 Brandon J. Converse
Brandon J. Converse James P. McKinley
James P. McKinley Charles T. Resch
Charles T. Resch Eric E. Roden
Eric E. Roden- 1Department of Geoscience, University of Wisconsin-Madison, Madison, WI, USA
- 2Pacific Northwest National Laboratory, Richmond, WA, USA
This study employed 16S rRNA gene amplicon pyrosequencing to examine the hypothesis that chemolithotrophic Fe(II)-oxidizing bacteria (FeOB) would preferentially colonize the Fe(II)-bearing mineral biotite compared to quartz sand when the minerals were incubated in situ within a subsurface redox transition zone (RTZ) at the Hanford 300 Area site in Richland, WA, USA. The work was motivated by the recently documented presence of neutral-pH chemolithotrophic FeOB capable of oxidizing structural Fe(II) in primary silicate and secondary phyllosilicate minerals in 300 Area sediments and groundwater (Benzine et al., 2013). Sterilized portions of sand+biotite or sand alone were incubated in situ for 5 months within a multilevel sampling (MLS) apparatus that spanned a ca. 2-m interval across the RTZ in two separate groundwater wells. Parallel MLS measurements of aqueous geochemical species were performed prior to deployment of the minerals. Contrary to expectations, the 16S rRNA gene libraries showed no significant difference in microbial communities that colonized the sand+biotite vs. sand-only deployments. Both mineral-associated and groundwater communities were dominated by heterotrophic taxa, with organisms from the Pseudomonadaceae accounting for up to 70% of all reads from the colonized minerals. These results are consistent with previous results indicating the capacity for heterotrophic metabolism (including anaerobic metabolism below the RTZ) as well as the predominance of heterotrophic taxa within 300 Area sediments and groundwater. Although heterotrophic organisms clearly dominated the colonized minerals, several putative lithotrophic (NH4+, H2, Fe(II), and HS- oxidizing) taxa were detected in significant abundance above and within the RTZ. Such organisms may play a role in the coupling of anaerobic microbial metabolism to oxidative pathways with attendant impacts on elemental cycling and redox-sensitive contaminant behavior in the vicinity of the RTZ.
Introduction
Microbially driven redox cycling of iron (Fe) is an important environmental process that can influence the fate of various subsurface constituents, including carbon, sulfur, nitrate, oxygen, as well as organic and metal/radionuclide contaminants (Tebo and He, 1999; Roden et al., 2004). In circumneutral pH environments, Fe(II)-oxidizing and Fe(III)-reducing bacteria (FeOB and FeRB, respectively) typically thrive in a coupled manner along redox boundaries, where Fe(II) produced via FeRB activity provides an energy substrate for FeOB, and Fe(III) produced via FeOB activity regenerates Fe(III) phases utilized by FeRB (Straub et al., 2001; Roden, 2012). Although aqueous Fe(II) is usually viewed as the main form of Fe(II) utilized by FeOB within redox interfacial environments, recent studies indicate that insoluble primary Fe(II)-bearing mineral phases such as almandine (Chaudhuri et al., 2001) and biotite (Shelobolina et al., 2012b), and secondary Fe-phyllosilicates such as smectite (Shelobolina et al., 2003, 2012a; Benzine et al., 2013) can serve as substrates for FeOB. In addition, the oxidized, Fe(III)-bearing forms of these phases may serve as electron acceptors for FeRB (Kostka et al., 1996, 2002; Dong et al., 2003; Brookshaw et al., 2014), setting up the potential for “solid-state” microbial Fe redox cycling in the vicinity of redox boundaries (Roden, 2012).
A recent study conducted at the Hanford 300 Area site in eastern Washington state recovered several novel subsurface FeOB which are capable of utilizing solid-phase Fe(II) contained in biotite and smectite as energy sources for chemolithotrophic growth (Benzine et al., 2013). The Hanford 300 Area (referred to hereafter as the “300 Area”) subsurface is contaminated with radionuclides (mainly uranium) and nitrate from Cold War-era Pu production (Peterson and Connelly, 2001; Christensen et al., 2004), and as a result there is strong interest in the potential impacts of Fe redox-associated processes on contaminant mobility (Peretyazhko et al., 2012). The 300 Area contains a redox discontinuity at ca. 15–18 m depth, just below the boundary between the coarse-grained Pleistocene Hanford formation (primarily Ice Age cataclysmic flood deposits) and the fine-grained Miocene/Pliocene Ringold formation (primarily of ancestral Columbia River deposits; Lindsey and Gaylord, 1990; Zachara et al., 2007). This redox transition zone (RTZ) has been the subject of a variety of recent work focused on geochemical and microbial interactions and their implications for contaminant mobility and transport (Lee et al., 2012; Lin et al., 2012a,b; Peretyazhko et al., 2012), and was the source of organisms for the silicate mineral-utilizing FeOB study of Benzine et al. (2013). A parallel study from our group (Percak-Dennett and Roden, 2014) examined the hypothesis that microorganisms within and below RTZ could participate in the oxidation of native Fe(II)-bearing phases (primarily dioctahedral smectite with traces of chlorite, Peretyazhko et al., 2012). Contrary to expectations, Fe(II) oxidation was only observed in aerobic, sterile sediments; all other inoculated and sterile reactors amended with oxygen or nitrate showed no Fe(II) loss. Non-sterile sediments consumed significant amounts of nitrate, and were dominated by heterotrophic microbes consuming residual sediment organic carbon. These results indicated that heterotrophic metabolism consumed available oxidants, thereby preventing microbial Fe(II) oxidation. This inference was consistent with previous observations that Ringold Formation sediments contain active heterotrophic microbial communities (Lin et al., 2012a).
Although the above results argued against Fe(II)-driven chemolithotrophy as a major control on redox balance within the 300 Area RTZ, there is nevertheless ongoing interest in the potential for the RTZ to support novel FeOB. This study sought to gain insight into potential 300 Area RTZ FeOB populations by incubating a mixture of quartz sand with Fe(II)-bearing silicate mineral biotite (in comparison to quartz sand alone) in situ across (above, within, and below) the RTZ for several months. Our hypothesis was that the sand+biotite mixture would be preferentially colonized by lithotrophic FeOB capable of utilizing Fe(II) in biotite as an energy source. Both the sand+biotite and sand-only in situ incubations were free of particulate organic carbon, such that lithotrophic organisms could potentially compete successfully with the heterotrophic communities that otherwise dominate RTZ microbial energy metabolism. In situ incubation of the minerals across the RTZ tested the secondary hypothesis that differential colonization of the minerals would not occur below the RTZ where oxidants (oxygen and nitrate) are not available to support FeOB growth. More broadly, the deployment strategy allowed us to examine basic patterns of microbial community composition on the colonized materials in relation to redox and chemical gradients across the RTZ. In addition, the experiment provided an opportunity to evaluate the relative importance of heterotrophic vs. lithotrophic pathways in situ within this unique geochemical environment.
Materials and Methods
Multi-Level Sampler (MLS) Deployments
In August 2012, a series of 38-mL polypropylene cartridges were loaded with 55 g of either commercial (Accusand, AGSCO Corp., Wheeling, IL, USA) silica sand (ca. 1 mm diameter), or a mixture (1:10 mass ratio) of sand plus fine-grain (ca. 20 μm diameter flakes) biotite, or silica sand only. The biotite was obtained from Ward Scientific and prepared as previously described (Shelobolina et al., 2012b). Biotite was chosen as a Fe(II)-bearing mineral substrate because it is not susceptible to abiotic oxidation by oxygen (or nitrate) on a time scale of weeks to months, and because it was used successfully in a previous study to enrich for FeOB in 300 Area groundwater (Benzine et al., 2013). The cartridges were capped with permeable nylon net filters (20 μm), and deployed in multi-level sampling (MLS) sampling arrays suspended in two different wells at the Hanford 300 Area Integrated Field Research Site (IFRC) as described previously (Lee et al., 2014). Fourteen cartridges were deployed in borehole well 3–24, and 14 cartridges were deployed in well 3–27 [detailed descriptions of these wells can be found in Bjornstad et al. (2009)]. Cartridges were deployed at 10 or 20 cm intervals over depths ranging from 15.3–17.1 m in well 3–24 and 15.5–17.2 m in well 3–27, with sand+biotite or sand-only cartridges alternating with increasing depth. In January 2013, the cartridges were removed, and the contents were transferred to Whirl-Pack bags on ice. The samples were frozen at -80°C upon return to the lab and remained frozen until DNA extraction.
Groundwater samples were collected from MLS arrays deployed prior to the in situ mineral deployments, and concentrations of nitrate, total Fe, and total Mn (μM), as well as CH4, and H2 (ppmv) were determined as previously described (Lin et al., 2012a).
DNA Extraction and Analysis
DNA Extraction from MLS Cartridges
The Whirl-Pack® bags containing the samples were removed from the -80°C freezer and thawed at room temperature. After thawing, the entire content of the bags was transferred to sterile 50 ml Falcon tubes, and the samples were centrifuged at 5000 rpm for 10 min. The supernatant was discarded and the solid-phase material was mixed vigorously to homogenize the samples. Approximately 0.7 g of material was removed for DNA extraction, and the remaining samples were returned to the -80°C freezer.
DNA was extracted from samples using a Mo-Bio PowerSoil® DNA extraction kit (Mo-Bio, Carlsbad, CA, USA) following the manufacturer’s protocol with some modifications. The bead beating tubes (containing the proprietary bead-beating solution) were heated to 70°C prior to the addition of any samples. After heating and sample addition, 80 μl solution C1 was added, and the tubes were heated at 70°C for 10 min with vigorous shaking (14,000 rpm) to aide in cell lysis. As recommended by Mo-Bio, the precipitation steps using solutions C2 and C3 were combined into a single step, using only 100 μL of each, and mixing thoroughly after adding each solution. This procedure is recommended for low-biomass samples to reduce the volume of liquid and the potential for DNA loss. DNA was eluted in a total of 25 μL of Solution C5 and quantified using a Qubit® fluoromoeter (version 1.0, Life Technologies, Inc., Madison, WI, USA). The DNA extracts were stored at -20°C.
DNA Extraction from Groundwater Samples
Bulk groundwater samples were collected as previously described (Lin et al., 2012c) before and after the in situ mineral deployments for DNA extraction. The pre-MLS deployment groundwater samples were passed through a 47 mm diameter 0.2-μm polyethersulfone filter, and genomic DNA was extracted from half of each membrane filter using a Mo-Bio PowerSoil-HTP 96-well DNA isolation kit according to the manufacturer’s protocol (MoBio Laboratories, Carlsbad, CA, USA). Post-MLS deployment samples were filtered through 0.2 μm Sterivex in-line filters, and DNA was extracted from segments of filters using a Mo-Bio PowerSoil® DNA extraction kit with the modifications described above.
Primers and 16S rRNA Gene Amplification
16s rRNA amplicons from each sample were generated using the previously described universal primer set 515f/806r (Bates et al., 2011). Primers containing the template-specific forward or reverse primer, the Roche A (515f primers: 5′-CCATCTCATCCCTGCGTGTCTCCGAC-3′) or B (806r primer only: 5′-CCTATCCCCTGTGTGCCTTGGCAGTC-3′) sequencing adapters, and linker sequence (TCAG) were added to all primers. Appropriate Roche 10-bp MID tags were only added to the 515f primer sets. The final fusion primers were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA, USA).
