 Thi Thu Hao Van1
Thi Thu Hao Van1 Jake A. Lacey
Jake A. Lacey Canh Phung
Canh Phung Robert J. Moore
Robert J. Moore- 1School of Science, RMIT University, Bundoora, VIC, Australia
- 2Doherty Department, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Melbourne, VIC, Australia
- 3Scolexia Pty Ltd., Moonee Ponds, VIC, Australia
Chickens infected with Campylobacter jejuni or Campylobacter coli are largely asymptomatic, however, infection with the closely related species, Campylobacter hepaticus, can result in Spotty Liver Disease (SLD). C. hepaticus has been detected in the liver, bile, small intestine and caecum of SLD affected chickens. The survival and colonization mechanisms that C. hepaticus uses to colonize chickens remain unknown. In this study, we compared the genome sequences of 14 newly sequenced Australian isolates of C. hepaticus, isolates from outbreaks in the United Kingdom, and reference strains of C. jejuni and C. coli, with the aim of identifying virulence genes associated with SLD. We also carried out global comparative transcriptomic analysis between C. hepaticus recovered from the bile of SLD infected chickens and C. hepaticus grown in vitro. This revealed how the bacteria adapt to proliferate in the challenging host environment in which they are found. Additionally, biochemical experiments confirmed some in silico metabolic predictions. We found that, unlike other Campylobacter sp., C. hepaticus encodes glucose and polyhydroxybutyrate metabolism pathways. This study demonstrated the metabolic plasticity of C. hepaticus, which may contribute to survival in the competitive, nutrient and energy-limited environment of the chicken. Transcriptomic analysis indicated that gene clusters associated with glucose utilization, stress response, hydrogen metabolism, and sialic acid modification may play an important role in the pathogenicity of C. hepaticus. An understanding of the survival and virulence mechanisms that C. hepaticus uses will help to direct the development of effective intervention methods to protect birds from the debilitating effects of SLD.
Introduction
Spotty Liver Disease (SLD) causes significant egg production losses and mortality in layer birds (Crawshaw and Young, 2003; Grimes and Reece, 2011). It has been sporadically reported over the last 60 years, first from the United States then from Canada, New Zealand, Estonia, the United Kingdom, Austria, Germany and Australia (Tudor, 1954; Bertschinger, 1965; Leesment and Parve, 1965; Truscott and Stockdale, 1966; Kölbl and Willinger, 1967; Pohl et al., 1969; Crawshaw and Irvine, 2012). The disease has become increasingly common in Australia over the last decade and is now considered one of the most significant health challenges in the egg industry (Grimes and Reece, 2011). However, it was only in 2015 that a novel Campylobacter species was isolated from SLD cases in the UK and in 2016 Campylobacter hepaticus was identified and characterized from Australian cases of SLD (Crawshaw et al., 2015; Van et al., 2016). In 2017 C. hepaticus was definitively shown to be the cause of SLD (Van et al., 2016, 2017a,b).
As C. hepaticus has only recently been identified, the study of its biology is just beginning. The draft genomes of the type strain, C. hepaticus HV10, isolated from the liver of an Australian SLD affected chicken, and a series of British isolates are available (Van et al., 2016; Petrovska et al., 2017). C. hepaticus is most closely related to the foodborne pathogens C. jejuni and C. coli. However, C. hepaticus lacks some of the well-identified virulence genes found in C. jejuni, such as the cytolethal distending toxin (CDT) genes. It is anticipated that C. hepaticus must harbor a set of genes responsible for the pathogenesis observed in SLD affected chickens. These genes must encode products that lead to damage to the liver, as well as mortality and egg production losses. C. hepaticus has been isolated from liver and bile of SLD affected birds and has also been shown to be present in the gastrointestinal tract (Van et al., 2017b, 2018). Bile is a challenging environment and presumably C. hepaticus must orchestrate the expression of certain genes to help them survive within this niche.
High-throughput next-generation sequencing (NGS) has revolutionized transcriptomics by allowing global expression studies through RNA sequencing or RNA-Seq, through the sequencing of complementary DNA (cDNA) (Kukurba and Montgomery, 2015). RNA-Seq has rapidly taken the place of previous methods of genome-wide quantification of gene expression (transcriptomics) including hybridization-based microarrays and Sanger sequencing-based approaches. RNA-Seq has proven to be a fast, sensitive and reliable method because of the high sequence coverage. This approach has been used widely to study bacterial transcriptomics (Taveirne et al., 2013; Rao et al., 2015).
Genes that encode products required for niche adaptation, colonization, and virulence are yet to be identified in C. hepaticus. The aim of this study was to investigate potential virulence factors that could explain the pathogenic nature of C. hepaticus in poultry and identify strategies that C. hepaticus uses to colonize and survive in the host. We compared the genome sequences of 14 newly sequenced Australian isolates of C. hepaticus to nine C. hepaticus isolates from outbreaks in the United Kingdom that had previously been sequenced (Petrovska et al., 2017) and 10 reference strains of C. jejuni and C. coli, with the aim of identifying potential virulence genes in C. hepaticus. Furthermore, we studied the differential gene expression of C. hepaticus HV10; comparing the transcriptomes of in vivo (recovered from chicken bile samples) and in vitro (cultured on horse blood agar plates) grown bacteria. These results were combined with the comparative genomics analysis to investigate the mechanisms that C. hepaticus may use to adapt to the challenging bile environment and cause disease in chickens.
Materials and Methods
Whole Genome Analysis of C. hepaticus Australian and UK Isolates
Campylobacter hepaticus HV10 was published as a draft genome (Van et al., 2017b). In this study, the complete closed genome of HV10 was obtained by combining short Illumina reads, and long PacBio reads and a series of bioinformatics pipelines as described in Lacey et al. (2018). The closed C. hepaticus HV10 genome was deposited in the NCBI database (accession number: CP031611.1). Fourteen Australian C. hepaticus isolates, each from an independent SLD outbreak event, were sequenced (methods as described in Van et al. (2016) and compared with the publicly available whole genome sequences of nine C. hepaticus isolates from the United Kingdom, five C. jejuni, and five C. coli representative genomes extracted from the NCBI database (Table 1). Genomes were assembled using the A5MiSeq pipeline version 20150522 (Coil et al., 2015) and they were annotated using both Prokka 1.14-dev and RAST version 2.0 (Aziz et al., 2008; Seemann, 2014). All assemblies and read sets were deposited in NCBI (Bioproject PRJNA485661).
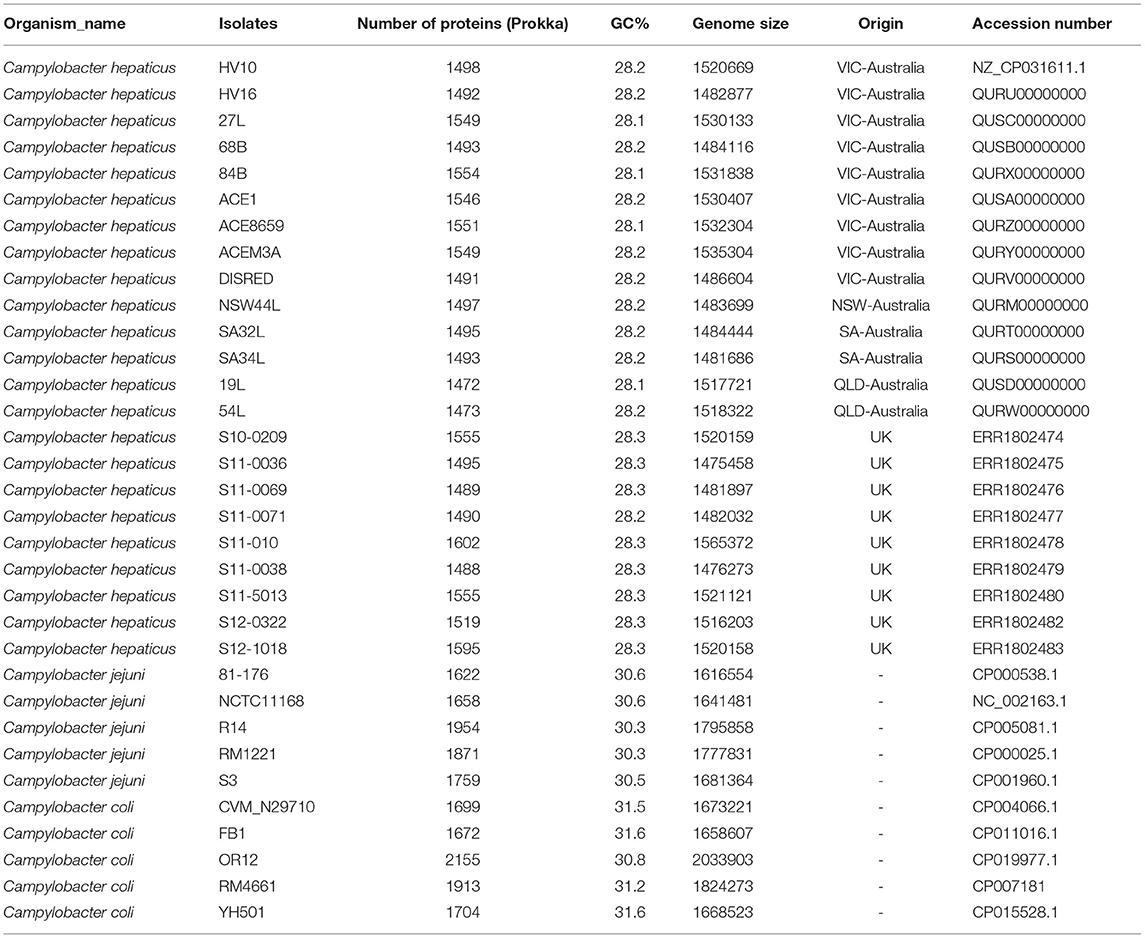
Table 1. Isolates used in this study.
