Erratum: Large-Scale Genomic Analyses and Toxinotyping of Clostridium perfringens Implicated in Foodborne Outbreaks in France
 Abakabir Mahamat Abdelrahim
Abakabir Mahamat Abdelrahim Nicolas Radomski
Nicolas Radomski Sabine Delannoy
Sabine Delannoy Sofia DjellalMarylène Le Négrate
Sofia DjellalMarylène Le Négrate Katia Hadjab
Katia Hadjab Patrick Fach
Patrick Fach Jacques-Antoine Hennekinne
Jacques-Antoine Hennekinne Michel-Yves Mistou
Michel-Yves Mistou Olivier Firmesse*
Olivier Firmesse*- Université PARIS-EST, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), Laboratory for Food Safety, Maisons-Alfort, France
Clostridium perfringens is both an ubiquitous environmental bacterium and the fourth most common causative agent of foodborne outbreaks (FBOs) in France and Europe. These outbreaks are known to be caused by C. perfringens enterotoxin (CPE) encoded by the cpe gene. However, additional information on the toxin/virulence gene content of C. perfringens has become available in the last few years. Therefore, to understand the enteropathogenicity of this bacterium, we need to describe the toxin and virulence genes content of strains involved in FBOs. In this study, we used a new real-time PCR typing technique based on a comprehensive set of 17 genes encoding virulence factors. The analysis was performed on a collection of 141 strains involved in 42 FBOs in the Paris region. It was combined with whole genome sequence (WGS) phylogenomic reconstruction, based on the coregenome single nucleotide polymorphisms (SNPs) of 58 isolates, representatives of the identified virulence gene profiles. Two or three different virulence gene profiles were detected in 10 FBOs, demonstrating that C. perfringens FBOs may be associated with heterogeneous strains. cpe-positive strains were isolated in 23 outbreaks, confirming the prominent role of CPE in pathogenicity. However, while C. perfringens was the sole pathogen isolated from the incriminated food, the cpe gene was not detected in strains related to 13 outbreaks. This result indicates either that the standard method was not able to isolate cpe+ strains or that the cpe gene may not be the only determinant of the enterotoxigenic potential of C. perfringens strains. Using phylogenomic reconstruction, we identified two clades distinguishing chromosomal cpe-positive from cpe-negative and plasmid-borne cpe. Important epidemiological information was also garnered from this phylogenomic reconstruction that revealed unexpected links between different outbreaks associated with closely related strains (seven SNP differences) and having common virulence gene profiles. This study provides new insight into the characterization of foodborne C. perfringens and highlights the potential of WGS for the investigation of FBOs.
Introduction
Clostridium perfringens is a Gram-positive, spore-forming, anaerobic, rod-shaped bacterium, known as an important causative agent of foodborne and non-foodborne gastroenteritis (Grass et al., 2013). The ability of this bacterium to form resistant spores contributes to its survival in many environmental niches, including soil, sewage, foods, and the intestinal microbiota of humans and animals (Xiao, 2014; Li et al., 2016). C. perfringens can cause necrotic enteritis, enterotoxemia, and gastroenteritis in animals (Keyburn et al., 2008; Uzal et al., 2015). It is also a foodborne pathogen that causes diarrhea and abdominal pain in humans (Scallan et al., 2011). C. perfringens is commonly found as a contaminant in meat and poultry products, as well as in vegetables and crops. This bacterium is an important pathogen involved in foodborne outbreaks (FBOs) and ranks as the fourth most common causative agent of foodborne illness due to bacterial toxins in France and Europe, with more than 1,500 human cases each year (EFSA, 2016). The pathogenicity of this bacterium is largely attributable to its ability to produce a variety of virulence factors. These virulence factors include well-characterized pathogenic toxins and hydrolytic enzymes. Their roles in FBO remain to be defined but their presence is an aggravating factor (Li et al., 2016). Toxin production varies among C. perfringens strains and is the basis for the classification system, which is based on the production of four major toxins, namely CPA, CPB, ETX, and ITX that divide C. perfringens strains into toxinotypes A to E (McClane et al., 2013). This classification was recently revised to include two additional toxinotypes, i.e., C. perfringens toxinotype F strains producing enterotoxin (CPE) but not CPB, ETX or ITX, and C. perfringens toxinotype G strains producing NetB (Rood et al., 2018) (Table 1). The number of characterized virulence factors is constantly increasing, with more than 20 toxins and hydrolytic enzymes identified to date (Li et al., 2016; Kiu and Hall, 2018). However, there is considerable variability in the toxin armamentarium, and a single strain cannot produce all these virulence factors (Freedman et al., 2016). The virulence factors of C. perfringens can be classified functionally as membrane-damaging enzymes, pore-forming toxins, intracellular toxins, and hydrolytic enzymes (Revitt-Mills et al., 2015). Genes encoding these virulence factors may be located on the chromosome, and on the large plasmid (Freedman et al., 2016). Chromosomal virulence factors include the cpa gene (encoding α-toxin), colA (κ-toxin), pfoA (θ-toxin), and nagH (hyaluronidase or μ-toxin). These genes are located on the variable region of the chromosome (Chalmers et al., 2008a). The nanH, nanI, and nanJ genes encoding different sialidases, and cadA encoding ν-toxin are located on a conserved region of the chromosome (Obana and Nakamura, 2011). The genes cpb (encoding CPB-toxin), cpb2 (CPB2-toxin), etx (ETX-toxin), ia/ib (ITX-toxin), netB (NetB-toxin), tpeL (TpeL-toxin), ureABC (ureases), cpd (δ-toxin), lam (λ-toxin), and becA/becB encoding the binary enterotoxin for C. perfringens are located on large plasmids of variable size ranging from 65 to 110 kb (Li et al., 2013). Recently, partial genome sequencing of the canine isolate revealed three novel putative toxin genes encoding proteins related to the pore-forming leucocidin/hemolysin family. These putative toxin genes were designated netE, netF, and netG (Mehdizadeh Gohari et al., 2015). netE and netF are located on a large conjugative plasmid, and netG is located on another large plasmid (Mehdizadeh Gohari et al., 2017). The C. perfringens enterotoxin gene (cpe) can be located on either the chromosome or on the plasmid (Gao and McClane, 2012). A strain harboring both a chromosomal and plasmid-borne cpe gene has not yet been observed (Freedman et al., 2016). C. perfringens enterotoxin (CPE) is one of the toxins associated with sporulation and has been implicated in FBOs and non-foodborne gastroenteritis illnesses (Lukinmaa et al., 2002).
TABLE 1
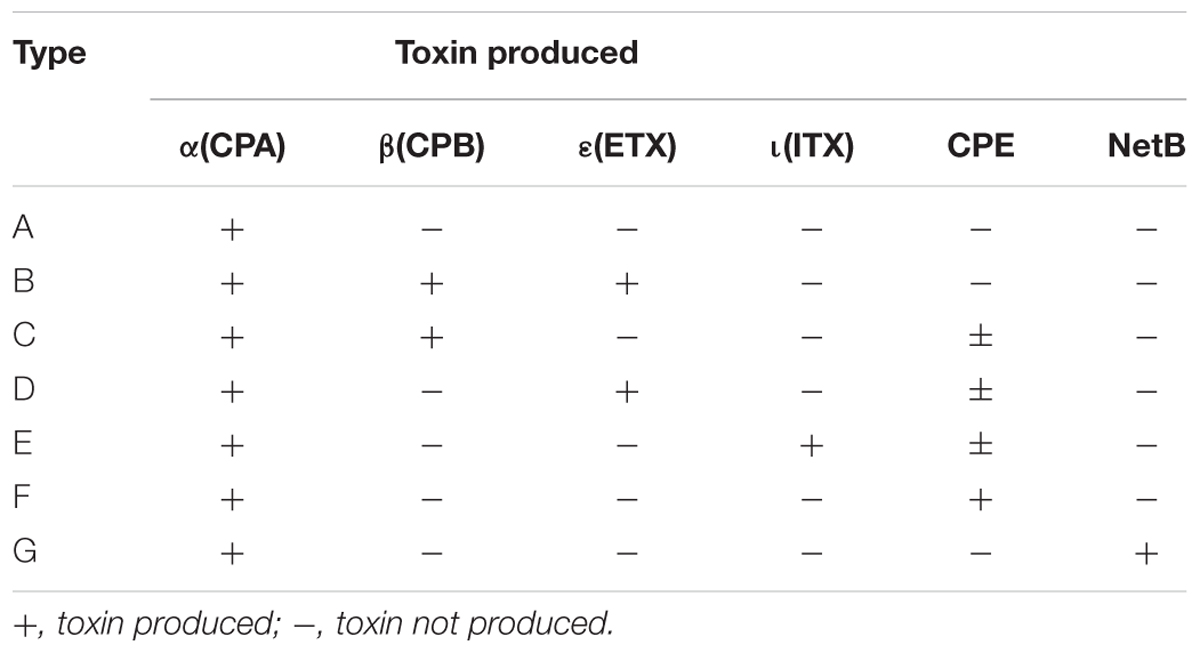
Table 1. Revised classification of C. perfringens based on the production of six major toxins (Rood et al., 2018).
