beech, all suggest a mechanistic and novel role for Mn as a control on organic carbon dynamics in aquatic ecosystems.
While the SW treatment was clearly different in metaproteome composition, phylogenetic origin of proteins, and activity of the oxidative enzymes, it is not clear how those metrics were related to the increased C:N of microbial biomass we observed in SW relative to the other treatments. We propose two mechanisms that could explain such a shift. Stimulation by Mn or organic chemistry could have resulted in an up-regulation of oxidative enzyme production in the SW treatment as was seen in the Basidiomycetes study (Steffen et al., 2002). Increased enzyme production may have resulted in increased cellular N demands to synthesize N-rich enzymes. This would result in a decrease in biomass N and an increase in biomass C:N relative to the other treatments. Similarly, increased oxidative enzyme activity could have resulted in an increase in labile organic carbon, enriching the microbial biomass in C thus increasing microbial C:N. With the current study it is not clear which mechanism, if either, was responsible for the increase in microbial C:N. Regardless, our biomass stoichiometry fell within the range of microbial communities reported elsewhere (Cleveland and Liptzin, 2007; Cotner et al., 2010) and small differences in biomass stoichiometry have been shown to result in large difference nutrient recycling (see Hall et al., 2011, Figure 3). This implies that chemical control of biomass stoichiometry by elements other than the limiting nutrients may result in differential nutrient recycling of limiting nutrient (e.g., N) among communities accessing distinct resource pools.
Microbial ecology of streams has received significantly more attention over the last years (Findlay, 2010). While benthic community composition becomes increasingly studied (e.g., Hullar et al., 2006; Besemer et al., 2009; Vishnivetskaya et al., 2010), the community composition of hyporheic biofilms remains poorly addressed (e.g., Feris et al., 2003). Furthermore, most of these studies do not link microbial community structure to function, although this has been recognized as a major area of potential advancement of stream microbial ecology (Findlay, 2010). Our findings on differences in resource quality inducing shifts in community structure and function of hyporheic biofilms add to this rapidly growing field of stream microbial ecology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Gerti Steniczka and Margarete Watzka, Claudia Hinterleitner, and Ieda Häemmerle for assistance in the laboratory. Financial support came from the Austrian Science Fund (FWF) projects START (Y420-B17) and MICDIF (S10005-B17) to T. J. Battin. The manuscript was greatly improved by addressing a series of specific critiques from three reviewers and an additional comment from Claudia Michaela Boot.
References
Battin, T., Butturini, A., and Sabater, F. (1999). Immobilization and metabolism of dissolved organic carbon by natural sediment biofilms in a Mediterranean and temperate stream. Aquat. Microb. Ecol. 19, 297–305.
Battin, T., Kaplan, L. A., Findlay, S., Hopkinson, C. S., Marti, E., Packman, A. I., Newbold, L., and Sabater, F. (2008). Biophysical controls on organic carbon fluxes in fluvial networks. Nat. Geosci. 1, 95.
Battin, T., Kaplan, L. A., Newbold, L., Cheng, X., and Hansen, C. (2003). Effects of current velocity on the nascent architecture of stream microbial biofilms. Appl. Environ. Microbiol. 69, 5443–5452.
Battin, T. J., Luyssaert, S., Kaplan, L. A., Aufdenkampe, A. K., Richter, A., and Tranvik, L. J. (2009). The boundless carbon cycle. Nat. Geosci. 2, 598–600.
Beaulieu, J. J., Tank, J. L., Hamilton, S. K., Wollheim, W. M., Hall, R. O. Jr., Mulholland, P. J., Peterson, B. J., Ashkenas, L. R., Cooper, L. W., Dahm, C. N., Dodds, W. K., Grimm, N. B., Johnson, S. L., McDowell, W. H., Poole, G. C., Valett, H. M., Arango, C. P., Bernot, M. J., Burgin, A. J., Crenshaw, C. L., Helton, A. M., Johnson, L. T., O’Brien, J. M., Potter, J. D., Sheibley, R. W., Sobota, D. J., and Thomas, S. M. (2010). Nitrous oxide emission from denitrification in stream and river networks. Proc. Natl. Acad. Sci. U.S.A. 108, 214–219.