Samples were amplified with InvitrogenTM Platinum® Taq Supermix (Life Technologies, Grand Island, NY, USA), which contains dNTPs. Each reaction was set up with 5 μL template DNA, 0.1 μL forward and reverse primer (200 nm final concentration), and 45 μL Platinum® Taq Supermix. The following reaction protocol was used to amplify the samples: 94°C for 2 min (initial denaturation) followed by 30 cycles of 94°C for 15 s (denaturing), 55°C for 30 s (annealing), and 72°C for 15 s (extension). A final extension step was conducted at 72°C for 6 min, and the samples were held overnight at 4°C. Each reaction was run on a 1% agarose gel to verify the presence of the amplicon, and to check for spurious amplification products.
Sample Pooling and Titanium 454 FLX+ Amplicon Sequencing
Amplicons were pooled in an equimolar ratio, and cleaned using Agencourt® AMPure® XP magnetic beads (Beckman Coulter, Inc., Indianapolis, IN, USA) following the manufacturer’s instructions. The cleanup procedure was repeated six times, as recommended by the University of Wisconsin-Madison Biotechnology Center (UWMBC, James Speers, personal communication). The clean pool was submitted to the UWMBC for quality analysis and Titanium 454 FLX+ sequencing.
Sequence Analysis
Raw sequence data was processed using the Quantitative Insights into Microbial Ecology (QIIME) pipeline (http://www.qiime.org, version 1.7.0; Bates et al., 2011). A.sff.txt file was not supplied by the UWMBC, so the mothur script was used to generate it from the raw.sff file (Schloss et al., 2009). This file was used with the QIIME default denoising software (Caporaso et al., 2010). Data processing was conducted using the default QIIME settings: sequences less than 200 bp, with quality scores less than 25, and more than six ambiguous bases were discarded. After quality filtering, the samples were denoised, and operational taxonomic units (OTUs) were picked de novo with either 94, 97, or 99% similarity (nominally family-, genus-, and species-level phylogenetic resolution Gillis et al., 2001) using uclust (Edgar, 2010). Putative chimeras were identified and removed with ChimeraSlayer (Edgar et al., 2011), and taxonomy was assigned based on the Greengenes database (Brodie et al., 2006) using the Ribosomal Database Pipeline (RDP) classifier (Wang et al., 2007). The full set of sequences and taxonomic assignments (based on 97% sequence similarity) are provided in Data Sheet 1 of the on-line supplementary material. In addition, the sequences and their taxonomic assignments are available in GenBank submission SUB1010609, accession numbers KT429935 – KT437631 (note that 505 out of the original 8202 OTU sequences were identified as chimeric by NCBI and removed from the list of sequences added to Genbank; the entire set of OTU sequences is included in the supplementary file).
Sequences were aligned using PyNAST (Bates et al., 2011) and a phylogenetic tree for alpha and beta diversity analysis was constructed with FastTree 2 (Price et al., 2010). OTU tables were rarefied at a range of 100–2000 sequences/sample, and ten iterations (without replacement) were conducted. Each rarefaction increased by 100 sequences/sample. OTUs with less than 100 sequences were removed before conducting rarefaction the to smooth the plots. Whole-tree phylogenetic diversity (PD), Chao1, and Observed Species alpha diversity metrics were calculated for each sample, and rarefaction curves were generated in QIIME. The PD-based rarefaction curves were fit by non-linear least-squares regression (Prism GraphPad software, La Jolla, CA, USA) to an equation describing a rectangular hyperbola [y = ax/(b + x)], and the estimated maximum number of OTUs (a in the fitting equation) was compared to the total number of identified OTUs in order to calculate the degree of saturation for each sample.
Within QIIME, a jackknifed beta diversity analysis was performed after rarefying each sample to 2,000 sequences. Unifrac (Lozupone and Knight, 2005) was used to construct weighted and unweighted Unifrac matrices, and principal coordinates (PCoAs) plots from the results. When further examining abundant taxa, only sequences comprising the 10 most abundant OTUs were considered. To obtain more detailed information about these abundant OTUs, their representative sequences were submitted to BLAST, using the blast-n algorithm (Altschul et al., 1997) with environmental sequences excluded from the database. These results were compared to those obtained when classifying with the RDP pipeline via the Greengenes database.
Statistical Analyses
To determine if statistically significant differences or similarities existed among the bacterial communities observed at different depths, or if there were significant differences between deployment types, a Bray–Curtis similarity matrix was constructed from the OTU table using PRIMER for Windows (v.6; Clarke et al., 2006). One-way analysis of similarity (ANOSIM) tests were conducted with 9999 permutations on the untransformed similarity matrix (Clarke, 1993).
CANOCO for Windows (version 4.51) was used to conduct a canonical correspondence analysis (CCA) to determine if geochemical gradients of Fe, NO3-, and Mn, CH4, or H2 were responsible for driving the microbial communities observed at different depths as previously described (Beversdorf et al., 2013).
To check for statistical difference in alpha diversity scores (PD, Chao1 matrices), the QIIME script compare_alpha_ diversity.py was used to conduct a Student’s t-test and to calculate the P-values between groups of mean alpha diversity scores.
Results
Groundwater Geochemistry
Groundwater samples collected in MLS arrays prior to the in situ mineral deployments revealed redox gradients comparable to those observed previously (Lin et al., 2012a), with nitrate decreasing to near zero values at the base of the RTZ, and dissolved Fe2+ and Mn2+ accumulating below that depth (Figures 1A,C). Although dissolved oxygen concentrations were not measured in this study, previous studies have shown that oxygen approaches zero ca. 1 m above the depth where nitrate is exhausted (Lin et al., 2012a). The redox gradient was generally steeper in well 3–27, where elevated levels of dissolved methane and hydrogen were also observed below the RTZ (Figure 1D).
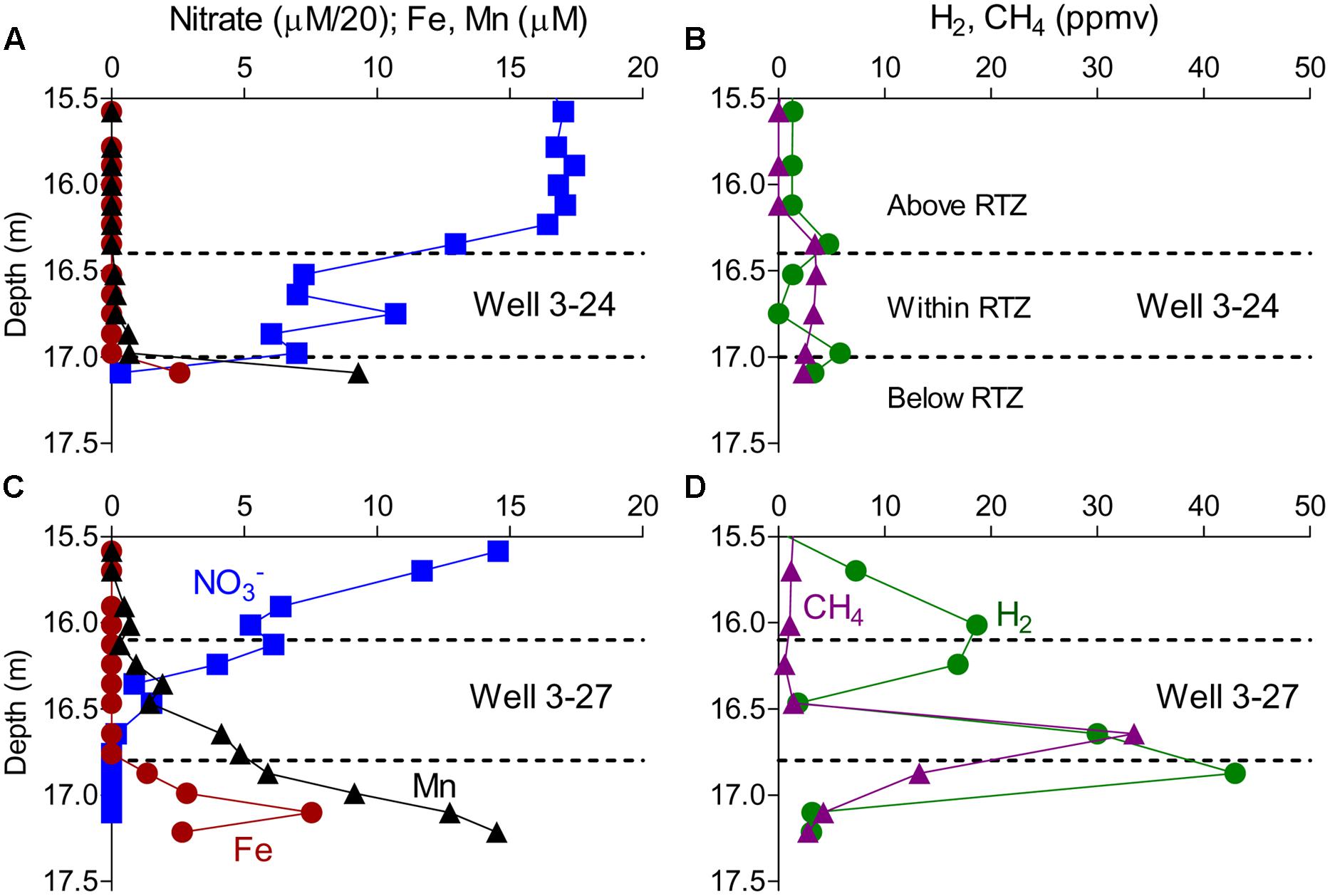
FIGURE 1. Concentrations of nitrate, Fe, Mn, CH4, and H2 measured in wells 3–24 (A,B) and 3–27 (C,D) measured in August 2012 prior to the in situ mineral deployment. The dashed lines demarcate the depth used to group samples as above, within, or below the RTZ.
16S rRNA Gene Amplicon Libraries
Thirty-two 16S rRNA gene amplicon libraries were constructed from DNA extracted from 14 MLS cartridges (seven with sand+biotite, seven with sand only) from each of the two wells, plus groundwater samples collected from the two wells before and after in situ mineral deployment. After quality filtering and removal of chimeras, a total of 151,477 sequences remained out of a total of 529,551 raw reads, with an average read length of 262 bp. The number of reads per sample ranged from 2235 to 7792, with a mean of 4734. Together the sequences clustered into 8202 OTUs at 97% similarity. The amplicon libraries were grouped by deployment type to explore differences between colonization of sand+biotite or sand alone. In addition, to explore the microbial communities as a function of depth, the samples within each well were grouped into three depth intervals: above the redox transition (15.4–16.4 m in well 3–24, 15.5–16.1 m in well 3–27), within the redox transition (16.5–17.1 m in well 3–24, 16.2–16.8 m in well 3–27) and below the redox transition [17.2 m 3–24 (note: only one sample), 16.8–17.0 m 3–27 in well 3–27]. The groupings were made based on the geochemical gradients as indicated in Figure 1. The results of various analyses performed on the grouped data sets are summarized below.