The Standardized method NF EN ISO 7937 is the conventional method for detecting C. perfringens strains in foods by the enumeration of bacterial colonies on specific agar media (Anonymous, 2005). With this robust method, it is possible to detect and enumerate viable C. perfringens in food samples and to obtain isolates that can be characterized further. This reference method therefore makes it possible to isolate the bacterium but does not provide any phenotypic information distinguishing the strains from one another, particularly with regards to their virulence (Law et al., 2015). While the detection of CPE in the stools of patients is a marker for implicating C. perfringens as the etiological agent in foodborne illnesses, CPE is produced in the intestine during sporulation of vegetative cells and is consequently absent from the contaminated food (Lin and Labbe, 2003). Moreover, isolation of cpe-positive C. perfringens in suspected foods and from stools of affected individuals may be accompanied by the presence of cpe-negative C. perfringens from the environment and microbiota (Scallan et al., 2011). This situation makes toxin detection hazardous to link isolates from patients to food vehicles (Lindström et al., 2011). The common assumption that the intestinal pathogenicity of C. perfringens relies on a specific toxin was recently challenged by findings suggesting that additional virulence factors may be involved in cytotoxicity (Yasugi et al., 2015). This prompted us to explore the virulence potential of C. perfringens, taking into account the more recently described virulence factors and phylogenomic inference presenting high discrimination power.
To improve epidemiologic knowledge about C. perfringens involved in FBOs, we developed a new real-time PCR typing technique targeting 17 genes encoding 15 virulence factors. The method was applied to 141 C. perfringens FBO strains isolated in the Paris region between 2013 and 2017. The virulence gene profiles obtained by this method would make it possible to describe strain diversity with regards to the cpe gene among these outbreaks, and robust phylogenomic analysis based on whole genome sequencing (WGS) data (Felten et al., 2017) would enable us to determine phylogenomic diversity at a large genomic scale, as recently emphasized (Kiu et al., 2017).
While many food pathogens have been sequenced and studied extensively, limited genome sequence data for C. perfringens are available in public databases, and very few genomic studies have been conducted on food isolates (Shimizu et al., 2002; Myers et al., 2006; Mehdizadeh Gohari et al., 2017; Lacey et al., 2018). To our knowledge, the largest genomic study conducted on 56 C. perfringens genomes contained only three foodborne strains (Kiu et al., 2017). Genomic comparison previously conducted on 12 strains from different toxinotypes, isolated from different sources, highlighted a lack of features differentiating toxinotype A from the other isolates, indicating that toxinotype A can easily shift to other types by acquisition of toxin-encoding plasmids (Hassan et al., 2015). A recent genomic study conducted on necrotic enteritis-causing isolates of C. perfringens demonstrated that pathogenicity does not correlate with coregenome content, but rather with accessory gene content located on the chromosome and plasmids (Lacey et al., 2018). To better understand the diversity among C. perfringens implicated in FBOs, we performed whole genome analysis on 58 C. perfringens strains selected on the basis of epidemiologic information and diversity of virulence gene profiles.
Materials and Methods
Microbiological and Epidemiological Data on Foodborne Outbreaks Associated With C. perfringens in the Paris Region
The microbiological and epidemiological data concerning each FBO (i.e., number of human cases, location, symptoms, and type of incriminated food) were collected by interviews or questionnaires managed by the local health authorities. The incriminated food was collected and sent to laboratories at ANSES (French Agency for Food, Environmental and Occupational Health and Safety) for microbiological analysis. We studied a collection of 141 isolates of C. perfringens involved in 42 FBOs from 2013 to 2017, including 1,267 human cases. Bacterial strains were isolated from suspected food by plating on selective Tryptose Sulfite Cycloserine (TSC) media according to the EN NF ISO 7937 standard method. The level of contamination by C. perfringens in the implicated foods was between 3.0 × 101 and 1.0 × 106 cfu/g. C. perfringens was isolated from 10 outbreaks in association with other bacterial species (including B. cereus and S. aureus) during microbiological investigations. Overall, vegetable dishes were the most common food reported (15 outbreaks, 36%), followed by poultry (10 outbreaks, 24%). Pork and beef sources were involved in practically the same proportion (six outbreaks, 14% for each) as other sources (five outbreaks, 12%) (Table 2).
TABLE 2
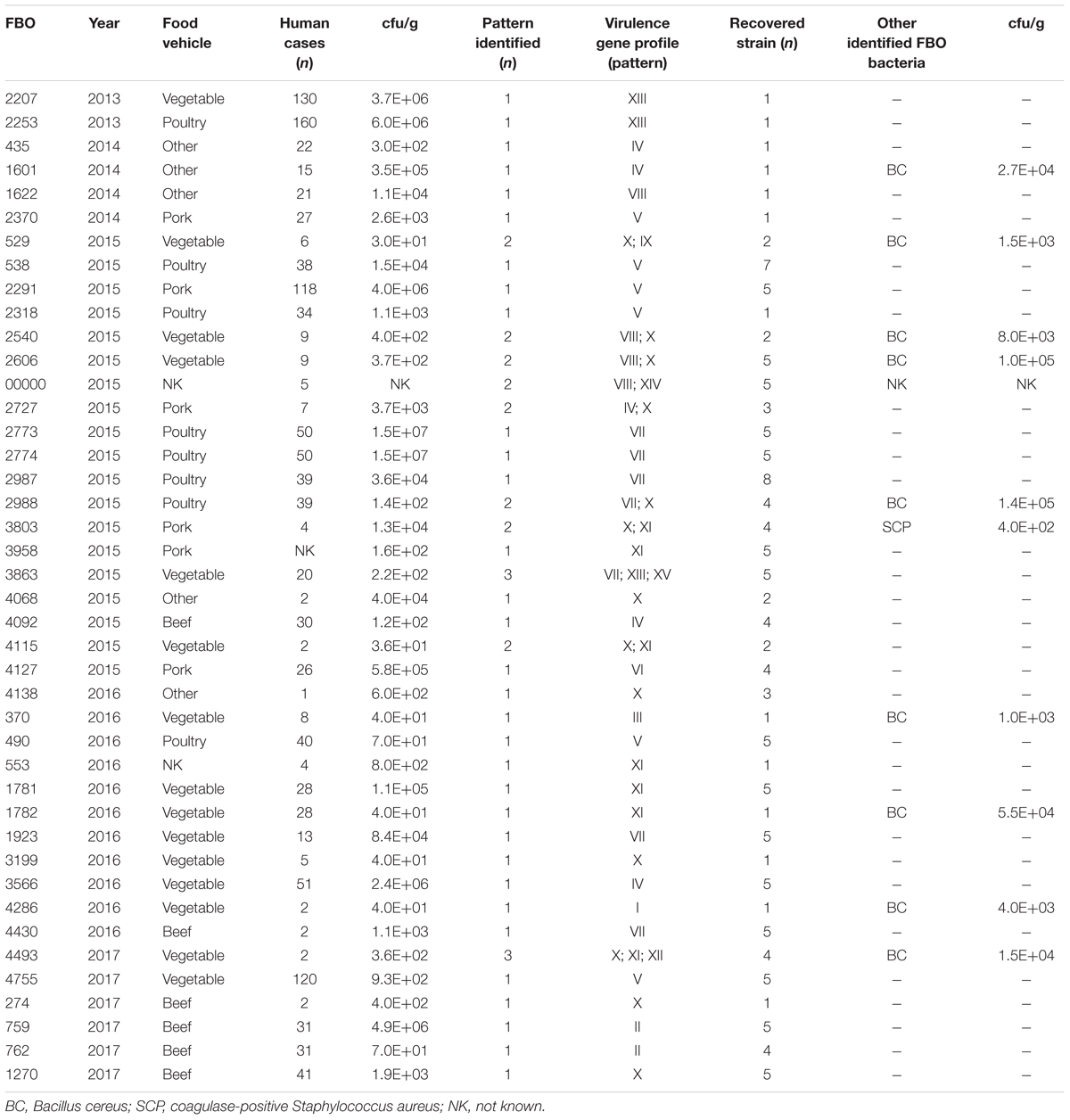
Table 2. Microbiologic data for FBOs associated with C. perfringens from the Paris region between 2013 and 2017.