Homolog identification and pan genome investigation of all predicted coding sequences was performed using two independent methods. Clustering using USEARCH v10.0 at 70% similarity across 70% protein length within the Bacterial Pan genomes analysis (BPGA v1.3) tool pipeline (Chaudhari et al., 2016), and with Roary v3.12 at a protein identity of 70% (-i70) and no splitting of paralogs (-s) (Page et al., 2015).
For phylogenetic inferences, single nucleotide variants (SNVs) were called by aligning reads to a reference genome, C. hepaticus strain HV10, using Snippy v3.2 (https://github.com/tseemann/snippy). Gubbins v2.3.4 (Croucher et al., 2015) was used for the detection and removal of recombinogenic regions, and PHASTER was used to screen for prophage integrations that would be outside the clonal frame (Zhou et al., 2011). A Maximum-likelihood tree was built in RAxML v8.2.12 (Stamatakis, 2014) using the general-time reversible model (GTRCAT) with 1,000 bootstrap replicates. Clustering of strains was performed using RAMI at a patristic distance threshold of 0.05 divergence (Pommier et al., 2009).
The map of the DNA features of C. hepaticus reference strain HV10 was produced in DNAplotter v16.0.0 (Carver et al., 2009). The core and pan genome plots of the 24 C. hepaticus isolates and COG distribution plot of functional categories for coding sequences within these 24 genomes were produced using BPGA.
Identification of Virulence Associated Genes of C. hepaticus
The C. hepaticus HV10 genome was examined for potential virulence genes by searching against the Virulence Factor Database (http://www.mgc.ac.cn/VFs/main.htm) in ABRicate (https://github.com/tseemann/abricate) (data assessed June 2018) and by inspecting the annotated genome manually. To elucidate the genetic potential of C. hepaticus to cause SLD a pan genome wide association study (PGWAS) was performed using Scoary v1.6.10 (Brynildsrud et al., 2016), and each gene in the C. hepaticus pan genome for association to SLD was screened. Genes of interest were identified as specific to the C. hepaticus genomes (present in 100% of isolates and absent in all reference C. coli and C. jejuni strains). The functionality of these unique genes was inferred from matches to the Pfam database (Finn et al., 2014), Interproscan (Jones et al., 2014), Swiss-Prot (Bairoch and Apweiler, 1997) and Uniprot (UniProt Consortium, 2015). Product descriptions were assigned with homologs of 70% similarity across 90% of protein length. CRISPRFinder (v2017-05-09) (Grissa et al., 2007) was used to analyse CRISPRs.
Investigation of C. hepaticus Horizontally Acquired Elements
The annotated genomes of the 23 C. hepaticus isolates were first manually inspected for any potential acquired genetic materials. Contigs with genes annotated as suspected plasmid elements were Blasted against the NCBI database. Significant matches were determined by matches >90% coverage and identity. ABRicate v0.8.7 was used to screen for antibiotic resistance genes.
RNA-Seq Analysis of C. hepaticus During in vivo Colonization and in vitro Growth
C. hepaticus HV10 (Van et al., 2016) were grown on Brucella agar (Becton Dickinson) with 5% horse blood (HBA) and incubated at 37°C in microaerobic conditions using CampyGen gas packs (Oxoid).
SLD in chickens was induced by challenge with C. hepaticus HV10. The animal experimentation was approved by the Wildlife and Small Institutions Animal Ethics Committee of the Victorian Department of Economic Development, Jobs, Transport and Resources (approval number 14.16). Hy-Line layer hens (26-weeks old, sourced from a farm that had not observed any SLD in their flocks for several years) were used in the study. Birds were also tested for C. hepaticus to ensure they were C. hepaticus negative before the trial by using the specific PCR developed by Van et al. (2017b) on the cloacal swab samples. Experimental chickens were challenged as previously described (Van et al., 2017a). Briefly, birds were challenged by direct oral gavage with 1x109 CFU of C. hepaticus HV10 strain in 1 ml of Brucella broth, whereas the control chickens were given 1 ml of Brucella broth. The birds were sacrificed 5 days post-challenge and the livers were examined for lesions. Bile samples from all chickens were taken aseptically from the gall bladder and placed in tubes containing RNAlater (Qiagen) for RNA isolation. Samples were kept on ice, transported to the laboratory and processed immediately.
For RNA isolation from C. hepaticus grown in HBA (in vitro), C. hepaticus was harvested from HBA plates and resuspended in Brucella broth to an OD600 of 0.5 then centrifuged. The cell pellet was resuspended in RNAlater to stabilize RNA. RNA was extracted using the ScriptSeq Complete kit (Epicenter) following the manufacturer's instructions. The RNA was treated with DNase I (NEB) to remove DNA contamination. The quality of the total RNA in the samples was checked using a Nanodrop spectrophotometer (Thermofisher). RNA was also electrophoresed on a 1% agarose gel and PCR amplified using SLD specific primers (to check genomic DNA contamination) as previously described (Van et al., 2017b). RNA concentration was measured using the Qubit RNA Assay Kit (Life Technologies). The RNA samples were stored at −80°C. Both in vitro and in vivo samples were done in triplicate.
Ribosomal RNA was first removed from the RNA samples using a Ribo-Zero Magnetic Kit (Bacteria) (Illumina). Libraries for Illumina sequencing were prepared using a ScriptSeq v2 RNA-Seq Library Preparation Kit (Illumina) from the rRNA-deleted RNA. The libraries were sequenced on an Illumina MiSeq platform using 300 bp paired end reads.
The Illumina reads were mapped to the reference genome and differentially expressed genes (DEGs) were identified. Raw reads were quality trimmed using Trimmomatic version 0.36 (Bolger et al., 2014), and the trimmed reads were aligned against the C. hepaticus HV10 reference genome using BWA (Li and Durbin, 2010). The SAM files were imported into Blast2Go version 4.1.9 for unique read counts and differential expression analysis (Conesa and Götz, 2008). Parameters for classifying significantly expressed genes (DEGs) were ≥2-fold differences in the transcript abundance and ≤ 0.5% false discovery rate (FDR). The list of up-regulated/down-regulated genes were motif scanned to investigate their biological significances and SEED viewer was used for subsystem functional categorization of the predicted open reading frames (ORFs) from RAST annotation. DEGs were further examined by determining the KEGG Biosynthesis pathway to which they belonged.
Confirmation of the Glucose Utilization Ability of C. hepaticus
Campylobacter jejuni strain 81116 (NCTC11828), C. coli NCTC 11366 and three C. hepaticus isolates, C. hepticus HV10, C. hepaticus 19L and C. hepaticus 44L were used in glucose utilization studies. After cultures were grown in HBA for 3 days, cells were collected and resuspended in physiological saline (0.9% NaCl) to an OD600 of 1.0. The medium used to test the ability of C. hepaticus to utilize glucose consisted of inorganic salts (IS) as described previously (Alazzam et al., 2011). L-cysteine (0.2 mM) was used as a nitrogen source, and α-D-glucose (10 mM) (Sigma) was used as the sole carbon source. The experiment was carried out in 24-well plates. Each well-contained 100 μl of culture (OD600 = 1). Controls included culture in IS plus L-cysteine only and IS plus α-D-glucose only. 2,3,5 tetrazolium chloride (TTC) (Sigma) (0.0665 g/L) was used as an indicator in all wells (Menolasino, 1959). A color change is observed in growing cultures, indicating utilization. The color change results were read after 36 h of incubation at 37°C. C. hepaticus, C. jejuni, and C. coli were also grown in Brucella broth to confirm their viability. The experiment was repeated twice, each time in biological triplicate.
Results
C. hepaticus Has a Closed Pan Genome
By combining short Illumina reads and long PacBio reads, the complete closed genome of C. hepaticus HV10 strain was obtained in this study (NCBI accession number CP031611). The genome size of C. hepaticus HV10 is 1,520,669 bp with a GC content of 28.2%. It has been used as the reference genome to compare with the other draft genome sequences.
Genome sizes of the C. hepaticus isolates ranged from 1.475 to 1.565 Mb whereas the representative C. jejuni genomes ranged between 1.617 and 1.800 Mb and C. coli genome sizes ranged between 1.668 and 2.034 Mb. There were between 1472 and 1595 annotated protein-coding genes predicted to be encoded by the 23 C. hepaticus isolates, whereas that were 1622–1954 for five C. jejuni isolates and 1672–2155 for five C. coli isolates (Table 1). A total of 1,059 core genes were conserved in all 33 genomes (fourteen Australian C. hepaticus isolates, nine United Kingdom C. hepaticus isolates, five C. jejuni and five C. coli). Maximum likelihood (ML) phylogeny as produced from RAxML was inferred from 33,157 SNVs. The core genome tree of the 33 genomes comprised 3 phylogenetically distinct lineages corresponding to each of the 3 species (Figure 1A).