Phylogenetic Diversity
Faith’s PD (Faith and Baker, 2006) diversity metrics were calculated in QIIME for each of the sample groups after rarefaction (2000 OTUs per sample; and Table 1). The post-deployment groundwater samples were the most phylogenetically diverse, and the pre-mineral deployment sample from well 3–24 was also more diverse than any of the samples from the in situ mineral deployments. The pre-mineral deployment groundwater sample taken from the 3–27 well was the least phylogenetically diverse of all the samples. There was no difference in PD among the deployment types (sand+biotite or sand only; data not shown). However, there was a decrease in diversity between the mineral deployments above the RTZ and those taken within the RTZ in both wells, and samples from below the RTZ were the least phylogenetically diverse.
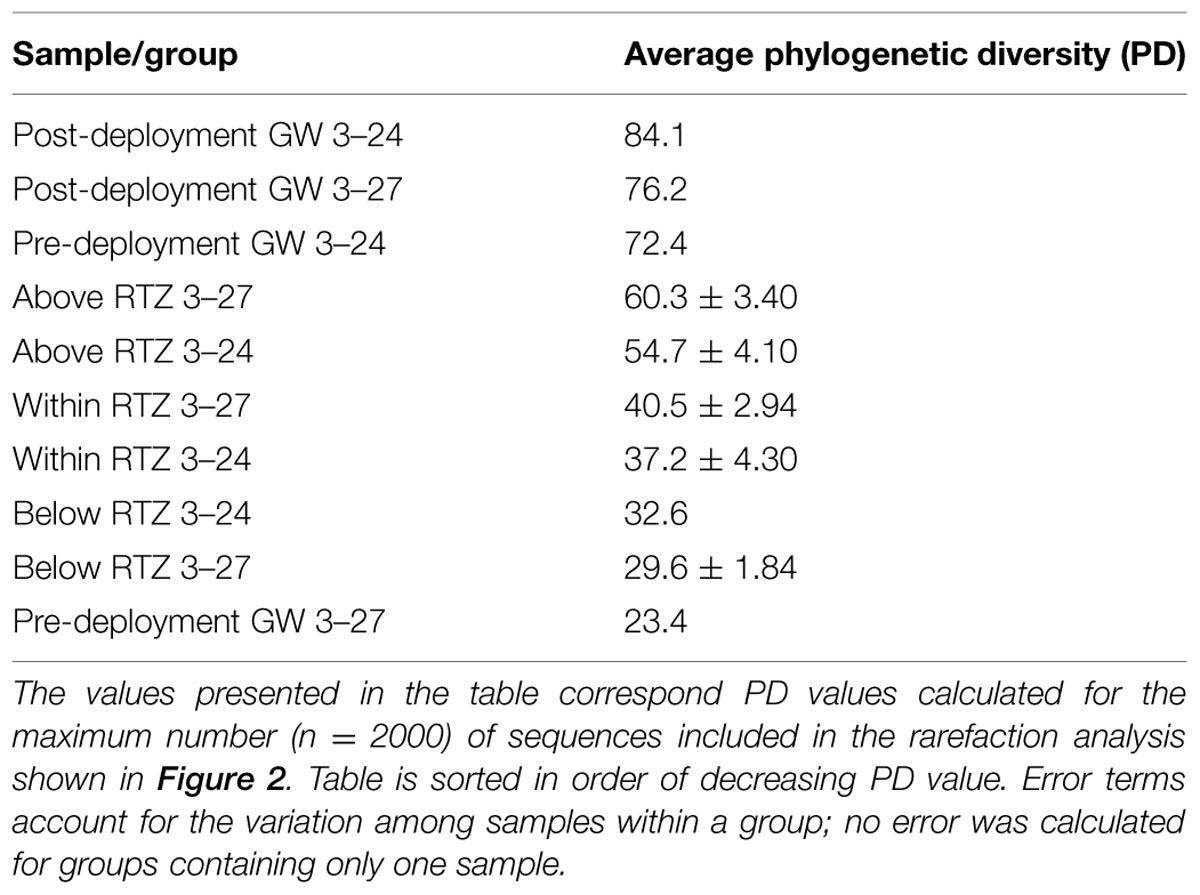
TABLE 1. Average Faith’s (Faith and Baker, 2006) phylogenetic diversity (PD) calculated for groundwater and grouped mineral deployment 16S rRNA gene amplicon libraries.
Rarefaction curves were generated for each of the libraries using PD-based OTU assignments made with either 94, 97, or 99% sequence similarity (Figure 2). The estimated degree of saturation for the 28 mineral-colonized samples was 75.4 ± 2.9, 71.9 ± 4.1, and 69.0 ± 3.0 for the three levels of similarity. There were no systematic differences in the degree of saturation between the biotite+sand vs. biotite-only samples, or with deployment depth (data not shown). These relatively high levels of saturation permit robust comparisons of mineral-colonized community composition across deployment type and depth. For simplicity, these comparisons were made using the 97% similarity OTU assignments.
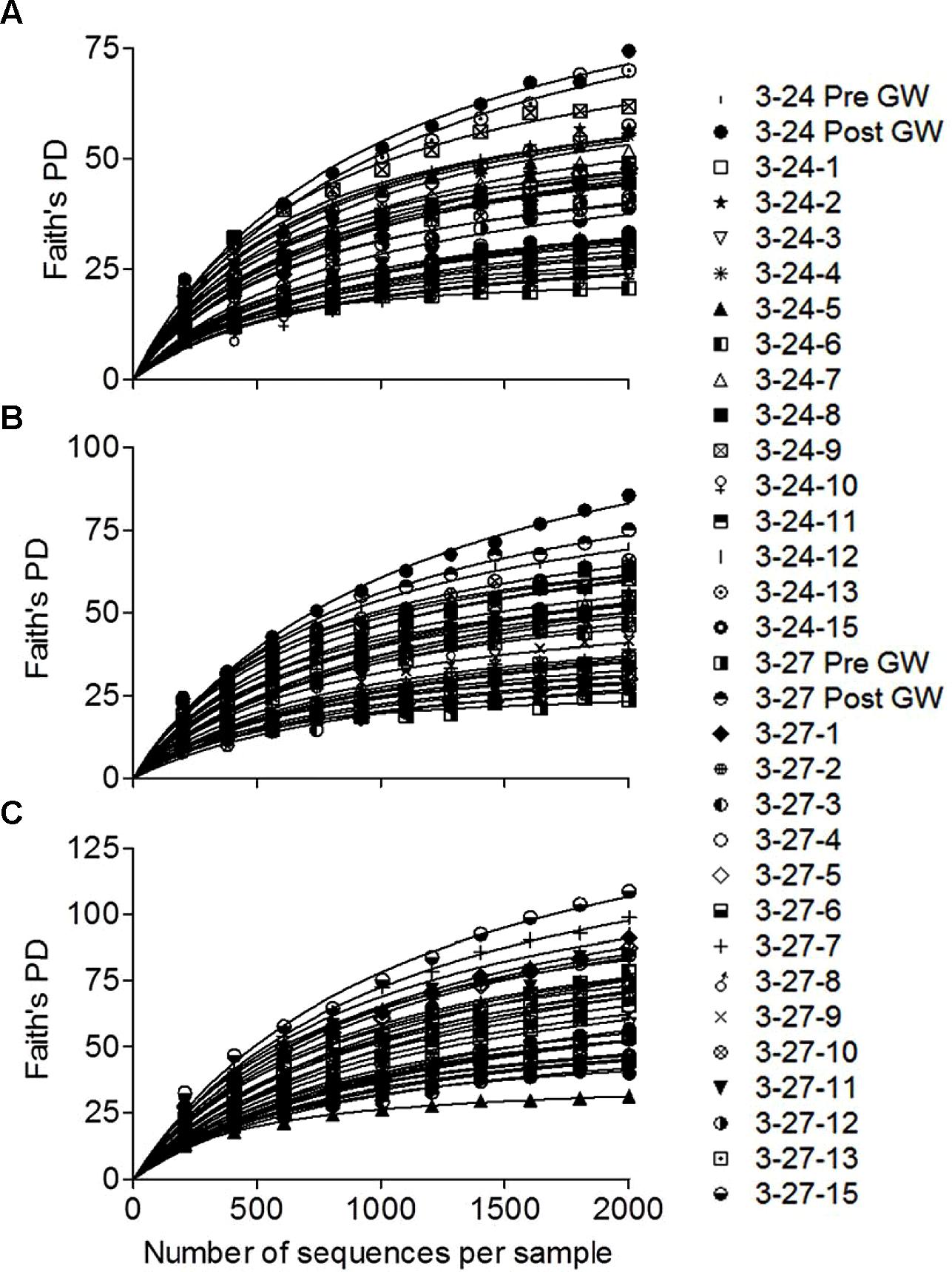
FIGURE 2. Rarefaction curves for each of the 16S rRNA gene amplicon libraries generated using PD-based OTU assignments made with either 94 (A), 97 (B), or 99% (C) sequence similarity. Lines represent non-linear least-squares regression fits of the data to an equation describing a rectangular hyperbola y = ax/(b + x), where a is the estimated maximum number of OTUs.
Community Composition across Deployment Type and Depth
Weighted and unweighted Unifrac (Caporaso et al., 2010) matrices were constructed to determine if the microbial communities were similar across deployment type (sand+biotite vs. sand only) or depth (above, below, or within the RTZ). The OTU table was rarefied to a depth of 2000 sequences prior to calculating Unifrac distances, and PCoA plots were constructed from the weighted Unifrac matrices. The PCoA plot of the weighted Unifrac matrix (Figure 3) indicated that there was no difference between the microbial communities associated with mineral deployment type, indicating that differential colonization of cartridges containing biotite did not occur or was below the resolution limits of our data set.