DNA Extraction
With the aim of obtaining DNA for PCR and WGS-based applications, isolates of C. perfringens were revived from a frozen stored culture (-20°C) and anaerobically inoculated for 24 h at 37°C in Thioglycolate Broth with Resazurin® (bioMérieux, Marcy l’Etoile, France). The subsequent culture was streaked on Columbia agar with 5% sheep blood (bioMérieux, Marcy l’Etoile, France) and incubated for 24 h at 37°C under anaerobic conditions using a Genbox anaer sachet® (bioMérieux, Marcy l’Etoile, France).
Concerning PCR-based applications, DNA extraction was performed using the InstaGene kit (Bio-Rad Laboratories, Marnes-la-Coquette, France), following the manufacturer’s instructions for Gram-positive bacteria. For each culture, cells were washed by centrifugation at 17,000 × g for 3 min in 1 mL of phosphate buffer solution (PBS). The cell pellet was resuspended in 200 μL of Chelex-based solution and heated for 20 min at 56°C and then at 100°C for 10 min. The DNA solution was cooled for 5 min, and centrifuged at 17,000 × g for 3 min to eliminate cell debris. The purity of the extracted DNA solution was assessed using NanoDrop 2000 (Thermo Scientific, Villebon, France), according to the instructions provided by the manufacturer. The ratio of absorbance at 260 nm and 280 nm was used to assess protein contamination, while the ratio of absorbance at 260 nm and 230 nm was estimated to assess phenol contamination. The standard for purity was an A260/A280 ratio between 1.8 and 2.2, and an A260/A230 ratio between 2.0 and 2.2. The DNA was then diluted to obtain 3.3 × 102 ng.μL-1, corresponding to 105 genome equivalents. DNA solutions were stored at -20°C for further PCR analysis.
Concerning WGS-based applications, DNA was extracted using the Wizard Genomic DNA Purification kit (Promega, Charbonnières-les-Bains, France), according to the manufacturer’s instructions. Culture conditions and assessment of the quality and quantity of extracted DNA were performed as described above. Before library preparation, 200 ng of genomic DNA were analyzed on 0.8% agarose gel to ensure the absence of RNA contamination and DNA degradation.
PCR Conditions
The BioMarkTM real-time PCR system (Fluidigm, San Francisco, CA, United States) was used for high-throughput microfluidic real-time PCR amplification using the 192×24 dynamic array (Fluidigm, San Francisco CA, United States). This chip dispenses 24 PCR mixes and 192 samples into individual wells, after which on-chip microfluidics assemble PCR reactions in individual chambers prior to thermal cycling resulting in 4,608 reactions.
Amplifications were performed using EvaGreen DNA binding dye (Biotium Inc., Hayward, CA, United States) with Perfecta qPCR tough mix (Quanta bio, Beverly, MA, United States), in accordance with the manufacturer’s recommendations. Three microliters of sample mix (containing 1.47 μL perfecta qPCR tough mix, 0.15 μL DNA binding dye sample loading reagent, 0.15 μL EvaGreen, 0.06 μL Rox reference dye, 0.76 μL DNA, and 0.41 μL DNase free water) were loaded into each sample inlet of the dynamic chip. Three μL of primer mix (containing 1.5 μL assay loading reagent, 0.75 μL primer stock containing 20 μM of each primer and 0.75 μL DNase free water) were loaded into each assay inlet of the dynamic chip. The thermal profile comprised 10 min at 95°C (Taq polymerase activation), followed by 35 cycles of 95°C for 15 s, and 60°C for 1 min (amplification steps), followed by melting curve analysis. The assays were performed in duplicate. Data were acquired on the BioMarkTM Real-Time PCR system and analyzed using Fluidigm Real-time PCR Analysis software to obtain crossing point (Ct) values. Two negative controls (no template controls) were included per chip and a pool of DNA for reference strains. CIP106156 known to be positive for the cpa, cpe, ia, ib, pfoA, colA, lam, nagH, nanH, nanI, nanJ, and cadA genes, CIP106527 positive for the cpb1 and tpeL genes, and CIP104612 positive for the cpb2 gene were used as positive controls. Targeted genes and corresponding primers are listed in Table 3.
TABLE 3
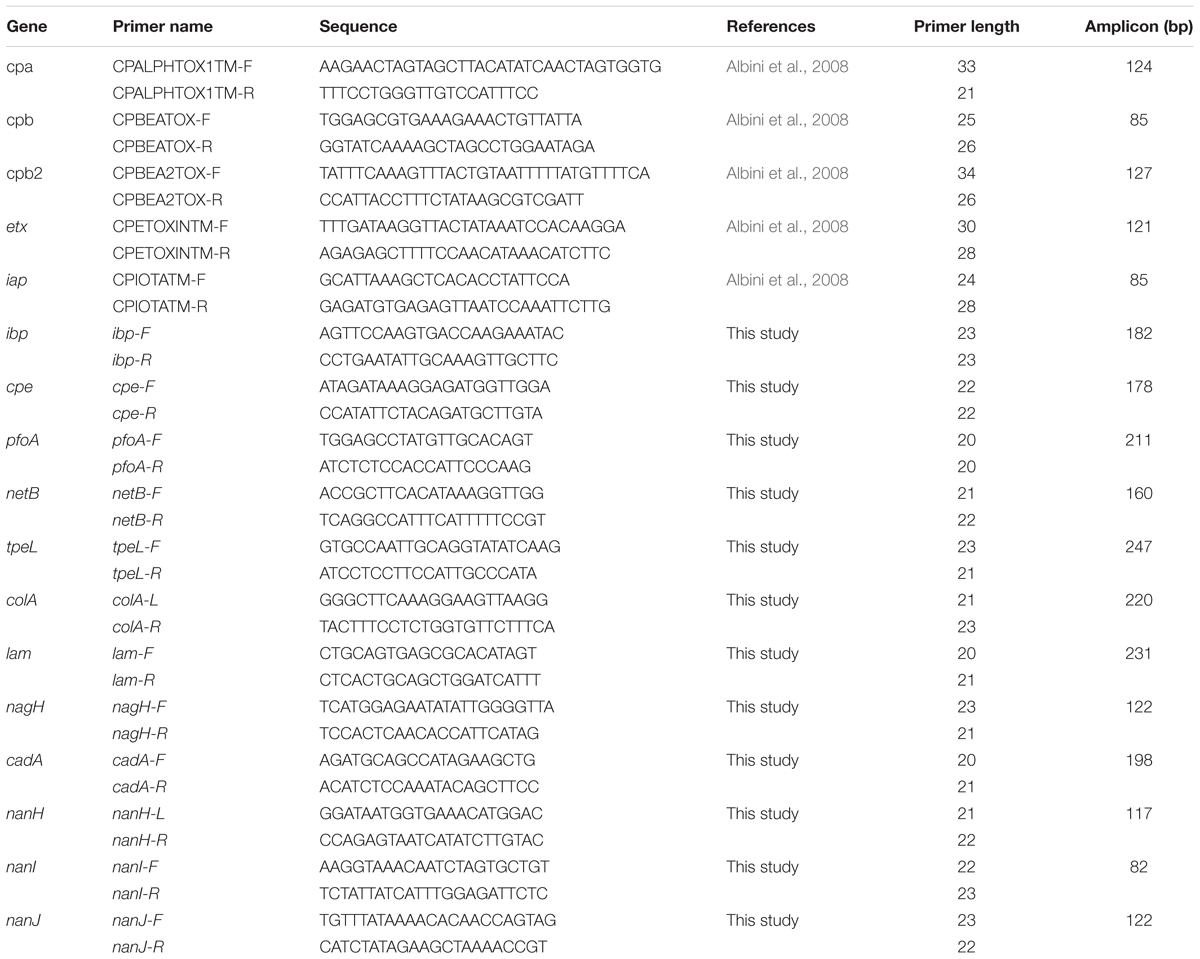
Table 3. Primers used for the detection of 17 genes encoding 15 virulence factors.
Strain Selection for WGS
Fifty-eight strains representative of all toxin gene profiles in all FBOs were selected for WGS. Additional isolates were selected to confirm the specificity and sensitivity of the PCR method to detect the 17 virulence genes. Three contaminated DNA samples of profile I (FBO 4286), profile X (FBO 2606), and profile XIV (FBO, NK) were removed from WGS analysis.