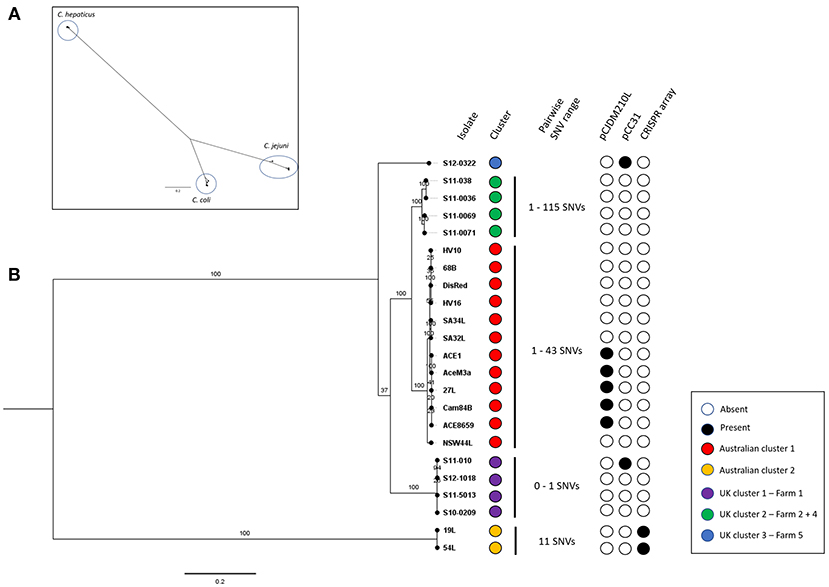
Figure 1. (A) Campylobacter spp. core genome phylogeny. Core genome alignment to the C. hepaticus reference strain HV10 of all C. hepaticus isolates and 5 isolates of C. coli and C. jejuni was performed. A maximum likelihood tree was inferred from 33,157 SNVs and built using RAxML using the general-reversible model (GTRCAT) with 1,000 bootstrap replicates. Scale bar shows nucleotide divergence. Three clusters corresponding to each of the species were circled. (B) C. hepaticus core genome phylogeny of Australian and UK isolates against the HV10 reference strain. Maximum-likelihood tree was inferred from 4,812 SNVs and built in RAxML using the general-reversible model (GTRCAT) with 1,000 bootstrap replicates. Scale bar shows nucleotide divergence. Clustering was performed using RAMI at a patristic distance threshold of 0.05, corresponding phylogroups were colored blue, green, red, purple, and yellow. The range of pairwise SNVs within each phylogroup is shown (minimum value—maximum value). The presence key accessory genome elements including the CRISPR-array and the two plasmids pCC31 and pCJDM210L are shown.
Within each phylogroup the mean pairwise nucleotide divergence between genomes was ~3.58%, but the C. hepaticus genomes showed much less divergence (0.46%) than the C. coli (6%) and C. jejuni (4.29%) genomes. Nucleotide divergence from C. hepaticus to C. coli and C. jejuni was ~68 and 63.8%, respectively (calculated across core sequence alignments). The nucleotide divergence between C. coli and C. jejuni was 23.16%, indicating that C. coli and C. jejuni are more closely related to each other than to C. hepaticus. Gubbins did not detect any recombinogenic regions, showing there is no evidence of recombination between the phylogroups, adding further support to the clear separation of C. hepaticus from C. jejuni and C. coli. Accessory genome variation also supports the separation of C. hepaticus from the other species, demonstrating that each phylogroup is a discrete bacterial population that is evolving independently, with limited homologous recombination between groups.
Comparison of the genome sequences of the 23 C. hepaticus isolates revealed a total of 1,360 core genes conserved across all genomes. Maximum Likelihood (ML) phylogeny was produced from 4,812 core genome SNVs and revealed a shallow branching population structure with high bootstrapping support. There is a high level of conservation within the genomes (median of 95.19% coverage of the reference strain HV10), possibly due to the specific niche adaption of the species. The C. hepaticus genomes clustered into five phylogenetic lineages based on the core genome ML tree using a patristic distance of 0.05 (Figure 1B).
The Australian isolates formed two lineages, which differed by on average ~4,429 SNPs. UK isolates formed three lineages. The Australian HV10 phylogroup differ from the UK isolates by between ~500 and 1300 SNPs. The UK lineages, represented by UK cluster 1 and UK cluster 2, differ by ~1,100 SNPs while UK lineage 3 (single isolate) differs to the rest of the UK isolates by ~1,100 to ~1,300 SNPs. Within each phylogroup the SNP frequency was very low with an average of ~41 SNPs (range 0–115 SNPs).
A pan genome of 1,709 unique protein-coding sequences was identified across the 23 C. hepaticus isolates. A map of C. hepaticus HV10 DNA features is presented in Figure 2A. The rapid plateauing of the gene accumulation curve (Figure 2B) revealed an almost closed pan genome, suggesting most of the genetic diversity has been discovered, despite the relatively small sample size of sequenced genomes. Each genome carried on average 103 accessory genes (range = 70–192), and most of those genes were associated with multiple strains, with very few rare/unique (single isolate) accessory genes. This supports the hypothesis of genome reduction/speciation events in C. hepaticus as suggested by Petrovska et al. (2017). Isolates with >100 accessory genes were found to have sequences associated with mobilizable tetracycline resistance plasmids, highly similar to Campylobacter plasmids pCJDM210L and pCC31. The COG distribution plot of functional categories for coding sequences within the 24 C. hepaticus genomes (Figure 2C) showed that transport and metabolism categories of various substrates are mostly encoded in the core genome, while most of the accessory and unique genome variation is categorized as replication, signal transduction, cell wall biogenesis, and motility.
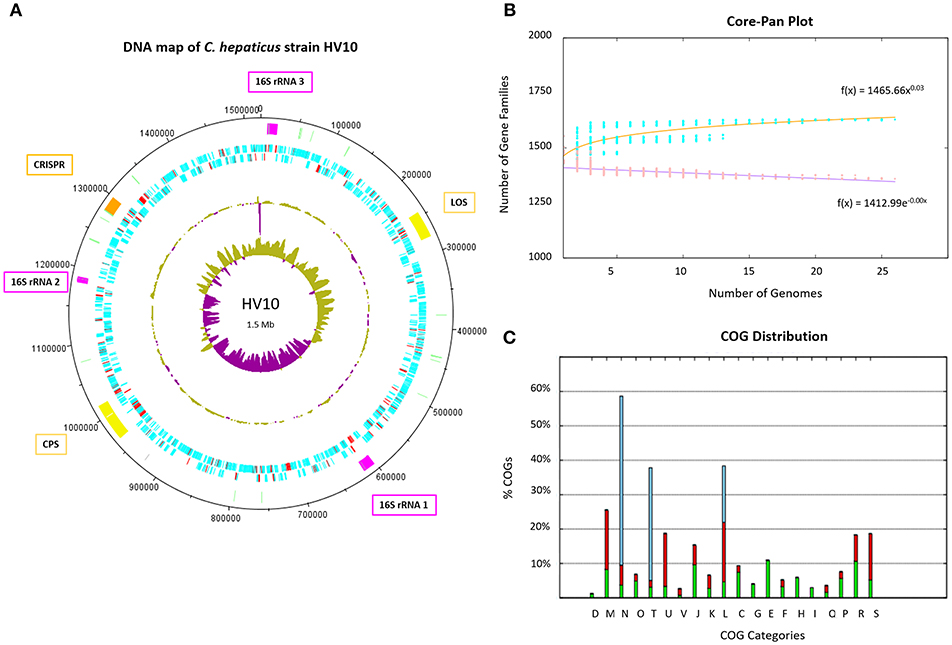
Figure 2. C. hepaticus genomes. (A) DNA map of the C. hepaticus reference strain HV10 produced in DNAplotter (Carver et al., 2009). From the most inner to outer rings shows: GC-skew, GC-content, Coding sequence (CDS) on reverse strains, CDS on forward strand, and key loci of interest. The CDS are colored blue and red, where red unique to C. hepaticus but not present in C. jejuni or C. coli. The loci of interest are colored as follows; pink for the three 16S ribosomal RNA operons including gene insertions, yellow for the lipooligosaccharide (LOS) and capsule (CPS) biosynthesis loci, pale green for tRNA, and orange for the CRISPR-cas machinery including gene insertions. Major ticks are observed at every 100 kb and minor ticks at 50 kb. The ribosomal operon numbering refers to the variants shown in Figure 3. (B) Core and pan genome plot of the 24 C. hepaticus genomes. The gene accumulation curves revealed an almost closed pan genome for the species. (C) COG distribution plot of functional categories for coding sequences within the 24 C. hepaticus genomes. Green represents core genes, red accessory genes and blue unique genes. X-axis letters; (D) Cell cycle control, (M) Cell wall biogenesis, (N) cell motility, (O) Post-translational modification, (T) Signal transduction, (U) Intracellular trafficking, (V) defense mechanisms, (J) Translation, (K) Transcription, (L) Replication, (C) Energy production, (G) Carbohydrate transport and metabolism, (E) Amino acid transport and metabolism, (F) Nucleotide transport and metabolism, (H) Coenzyme transport and metabolism, (I) lipid transport and metabolism, (Q) Secondary metabolites, (P) inorganic ion transport and metabolism, (R) General function and (S) function unknown. It can be observed transport and metabolism categories of various substrates are mostly encoded in the core genome (G and E), while most of the accessory and unique genome variation is categorized as replication (L), signal transduction (T) cell wall biogenesis (M), and motility (N).
Genetic Determinants of C. hepaticus—Niche Adaption and Virulence
Initial annotation and characterization of the C. hepaticus isolates, using the type strain HV10 as the reference sequence for the species, showed the typical structure for a Campylobacter genome with many genes encoding chemotaxis (11 genes), motility (47 genes), adherence/surface protein (59 genes), as well as various metabolism loci for acquisition of metals and carbohydrates (Table 2).