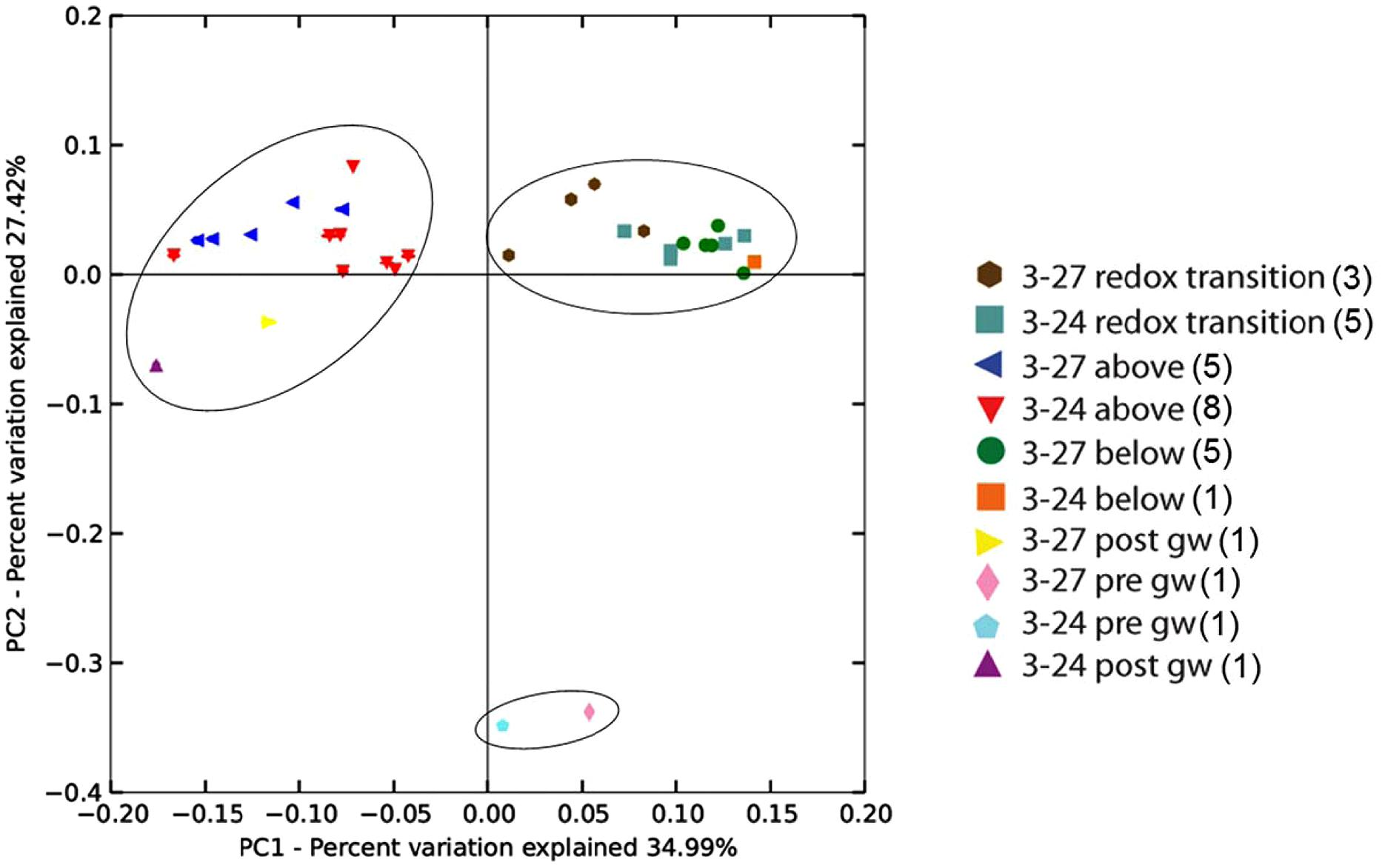
FIGURE 3. Weighted Unifrac principal components analysis of 16S rRNA gene sequences from groundwater and grouped mineral deployments constructed from jackknifed beta diversity analysis. The number of sequences per sample was normalized to n = 2000. Values in parentheses in the symbol legend indicate the number of samples included in that groupling. The circles indicate the three clusters of samples, which were determined to be statistically different with ANOSIM (R = 0.818). There was no significant similarity between samples from different deployment types (R = 0.314).
Principal coordinate analysis revealed two distinct sample clusters based on depth, together with one loose cluster comprising the pre-mineral deployment groundwater sample. Samples from above the RTZ in both wells formed a cluster with the post-deployment groundwater samples, and samples from within and below the RTZ formed a distinct cluster, although samples from within the RTZ in well 3–27 (brown hexagons in Figure 3) did not cluster as tightly as the other samples, suggesting some similarity with the samples from above the RTZ. The slight difference in RTZ samples from each well is likely due to the differences in sampling depths within each well, as some of the RTZ samples in well 3–24 were from the same depth as the below RTZ samples in well 3–27. There was no significant difference between the communities between each well.
Abundant Taxa Present in all Depth Intervals
The 10 most abundant (percentage-wise) OTUs from each sample were pooled within their respective depth intervals (above, within, or below the RTZ), and the wells were evaluated independently due to the slight differences in deployment depth in each well. The 10 most abundant OTUs accounted for an average of 88.0 ± 6.1% and 89.7 ± 4.5% of total OTUs (averaged across all depths) for wells 3–24 and 3–27, respectively. In some cases, RDP classified independent OTUs at the same level, and these OTUs were grouped together. Figure 4 highlights the taxa that were found in all depth intervals.
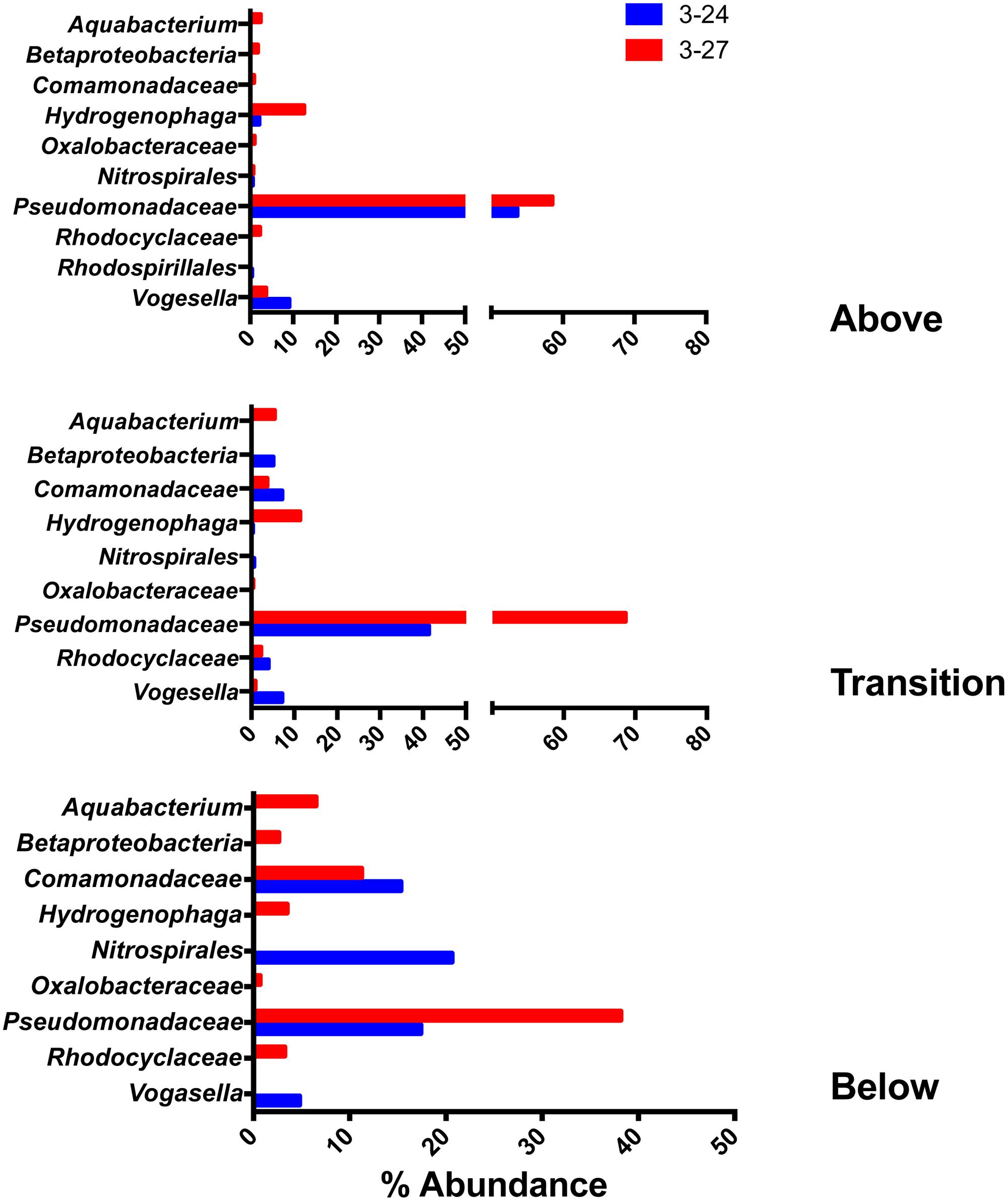
FIGURE 4. Top 10 most abundant taxa detected in all three of the depth interval groupings (above, within, and below the RTZ).
Members of the Pseudomonadaceae were the most abundant taxa at all depth intervals in both wells. They were also the only taxa detected in both wells at all depth intervals. Two different OTUs (see Table 2) comprised anywhere from 17% (well 3–24, below the RTZ) to 69% (well 3–27, within the RTZ) of the reads within a given depth interval. Other abundant OTUs detected at all depths (in one or the other of the two wells) included Aquabacterium, Betaproteobacteria, Comamonadaceae, Hydrogenophaga, Nitrospirales, Oxalobacteraceae, Rhodocyclaceae, and Vogesella. The abundance of Hydrogenophaga was considerably lower at all depths compared to Pseudomonadaceae, with a maximum abundance of 2.8% in well 3–24, and 12.5% in well 3–27. The abundance percentages of this group decreased with depth. Vogesella abundance also decreased in abundance with depth, from 9.1% to 4.8% in 3–24, and 3.7% to 0% in 3–27. Aquabaterium and Oxalobacteraceae were only detected in well 3–27. At the shallowest depths, Aquabaterium comprised only 2.4% the reads, but this increased to 6.5% below the RTZ. Oxalobacteraceae comprised ca. 1% abundance at all depths.
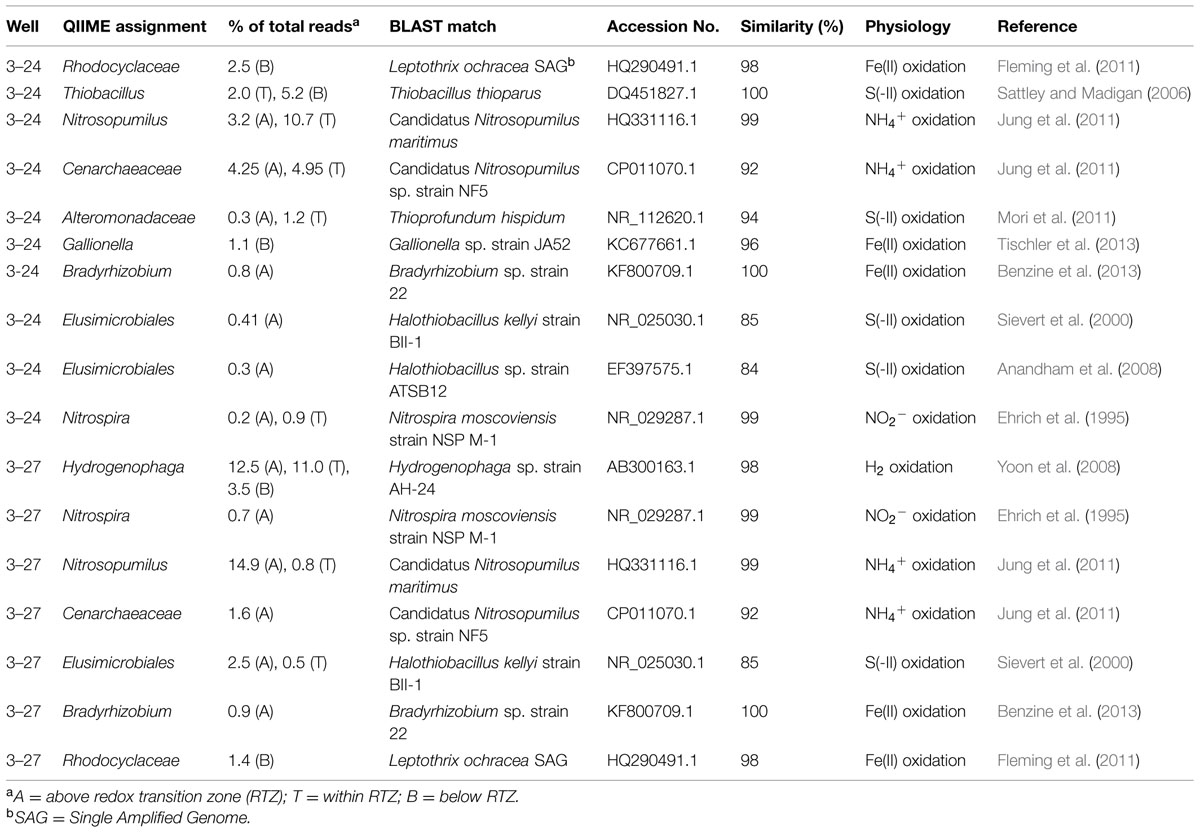
TABLE 2. Potential chemolithotrophic taxa identified in the 16S rRNA gene amplicon libraries.