Read Quality Check and Assembly
The genome sequencing was performed by the ‘Institut du Cerveau et de la Moelle épinière’ (ICM)1 (Hôpital de la Pitié-Salpêtrière, Paris) using NextEra XT libraries (Illumina, San Diego, CA, United States), indexed following the manufacturer’s recommendations (Illumina, San Diego, CA, United States), purified with the Agencourt AMPure XP system (Beckman Coulter), and controlled with Microfluidic Labchip GX (PerkinElmer, Villebon-sur-Yvette). These libraries were sequenced with a NextSeq 500 sequencer (Illumina, paired-end reads of 150 bases each). Assembly and variant calling of genomes were performed with an in-house workflow called ARtWork. Briefly, Paired-end reads stored as fastq.gz files were decompressed and BBMap (Bushnell, 2014) was used in order to estimate deep coverage. The reads with depth coverage lower than 20× were discarded from the workflow. Those higher than 100× were normalized at 100× using BBNorm (Xu et al., 2015). The quality of paired-end reads was evaluated with FASQC software v0.11.2. FASQC reports were produced for each sample of retained reads. Trimmomatic was used to trim reads less than 50 bp in length and bases presenting a phred score less than 20 (Bolger et al., 2014). The closest related reference genome of each sample was identified by estimating the Jaccard index (Ondov et al., 2016) against a collection of closest reference genomes (Supplementary Material) and was further used to perform scaffolding with MeDuSa (Bosi et al., 2015) and gap filling with GMcloser (Kosugi et al., 2015). The scaffolds of less than 200 bp in length were trimmed with Biopython (Cock et al., 2009). The quality of assemblies was assessed using QUAST metrics (Gurevich et al., 2013). Overall, the genomes showed the expected GC content of 28% and most of the contigs could be organized into single scaffolds. The sizes of the largest scaffolds ranged from 2.6 to 3.33 Mbp, with an average of 3.0 Mbp, in perfect agreement with the expected 3.0 Mbp size of the C. perfringens genome (Supplementary Material).
Variant Calling Analysis
Also implemented in the ARtWork workflow, a variant calling analysis was performed using the iVARCall2 workflow (Felten et al., 2017), an in-house variant caller previously published and based on the HaplotypeCaller algorithm able to call single nucleotide polymorphisms (SNPs) and small insertions/deletions (InDels) at the coregenome scale simultaneously, and for each genome independently. The iVARCall2 workflow produces g.VCF files for each genome, which can be filtered and merged in order to produce high-quality variants and related pseudogenomes (i.e., VCFtoPseudogenome script). This pseudogenome corresponds to the reference genome where the genotypes of detected variants are replaced in each genome of the dataset. At the end of the iVARCall2 workflow, reports on breadth and depth coverage were produced, as well as matrices of pairwise distances. To assess quality of variant calling (Sandmann et al., 2017), mapping against the reference genome ATCC13124 was performed and resulted in breadth and depth coverages of 84 ± 7% and 389×, respectively.
Coregenome Phylogenomic Inference
A maximum likelihood (ML) coregenome phylogenomic tree based on the pseudogenomes was constructed using the general time-reversible model with gamma distributed sites (GTRGAMMA), the secondary structure 16-state model, and a bootstrap analysis (n = 100) implemented in RaxML (Stamatakis, 2015). A total of 59,593 coregenome SNPs within 902 conserved core genes in all 58 genomes were identified in the dataset. Interactive Tree of Life (iTOL) was used to visualize the tree and annotate metadata (Letunic and Bork, 2016). In order to estimate the number of replicates necessary to yield accurate confidence values, a posterior bootstrap convergence test was computed with RAxML (Pattengale et al., 2010).
In silico Detection of Virulence Factors
The coding sequences (CDSs) of 24 genes (including becA, becB, ureABC, and cpd) encoding virulence factors were retrieved from NCBI and queried against the 58 assembled genomes using nucleotide BLAST (identity = 90%; minimum coverage = 85%) to investigate the presence of these genes and confirm the results obtained by real-time PCR amplification.
Statistical Analyses
Statistical analyses were conducted in R studio (version 2017-03-06). A Shapiro–Wilk test was performed to assess the normality of data, and a non-parametric Kolmogorov–Smirnov test was used to compare the diversity between phylogroups (Veshnyakova et al., 2012). The associations between different genes were tested using a Spearman’s rank correlation test (Myers and Sirois, 2004). The difference between the number of genes detected in cpe-positive strains and cpe-negative strains was tested using the Wilcoxon Rank Sum test (Rey and Neuhäuser, 2011).
Results
Distribution of Genes Encoding Virulence Factors Among C. perfringens Food Isolates
We successfully applied real-time PCR targeting 17 genes encoding 15 toxins or virulence factors on all 141 C. perfringens strains from 42 FBOs in order to discriminate virulence gene profiles. The distribution of genes encoding virulence factors shown in Table 4 was diverse among C. perfringens food strains. Four genes encoding virulence factors including cpa, colA, nanH and cadA were conserved in all tested isolates and were not included in Table 4. Similarly, the cpb, etx, netB and tpeL genes were not detected in any of the analyzed strains and were therefore excluded from Table 4. According to the new toxin-based classification system (Rood et al., 2018), toxinotype F was the most prevalent and represented 79 strains (56%). Sixty-one strains were classified as toxinotype A and only one strain belonged to toxinotype E. However, our analysis provided a much more contrasted view of the foodborne strains. In total, 15 virulence gene profiles from I to XV were obtained. The strains harbored 4 (profile XV) to 11 (profile I) virulence genes. Six profiles of virulence genes (I, III, IX, XII, XIV, and XV) were represented by only one strain. Interestingly, profile I was represented by a strain belonging to C. perfringens type E (i.e., presence of the ia and ib genes encoding iota toxin).
TABLE 4
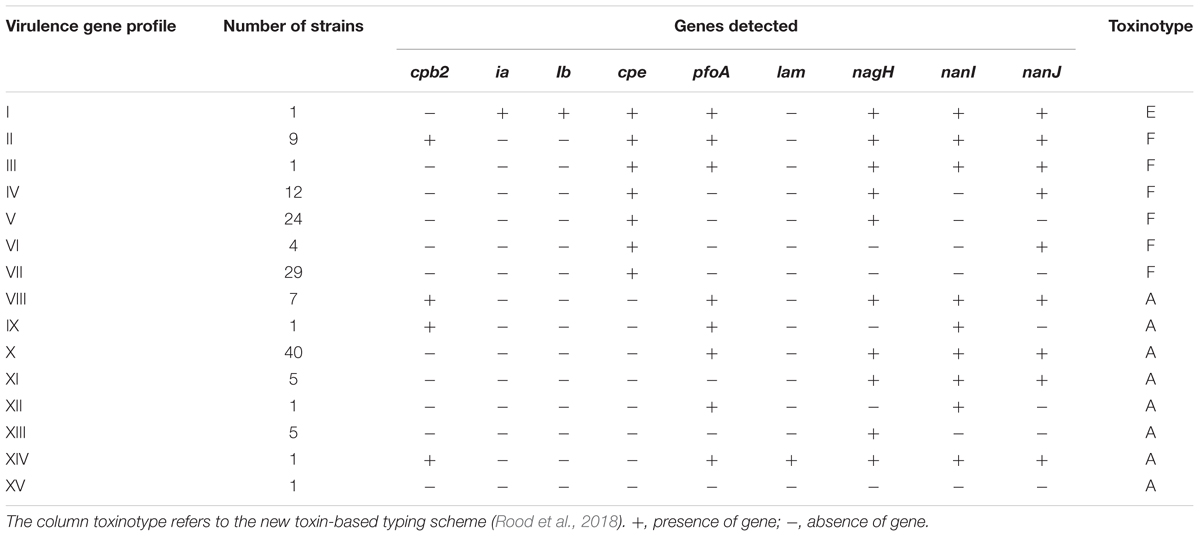
Table 4. Profiles of virulence genes identified in C. perfringens strains isolated in the Paris region from 2013 to 2017 (n = 141).
CPE is considered by many authors as the sole toxin responsible for the symptoms occurring during foodborne illness caused by C. perfringens (Brynestad and Granum, 2002; Lindström et al., 2011; Chon et al., 2012; Freedman et al., 2016). Consequently, we paid particular attention to the distribution of the cpe gene in our collection of strains (Table 4). In total, 80 strains were positive for the cpe gene and 61 were cpe-negative. The distributions of virulence genes were different according to the presence or absence of the cpe gene. Four profiles (IX, XI, XII, and XIV) with a combination of genes encoding virulence factors were detected only in cpe-negative strains. Conversely, three profiles (I, IV, and VI), which contain a combination of genes encoding virulence factors, were only detected in cpe-positive strains. Four profiles (II/VIII, III/X, V/XIII and VII/XV) were identical between cpe-negative and cpe-positive strains.