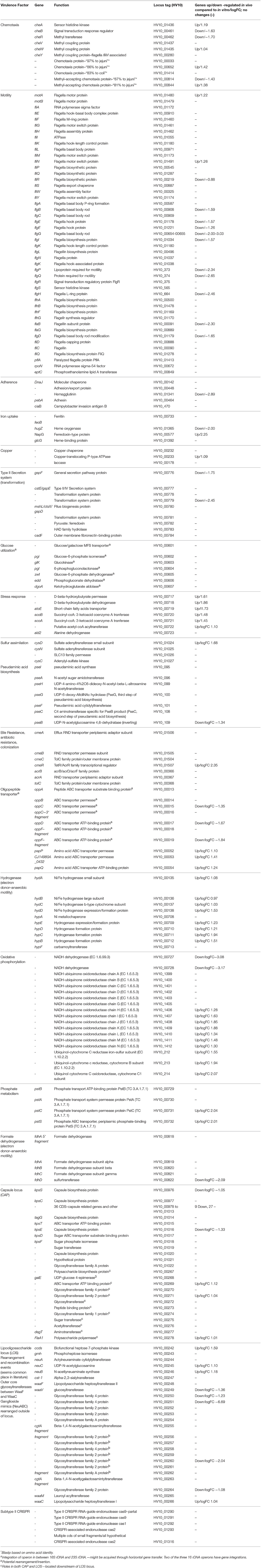
Table 2. Putative virulence genes and the changes in gene regulation in vivo compared to in vitro.
Various methods of genome comparison have been used to investigate coding sequences that are unique to C. hepaticus. The strongest associations with high specificity and selectivity to C. hepaticus were genes with predicted roles in chemotaxis, capsule and lipooligosaccharide synthesis and metabolism. Four chemotaxis proteins with < 88% homology to known chemotaxis proteins were characterized, which could play a role in the movement of C. hepaticus from the gastrointestinal tract to the liver (Table 2). Significant variation was also characterized in the lipooligosaccharide locus (LOS), a region of the Campylobacter chromosome known to undergo rearrangements and recombination events (Parker et al., 2005; Revez and Hänninen, 2012). Two points of interest in this locus were exclusive to C. hepaticus. Firstly the ganglioside mimics (NeuABC) are rearranged outside of the locus as normally seen in C. jejuni (no longer located between the Waac/WaaM to WaaV/WaaF). Secondly, there was an apparent ~6.6 kb insertion of seven CDS into the cst-II gene, all with functions predicted as various glycosyltransferases. This insertion in the middle of the locus resulted in the truncation of cgtA (Table 2). Roughly 2 kb of the inserted sequence is unique to C. hepaticus, with the remaining 4.6 kb showing high sequence divergence to C. jejuni isolates.
A glucose utilization operon was found to be associated with SLD and is discussed in detail in a later section. All C. hepaticus isolates encode a region of CRISPR-cas genes (type II-cas9 CRISPR), however a CRISPR array (section of repeats and spacers) was only found (CRISPR-finder) in two isolates from the divergent Australian Cluster 2 (three direct repeats and 2 spacers, isolates 19L and 54L). The remaining 22 isolates did not encode a complete CRISPR array, just the cas genes (cas9, cas1 and a fragmented cas2). A region of 13 kb is inserted within the two cas2 CDS of the 22 remaining isolates; ~7kb is unique to C. hepaticus. The GC content of this region is similar to that of the rest of the chromosome (28.03%), with most genes (11 CDS) having unknown functions. Within this region, three CDS encoding for luxA repressor, XRE family transcriptional regulation and type II toxin-antitoxin system mRNA interferase are present. A screen for prophage using PHASTER did not identify any complete prophage integrations within any of the genomes.
Horizontally Acquired-Elements: Plasmids
Plasmids are present in five out of fourteen C. hepaticus Australian isolates (Table 3). Using ABRicate to screen for antibiotic resistance genes, a single antibiotic resistance gene, tetO, was found in eight isolates (5 from Australia and 3 from the UK), which correlated directly to the presence of plasmid elements. Distinct plasmids were found based on the country of origin of the isolates. UK isolates contained a plasmid highly homologous to the previously characterized C. coli plasmid pCC31 (99% coverage and identity), while the Australian isolates contain plasmids homologous to the C. jejuni pCJDM210L plasmid (93% coverage and 99% identity). This plasmid harbored a type IV secretion system along with a tetracycline-resistant gene. Five of the Australian isolates within this study (27L, 84B, Ace1, Ace8659, and AceM3a) carry the plasmid and it accounts for roughly half of the gene content within the accessory genome of these isolates. As short-read sequence data was used it was not possible to assemble the plasmid in its entirety. At least three contigs from each of these genomes were highly conserved and carried plasmid elements.
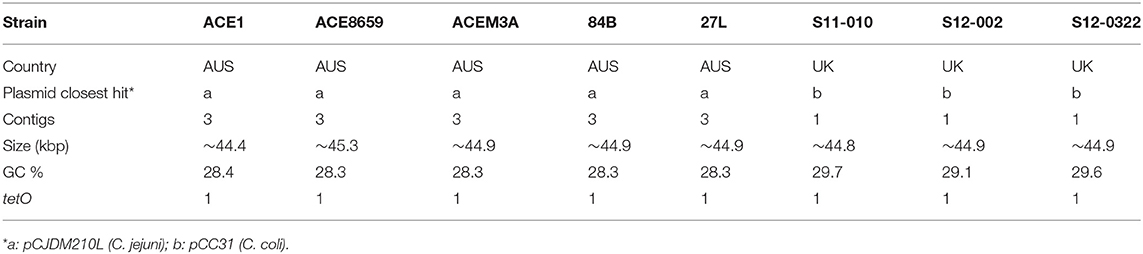
Table 3. Plasmid contents of C. hepaticus Australian and UK isolates.
Horizontally Acquired-Elements: Insertions in Ribosomal RNA Operons
C. hepaticus encodes three ribosomal RNA operons, however two have been disrupted by the insertion of multiple CDS between the 16S and the 23S genes. A glucose utilization operon and an oligopeptide transporter operon were located within ribosomal RNA operons (Figure 3). Analysis of the insertions showed that the glucose utilization and oligopeptide transporter regions have GC content of 28.17 and 27.56% respectively, which is similar to the average GC content of the HV10 genome, 28.2%.
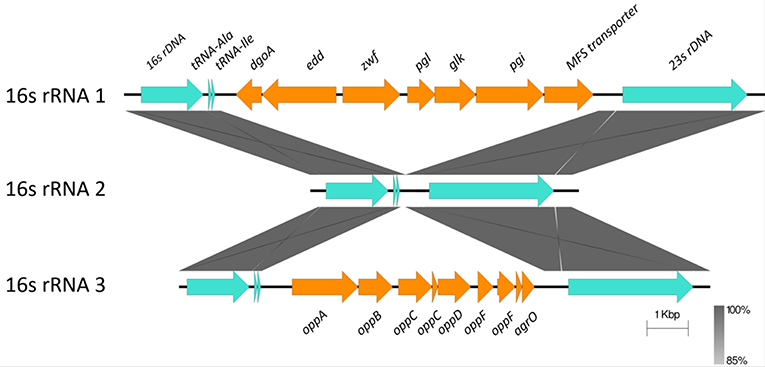
Figure 3. Sequence alignment of the three ribosomal RNA gene operons from C. hepaticus HV10. The conserved 16S, 23S, and tRNA are colored in pale blue, while the insertion regions are colored orange. The blastn and sequence alignment figure were produced in Easyfig v2.2.2 (Sullivan et al., 2011).
Confirmation of Glucose Utilization Ability in C. hepaticus
C. hepaticus cultures incubated for 24 h in IS media containing L-cysteine and D-glucose-6-phosphate, or L-cysteine and D-glucose showed color development (due to TTC) and therefore demonstrated utilization of the substrates, whereas C. jejuni and C. coli cultures did not. There was no color development in C. hepaticus cultures incubated in IS plus L-cysteine only or IS plus D-glucose or D-glucose-6–phosphate. This was due to a lack of carbon source and nitrogen source, respectively. The color change was observed in all cultures grown in Brucella broth, demonstrating the viability of the inoculated cultures.
Gene Expression in C. hepaticus Recovered From Bile
In C. hepaticus recovered from the gall bladder of SLD experimentally infected birds, 410 genes were differentially expressed (False Discovery Rate (FDR) < 0.05) when compared to in vitro grown bacteria. There were 164 up-regulated genes ((log2-fold-changes) > 1.0) in vivo and 246 down-regulated genes (logFC < −1.0). Functional gene categorization assessed using the SEED Viewer, showed that the 410 differentially expressed genes belonged to 56 subcategories (Figure 4). Notably, all genes associated with polyhydroxybutyrate (PHB) metabolism (Figure 5) were up-regulated (EC 1.1.1.30: D-beta-hydroxybutyrate, EC 2.3.1.9: Acetyl-CoA acetyltransferase, EC 2.8.3.5: Succinyl-CoA:3-ketoacid-coenzyme A transferase, and genes encoding D-beta-hydroxybutyrate permease, short chain fatty acids transporter and 3-ketoacyl-CoA thiolase). These genes may play a role in stress response in C. hepaticus and are putative virulence factors (Table 2).