Nitrospirales were the only other group detected in all depth intervals (aside from the Pseudomonadaceae) that were present at >20% abundance in at least one depth interval. They were detected in both wells above the RTZ, and in one sample from below the RTZ in well 3–24 where their relative abundance was ca. 20%. At all other depths, their abundance remained relatively low (≤2%). Comamonadaceae were detected both within and below the RTZ in both wells, and in one sample above the RTZ in well 3–27. A group of taxa classified by RDP only at the phylum level as Betaproteobacteria were found above and below the RTZ in well 3–27, and was present in both wells within the RTZ. This group of OTUs comprised ca. 0–5% of the abundance in both wells, but was most abundant within the RTZ in well 3–24 RT at 5%. BLAST searches indicated that this group was a 95% match to Burkholderia sp. The Rhodocyclaceae group was present in the same pattern of depth intervals at the Betaproteobacteria group, and, as above, there was no relationship between abundance and depth, with percentages ranging from 0 to 4%.
Taxa Abundant in some Depth Intervals
The remaining abundant taxa were detected in one or two of the depth interval groupings, but not all three. The abundance of these taxa are presented in Figure 5. None of these groups, with the exception of a group classified by RDP as Desulfobulbaceae, were detected at a greater than 20% abundance.
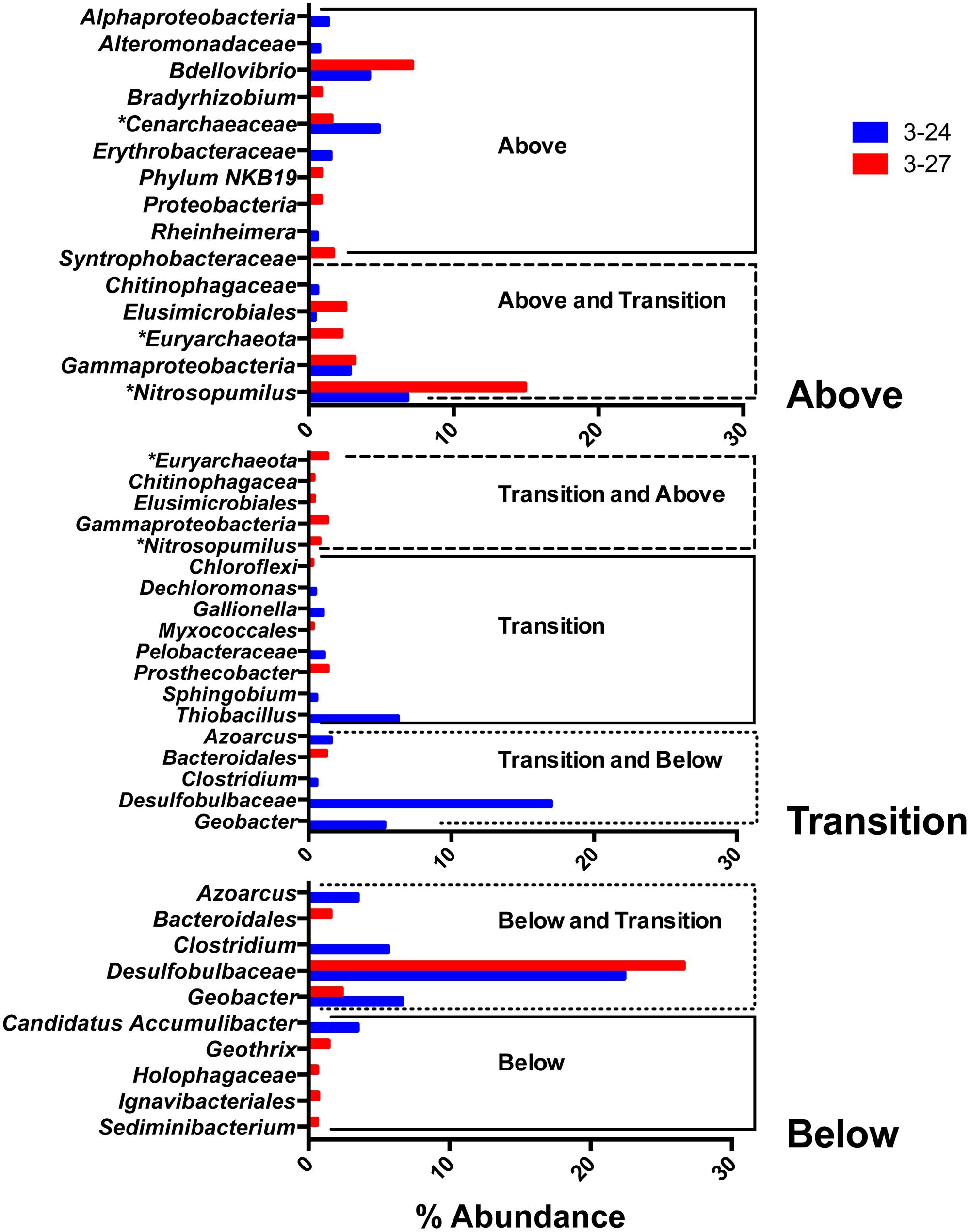
FIGURE 5. Top 10 most abundant taxa detected in one or two of the depth interval groupings (above, within, and below the RTZ).
Taxa above the RTZ
OTUs related to the predatory bacterial genus Bdellovibrio were abundant in colonized minerals from above the RTZ. Other taxa detected solely above the RTZ included groups classified as Bradyrhizobium and the candidate phylum NKB19. These groups were only detected in well 3–27 at low abundance.
Taxa above and within the RTZ
Of the taxa detected both above and within the RTZ, the most abundant were a collection of OTUs classified by RDP as Gammaproteobacteria, at 2.85% in well 3–24 and 3.15% in well 3–27. A number of archaeal taxa were detected above and within the RTZ, including a group classified as Cenarchaeaceae, which was present above the RTZ in both wells. Two other archaeal taxa belonged to the Euryarchaeota family and the Nitrosopumilus genus. Euryarchaeota were only present in well 3–27, whereas Nitrosopumilus was detected in both wells above the RTZ, and in 3–27 within the RTZ. Of the organisms detected only above the RTZ, Nitrosopumilus was detected at the greatest abundance at 15% in well 3–27. No archaeal taxa were detected below the RTZ.
Taxa within the RTZ
Among the taxa detected exclusively within the RTZ, only one group, classified as Thiobacillus, was detected at an abundance >5% (well 3–24 only). In well 3–27, the most abundant organism was a group classified as Prosthecobacter, at 1.35%.
Taxa within and below the RTZ
Of the taxa detected within and below the RTZ, all but one group were detected exclusively in well 3–24, including the most abundant taxa detected within the RTZ, a group classified as Desulfobulbaceae (16.7%). Other abundant groups detected in both these depth intervals include Clostridium and Geobacter, at 0.56 and 5.32%, respectively. The only taxa detected both within and below the RTZ in well 3–27 was a group classified as Bacteroidales (1.23%).
Taxa below the RTZ
The Desulfobulbaceae group described in the previous paragraph was the most abundant group detected below the RTZ at 27 and 22.4% in wells 3–27 and 3–24, respectively. The Clostridium and Geobacter groups described above were present in well 3–24 at 5.6 and 6.6%, and the Geobacter group was also present in well 3–27 at 2.25%. Among the taxa unique to below the RTZ, Geothrix, Holophagaceae, Ingavibacteriales, and Sediminibacterium were detected in well 3–27, and a group classified as Candidatus Accumulibater was detected in well 3–24.
Abundant Taxa in Groundwater Samples
The most abundant OTUs present in the groundwater samples were considerably different than the abundant taxa observed in the colonized minerals. In addition, there were no overly dominant taxa, with the most abundant OTUs in each sample representing less than 20% of the reads (Figure 6), and the top 10 most abundant OTUs accounting for only 39.5 ± 15.9% of total OTUs as compared to ≥88% for the mineral deployments. Although the post-mineral deployment groundwater communities clustered loosely with those that colonized minerals above RTZ, the pre-mineral deployment communities were distinct from all other samples. The pre-mineral deployment sample from well 3–24 was the most phylogenetically diverse sample recovered (Table 1), and contained organisms (e.g., Bacillales, Desulfosporosinus, Gemmataceae, Isosphaeraceae, Pirellulaceae, and Planctomycetia) that were rare or not present in the other libraries.