A significant trend emerged from the comparative analysis of cpe-positive vs. cpe-negative strains: 80% (49/61) of cpe-negative strains carried 8 to 10 virulence genes, while 86% (69/80) of cpe-positive strains carried only five to seven virulence genes (p < 0.05). The biological relevance of this observation can be assessed by performing in vitro cytotoxicity tests. Profile VII was the most common among cpe-positive strains (36%) and lacks all the other genes encoding virulence factors. Importantly, C. perfringens was the only pathogen identified in five of seven outbreaks where profile VII was detected, confirming the prominent role of CPE in pathogenicity. On the other hand, virulence gene profile X was the most prevalent among cpe-negative C. perfringens strains (66%, 40/61). Profile X carried the pfoA, nagH, nanI, and nanJ genes. The gene encoding perfringolysin O (pfoA), a well characterized pore-forming toxin, was found in 82% (50/61) of cpe-negative C. perfringens strains and was associated with hyaluronidases genes (nagH) in 48 of the 50 cpe-negative pfoA-positive strains. The Sialidase genes (nanI and nanJ) were also found predominantly in cpe-negative strains (87%, 53/61). An interesting finding is shown in Table 4: nagH is the only virulence gene present in profile XIII, which was the only strain isolated in large FBOs (outbreaks 2207 and 2253), suggesting that NagH could be a marker of the enterotoxigenic potential of cpe-negative strains. This hypothesis is compatible with the epidemiologic characteristics of nagH cpe-negative strains (profiles IX, XII, and XV), which were always found to be associated with nagH-positive profiles in the investigated outbreaks. However, this result could also be due to a fastest growth rate in food that could make cpe+ strains undetectable. This hypothesis could be tested experimentally in competition growth assays. In addition and to clarify the potential pathogenicity of cpe- strains it would be interesting to perform PCR targeting the cpe gene on DNA extracted directly from the food mother suspension. The development of such a PCR test made on food suspension would complement the standard microbiological detection method.
Profiles of Virulence Genes per Foodborne Outbreak
To explore the genetic diversity within the strains isolated in the same outbreak, we considered the 29 FBOs for which at least two to eight C. perfringens isolates per outbreak were recovered. A FBO is considered to be heterogeneous when two or more recovered isolates show different virulence gene profiles. Several profiles of virulence genes (i.e., up to three different profiles) were retrieved from 10 outbreaks. In addition, the cpe gene was not detected in seven of the 10 heterogeneous FBOs (outbreaks 2540, 2606, NK, 3803, 4115, 4493, and 529) (Table 2). C. perfringens was the sole pathogen identified in two outbreaks.
The isolated strains had homogeneous virulence gene profiles in 19 FBOs. The cpe gene was found to be present in 14 of the 19 homogeneous FBOs. In addition, C. perfringens was the only pathogen isolated in all homogeneous FBOs in which the cpe gene was detected. Among the homogeneous FBOs with cpe-positive strain profiles, profiles V and VII were the most commonly detected in nine out of 14 FBOs. Conversely, the cpe gene was not detected in 5 FBOs with homogeneous profiles of virulence genes.
Finally, only cpe-negative C. perfringens isolates were detected in 12 out of 42 FBOs using the horizontal method EN NF ISO 7937 for the enumeration of C. perfringens (Anonymous, 2005). This observation does not prove the pathogenicity of these strains since other pathogens may have escaped detection. However, we cannot exclude the possibility that cpe-negative C. perfringens isolates may have played a role in the diseases.
Phylogenomic Inference
Our PCR-based analysis focusing on the virulence gene profiles shows that our collection of FBO-associated C. perfringens strains is segmented into two genetically distinct clusters, namely cpe-positive and cpe-negative strains. In order to check the results obtained by PCR, we performed a whole genome analysis of strains representative of the diversity of the dataset. The study of genetic relationships was based on coregenome SNPs and the maximum-likelihood method to provide a strong phylogenetic analysis. The tree was robust with most branches of the backbone presenting high bootstraps values. Strikingly, two distinct clades were identified that distinguish most cpe-positive strains (24/27) from cpe-negative strains (27/30) (Figure 1). A closer examination of the genomic environment of the cpe locus in the 3 cpe-positive strains (16SBCL585, 17SBCL79, and 17SBCL85), which clustered in the cpe-negative clade, revealed that it is flanked by a putative cytosine methyltransferase gene (dcm) and two insertion sequences IS1469 and IS1470-like found in a 70 kb scaffold (Figure 2). By BLAST querying against plasmid NCBI database, we found 99.9% identity with plasmid pCPF4969 (Adams et al., 2015, 2018). This result indicates that the cpe gene is carried on a the pCPF4969 plasmid (Li et al., 2013).
FIGURE 1
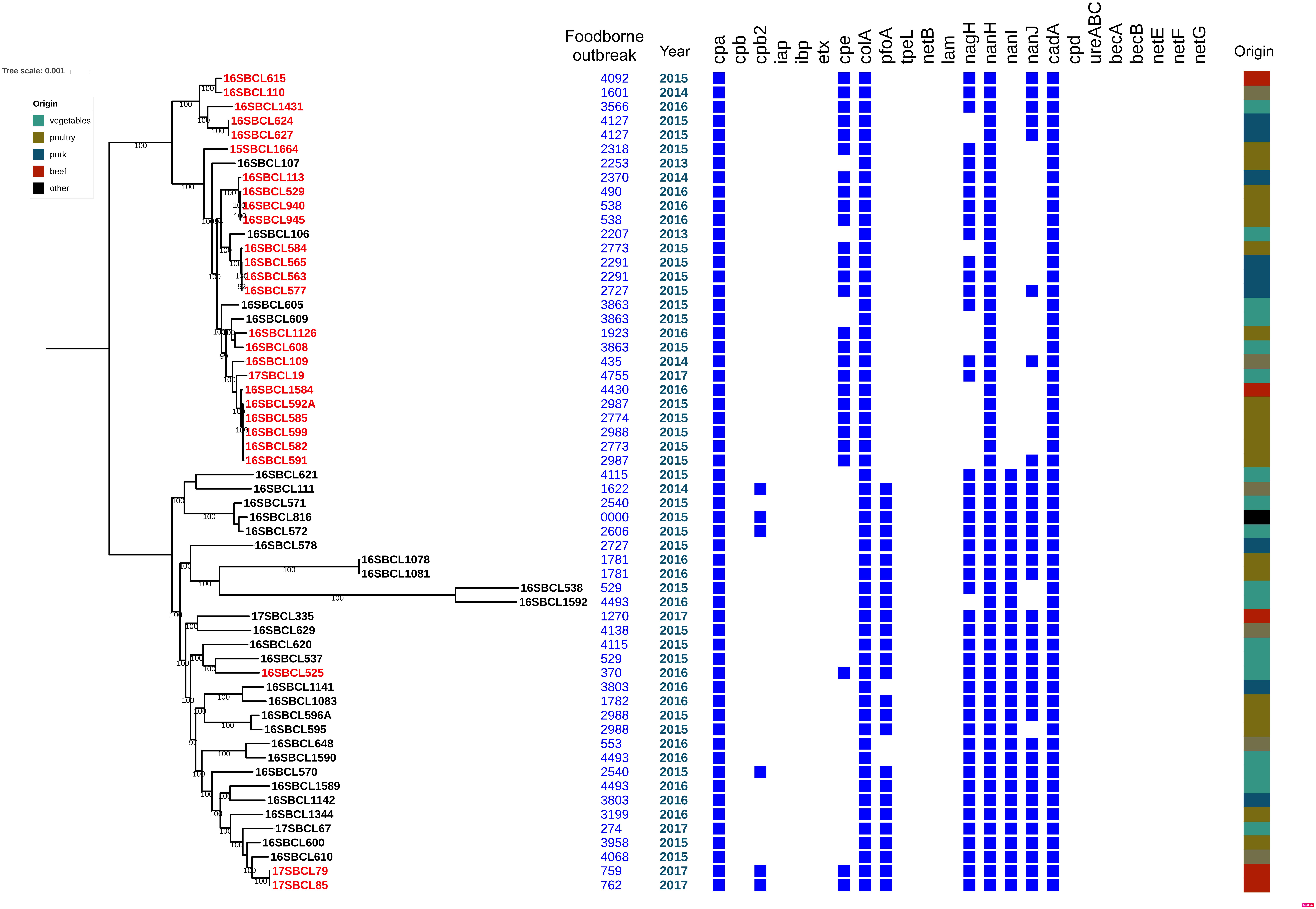
Figure 1. Coregenome phylogenomic inference of 58 C. perfringens isolates. The maximum likelihood tree was constructed using coregenome SNPs identified with iVARCall2. The phylogenomic history was inferred using RAxML and branches were supported by bootstrap analysis (n = 100). The phylogenetic inference converged after 50 bootstrap replicates. Metadata were visualized using iTOL. The presence of virulence genes is indicated with blue squares. The cpe-positive strains are in red. The large multi-colored strip indicates different food matrices (origin).