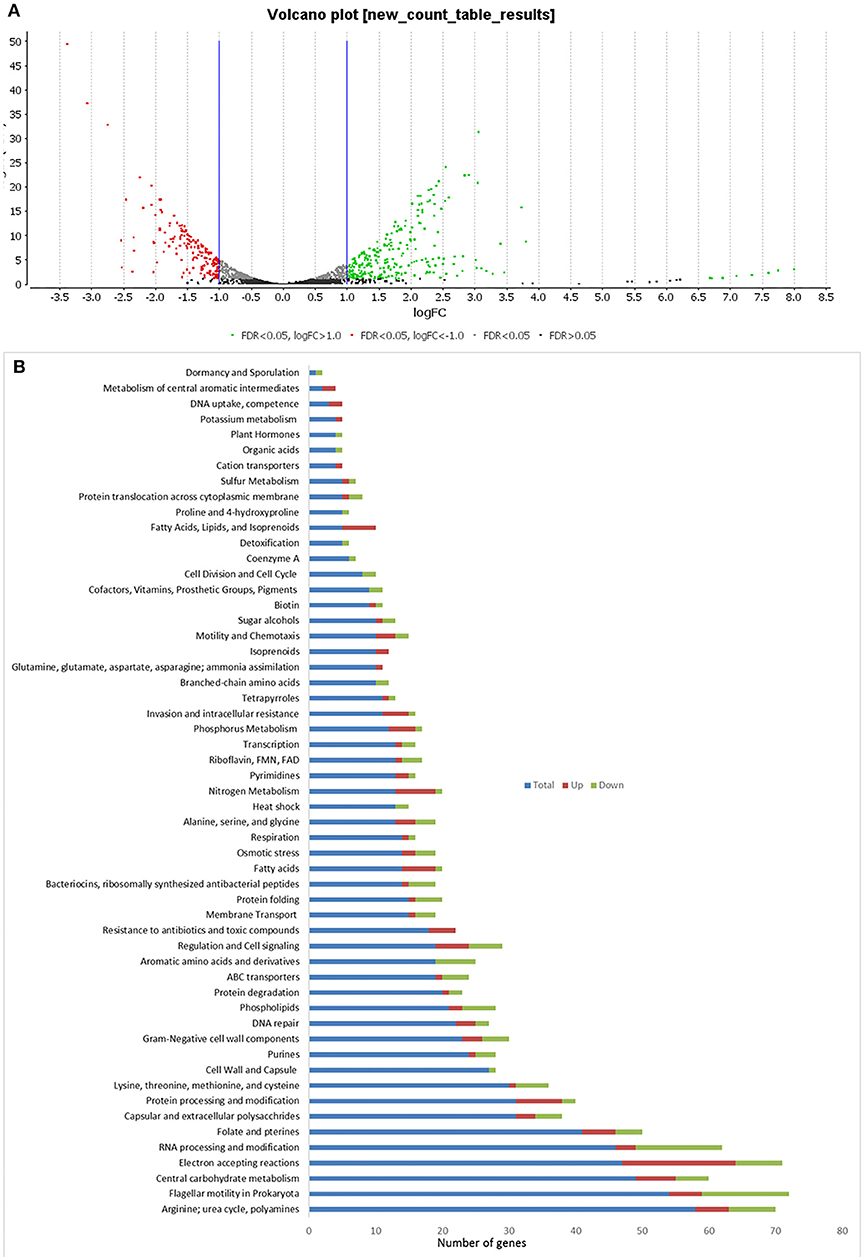
Figure 4. Comparison of differentially expressed genes identified between in vitro and in vivo conditions. (A) Volcano plots analysis of differentially expressed genes DEGs. The green dots represent DEGs up regulated in bile samples, the red dots represent DEGs down-regulated in bile samples, and the black/gray dots represent no DEGs. (B) Sub-categories of DEGs were as defined by the SEED viewer from the RAST annotations. Total: number of CDSs assigned to each subcategory, Up: DEGs up regulated while C. hepaticus in bile samples compared to in vitro samples.
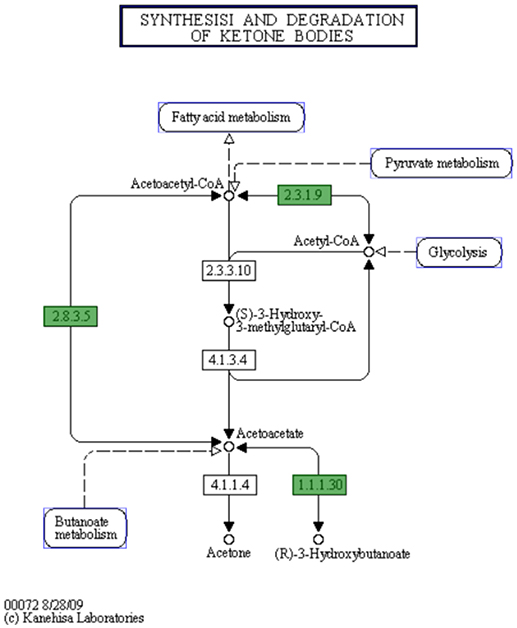
Figure 5. The putative biosynthetic pathway for PHB metabolites in C. hepaticus (KEGG map generated from the SEED Viewer). Genes in green boxes are present in C. hepaticus. EC 1.1.1.30: D-beta-hydroxybutyrate dehydrogenase. EC 2.3.1.9: Acetyl-CoA acetyltransferase. EC 2.8.3.5: Succinyl-CoA:3-ketoacid-coenzyme A transferase.
The gene clusters encoding Ni-Fe-hydrogenase were up-regulated in the cells recovered from bile (Table 2). Six of out eight genes associated with nitrate and nitrite ammonification (nitrogen metabolism system) were also up-regulated and only one was down-regulated in bile samples (Figure 4B). Transcripts from the phosphate transport system of pstS and pstC were increased in abundance in vivo compared to in vitro (Table 2). RNA-Seq identified increased abundance of many transcripts associated with copper homeostasis and up-regulation of pathogenesis-associated glutamine ABC transporters, papP and papQ (Table 2). The neuB (N-acetylneuraminate synthase) and neuC (UDP-N-acetylglucosamine) genes, necessary for sialic acid synthesis, were both up-regulated in the bile-derived bacteria. Thirteen genes associated with flagella motility were down-regulated, and only five were up-regulated. Increased expression of flagella associated genes included genes encoding flagella motor protein (MotA) and flagella switch motor protein (FliN). Down-regulated genes included a putative lipoprotein required for motility, a motility integral membrane protein and flagella-associated genes including flgB, flgD, flgF, flgG, flgH, and flgI. In addition, many genes in the aromatic amino acids and derivatives category were down-regulated (Figure 4B), including genes involved in common pathways for synthesis of aromatic compounds, tryptophan synthesis, and chorismate synthesis (intermediate for synthesis of tryptophan) and none of the genes in this category were up-regulated. Similarly, genes associated with the production of methionine (lysine, threonine, methionine, and cysteine subcategory, Figure 4B) were down-regulated. RNA-Seq identified decreased abundance of transcripts associated with tRNA processing, RNA methylation, and RNA pseudouridine syntheses. On the other hand, there was variation in the expression of genes involved in the oxidative phosphorylation pathway, as genes encoding the enzyme NADH dehydrogenase (EC 1.6.99.3) were down-regulated, while many genes encoding enzymes NADH ubiquinone oxidoreductase (EC 1.6.5.3) and ubiquinol-cytochrome C reductase (EC 1.10.2.2) were up-regulated (Table 2).
Discussion
Campylobacter hepaticus core genome phylogeny showed five phylogroups, two from Australian isolates and three from UK isolates. Interestingly, the main Australian phylogroup includes all the isolates from southern Australia (Victoria, South Australia, New South Waves) while the two Queensland isolates (northern Australia) formed a separate phylogroup. This indicates that C. hepaticus clonal populations are geographically confined.
The comparison of C. hepaticus genomes with those of representative isolates of C. jejuni and C. coli indicated that there are barriers to gene flow among these related populations, even though these species are known to be common colonizers of commercial poultry and are naturally transformable (Vegge et al., 2012). This suggests a mechanistic barrier to homologous recombination or an adaptive selection against hybrid genotypes, possibly influenced by the reduced genome size of C. hepaticus (0.2–0.4 Mbp reduction), reduced metabolic capabilities, and a reduced GC content (2–3.5% lower than C. jejuni and C. coli). The genetic divergence of C. hepaticus from other Campylobacter spp. is likely due to its adaptation to colonize and infect the bile and liver in chickens.
In C. jejuni, the cytolethal distending toxin (CdtA, B, C) has been recognized as a major virulence factor and is believed to induce host cell apoptosis (Dasti et al., 2010). However, the Cdt is not encoded by C. hepaticus and the genomic analysis has not identified any other candidate toxin genes. C. hepaticus has a large number of genes associated with chemotaxis (11 genes), motility (47 genes), and adherence/antigen presentation (45 genes); genes similar to many that have been shown to be required for the colonization and infection of other bacteria. In addition, C. hepaticus encodes Campylobacter invasion antigens (CiaB), presumably secreted from the flagella export apparatus. In the case of C. jejuni, this protein has been demonstrated to be delivered to the host cell cytoplasm, which stimulates host cell signaling and prompts bacterial internalization (Konkel et al., 2004). The CiaB antigen plays a major role in the invasion of chicken epithelial cells. Mutants which lack the ciaB gene were shown to have reduced virulence (Ziprin et al., 2001; Biswas et al., 2007). In vitro assays using chicken epithelial cells have demonstrated that C. hepaticus is invasive, probably more so than C. jejuni (Van et al., 2017a). C. hepaticus also encodes a set of genes involved in pseudaminic acid biosynthesis (Pse). The structural flagellin proteins of Helicobacter pylori and Campylobacter jejuni are glycosylated with Pse and this glycosylation is essential for flagella filament assembly and consequent motility, therefore Pse is considered to be a key virulence factor (Ménard et al., 2014).