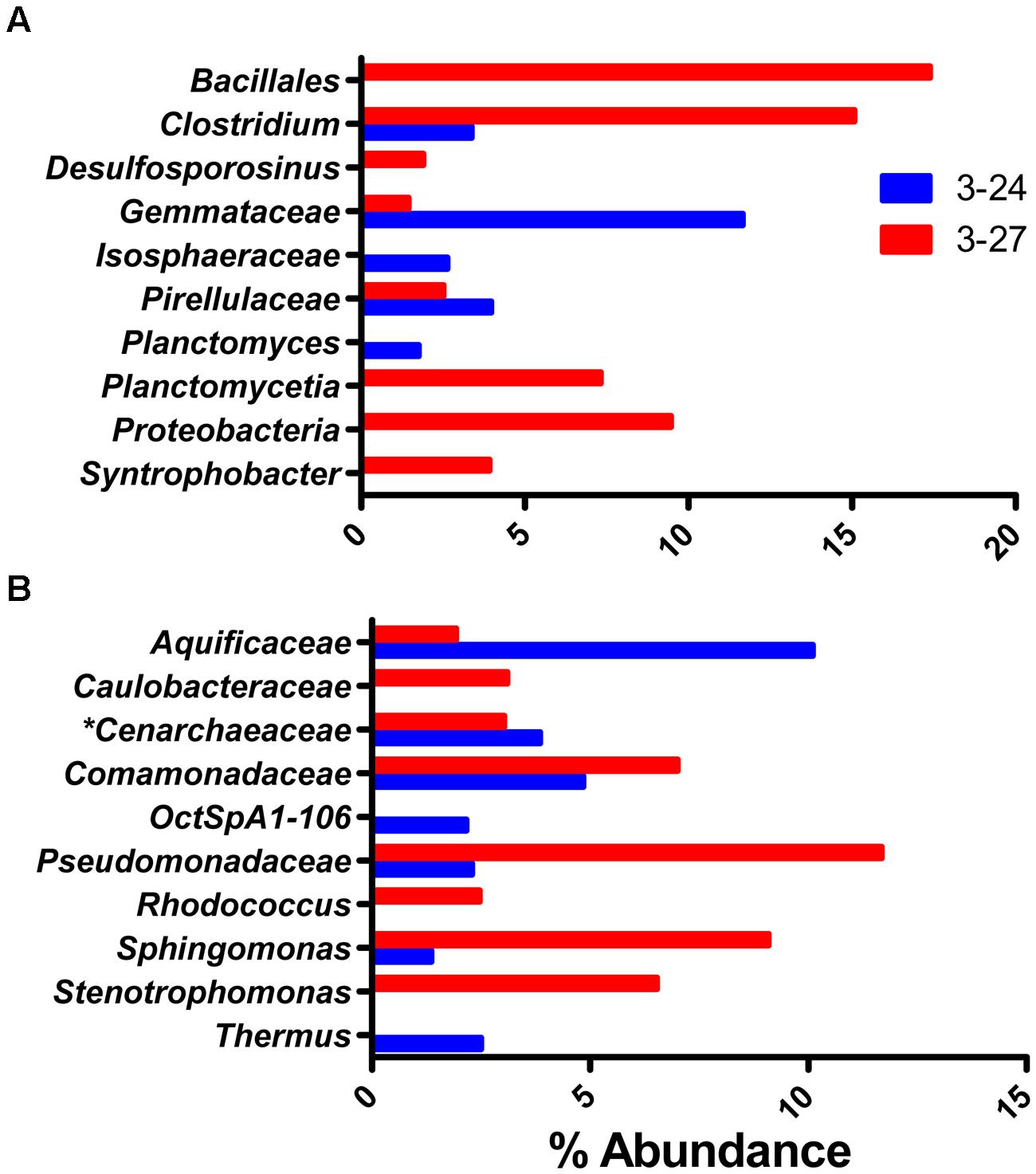
FIGURE 6. Top 10 most abundant taxa detected in one or two of the groundwater wells prior to (A) or after (B) the in situ mineral incubation experiment.
Discussion
The original goal of this study was to evaluate the hypothesis that organisms related to known FeOB would preferentially colonize mixtures of sand and the Fe(II)-bearing mineral biotite compared to sand alone when the minerals were suspended in Hanford Area 300 groundwater. This hypothesis arose from previous in situ biotite incubation studies which resulted in isolation of several different FeOB from the RTZ at the 300 Area site (Benzine et al., 2013). A secondary hypothesis was that differential colonization of the sand+biotite vs. sand-only deployments would disappear going across the RTZ, since oxidants (oxygen and nitrate) capable of supporting FeOB growth are absent below the RTZ. Contrary to these hypotheses, the 16S rRNA gene amplicon sequencing results indicated no difference in microbial community composition between the sand+biotite vs. sand-only treatments regardless of the depth at which the minerals were suspended in the groundwater (Figure 3). Instead, the sequence data demonstrated a predominance of heterotrophic taxa at all depth intervals. In addition, distinct depth-dependent changes in certain components of the microbial community were observed, indicating proliferation of anaerobic taxa within and below the RTZ, and the presence of chemolithotrophic taxa in the vicinity of the RTZ. Details related to these findings are presented below. Before doing so, however, it is important to acknowledge the possibility that the lack of difference between the sand+biotite vs. sand-only microbial communities is an artifact of the relatively short deployment interval, i.e., that preferential colonization of the biotite by lithotrophic organisms might have occurred if the minerals had undergone more lengthy in situ incubation. Put another way, it seems possible that the observed colonization represented an evolving response to environmental conditions, where the sampled community responded to the presence of readily available heterotrophic energy sources (e.g., DOC; see below) rather than the long-term presence of biotite as a lithotrophic energy source. A related issue (kindly raised by a reviewer of this paper) is that there could have been competition for mineral surface area as a resource for growth and colonization, with the heterotrophs having an energetic advantage (e.g., in terms of growth yield) compared to chemolithotrophs. This argument provides a simple explanation for why Fe(II)-oxidizers were not dominant members of the communities established on the emplaced minerals.
Heterotrophic Microbial Communities on Colonized Minerals
Members of the Pseudomonadaceae were the most abundant taxa detected in the colonized mineral samples (Figure 4). BLAST searches revealed that all of the Pseudomonadaceae assignments from the QIIME pipeline belong to the genus Pseudomonas. Pseudomonas sp. are known for their propensity to adhere to surfaces (Moore et al., 2006), and this together with their significant abundance in 300 Area groundwater [Figure 6B; see also Lin et al. (2012c)] and capacity to utilize a wide variety of organic substrates (Moore et al., 2006) may account for their predominance on the colonized materials. Other predominant taxa associated with the colonized minerals included known aerobic and/or facultatively anaerobic taxa such as Aquabacterium, Comamonadaceae, Vogesella (Figure 4); and known anaerobic taxa such as Clostridium, Desulfobulbaceae, Geobacter, Bacteroidales within and below the RTZ (Figure 5).
The observation that heterotrophic, rather than lithotrophic taxa, were most abundant on the incubated minerals is consistent with previous results indicating the capacity for heterotrophic metabolism within Hanford 300 Area sediments and groundwater (Lee et al., 2012; Lin et al., 2012a; Konopka et al., 2013; Percak-Dennett and Roden, 2014), as well as the predominance of heterotrophic taxa within 300 Area sediments and groundwater (Lin et al., 2012b,c). Hanford 300 Area groundwater contains low (≤1 ppm) but detectable quantities of dissolved organic carbon (DOC levels measured in bulk samples pumped from wells 3–24 and 3–27 prior to the in situ mineral deployment were approximately 0.6 ppm) which likely supported heterotrophic colonization of the mineral substrates. A recent single-cell genomic study of Pedobacter sp. present in 300 Area groundwater demonstrated the presence of a wide range of both intra and extracellular carbohydrate-active enzymes that may enable aerobic degradation of polymeric substrates as well as utilization of more labile sugars such as mannose and fucose (Wilkins et al., 2014). Although only a few OTUs related to Pedobacter were present in our libraries, it seems reasonable to assume that the various heterotrophic taxa that colonized the minerals possessed analogous metabolic capabilities.
The relatively high abundance of the aerobic predatory bacterium Bdellovibrio in samples from above the RTZ is consistent with the presence of a robust aerobic heterotrophic microbial community. Reports of Bdellovibrio in aquifer environments are rare, although Hutchens et al. (2004) found evidence for Bdellovibrio predation on methanotrophic bacteria in a groundwater-fed cave. The low abundance of Bdellovibrio in the groundwater samples (data not shown) compared to the colonized minerals suggests that these organisms became enriched in the MLS cartridges via predation on organisms attached to the minerals. More broadly, the large taxonomic differences between the most abundant mineral-associated vs. groundwater microbiota indicates that the presence of the particle attachment sites fundamentally altered microbial community structure. It should be noted, however, that fluid advection could have had an independent impact on groundwater community composition during the in situ mineral incubation period, i.e., by driving dispersal of taxa among differing depths or locations within the riparian corridor aquifer (Stegen et al., 2013).
Shifts in Microbial Community Composition across the RTZ
Principal coordinate plots of weighted Unifrac data revealed two distinct clusters of samples based on deployment depth, where communities above the RTZ were distinct from those within and below the RTZ (Figure 3). The observed shift in community composition going across the RTZ is consistent with alpha diversity analyses that indicated PD decreased with depth (Figure 2 and Table 1). A previous study noted differences in microbial communities at depths near the RTZ in 300 Area sediments (Lin et al., 2012b), likely due to the redox gradients present in the upper Ringold formation. Our results clearly demonstrate proliferation of the Fe(III)- and/or sulfate-reducing taxa Desulfobulbaceae (Holmes et al., 2004) and Geobacter (Lovley et al., 2011) [as well as other less abundant Fe(III)-reducing taxa such as Geothrix (Coates et al., 1999), Ignavibacteriales (Podosokorskaya et al., 2013), and Pelobacteraceae (Lovley et al., 1995)] on the colonized minerals within and below the RTZ. These results are consistent with qPCR studies which showed a peak in Geobacteraceae 16S rRNA and dsrA genes in the vicinity of the RTZ (Lin et al., 2012a), as well as with the appearance of the minerals upon recovery from the MLS arrays: materials from within and (more notably) below the RTZ had a distinct black color indicating accumulation of iron monosulfides (FeS). It is likely that Fe(III) oxides were produced from oxygen or nitrate-driven Fe(II) oxidation (see below) in the vicinity of the RTZ (see Figure 1), and that these were subsequently utilized by Fe(III)-reducers, generating Fe(II) that reacted with dissolved sulfide produced by sulfate-reducers to form the black FeS coatings on the sand grains. As observed previously (Lin et al., 2012a), dissolved sulfide concentrations were at or below detection (a few μM) in MLS groundwater samplers deployed prior to the in situ mineral incubation experiment, which is consistent with rapid scavenging of sulfide through reaction with Fe(II). Relatively low but detectable quantities of iron-sulfide minerals are present below the RTZ in Hanford 300 Area sediments (Peretyazhko et al., 2012; Percak-Dennett and Roden, 2014), and sediment incubation studies have demonstrated the capacity for Fe(III) and sulfate reduction driven by oxidation endogenous organic carbon sources in fine-grained 300 Area sediments (Lee et al., 2012; Percak-Dennett and Roden, 2014). Hence, it appears that the microbial communities that arose on the in situ-incubated minerals mirrored the composition and metabolic activity of communities in the sediments themselves.
Potential for Lithotrophic Microbial Metabolism within and above the RTZ
Although heterotrophic organisms clearly dominated the colonized minerals, several putative lithotrophic taxa were detected in significant abundance above, within, and in a few cases below the RTZ (Table 2). BLAST searches were conducted on these OTUs to gain further insight into the potential physiological capacities of the specific taxa detected in the libraries. Although caution must exercised in inferring physiology based on 16S rRNA gene similarity (Achenbach and Coates, 2000), this basic approach pioneered by Pace (1996, 1997) remains standard practice in microbial ecology. We focused on pure culture relatives of the clone sequences, for which reasonable physiological inferences could be made.
As expected given the requirement for oxidants such as oxygen and/or nitrate, the most abundant lithotrophic taxa were detected above and within the transition, including Nitrosopumilus, an aerobic, chemolithoautotrophic, NH4+ oxidizing Crenarchaeota (Thaumarchaeota) known to occur in both marine (Könneke et al., 2005) and soil (Jung et al., 2011) environments; and Hydrogenophaga, an aerobic, chemolithoautotrophic, H2-oxidizing Betaproteobacterial taxon (Willems et al., 1989; Yoon et al., 2008) that is present in a variety of natural and engineered (e.g., waste water treatment) environments (Schwartz and Friedrich, 2006). Although NH4+ measurements are not available for the Hanford 300 Area groundwaters, the presence of substantial nitrite in the vicinity of the RTZ is consistent with nitrification activity, as is the detection of nitrite-oxidizing relatives of Nitrospira (Table 2). It must acknowledged, however, that the presence of nitrite could be the result of partial denitrification rather than nitrification. In the case of H2 oxidizers, the presence of Hydrogenophaga makes sense as the geochemical data suggested an overall upward flux of dissolved H2 into the RTZ (Figure 1). A recent study recovered an autotrophic H2-oxidizing, nitrate-reducing Acidovorax sp. from in situ-incubated sand (using the same methods employed in this study) in 300 Area groundwater (Lee et al., 2014), which is consistent with the likely role of H2 metabolism in energy metabolism in the vicinity of the RTZ. A recent study demonstrated the ability of nitrite-oxidizing Nitrospira to grow autotrophically with H2 (Koch et al., 2014), and thus it seems possible that some of the Nitrospira relatives detected in 16S libraries could also have participated in H2 metabolism.