Berg, B., Davey, M. P., De Marco, A., Emmett, B., Faituri, M., Hobbie, S. E., Johanssom, M. B., Liu, C., McClaugherty, C., Norell, L., Rutigliano, F. A., Vesterdal, L., and De Santo, A. V. (2010). Factors influencing limit values for pine needle litter decomposition: a synthesis for boreal and temperature pine forest systems. Biogeochemistry 100, 57–73.
Berg, B., Steffen, K. T., and McClaugherty, C. (2007). Litter decomposition rate is dependent on litter Mn concentrations. Biogeochemistry 82, 29–39.
Bernhardt, E. S., Likens, G. E., Buso, D. C., and Driscoll, C. T. (2003). In stream uptake dampens the effect of major disturbance on watershed nitrogen export. Proc. Natl. Acad. Sci. U.S.A. 100, 10304–10308.
Besemer, K., Singer, G., Hödl, I., and Battin, T. J. (2009). Bacterial community composition of stream biofilms in spatially variable-flow environments. Appl. Environ. Microbiol. 75, 7189–7195.
Boulton, A. J., Findlay, S., Marmonier, P., Stanley, E. H., and Valett, H. M. (1998). The functional significance of the hyporheic zone in streams and rivers. Annu. Rev. Ecol. Evol. Syst. 29, 59–81.
Cavicchioli, R., Ostrowski, M., Fegatella, F., Goodchild, A., and Guixa-Boixereu, N. (2003). Life under nutrient limitation in oligotrophic marine environments: an eco/physiological perspective of Sphingopyxis alaskensis (formerly Sphingomonas alaskensis). Microb. Ecol. 45, 203–217.
Cleveland, C. C., and Liptzin, D. (2007). C:N:P stoichiometry in soil: is there a “Redfield ratio” for the microbial biomass? Biogeochemistry 85, 235–252.
Condon, R. H., Steinberg, D. K., del Giorgio, P., Bouvier, T. C., Bronk, D. A., Graham, W. M., and Duckow, H. (2011). Jellyfish blooms result in a major microbial respiratory sink of carbon in marine systems. Proc. Natl. Acad. Sci. U.S.A. doi:10.1073/pnas.1015782108
Cotner, J. B., Hall, E. K., Scott, J. T., and Heldal, M. (2010). Freshwater bacteria are stoichiometrically flexible with a nutrient composition similar to seston. Front. Aquat. Microbiol. 1:132. doi:10.3389/fmicb.2010.00132
Eaton, A. D., Clesceri, L. S., Rice, E. W., and Greenberg, A. E. (eds). (2005). Standard Methods for the Examination of Water and Wastewater, 20th Edn. Washington: American Public Health Association.
Eiler, A., and Bertilsson, S. (2007). Flavobacteria blooms in four eutrophic lakes: linking population dynamics of freshwater bacterioplankton to resource availability. Appl. Environ. Microbiol. 73, 3511–3518.
Feris, K., Ramsey, P., Frazar, C., Moore, J. N., Gannon, J. E., and Holben, W. E. (2003). Differences in a hyporheic-zone microbial community structure along a heavy-metal contamination gradient. Appl. Environ. Microbiol. 69, 5563–5573.
Findlay, S., and Sinsabaugh, R. L. (2006). Large-scale variation in subsurface stream biofilms: a cross-regional comparison of metabolic function and community similarity. Microb. Ecol. 52, 491–500.
Findlay, S. E. G., Sinsabaugh, R. L., Sobczak, W. V., and Hoostal, M. (2003). Metabolic and structural response of hyporheic microbial communities to variations in supply of dissolved organic matter. Limnol. Oceanogr. 48, 1608–1617.