FIGURE 2
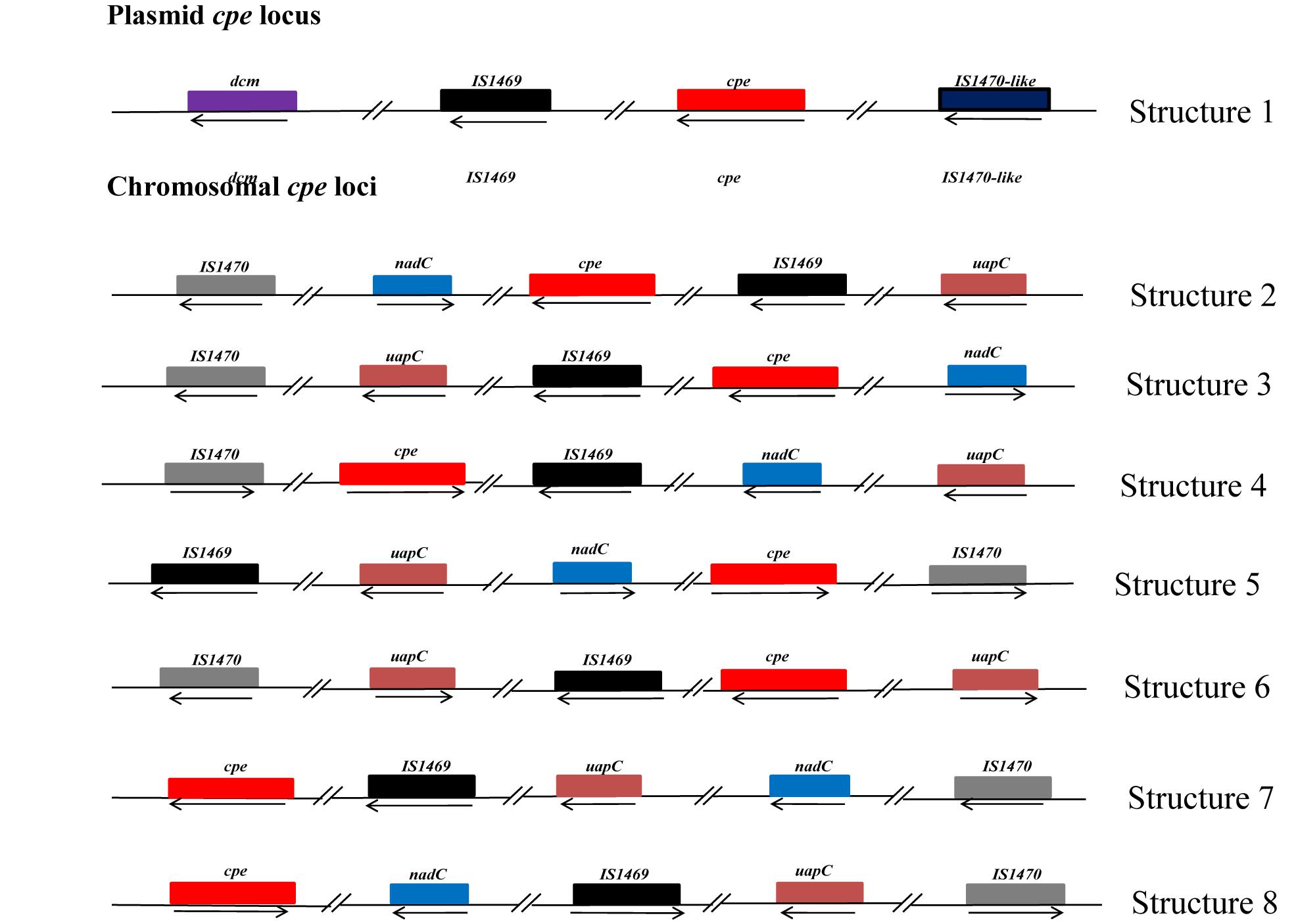
Figure 2. Description of the genetic organization of the cpe locus in C. perfringens strains. Each box represents an open reading frame. The plasmid-cpe locus (structure 1) includes the dcm gene and two characteristic insertions sequences, IS1469 and IS1470-like. The sequence analyses indicated 99% identity with 100% query coverage with plasmid pCPF4969 of C. perfringens strain F4969. Seven other different structures of the cpe locus are shown. Analysis of these structures revealed that these loci are located in 3 Mbp, corresponding to the chromosomal cpe locus. However, the identified structures in our study are different with respect to dual IS1470 flanking the cpe gene, as described by Miyamoto et al. (2002). Structure 2 corresponds to 16SBCL584, 16SBCL582, 16SBCL577, 16SBCL592A, 16SBCL592B, 16SBCL1584, 16SBCL1126, 16SBCL529, 16SBCL591, 16SBCL585, 16SBCL1664, 16SBCL945, and 16SBCL599. Structure 3 corresponds to 16SBCL565, 16SBCL563, 16SBCL110, and 16SBCL615, structure 4: 16SBCL627 and 16SBCL1431, structure 5: 16SBCL113, structure 6: 16SBCL109 and 16SBCL624, structure 7: 16SBCL940 and structure 8: 17SBCL19.
A similar analysis performed among 24 cpe-positive strains identified seven different cpe loci which were all found in the 3.0 Mb scaffold corresponding to the chromosome. The chromosomal organizations (Figure 2) of the cpe loci correspond to a variety of arrangements of the IS1469, IS1470, nadC, uapC, and cpe loci (Figure 2). To our knowledge, shuffling at the cpe locus has not previously been described and could have consequences on CPE production.
Genomic Analysis
Genomic analysis revealed unexpected links between different outbreaks. The pairwise SNP differences were used to establish the level of relatedness among the strains. A comparative analysis of intra-clade genetic diversity was performed. An average pairwise SNP difference of 8,494 ± 499 was calculated within chromosomal cpe-positive strains, compared to 21,718 ± 901 within cpe-negative and plasmid-cpe strains. The genetic diversities of these two clusters were significantly different in terms of pairwise SNP differences (p < 0.05, Figure 3).
FIGURE 3
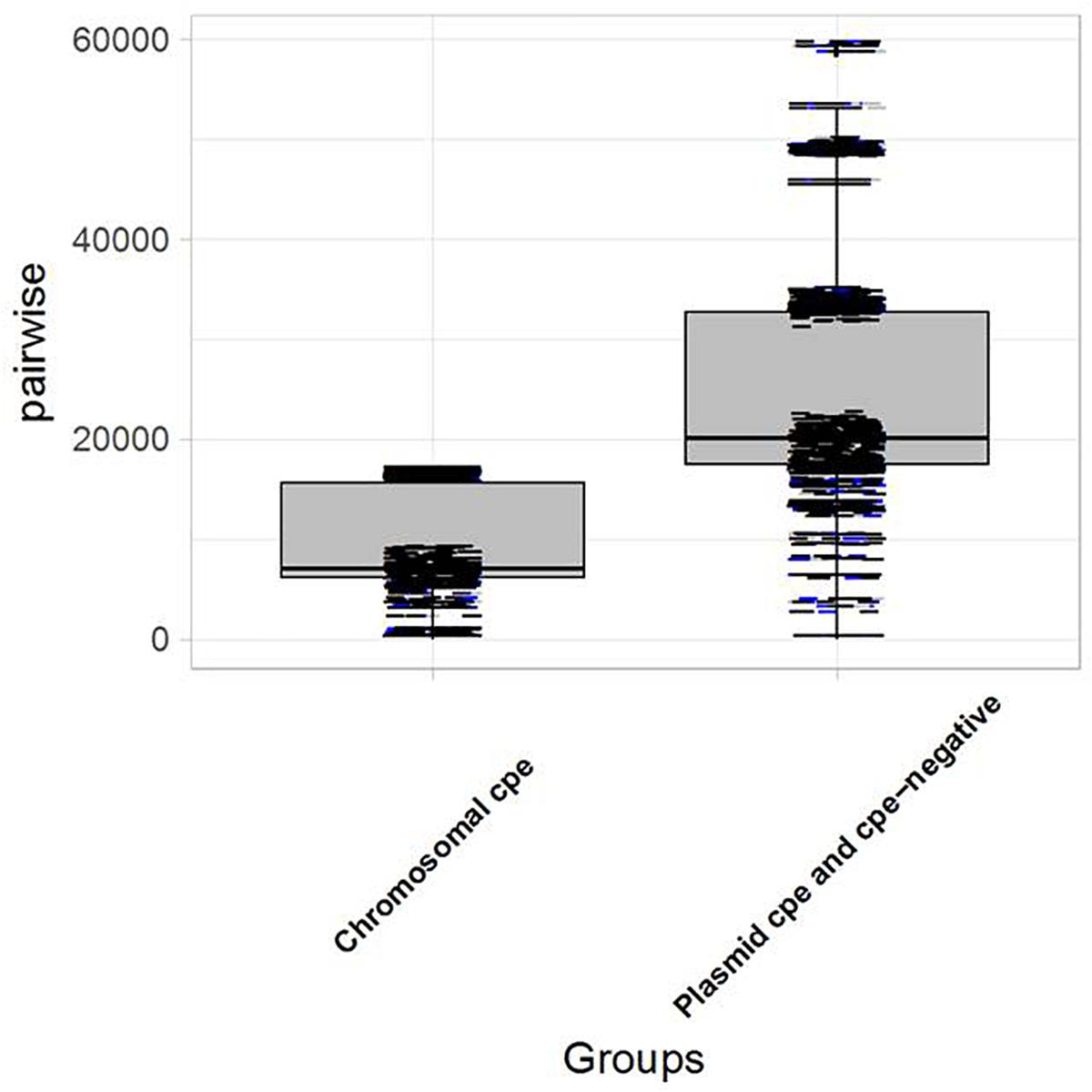
Figure 3. Boxplot showing the distribution of strains carrying the cpe gene on the chromosome vs. plasmid-cpe and cpe-negative strains. The Kolmogorov–Smirnov test yields D = 0.83266, and p-value < 2.2e-16, indicating a significant difference in terms of genetic diversity between the pairwise SNP differences of the two groups.