Amplicons related to three known chemolithotrophic FeOB were present at relatively low but easily detectable numbers in the 16S rRNA gene libraries, including sequences related to (1) a single amplified genome (SAG) of Leptothrix ochracea from a freshwater Fe seep in Maine, USA (Fleming et al., 2011); (2) Gallionella sp. strain JA52 isolated from mine water treatment plant in eastern Germany (Tischler et al., 2013); and (3) Bradyrhizobium sp. strain 22, one of the chemolithotrophic FeOB recently isolated from groundwater and sediments from the 300 Area RTZ (Benzine et al., 2013). It seems feasible that these organisms could thrive via oxidation of soluble Fe(II) produced below the RTZ (see Figures 1A,C).
Amplicons related to two reduced sulfur-oxidizing taxa were also detected in the 16S rRNA gene libraries, including strains of Thiobacillus thioparus isolated from stratified Lake Fryxell in Antarctica (Sattley and Madigan, 2006), and strains of Halothiobacillus sp. recovered from a shallow-water hydrothermal vent in the Aegean Sea and from various crop plant rhizosphere soils from Korea (Anandham et al., 2008). Although dissolved sulfide (HS-) levels were below detection in our MLS samples, Lin et al. (2012a) documented substantial HS- (up to ca. 40 μM) in groundwaters a few m below the RTZ. Lithotrophic sulfide oxidation could potentially contribute (along with reactions with sediment Fe phases) to scavenging of sulfide entering the RTZ from deeper groundwaters.
Collectively the above findings indicate that chemolithotrophic pathways, fueled by reduced compounds generated via anaerobic heterotrophic metabolism within and below the RTZ, are likely to play a significant role in elemental cycling in the vicinity of the RTZ.
Conclusion
An in situ mineral incubation experiment was conducted in the vicinity of a subsurface RTZ at the Hanford 300 Area site (Richland, WA, USA) to examine the hypothesis that FeOB would preferentially colonize mixtures of sand and the Fe(II)-bearing mineral biotite compared to sand alone. The study was motivated by previous work that documented the presence of Fe(II)-oxidizing bacteria capable of oxidizing structural Fe(II) in primary silicate and secondary phyllosilicate minerals in sediments and groundwater from the 300 Area (Benzine et al., 2013). In contrast to expectations, pyrosequencing of 16S rRNA gene amplicons from the colonized minerals showed no significant difference in community composition between the two treatments. However, the culture-independent analysis revealed interesting aspects of microbial community composition in the vicinity of the RTZ, including an overall predominance of heterotrophic taxa, and a clear shift toward anaerobic Fe(III)- and sulfate-reducing taxa below the RTZ. These results are consistent with previous studies of microbial community composition and activity in the vicinity of the RTZ, and reinforce the view that heterotrophic metabolism is likely to play a key role in maintenance of the redox boundary over long time scales in Area 300 sediments (Percak-Dennett and Roden, 2014). Despite the predominance of heterotrophic taxa, significant numbers of OTUs related to known NH4+, H2, Fe(II), and HS- oxidizing taxa were detected in the 16S rRNA gene libraries. The activity of these organisms, fueled by reduced compounds generated via anaerobic heterotrophic metabolism within and below the RTZ, could play a significant role in elemental cycling in the vicinity of the RTZ. The inferred coupling of heterotrophic and lithotrophic metabolic pathways has important implications for the fate and transport of redox-sensitive metals and radionuclides such as U and Tc across the Area 300 RTZ and other analogous environments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the US Department of Energy, Office of Biological and Environmental Research, Subsurface Biogeochemical Research Program through the SBR Scientific Focus Area at the Pacific Northwest National Laboratory (PNNL). We thank David Kennedy (PNNL) for help with collection and processing of the MLS samples.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00858
References
Achenbach, L. A., and Coates, J. D. (2000). Disparity between bacterial phylogeny and physiology. ASM News 66, 1–4.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J. H., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Anandham, R., Gandhi, P. I., Madhaiyan, M., and Sa, T. (2008). Potential plant growth promoting traits and bioacidulation of rock phosphate by thiosulfate oxidizing bacteria isolated from crop plants. J. Basic Microbiol. 48, 439–447. doi: 10.1002/jobm.200700380
Bates, S. T., Berg-Lyons, D., Caporaso, J. G., Walters, W. A., Knight, R., and Fierer, N. (2011). Examining the global distribution of dominant archaeal populations in soil. ISME J. 5, 908–917. doi: 10.1038/ismej.2010.171
Benzine, J., Shelobolina, E., Xiong, M. Y., Kennedy, D. W., Mckinley, J. P., Lin, X., et al. (2013). Fe-phyllosilicate redox cycling organisms from a redox transition zone in Hanford 300 Area sediments. Front. Microbiol. 4:388. doi: 10.3389/fmicb.2013.00388
Beversdorf, L. J., Miller, T. R., and Mcmahon, K. D. (2013). The role of nitrogen fixation in cyanobacterial bloom toxicity in a temperate, eutrophic lake. PLoS ONE 8:e56103. doi: 10.1371/journal.pone.0056103
Bjornstad, B. N., Horner, J. A., Vermeul, V. R., Lanigan, D. C., and Thorne, P. D. (2009). Borehole Completion and Conceptual Hydrolgeologic Model for the IFRC Well Field, 300 Area, Hanford Site. Richland, WA: Pacific Northwest Laboratory.
Brodie, E. L., Desantis, T. Z., Joyner, D. C., Baek, S. M., Larsen, J. T., Andersen, G. L., et al. (2006). Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl. Environ. Microbiol. 72, 6288–6298. doi: 10.1128/AEM.00246-06
Brookshaw, D. R., Lloyd, J. R., Vaughan, D. J., and Pattrick, R. A. D. (2014). Bioreduction of biotite and chlorite by a Shewanella species. Am. Miner. 99, 1746–1754. doi: 10.2138/am.2014.4774CCBY
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chaudhuri, S. K., Lack, J. G., and Coates, J. D. (2001). Biogenic magnetite formation through anaerobic biooxidation of Fe(II). Appl. Environ. Microbiol. 67, 2844–2848. doi: 10.1128/AEM.67.6.2844-2848.2001
Christensen, J. N., Dresel, P. E., Conrad, M. E., Maher, K., and Depaolo, D. J. (2004). Identifying the sources of subsurface contamination at the Hanford Site in Washington using high-precision uranium isotopic measurements. Environ. Sci. Technol. 38, 3330–3337. doi: 10.1021/es034700q
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Clarke, K. R., Clarke, R. K., and Gorley, R. N. (2006). Primer V6: User Manual – Tutorial. Plymouth: Plymouth Marine Laboratory.
Coates, J. D., Ellis, D. J., Gaw, C. V., and Lovley, D. R. (1999). Geothrix fermentans gen. nov., sp. nov., a novel Fe(III)-reducing bacterium from a hydrocarbon-contaminated aquifer. Int. J. Syst. Evol. Microbiol. 49, 1615–1622. doi: 10.1099/00207713-49-4-1615
Dong, H. L., Kukkadapu, R. K., Fredrickson, J. K., Zachara, J. M., Kennedy, D. W., and Kostandarithes, H. M. (2003). Microbial reduction of structural Fe(III) in illite and goethite. Environ. Sci. Technol. 37, 1268–1276. doi: 10.1021/es020919d
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Ehrich, S., Behrens, D., Lebedeva, E., Ludwig, W., and Bock, E. (1995). A new obligately chemolithoautotrophic, nitrite-oxidizing bacterium, Nitrospira moscoviensis sp. nov. and its phylogenetic relationship. Arch. Microbiol. 164, 16–23. doi: 10.1007/BF02568729
Faith, D. P., and Baker, A. M. (2006). Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol. Bioinform. 2, 121–128.
Fleming, E. J., Langdon, A. E., Martinez-Garcia, M., Stepanauskas, R., Poulton, N. J., Masland, E. D. P., et al. (2011). What’s new is old: resolving the identity of Leptothrix ochracea using single cell genomics, pyrosequencing and FISH. PLoS ONE 6:e17769. doi: 10.1371/journal.pone.0017769
Gillis, M., Vandamme, P., Devos, P., Swings, J., and Kersters, K. (2001). “Polyphasic taxonomy,” in Bergey’s Manual of Systematic Bacteriology, 2nd Edn, eds D. R. Boone and R. W. Castenholz (New York, NY: Springer), 43–48. doi: 10.1007/978-0-387-21609-6_7
Holmes, D. E., Bond, D. R., and Lovley, D. R. (2004). Electron transfer by Desulfobulbus propionicus to Fe(III) and graphite electrodes. Appl. Environ. Microbiol. 70, 1234–1237. doi: 10.1128/AEM.70.2.1234-1237.2004
Hutchens, E., Radajewski, S., Dumont, M. G., Mcdonald, I. R., and Murrell, J. C. (2004). Analysis of methanotrophic bacteria in Movile Cave by stable isotope probing. Environ. Microbiol. 6, 111–120. doi: 10.1046/j.1462-2920.2003.00543.x
Jung, M. -Y., Park, S. -J., Min, D., Kim, J. -S., Rijpstra, W. I. C., Sinninghe Damsté, J. S., et al. (2011). Enrichment and characterization of an autotrophic ammonia-oxidizing Archaeon of mesophilic Crenarchaeal Group I.1a from an agricultural soil. Appl. Environ. Microb. 77, 8635–8647. doi: 10.1128/AEM.05787-11
Koch, H., Galushko, A., Albertsen, M., Schintlmeister, A., Gruber-Dorninger, C., Lucker, S., et al. (2014). Growth of nitrite-oxidizing bacteria by aerobic hydrogen oxidation. Science 345, 1052–1054. doi: 10.1126/science.1256985
Könneke, M., Bernhard, A. E., Torre, J. R. D. L., Walker, C. B., Waterbury, J. B., and Stahl, D. A. (2005). Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437, 543–546. doi: 10.1038/nature03911
Konopka, A., Plymale, A. E., Carvajal, D. A., Lin, X. J., and Mckinley, J. P. (2013). Environmental controls on the activity of aquifer microbial communities in the 300 Area of the Hanford site. Microb. Ecol. 66, 889–896. doi: 10.1007/s00248-013-0283-3
Kostka, J. E., Dalton, D. D., Skelton, H., Dollhopf, S., and Stucki, J. W. (2002). Growth of iron(III)-reducing bacteria on clay minerals as the sole electron acceptor and comparison of growth yields on a variety of oxidized iron forms. AEM 68, 6256–6262. doi: 10.1128/AEM.68.12.6256-6262.2002
Kostka, J. E., Stucki, J. W., Nealson, K. H., and Wu, J. (1996). Reduction of structural Fe(III) in smectite by a pure culture of Shewanella putrefaciens strain MR-1. Clays Clay Miner. 44, 522–529. doi: 10.1346/CCMN.1996.0440411
Lee, J. H., Fredrickson, J. K., Kukkadapu, R. K., Boyanov, M. I., Kemner, K. M., Lin, X. J., et al. (2012). Microbial reductive transformation of phyllosilicate Fe(III) and U(VI) in fluvial subsurface sediments. Environ. Sci. Technol. 46, 3721–3730. doi: 10.1021/es204528m
Lee, J. H., Fredrickson, J. K., Plymale, A. E., Dohnalkova, A. C., Resch, C. T., Mckinley, J. P., et al. (2014). An autotrophic H2-oxidizing, nitrate-respiring, Tc(VII)-reducing Acidovorax sp. isolated from a subsurface oxic-anoxic transition zone. Environ. Microb. 7, 395–403. doi: 10.1111/1758-2229.12263
Lin, X., Kennedy, D., Peacock, A., Mckinley, J., Resch, C. T., Fredrickson, J., et al. (2012a). Distribution of microbial biomass and potential for anaerobic respiration in Hanford Site 300 Area subsurface sediment. Appl. Environ. Microbiol. 78, 759–767. doi: 10.1128/AEM.07404-11
Lin, X. J., Kennedy, D., Fredrickson, J., Bjornstad, B., and Konopka, A. (2012b). Vertical stratification of subsurface microbial community composition across geological formations at the Hanford Site. Environ. Microbiol. 14, 414–425. doi: 10.1111/j.1462-2920.2011.02659.x
Lin, X. J., Mckinley, J., Resch, C. T., Kaluzny, R., Lauber, C. L., Fredrickson, J., et al. (2012c). Spatial and temporal dynamics of the microbial community in the Hanford unconfined aquifer. ISME J. 6, 1665–1676. doi: 10.1038/ismej.2012.26
Lindsey, K. A., and Gaylord, D. R. (1990). Lithofacies and sedimentology of the Miocene-Pliocene Ringold Formation, Hanford Site, south-central Washington. Northwest Sci. 64, 165–180.