Fisher, S. G., Sponseller, R. A., and Heffernan, J. B. (2004). Horizons in stream biogeochemistry: flowpaths to progress. Ecology 85, 2369–379.
Florens, L., Carozza, M. J., Swanson, S. K., Fournier, M., Coleman, M. K., Workman, J. L., and Washburn, M. P. (2006). Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 40, 303–311.
Hall, E. K., Maixner, F., Franklin, O., Daims, H., Richter, A., and Battin, T. (2011). Linking microbial and ecosystem ecology using ecological stoichiometry: a synthesis of conceptual and empirical approaches. Ecosystems 14, 261–273.
Hamady, M., Walker, J. J., Harris, J. K., Gold, N. J., and Knight, R. (2008). Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5, 235–237.
Hullar, M. A. J., Kaplan, L. A., and Stahl, D. A. (2006). Recurring seasonal dynamics of microbial communities in stream habitats. Appl. Environ. Microbiol. 72, 713–722.
Jones, S. E., and McMahon, K. D. (2009). Species-sorting may explain an apparent minimal effect of immigration on freshwater bacterial community dynamics. Environ. Microbiol. 11, 905–913.
Keller, A., Nesvizhskii, A. I., Kolker, E., and Aebersold, R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685.
Moeseneder, M. M., Arrieta, J. M., Muyzer, G., Winter, C., and Herndl, G. J. (1999). Optimization of terminal-restriction fragment length polymorphism analysis for complex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 65, 3518–3525.
Mulholland, P. J., Helton, A. M., Poole, G. C., Hall, R. O. J., Hamilton, S. K., Peterson, B. J., Tank, J. L., Ashkenas, L. R., Cooper, L. W., Dahm, C. N., Dodds, W. K., Findlay, S., Gregory, S. V., Grimm, N. B., Johnson, S. L., McDowell, W. H., Meyer, J. L., Valett, H. M., Webster, J. R., Arango, C. P., Beaulieu, J. J., Bernot, M. J., Burgin, A. J., Crenshaw, C. L., Johnson, L. T., Niederlehner, B. R., O’Brien, J. M., Potter, J. D., Sheibley, R. W., Sobota, D. J., and Thomas, S. M. (2008). Stream denitrification across biomes and its response to anthropogenic nitrate loading. Nature 452, 202–205.
Nesvizhskii, A. I., Keller, A., Kolker, E., and Aebersold, R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658.
Olapade, O. A., and Leff, L. G. (2005). Seasonal response of stream biofilm communities to dissolved organic matter and nutrient enrichments. Appl. Environ. Microbiol. 71, 2278–2287.
Osborne, T. Z., Inglett, P. W., and Reddy, K. R. (2007). The use of senescent plant biomass to investigate relationships between potential particulate and dissolved organic matter in a wetland ecosystem. Aquat. Bot. 86, 53–61.
Phillippot, L., Andersson, S. G., Battin, T. J., Prosser, J. I., Schimel, J. P., Whitman, W. B., and Hallin, S. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat. Rev. Microbiol. 8, 523–529.
Quince, C., Curtis, T. P., and Sloan, W. T. (2008). The rational exploration of microbial diversity. ISME J. 2, 997–1006.
Quince, C., Lanzen, A., Davenport, R., and Turnbaugh, P. J. (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12, 38. doi:10.1186/1471-2105-12-38
Salinero, K. K., Keller, K., Feil, W. S., Feil, H., Trong, S., Di Bartolo, G., and Lapidus, A. (2009). Metabolic analysis of the soil microbe Dechloromonas aromatica str. RCB: indication of a surprisingly complex life-style and cryptic anaerobic pathways for aromatic degradation. BMC Genomics 10, 351. doi:10.1186/1471-2164-10-351
Schellekens, J., Buurman, P., and Pontevedra-Pombal, X. (2009). Selecting parameters for the environmental interpretation of peat molecular chemistry – a pyrolysis-GC/MS study. Org. Geochem. 40, 678–691.