To elucidate the genetic potential of C. hepaticus to cause SLD various genome comparison tools were used to screen each gene in the C. hepaticus pan genome for association to SLD. An association study was used to search for genes or markers associated with SLD, and genes with predicted roles in chemotaxis, capsule and lipooligosaccharide synthesis and metabolism were identified (Table 2). Four chemotaxis proteins with low identity to known chemotaxis proteins (< 88%) were identified and two of these genes were up-regulated in vivo, in the C. hepaticus recovered from bile. These genes could play a role in the movement of C. hepaticus from the gastrointestinal tract to the liver and bile and are priority gene targets for further study.
A screen for prophage insertions into the genome using PHASTER failed to identify any prophage integrations within the genomes. The lack of a CRISPR spacer array suggests the CRISPR region is not actively used as an immune system for C. hepaticus. Type II cas9 systems in C. jejuni and Neisseria meningititis are required for the ability to invade, attach to and replicate within epithelial cells (Sampson and Weiss, 2013), although mechanisms are currently unknown. Cas9 has been correlated with strains producing sialylated lipooligosaccharide structures in the outer envelope (Sampson and Weiss, 2013). However, there is a unique ~7kb insertion within the two cas9 CDS, with found exclusively with C. hepaticus isolates. This insertion encodes many genes with unknown function including three CDS encoding for luxA repressor, XRE family transcriptional regulation and type II toxin-antitoxin system mRNA interferase. This indicates this region may play a regulatory role in C. hepaticus, possible affecting virulence.
Although the C. hepaticus genomes do not appear to be highly influenced by horizontal gene transfer and acquisition of genetic material, there are three regions, two chromosomal and one plasmid, associated with lateral gene transfer events. Glucose utilization and oligopeptide transporter operons were located within two of the three ribosomal RNA operons (Figure 3). It is unusual to have a large operon inserted between the 16S and 23S rRNA genes, although strains C. coli CHW470 and C. jejuni subsp. Doylei 269.97 were found to have glucose utilization operons inserted between 16S rRNA and 23S rRNA genes (Vorwerk et al., 2015). The region which lays between the 16S rRNA and 23S rRNA genes, called the Internal Transcribed Spacer (ITS) region, of other Campylobacter species were highly variable in % GC content and length, with an average size of 880 bp, and the longest was 1,646 bp in C. hominis ATCC BAA 381 (Man et al., 2010).
Typically, Campylobacter species are characterized as non-glycolytic bacteria. C. hepaticus contain many more genes in carbohydrate utilization pathways than C. jejuni and this may help C. hepaticus to survive in the carbohydrate-rich environment of the chicken liver (Petrovska et al., 2017). The presence of the glucose utilization operon enables the metabolism of glucose through the glycolytic (Entner-Doudoroff, ED) pathway and has previously been found in other bacteria such as Helicobacter (Hofreuter, 2014). Most C. jejuni and C. coli genomes do not have genes encoding glucokinase (EC.2.7.1.2), glucose-6-phosphate dehydrogenase (EC 1.1.1.49) and 6-phosphogluconolactonase (EC 3.1.1.31) and are therefore mostly ED-negative. Vegge et al. found that only 1.7% of >6,000 genomes of C. coli and C. jejuni encoded a complete ED pathway (Vegge et al., 2016). From the fully closed and finished genome of C. hepaticus HV10, three rRNA were identified, two of which were disrupted by a glucose utilization oligopeptide transporter operons. All the genes in these two operons are present in all the other C. hepaticus isolates. The bioinformatics prediction of D-glucose utilization by C. hepaticus was experimentally confirmed and both C. jejuni NCTC 11828 and C. coli NCTC 11366 were shown to be unable to utilize D-glucose. Vorwekr et al. demonstrated the ED pathway of glucose-catabolising C. coli strains could be acquired by non-glycolytic C. coli isolates through natural transformation, showing that the ED pathway genes could be transferred by horizontal gene transfer (Vorwerk et al., 2015). In C. hepaticus, the GC content of these two regions (27.56% and 28.17%) are similar to the average GC content of the HV10 genome (28.2%), suggesting that these have been present within the genome for an extended period of time, or have been obtained from a close relative. In contrast, the GC content of ED pathway genes in C. coli CHW470 are 34.7–36.5% while the GC content of the isolate is 31.1%. These loci are present in all the C. hepaticus isolates and therefore it is suggested that these loci might provide a selective advantage. Carbon source utilization is characteristic of growth of intercellular gastrointestinal pathogens such as Listeria monocytogenes and Salmonella Typhimurium (Dandekar et al., 2012; Fuchs et al., 2012), therefore the high level of conservation of this locus in C. hepaticus may provide a new pathway for pathogenesis of SLD.
The presence of two distinct tetracycline resistant plasmids that appear to originate from two distinct species suggest that other Campylobacter species may act as a genetic reservoir for C. hepaticus and vice versa, which is likely due to the presence of the type II secretions systems (transformation locus) present in C. hepaticus. However, as two different plasmids are present in the C. hepaticus sampled here and only in 1/3 isolates (absent from HV10), this suggests that the genes encoded on plasmids do not play a role in SLD development. The C. coli pCC31 plasmid has been shown to be conjugative (Batchelor et al., 2004); therefore, the closely related plasmid found in some C. hepaticus isolates may also be transferable. This is concerning as this antibiotic resistance plasmid could be disseminate to other bacteria. This should be taken as an early warning sign that alternative treatments, other than antibiotic treatment, are needed for the control of SLD.
PHB is produced by microorganisms in responses to physiologically stressed conditions, especially when nutrients are limited (Ackermann et al., 1995; Batista et al., 2018). In C. hepaticus, PHB might be produced by the condensation of acetyl-CoA to acetoacetyl-CoA and is later converted to acetoacetate, and acetoacetate is then reduced by NADH to R-3-hydroxybutyrate where D-beta-hydroxybutyrate dehydrogenase enzyme catalyzes the reaction (Figure 5). In contrast, C. jejuni and C. coli lack this pathway. A Biolog Phenotype Microarray confirmed the metabolic activity of D-beta-hydroxybutyrate dehydrogenase as the color change was observed in the wells with acetoacetate substrate and C. hepaticus added but not in wells with C. jejuni (data not shown). All genes associated with PHB metabolism were up-regulated. PHB is accumulated by bacteria as a carbon and energy storage when carbon sources are freely available but limited for other nutrients (Ratcliff et al., 2008; Reusch, 2013).
Sialic acid has been demonstrated to shield pathogens from host immune responses by interacting with the sialic acid-binding proteins of the host. For example, Group B Streptococcus (GBS) can evade host responses and proliferate in blood due to capsular polysaccharide displaying sialic acid residues (Chang et al., 2014; Lewis et al., 2016). C. hepaticus harbors a sialic acid biosynthetic gene locus (UDP-GlcNAc converts to ManNAc, then converts to Neu5 Ac, followed by CMP-Neu5Ac, with the action of NeuC (EC 5.1.3.14), NeuB (EC 2.5.1.56) and NeuA (N-Acetylneuraminate cytidylyltransferase, EC 2.7.7.43, respectively). The RNA-Seq analysis showed up-regulation of neuB and neuC in the bile environment, suggesting the sialic acid biosynthetic genes may encode a host immune response avoidance mechanism.
The gene clusters encoding Ni-Fe-hydrogenase were up-regulated in the in vivo cells recovered from bile. Hydrogenases catalyze the reversible reaction: 2H+ + 2e− ⇔ H2 and they play an important role in dealing with fluctuations in energy and oxygen supply (Vignais et al., 2001). In H. pylori, H2 produced by the gastric microbiota serves as a respiratory substrate which substantially enhances its ability to colonize the stomach (Olson and Maier, 2002). Similarly, the hydrogenase may function as a virulence factor in C. hepaticus.
The pathogenesis-associated glutamine ABC transporter genes, papP and papQ were up-regulated in the in vivo cells. This was expected as glutamine is the only amino acid that contains an additional nitrogen molecule and the liver is the major site of nitrogen metabolism (Haüssinger, 1990). PaqP and PaqQ have been demonstrated to play a role in bacterial stress tolerance and pathogenesis of C. jejuni (Lin et al., 2009). Genes encoding products involved in copper homeostasis were up regulated in vivo. This may explain the survival ability of C. hepaticus in the high copper environment of bile.
Depending on cell growth and metabolism needed to adapt to a new environment, proteins are produced and mRNAs, tRNAs, and rRNAs are all orchestrated to accomplish their roles (Arraiano et al., 2010). It is no surprise that many genes involved in RNA metabolism and genes associated with synthesis of amino acids in C. hepaticus in bile samples were down-regulated, as it appears that the bacterium was in a somewhat quiescent, resting stage in bile.
Up-regulation of a phosphate transport system pstSCAB was observed in C. jejuni in vivo (caecum) compared to in vitro conditions (Taveirne et al., 2013). In our study, up regulation of pstS and pstC in bile samples compared to in vitro samples was also observed, suggesting that the bile environment is limited in phosphate. A study by Stintzi et al. found that the expression of genes encoding NADH dehydrogenase and succinate dehydrogenase were decreased in rabbit intestines. This is consistent with the oxygen-limited environment of the intestine (Stintzi et al., 2005). However, the situation seems to be more complex in bile. In the oxidative phosphorylation pathway, there was decreased expression of the genes encoding enzyme NADH dehydrogenase, while genes in this pathway encoding enzymes such as NADH ubiquinone oxidoreductase and ubiquinol-cytochrome C reductase were up-regulated.