Lovley, D. R., Phillips, E. J. P., Lonergan, D. J., and Widman, P. K. (1995). Fe(III) and S0 reduction by Pelobacter carbinolicus. AEM 61, 2132–2138.
Lovley, D. R., Ueki, T., Zhang, T., Malvankar, N. S., Shrestha, P. M., Flanagan, K. A., et al. (2011). “Geobacter: the microbe electric’s physiology, ecology, and practical applications,” in Advances in Microbial Physiology Physiol, ed. R. K. Poole (Amherst, MA: University of Massachusetts), 1–100. doi: 10.1016/b978-0-12-387661-4.00004-5
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microb. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Moore, E. R. B., Tindall, B. J., Santos, V. A. P. M. D., Pieper, D. H., Ramos, J. L., and Palleroni, N. J. (2006). “Nonmedical: Pseudomonas,” in The Prokaryotes Proteobacteria: Gamma Subclass, Vol. 6: eds M. Dworkin and S. Falkow (New York, NY: Springer), 646–703.
Mori, K., Suzuki, K.-I., Urabe, T., Sugihara, M., Tanaka, K., Hamada, M., et al. (2011). Thioprofundum hispidum sp. nov., an obligately chemolithoautotrophic sulfur-oxidizing gammaproteobacterium isolated from the hydrothermal field on Suiyo Seamount, and proposal of Thioalkalispiraceae fam. nov. in the order Chromatiales. Int. J. System. Evol. Microbiol. 61, 2412–2418. doi: 10.1099/ijs.0.026963-0
Pace, N. R. (1996). New perspective on the natural microbial world: molecular microbial ecology. ASM News 62, 463–470.
Pace, N. R. (1997). A molecular view of microbial diversity and the biosphere. Science 276, 734–740.
Percak-Dennett, E. M., and Roden, E. E. (2014). Geochemical and microbiological responses to oxidant introduction in reduced subsurface sediment from the Hanford 300 Area, WA. Environ. Sci. Technol. 48, 9197–9204. doi: 10.1021/es5009856
Peretyazhko, T. S., Zachara, J. M., Kukkadapu, R. K., Heald, S. M., Kutnyakov, I. V., Resch, C. T., et al. (2012). Pertechnetate (TcO4-) reduction by reactive ferrous iron forms in naturally anoxic, redox transition zone sediments from the Hanford Site, USA. Geochim. Cosmochim. Acta 92, 48–66. doi: 10.1016/j.gca.2012.05.041
Peterson, R. E., and Connelly, M. P. (2001). Zone of Interaction Between Hanford Site Groundwater and Adjacent Columbia River Pnnl-13674 Pacific. Richland, WA: Northwest National Laboratory.
Podosokorskaya, O. A., Kadnikov, V. V., Gavrilov, S. N., Mardanov, A. V., Merkel, A. Y., Karnachuk, O. V., et al. (2013). Characterization of Melioribacter roseus gen. nov., sp nov., a novel facultatively anaerobic thermophilic cellulolytic bacterium from the class Ignavibacteria, and a proposal of a novel bacterial phylum Ignavibacteriae. Environ. Microbiol. 15, 1759–1771. doi: 10.1111/1462-2920.12067
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Roden, E. E. (2012). Microbial iron-redox cycling in subsurface environments. Biochem. Trans. 40, 1249–1256. doi: 10.1042/BST20120202
Roden, E. E., Sobolev, D., Glazer, B., and Luther, G. W. (2004). Potential for microscale bacterial Fe redox cycling at the aerobic-anaerobic interface. Geomicrobiol. J. 21, 379–391. doi: 10.1080/01490450490485872
Sattley, W. M., and Madigan, M. T. (2006). Isolation, characterization, and ecology of cold-active, chemolithotrophic, sulfur-oxidizing bacteria from perennially ice-covered Lake Fryxell, Antarctica. Appl. Environ. Microb. 72, 5562–5568. doi: 10.1128/AEM.00702-06
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schwartz, E., and Friedrich, B. (2006). “The H2-metabolizing prokaryotes,” in The Prokaryotes, Vol. 7, 3rd Edn, ed. S. F. M. Dworkin, E. Rosenberg, K .H. Schleifer and E. Stackebrandt (New York, NY: Springer), 496–563.
Shelobolina, E. S., Gaw-Vanpraagh, C., and Lovley, D. R. (2003). Use of ferric and ferrous iron containing minerals for respiration by Desulfitobacterium frappieri. Geomicrobiol. J. 20, 143–156. doi: 10.1080/01490450303884
Shelobolina, E. S., Konishi, H., Xu, H., Benzine, J., Xiong, M., Wu, T., et al. (2012a). Isolation of phyllosilicate–iron redox cycling microorganisms from an illite–smectite rich hydromorphic soil. Front. Microbiol. 3:134. doi: 10.3389/fmicb.2012.00134
Shelobolina, E. S., Xu, H., Konishi, H., Kukkadapu, R., Wu, T., Blothe, M., et al. (2012b). Microbial lithotrophic oxidation of structural Fe(II) in biotite. Appl. Environ. Microbiol. 78, 5746–5752. doi: 10.1128/AEM.01034-12
Sievert, S. M., Heidorn, T., and Kuever, J. (2000). Halothiobacillus kellyi sp. nov., a mesophilic, obligately chemolithoautotrophic, sulfur-oxidizing bacterium isolated from a shallow-water hydrothermal vent in the Aegean Sea, and emended description of the genus Halothiobacillus. Int. J. System. Evol. Microbiol. 50, 1229–1237. doi: 10.1099/00207713-50-3-1229
Stegen, J. C., Lin, X. J., Fredrickson, J. K., Chen, X. Y., Kennedy, D. W., Murray, C. J., et al. (2013). Quantifying community assembly processes and identifying features that impose them. ISME J. 7, 2069–2079. doi: 10.1038/ismej.2013.93
Straub, K. L., Benz, M., and Schink, B. (2001). Iron metabolism in anoxic environments at near neutral pH. FEMS Microbiol. Ecol. 34, 181–186. doi: 10.1111/j.1574-6941.2001.tb00768.x
Tebo, B. M., and He, L. M. (1999). “Microbially mediated oxidation precipitation reactions,” in Mineral-Water Interfacial Reactions, eds D. L. Sparks and T. J. Grundl (Washington, DC: Americal Chemical Society), 393–414. doi: 10.1021/bk-1998-0715.ch020
Tischler, J. S., Jwair, R. J., Gelhaar, N., Drechsel, A., Skirl, A.-M., Wiacek, C., et al. (2013). New cultivation medium for “Ferrovum” and Gallionella-related strains. J. Microbiol. Meth. 95, 138–144. doi: 10.1016/j.mimet.2013.07.027
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wilkins, M., Kennedy, D. W., Castelle, C. J., Field, E. K., Stepanauskas, R., Fredrickson, J. K., et al. (2014). Single-cell genomics reveals metabolic strategies for microbial growth and survival in an oligotrophic aquifer. Microbiology 160, 362–372. doi: 10.1099/mic.0.073965-0
Willems, A., Busse, J., Goor, M., Pot, B., Falsen, E., Jantzen, E., et al. (1989). Hydrogenophaga, a new genus of hydrogen-oxidizing bacteria that includes Hydrogenophaga flava comb. nov. (formerly Pseudomonas flava), Hydrogenophaga palleronii (formerly Pseudomonas palleronii), Hydrogenophaga pseudoflava (formerly Pseudomonas pseudoflava and “Pseudomonas carboxydoflava”), and Hydrogenophaga taeniospiralis (formerly Pseudomonas taeniospiralis). Int. J. System. Evol. Microbiol. 39, 319–333. doi: 10.1099/00207713-39-3-319
Yoon, K. S., Tsukada, N., Sakai, Y., Ishii, M., Igarashi, Y., and Nishihara, H. (2008). Isolation and characterization of a new facultatively autotrophic hydrogen-oxidizing Betaproteobacterium, Hydrogenophaga sp. AH-24. FEMS Microbiol. Lett. 278, 94–100. doi: 10.1111/j.1574-6968.2007.00983.x
Keywords: subsurface sediments, redox transition, minerals, colonization, amplicon sequencing
Citation: Converse BJ, McKinley JP, Resch CT and Roden EE (2015) Microbial mineral colonization across a subsurface redox transition zone. Front. Microbiol. 6:858. doi: 10.3389/fmicb.2015.00858
Received: 08 May 2015; Accepted: 06 August 2015;
Published: 28 August 2015.
Edited by:
Paul Bodelier, Netherlands Institute of Ecology, NetherlandsReviewed by:
Neil Duncan Gray, University of Newcastle, UKSteffen Kolb, Friedrich Schiller University Jena, Germany
Copyright © 2015 Converse, McKinley, Resch and Roden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric E. Roden, Department of Geoscience, University of Wisconsin-Madison, 1215 West Dayton Street, Madison, WI 53706, USA,ZXJvZGVuQGdlb2xvZ3kud2lzYy5lZHU=