We compared the genetic relatedness of strains isolated from the same FBOs and presenting the same profile of virulence genes. We found that the strains were indistinguishable in three out of four cases, indicating homogeneous contamination. On the other hand, two co-contaminated strains were detected with a highly unrelated pairwise distance of 6,484 SNPs (FBO2773). This result indicates that contamination with a heterogeneous profile can occur and that virulence gene profiling is not a sufficiently discriminatory criterion, in particular to differentiate strains during FBO investigations.
Nucleotide BLAST was used to identity and confirm virulence factors previously detected by real-time PCR, including the becA, becB, cpd, ureABC, netE, netF and netG genes. For the 58 sequenced C. perfringens isolates, the results obtained by BLAST were similar to those obtained by real-time PCR detection. These results confirm the specificity and robustness of our PCR systems. Our in silico analyses were negative for becA and becB encoding BEC toxin, ureABC encoding ureases, cpd encoding lambda, and the recently identified pore-forming toxin genes netE, netF and netG in the 58 genomes.
Discussion
C. perfringens has been recorded as the fourth most common cause of FBOs in France (Santé Publique France, 2016). FBO caused by C. perfringens results from the ingestion of foods that are contaminated with spore or vegetative cells (Brynestad et al., 1997). The true human burden of C. perfringens is likely much higher as many outbreaks are never investigated and many illnesses occur sporadically (Vaillant et al., 2005). Our study shows that FBOs were commonly due to meat and meat product foods (52%, 22/42), which is consistent with the epidemiology of C. perfringens (Wen and McClane, 2004; Grass et al., 2013). The contamination of meats may occur through contact of carcasses with feces, as well as via cross-contamination by other foods or contaminated surfaces during slaughtering. Although most C. perfringens FBOs are attributed to meat and poultry products (Grass et al., 2013), our study shows that in eight cases C. perfringens was the only pathogen isolated from vegetable dishes. This unexpected finding is likely linked to the ubiquitous nature of C. perfringens, which can be isolated in the environment and the soil (Uzal et al., 2014). Improper holding temperatures and incomplete cooking of foods are recognized as major factors contributing to the development of C. perfringens FBOs (Sarker et al., 2000a). Moreover, vegetables may initially be contaminated by spores of C. perfringens (Xiao et al., 2012), while the cooking temperature of vegetable dishes is too low to destroy spores (Talukdar et al., 2016). C. perfringens can grow at temperatures from 20 to 53°C and spores can survive high temperatures (up to 95°C for 1 h) (McClane et al., 2013). We observed that certain virulence gene profiles (V, VII, X, see Table 4) are prevalent in our collection. It would be of interest to measure the heat resistance of a selection of isolates to determine if this phenotypic trait can explain the prevalence of specific genomic profiles in food.
Symptoms due to foodborne C. perfringens are similar to those caused by other foodborne pathogens, especially B. cereus diarrheal strains (Tewari and Abdullah, 2015). Moreover, current methods for investigation of C. perfringens in FBOs are based on enumeration of characteristic colonies in specific media, followed by biochemical confirmation. These methods are useful to collect strains but do not provide any typing information. As mentioned above, the revised typing scheme classifies strains producing CPE as type F. However, understanding the role and virulence of C. perfringens strains isolated in the context of FBOs requires us to describe the genetic diversity of the implicated strains and to develop robust and novel typing methods. In the present work, we developed a new PCR-based technique on a comprehensive set of 17 genes encoding putative virulence and toxin factors. This new PCR-based technique was used to characterize the diversity of a collection of 141 strains implicated in 42 FBOs.
The cpa, colA, nanH, and cadA genes encoding α-toxin, κ-toxin, sialidase (i.e., a small intracellular sialidase with 43 kDa of molecular weight) and deoxyribonuclease were found to be conserved in all isolates analyzed. Previous genomic studies support this result (Goossens et al., 2017; Kim et al., 2017; Kiu et al., 2017). Consequently, these genes cannot be considered relevant markers to discriminate FBO isolates. Conversely, four genes were not identified in our collection. Consequently, our typing scheme was reduced to nine genes and was used to characterize the virulence profile of a collection of 141 C. perfringens strains implicated in 42 FBOs. Fifteen different profiles were recognized (Table 4), demonstrating the discriminatory power of this method. The epidemiological relationships between C. perfringens of different origins, including isolates from FBOs, have been investigated previously (Jost et al., 2006; Afshari et al., 2016). The previously published articles focusing on FBO isolates and animals concluded that isolates from the same outbreak have a similar pattern, while genetic diversity is high in non-outbreak isolates selected randomly (Johansson et al., 2006; Chalmers et al., 2008b; Fohler et al., 2016). Our results challenge this view and demonstrate that a range of virulence gene profiles can be found within the same FBO.
A more thorough examination of C. perfringens diversity revealed interesting patterns. Except for profile I, the presence of cpe (p < 0.05) was associated with a low number (5–7) of other virulence genes. More precisely, a strong association was observed between the presence of cpe and the lack of pfoA and nanI (r = -0.67, p < 2.2 × 10-16). This observation is consistent with previous studies showing that most cpe-positive C. perfringens strains lack the pfoA gene (Deguchi et al., 2009). The presence of the pfoA and nanI genes was found to be significantly associated (61/66; p < 2.2 × 10-16). The most predominant profiles in cpe-negative strains were the profiles X, with 40 of the 61 strains, and VIII, with seven of the 61 strains. In these two profiles, the pfoA gene was associated with sialidases and/or hyaluronidase. These toxins could contribute to the overall virulence of the bacterium (Revitt-Mills et al., 2015). Previous in vitro studies have demonstrated a synergic effect of perfringolysin O and α-toxin on the cytotoxicity of non-foodborne strains (Awad et al., 2001); the association between pfoA and nanI could have toxicologic consequences that would be worth studying in an appropriate experimental system.
Profiles IV, V, and VII were predominant and were found in 18 of the 29 FBOs with cpe-positive profiles. In addition, profile VII carries cpe as the sole toxin-encoding gene. These results confirm that CPE is a strong contributor to foodborne illness due to C. perfringens (Sarker et al., 2000b).
Two to three different profiles of virulence genes were identified in three FBOs where C. perfringens was the only isolated pathogen, demonstrating that Clostridium FBOs can be associated with heterogeneous contamination of food. This important result complicates the epidemiology of clostridial FBOs, and the contribution of the different gene profiles to the development of symptoms needs to be investigated further. A direct consequence is that pathogenic strains co-infecting foods with non-pathogenic strains can be overlooked. It is therefore important to couple a PCR-based method targeting genes with the NF EN ISO 7937 standard method and characterize more than five strains isolated in the same FBO.