Campylobacter jejuni can use a wide range of alternative electron acceptors to oxygen, including fumarate, nitrate, nitrite, and N- or S-oxides, under oxygen-restricted conditions in vitro (Sellars et al., 2002). C. hepaticus HV10 encodes a number of reductases including fumarate reductase and a nitrate reductase of the periplasmic Nap type. Our study showed up-regulation of all genes encoding nitrate reductase, suggesting a C. hepaticus response to the oxygen-limited environment found in bile.
Bacterial flagellum is a complex apparatus assembled of more than 20 different proteins (Haiko and Westerlund-Wikström, 2013). Flagella can play an essential role in colonization of many bacteria by facilitating bacterial motility. They also have adhesive and invasive properties and act as potential virulence factors. Many genes involved in flagella and chemotaxis were found to be down-regulated in bacteria recovered from bile. This indicates there may be no requirement for facilitating bacterial motility once C. hepaticus successfully colonizes the bile. Down regulation of flagella has also been observed in C. jejuni growth within the gastrointestinal tract. It has been suggested that Campylobacter might shut down flagellum production to evade the host immune system (Stintzi et al., 2005). C. jejuni flagella are required to pass the gastrointestinal tract of chickens but not for survival and persistence within the caeca (Wösten et al., 2004).
It is not currently possible to test the identified potential virulence genes, as the appropriate genetic tools for C. hepaticus have not yet been developed. However, this study adds a significant number of candidate gene targets for knockout and virulence-association assays due to the bioinformatics analyses performed. Utilizing the comparative genome approach, we have reduced the potential number of essential virulence genes from 1,709 to 1,059 and further in-depth genetic analysis has allowed us to generate a shortlist of likely virulence-associated genes (Table 2).
In conclusion, the in vivo transcriptome pattern of C. hepaticus found in this study was consistent with the nutrient-limited environment in bile. C. hepaticus harbors a wide range of potential virulence factors which we have identified using a comparative genomics and transcriptomics study. It appears that some of these genes play a key role in pathogenicity and adaptation of C. hepaticus to the low energy, low nutrient environments in chickens; in particular, gene clusters associated with glucose utilization, stress response, hydrogen metabolism and sialic acid biosynthesis. The virulence mechanisms that lead to the formation of liver lesions, mortalities and reduction in production in infected birds are yet to be elucidated but now a series of genes potentially involved in these processes have been identified.
Data Availability Statement
All genomic assemblies and read sets have been deposited at NCBI (Bioproject PRJNA485661). The closed C. hepaticus HV10 genome has accession number CP031611.1. The raw RNA-Seq data and PacBio long-read DNA data were submitted to NCBI and can be accessed with accession number SAMN04544305.
Author Contributions
RM, TV, and PS conceived and designed the experiments. TV, AA, and CP performed the experiments. TV, JL, and BV analyzed the data. TV and RM interpreted the data. TV, JL, and RM drafted the manuscript. All of the authors read and approved the final manuscript.
Conflict of Interest Statement
AA and PS were employed by company Scolexia Pty Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SLD related research at RMIT University and Scolexia Pty Ltd is supported by grants from Australian Eggs, Poultry Hub Australia and the Innovation Connections scheme of the Australian Commonwealth Government.
References
Ackermann, J., Müller, S., Lösche, A., Bley, T., and Babel, W. (1995). Methylobacterium rhodesianum cells tend to double the DNA content under growth limitations and accumulate PHB. J. Biotechnol. 39, 9–20. doi: 10.1016/0168-1656(94)00138-3
Alazzam, B., Bonnassie-Rouxin, S., Dufour, V., and Ermel, G. (2011). MCLMAN, a new minimal medium for Campylobacter jejuni NCTC 11168. Res. Microbiol. 162, 173–179. doi: 10.1016/j.resmic.2010.09.024
Arraiano, C. M., Andrade, J. M., Domingues, S., Guinote, I. B., Malecki, M., Matos, R. G., et al. (2010). The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 34, 883–923. doi: 10.1111/j.1574-6976.2010.00242.x
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bairoch, A., and Apweiler, R. (1997). The SWISS-PROT protein sequence data bank and its supplement TrEMBL. Nucleic Acids Res. 25, 31–36.
Batchelor, R. A., Pearson, B. M., Friis, L. M., Guerry, P., and Wells, J. M. (2004). Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiology 150, 3507–3517. doi: 10.1099/mic.0.27112-0
Batista, M. B., Teixeira, C. S., Sfeir, M. Z. T., Alves, L. P. S., Valdameri, G., Pedrosa, F., et al. (2018). PHB biosynthesis counteracts redox stress in Herbaspirillum seropedicae. Front. Microbiol. 9:472. doi: 10.3389/fmicb.2018.00472
Bertschinger, H. U. (1965). [Detection of Vibrio in hens with hepatitis]. Zentralblatt Veterinarmedizin Reihe B. 12, 33–40.
Biswas, D., Fernando, U. M., Reiman, C. D., Willson, P. J., Townsend, H. G. G., Potter, A. A., et al. (2007). Correlation between in vitro secretion of virulence-associated proteins of Campylobacter jejuni and colonization of chickens. Curr. Microbiol. 54, 207–212. doi: 10.1007/s00284-006-0295-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brynildsrud, O., Bohlin, J., Scheffer, L., and Eldholm, V. (2016). Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 17:238. doi: 10.1186/s13059-016-1108-8
Carver, T., Thomson, N., Bleasby, A., Berriman, M., and Parkhill, J. (2009). DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25, 119–120. doi: 10.1093/bioinformatics/btn578
Chang, Y.-C., Olson, J., Beasley, F. C., Tung, C., Zhang, J., Crocker, P. R., et al. (2014). Group B Streptococcus engages an inhibitory Siglec through sialic acid mimicry to blunt innate immune and inflammatory responses in vivo. PLoS Pathog. 10:e1003846. doi: 10.1371/journal.ppat.1003846
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Coil, D., Jospin, G., and Darling, A. E. (2015). A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589. doi: 10.1093/bioinformatics/btu661
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
Crawshaw, T., and Irvine, R. (2012). Spotty liver syndrome in poultry in Great Britain. Veterinary Record 170, 317–318. doi: 10.1136/vr.e2201
Crawshaw, T., and Young, S. (2003). Increased mortality on a free-range layer site. Vet. Rec. 153:664.
Crawshaw, T. R., Chanter, J. I., Young, S. C. L., Cawthraw, S., Whatmore, A. M., Koylass, M. S., et al. (2015). Isolation of a novel thermophilic Campylobacter from cases of spotty liver disease in laying hens and experimental reproduction of infection and microscopic pathology. Vet. Microbiol. 179, 315–321. doi: 10.1016/j.vetmic.2015.06.008
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Dandekar, T., Astrid, F., Jasmin, P., and Hensel, M. (2012). Salmonella enterica: a surprisingly well-adapted intracellular lifestyle. Front. Microbiol. 3:164. doi: 10.3389/fmicb.2012.00164
Dasti, J. I., Tareen, A. M., Lugert, R., Zautner, A. E., and Gro,ß, U. (2010). Campylobacter jejuni: a brief overview on pathogenicity-associated factors and disease-mediating mechanisms. Int. J. Med. Microbiol. 300, 205–211. doi: 10.1016/j.ijmm.2009.07.002
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Fuchs, T. M., Eisenreich, W., Kern, T., and Dandekar, T. (2012). Toward a systemic understanding of Listeria monocytogenes metabolism during infection. Front. Microbiol. 3:23. doi: 10.3389/fmicb.2012.00023
Grimes, T., and Reece, R. (2011). ““Spotty liver disease” - an emerging disease in free-range egg layers in Australia,” in Proceedings of the Sixtieth Western Poultry Disease Conference (Sacremento, CA), 53–56.
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Haiko, J., and Westerlund-Wikström, B. (2013). The role of the bacterial flagellum in adhesion and virulence. Biology 2, 1242–1267. doi: 10.3390/biology2041242
Haüssinger, D. (1990). Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem. J. 267, 281–290
Hofreuter, D. (2014). Defining the metabolic requirements for the growth and colonization capacity of Campylobacter jejuni. Front. Cell. Infect. Microbiol. 4:137. doi: 10.3389/fcimb.2014.00137
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kölbl, O., and Willinger, H. (1967). [Animal experiment studies on vibrio hepatitis in chickens]. Wien. Tierarztl. Monatsschr. 54, 85–91.
Konkel, M. E., Klena, J. D., Rivera-Amill, V., Monteville, M. R., Biswas, D., Raphael, B., et al. (2004). Secretion of virulence proteins from Campylobacter jejuni is dependent on a functional flagellar export apparatus. J. Bacteriol. 186, 3296–3303. doi: 10.1128/JB.186.11.3296-3303.2004
Kukurba, K. R., and Montgomery, S. B. (2015). RNA sequencing and analysis. Cold Spring Harb. Protoc. 2015, 951–969. doi: 10.1101/pdb.top084970
Lacey, J. A., Allnutt, T. R., Vezina, B., Van, T. T. H., Stent, T., Han, X., et al. (2018). Whole genome analysis reveals the diversity and evolutionary relationships between necrotic enteritis-causing strains of Clostridium perfringens. BMC Genomics 19:379. doi: 10.1186/s12864-018-4771-1
Leesment, L., and Parve, T. (1965). [On vibrio hepatitis in poultry in the Estonian SSR]. Veterinariia 42, 46–47.