Schneider, T., Gerrits, B., Gassmann, R., and Schmid, E. (2010). Proteome analysis of fungal and bacterial involvement in leaf litter decomposition. Proteomics 10, 1819–1830.
Schneider, T., Schmid, E., de Castro, J. V. Jr., Cardinale, M., Eberl, L., Grube, M., Berg, G., and Riedel, K. (2011). Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics 11, 2752–2756.
Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858.
Sinsabaugh, R. L. (2010). Phenoloxidase, peroxidase and organic matter dynamics of soil. Soil Biol. Biochem. 42, 391–404.
Sinsabaugh, R. L., Klug, M. J., Collins, H. P., Yeager, P. E., and Petersen, S. O. (1999). “Characterizing soil microbial communities” in Standard Soil Methods for Long-Term Ecological Research, eds G. P. Robertson, D. C. Coleman, C. S. Bledsoe, and P. Sollins (New York: Oxford University Press), 318–348.
Steffen, K. T., Hatakka, A., and Hofricther, M. (2002). Degradation of humic acids by the litter-decomposing basidiomycete Collybia dyophila. Appl. Environ. Microbiol. 68, 3442–3448.
Tringe, S. G., von Mering, C., Kobayashi, A., Salamov, A. A., Chen, K., Chang, H. W., Podar, M., Short, J. M., Mathur, E. J., Detter, J. C., Bork, P., Hugenholtz, P., and Rubin, E. M. (2005). Comparative metagenomics of microbial communities. Science 308, 554–557.
Trum, F., Titeux, H., and Delvaux, B. (2010). “Effects of manganese concentration on beech leaf litter decomposition: results from field and laboratory experiments,” in 19th World Congress of Soil Science, Soil Solutions for a Changing World, Brisbane, 40–43.
Valett, H. M., Morrice, J. A., Dahm, C. N., and Campana, M. E. (1996). Parent lithology, groundwater-surface water exchange and nitrate retention in headwater streams. Limnol. Oceanogr. 41, 333–345.
Vishnivetskaya, T. A., Mosher, J. J., Palumbo, A. V., Yang, Z. K., Podar, M., Brown, S. D., Brooks, S. C., Gu, B., Southworth, G. R., Drake, M. M., Brandt, C. C., and Elias, D. A. (2010). Mercury and other heavy metals influence bacterial community structure in contaminated Tennessee streams. Appl. Environ. Microbiol. 77, 302–311.
Wallace, J. B., Eggert, S. L., Meyer, J. L., and Webster, J. R. (1997). Multiple trophic levels of a forest stream linked to terrestrial litter inputs. Science 277, 102–104.
Keywords: biofilms, enzyme activity, leachate, stream ecology, microbial community composition, metaproteome
Citation: Hall EK, Besemer K, Kohl L, Preiler C, Riedel K, Schneider T, Wanek W and Battin TJ (2012) Effects of resource chemistry on the composition and function of stream hyporheic biofilms. Front. Microbio. 3:35. doi: 10.3389/fmicb.2012.00035
Received: 19 July 2011; Accepted: 23 January 2012;
Published online: 14 February 2012.
Edited by:
James Cotner, University of Minnesota, USAReviewed by:
Colleen Hansel, Harvard University, USAS. Leigh McCallister, Virginia Commonwealth University, USA
David Bastviken, Linköping University, Sweden
Copyright: © 2012 Hall, Besemer, Kohl, Preiler, Riedel, Schneider, Wanek and Battin. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: E. K. Hall, Natural Resources Ecology Laboratory, United States Geological Survey, Colorado State University, Fort Collins, CO 80523, USA. e-mail:ZWQuaGFsbEBjb2xvc3RhdGUuZWR1