Lewis, A. L., Robinson, L. S., Agarwal, K., and Lewis, W. G. (2016). Discovery and characterization of de novo sialic acid biosynthesis in the phylum Fusobacterium. Glycobiology 26, 1107–1119. doi: 10.1093/glycob/cww068
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Lin, A. E., Krastel, K., Hobb, R. I., Thompson, S. A., Cvitkovitch, D. G., and Gaynor, E. C. (2009). Atypical roles for Campylobacter jejuni amino acid ATP binding cassette transporter components PaqP and PaqQ in bacterial stress tolerance and pathogen-host cell dynamics. Infect. Immun. 77, 4912–4924. doi: 10.1128/IAI.00571-08
Man, S. M., Kaakoush, N. O., Octavia, S., and Mitchell, H. (2010). The internal transcribed spacer region, a new tool for use in species differentiation and delineation of systematic relationships within the Campylobacter genus. Appl. Environ. Microbiol. 76, 3071–3081. doi: 10.1128/AEM.02551-09
Ménard, R., Schoenhofen, I. C., Tao, L., Aubry, A., Bouchard, P., Reid, C. W., et al. (2014). Small-molecule inhibitors of the pseudaminic acid biosynthetic pathway: targeting motility as a key bacterial virulence factor. Antimicrob. Agents Chemother. 58, 7430–7440. doi: 10.1128/AAC.03858-14
Menolasino, N. J. (1959). Tetrazolium chloride as an indicator in bacteriophage typing of staphylococci. Am. J. Clin. Pathol. 32, 578–579. doi: 10.1093/ajcp/32.6_ts.578
Olson, J. W., and Maier, R. J. (2002). Molecular hydrogen as an energy source for Helicobacter pylori. Science 298, 1788–1790. doi: 10.1126/science.1077123
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Parker, C. T., Horn, S. T., Gilbert, M., Miller, W. G., Woodward, D. L., and Mandrell, R. E. (2005). Comparison of Campylobacter jejuni lipooligosaccharide biosynthesis loci from a variety of sources. J. Clin. Microbiol. 43, 2771–2781. doi: 10.1128/JCM.43.6.2771-2781.2005
Petrovska, L., Tang, Y., Rensburg, J., van, J, M., Cawthraw, S., Nunez, J., et al. (2017). Genome reduction for niche association in Campylobacter hepaticus, a cause of Spotty Liver Disease in poultry. Front. Cell. Infect. Microbiol. 7:354. doi: 10.3389/fcimb.2017.00354
Pohl, R., Marshall, R. M., and Pearson, R. (1969). Vibriosis in chickens in New Zealand. N. Z. Vet. J. 17, 51–52. doi: 10.1080/00480169.1969.33781
Pommier, T., Canbäck, B., Lundberg, P., Hagström, Å., and Tunlid, A. (2009). RAMI: a tool for identification and characterization of phylogenetic clusters in microbial communities. Bioinformatics 25, 736–742. doi: 10.1093/bioinformatics/btp051
Rao, R., Bing Zhu, Y., Alinejad, T., Tiruvayipati, S., Lin Thong, K., Wang, J., et al. (2015). RNA-seq analysis of Macrobrachium rosenbergii hepatopancreas in response to Vibrio parahaemolyticus infection. Gut Pathog. 7:6. doi: 10.1186/s13099-015-0052-6
Ratcliff, W. C., Kadam, S. V., and Denison, R. F. (2008). Poly-3-hydroxybutyrate (PHB) supports survival and reproduction in starving rhizobia. FEMS Microbiol. Ecol. 65, 391–399. doi: 10.1111/j.1574-6941.2008.00544.x
Reusch, R. N. (2013). The role of short-chain conjugated poly-(R)-3-hydroxybutyrate (cPHB) in protein folding. Int. J. Mol. Sci. 14, 10727–10748. doi: 10.3390/ijms140610727
Revez, J., and Hänninen, M.-L. (2012). Lipooligosaccharide locus classes are associated with certain Campylobacter jejuni multilocus sequence types. Eur. J. Clin. Microbiol. Infect. Dis. 31, 2203–2209. doi: 10.1007/s10096-012-1556-3
Sampson, T. R., and Weiss, D. S. (2013). Alternative roles for CRISPR/Cas systems in bacterial pathogenesis. PLoS Pathog. 9:e1003621. doi: 10.1371/journal.ppat.1003621
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sellars, M. J., Hall, S. J., and Kelly, D. J. (2002). Growth of Campylobacter jejuni supported by respiration of fumarate, nitrate, nitrite, trimethylamine-N-oxide, or dimethyl sulfoxide requires oxygen. J. Bacteriol. 184, 4187–4196. doi: 10.1128/JB.184.15.4187-4196.2002
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stintzi, A., Marlow, D., Palyada, K., Naikare, H., Panciera, R., Whitworth, L., et al. (2005). Use of genome-wide expression profiling and mutagenesis to study the intestinal lifestyle of Campylobacter jejuni. Infect. Immun. 73, 1797–1810. doi: 10.1128/IAI.73.3.1797-1810.2005
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Taveirne, M. E., Theriot, C. M., Livny, J., and DiRita, V. J. (2013). The complete Campylobacter jejuni transcriptome during colonization of a natural host determined by RNAseq. PLoS ONE 8:e73586. doi: 10.1371/journal.pone.0073586
Truscott, R. B., and Stockdale, P. H. G. (1966). Correlation of the identity of bile and cecal vibrios from the same field cases of avian vibrionic hepatitis. Avian Dis. 10, 67–73. doi: 10.2307/1588208
Tudor, D. C. (1954). A liver degeneration of unknown origin in chickens. J. Am. Vet. Med. Assoc. 125, 219–220.
UniProt Consortium (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–212. doi: 10.1093/nar/gku989
Van, T. T. H., Anwar, A., Scott, P. C., and Moore, R. J. (2018). Rapid and specific methods to differentiate foodborne pathogens, Campylobacter jejuni, Campylobacter coli, and the new species causing Spotty Liver Disease in chickens, Campylobacter hepaticus. Foodborne Pathog. Dis. 15, 526–530. doi: 10.1089/fpd.2017.2367
Van, T. T. H., Elshagmani, E., Gor, M.-C., Anwar, A., Scott, P. C., and Moore, R. J. (2017a). Induction of spotty liver disease in layer hens by infection with Campylobacter hepaticus. Vet. Microbiol. 199, 85–90. doi: 10.1016/j.vetmic.2016.12.033
Van, T. T. H., Elshagmani, E., Gor, M. C., Scott, P. C., and Moore, R. J. (2016). Campylobacter hepaticus sp. nov., isolated from chickens with spotty liver disease. Int. J. Syst. Evol. Microbiol. 66, 4518–4524. doi: 10.1099/ijsem.0.001383
Van, T. T. H., Gor, M.-C., Anwar, A., Scott, P. C., and Moore, R. J. (2017b). Campylobacter hepaticus, the cause of spotty liver disease in chickens, is present throughout the small intestine and caeca of infected birds. Vet. Microbiol. 207, 226–230. doi: 10.1016/j.vetmic.2017.06.022
Vegge, C. S., Brøndsted, L., Ligowska-Marzeta, M., and Ingmer, H. (2012). Natural transformation of Campylobacter jejuni occurs beyond limits of growth. PLoS ONE 7:e45467. doi: 10.1371/journal.pone.0045467
Vegge, C. S., Jansen van Rensburg, M. J., Rasmussen, J. J., Maiden, M. C. J., Johnsen, L. G., Danielsen, M., et al. (2016). Glucose metabolism via the Entner-Doudoroff pathway in Campylobacter: a rare trait that enhances survival and promotes biofilm formation in some isolates. Front. Microbiol. 7:1877. doi: 10.3389/fmicb.2016.01877
Vignais, P. M., Billoud, B., and Meyer, J. (2001). Classification and phylogeny of hydrogenases1. FEMS Microbiol. Rev. 25, 455–501. doi: 10.1111/j.1574-6976.2001.tb00587.x
Vorwerk, H., Huber, C., Mohr, J., Bunk, B., Bhuju, S., Wensel, O., et al. (2015). A transferable plasticity region in Campylobacter coli allows isolates of an otherwise non-glycolytic food-borne pathogen to catabolize glucose. Mol. Microbiol. 98, 809–830. doi: 10.1111/mmi.13159
Wösten, M. M., Wagenaar, J. A., and Putten, J. P. M. van (2004). The FlgS/FlgR two-component signal transduction system regulates the fla regulon in Campylobacter jejuni. J. Biol. Chem. 279, 16214–16222. doi: 10.1074/jbc.M400357200
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. doi: 10.1093/nar/gkr485
Keywords: comparative genomics, transcriptomics, Campylobacter hepaticus, glucose utilization, polyhydroxybutyrate, stress response
Citation: Van TTH, Lacey JA, Vezina B, Phung C, Anwar A, Scott PC and Moore RJ (2019) Survival Mechanisms of Campylobacter hepaticus Identified by Genomic Analysis and Comparative Transcriptomic Analysis of in vivo and in vitro Derived Bacteria. Front. Microbiol. 10:107. doi: 10.3389/fmicb.2019.00107
Received: 02 October 2018; Accepted: 18 January 2019;
Published: 05 February 2019.
Edited by:
Jörg Linde, Friedrich Loeffler Institut, GermanyReviewed by:
Martin Hölzer, Friedrich-Schiller-Universität Jena, GermanyHafez Mohamed Hafez, Freie Universität Berlin, Germany
Copyright © 2019 Van, Lacey, Vezina, Phung, Anwar, Scott and Moore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert J. Moore, rob.moore@rmit.edu.au