To further analyze the diversity of C. perfringens-associated FBOs, WGS of 58 isolates of C. perfringens was conducted to provide insight into the phylogenomic relationships and distribution of virulence factors among cpe-positive and cpe-negative strains. To the best of our knowledge, this study represents the first large genomic analysis focusing on C. perfringens food isolates, compared to other studies screening other sources and including few foodborne samples (Hassan et al., 2015; Kiu et al., 2017). The computed coregenome phylogenomic inference indicates that C. perfringens strains carrying the cpe gene on the chromosome have evolved independently from cpe-negative and plasmid carrying cpe strains. This argues for the possibility of its involvement in adaptation to specific niches, and supports previous research conducted on C. perfringens strains from different origins showing a clear partitioning of strains capable of inducing foodborne illness into lineages distinct from those of strains isolated from other environments (Hibberd et al., 2011). Previous studies (Miyamoto et al., 2002; Li et al., 2013) have localized the cpe gene near the dcm gene on both the pCPF4969 and pCPF5603 plasmids of type A isolates. Our investigation of the genetic environment of 16SBCL525, 17SBCL79, and 17SBCL85 is consistent with these previous studies and demonstrates that the dcm gene is proximal to the plasmid-borne cpe gene. The fact that these strains were phylogenetically clustered with cpe-negative strains supports the notion that the dcm region of plasmids represents a hot-spot for insertion of certain mobile genetic elements, including some carrying a cpe gene. An early study indicated that C. perfringens carrying the cpe gene on the chromosome is flanked by upstream and downstream IS1470 and, instead of a dcm gene, they carry the purine permease (uapC) and quinolinate phosphoribosyltransferase (nadC) genes on the chromosome (Brynestad et al., 1997). Our investigations support this assertion and, to our knowledge, this is the first study describing a diversity of genetic arrangements of the cpe locus in chromosomal cpe strains (Brynestad et al., 1997; Miyamoto et al., 2002; Li et al., 2010).
The coregenome phylogenomic reconstructions provide interesting epidemiologic information and indicate actual relationships between strains from different outbreaks. FBOs 2987, 2774, 2988, and 2773 were caused by closely related strains with regard to a limited average of pairwise distances between them (i.e., 7.0 SNPs) and common profiles of virulence genes. This similarity could be due to the same biotype that may reach food and cause FBOs. Due to wide biotype and ecological niches, genetically diverse strains of C. perfringens may be found in food matrices. Important epidemiologic findings have resulted from the high resolution of genome sequencing. For instance, WGS was used to demonstrate that many C. difficile cases could not be attributed to transmission from asymptomatic patients, but that there is high diversity of C. difficile in the environment (Tang et al., 2017). This approach could be used in our analysis. We have identified considerable genetic diversity within cpe-negative strains. The high genetic diversity may in part be explained by the broad distribution of this group in the environment. An earlier genomic comparison study conducted on three genomes of C. perfringens (ATCC13124, SM101, and strain 13) revealed considerable genomic diversity with more than 300 unique genomic islands identified (Myers et al., 2006). Our phylogenomic analysis supports this notion with regard to strains 16SBCL582 and 16SBCL584 from FBO 2273. However, a comparison of the virulence gene profiles of these distant strains (obtained by real-time PCR) indicates that they present the same virulence gene profile. This result suggests that the approaches based on the detection of virulence factors are not sufficient to efficiently discriminate C. perfringens strains.
Another interesting finding is that strains are grouped according to the distribution of virulence factors. In particular, the presence of the nanI, pfoA and nagH genes in cpe-negative strains is of interest, and it would be worthwhile to investigate the putative role of these virulence factors in cpe-negative disease.
Conclusion
The aim of our study was to characterize C. perfringens involved in FBOs from the Paris region between 2013 and 2017 by investigating profiles of virulence genes according to the presence or absence of 17 genes encoding 15 virulence factors screened by a new real-time PCR tool. High diversity was obtained between strains in different outbreaks. Heterogeneous profiles of virulence genes within the same outbreaks were highlighted for the first time, indicating that food may be contaminated with different C. perfringens strains that may or may not harbor the cpe gene. Our study emphasizes the importance of more detailed characterization of C. perfringens isolates involved in FBOs than simple enumeration on specific agar, as suggested by the EN ISO 7937 reference method and/or characterization of five random isolates.
We have shown that the reconstructed coregenome phylogenomic history of C. perfringens food isolates can identify two mains groups, both being able to contaminate food. Our research did not clearly associate food sources with a specific clade. However, future comparative genomic studies on strains of various origins and from different geographical areas should provide clues to understand C. perfringens pathogenicity and associations to ecological niches. Genomic studies combined with epidemiological data provide new leads to design toxicity assays that may renew our understanding of the molecular basis of C. perfringens pathogenicity, and ultimately identify new biomarkers to differentiate between pathogenic and non-pathogenic C. perfringens strains. Finally, it would be interesting to characterize C. perfringens strains implicated in the FBO with respect to their reservoir.
Author Contributions
AMA conceived the study, designed the experiments, and wrote the manuscript. NR approved bioinformatics analyses, provided advice for statistical analyses, and revised the manuscript. AMA and KH performed all bioinformatics analyses. MLN contributed to the microbiologic analyses. SaD, PF, and SoD conceived the methodology and performed high throughput real-time PCR analyses. OF, J-AH, and M-YM conceived and piloted the project and revised and approved the manuscript for publication.
Funding
This study was supported by the French Agency for Food, Environmental and Occupational Health and Safety (ANSES). AMA is the recipient of a doctoral scholarship funded by the Islamic Development Bank (IsDB). This article is also part of the European Joint Programme One Health EJP. This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 773830.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the LCSV (Laboratoire Central des Services Vétérinaires) for strain collection, the GAMeR team, and Deborah Merda for advice on bioinformatics analyses. We also like to thank Dr. Hélène Bergis for providing us with positive control strains for PCR, Pierre-Emmanuel Douarre for his valuable advice, and Craig Stevens, MA, ELS for English editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00777/full#supplementary-material
Footnotes
References
Adams, V., Han, X., Lyras, D., and Rood, J. I. (2018). Antibiotic resistance plasmids and mobile genetic elements of Clostridium perfringens. Plasmid 99, 32–39. doi: 10.1016/j.plasmid.2018.07.002
Adams, V., Li, J., Wisniewski, J. A., Uzal, F. A., Moore, R. J., McClane, B. A., et al. (2015). “Virulence plasmids of spore-forming bacteria,” in Plasmids: Biology and Impact in Biotechnology and Discovery, eds M. Tolmasky and J. Carlos (Washington, D.C: ASM Press), 533–557.
Afshari, A., Jamshidi, A., Razmyar, J., and Rad, M. (2016). Genomic diversity of Clostridium perfringens strains isolated from food and human sources. Iran. J. Vet. Res. 17, 160–164.
Albini, S., Brodard, I., Jaussi, A., Wollschläger, N., Frey, J., Miserez, R., et al. (2008). Real-time multiplex PCR assays for reliable detection of Clostridium perfringens toxin genes in animal isolates. Vet. Microbiol. 127, 179–185. doi: 10.1016/j.vetmic.2007.07.024
Anonymous (2005). NF EN ISO 7937, food microbiology-horizontal method for enumeration of C. perfringens. AFNOR V08–019, 17.
Awad, M. M., Ellemor, D. M., Boyd, R. L., Emmins, J. J., and Rood, J. I. (2001). Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 69, 7904–7910. doi: 10.1128/IAI.69.12.7904-7910.2001
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bosi, E., Donati, B., Galardini, M., Brunetti, S., Sagot, M. F., Lió, P., et al. (2015). MeDuSa: a multi-draft based scaffolder. Bioinformatics 31, 2443–2451. doi: 10.1093/bioinformatics/btv171
Brynestad, S., and Granum, P. E. (2002). Clostridium perfringens and foodborne infections. Int. J. Food Microbiol. 74, 195–202. doi: 10.1016/S0168-1605(01)00680-8
Brynestad, S., Synstad, B., and Granum, P. E. (1997). The Clostridium perfringens enterotoxin gene is on a transposable element in type A human food poisoning strains. Microbiology 143, 2109–2115. doi: 10.1099/00221287-143-7-2109
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner. Available at: https://escholarship.org/uc/item/1h3515gn (accessed May 11, 2018).
Chalmers, G., Bruce, H. L., Hunter, D. B., Parreira, V. R., Kulkarni, R. R., Jiang, Y. F., et al. (2008a). Multilocus sequence typing analysis of Clostridium perfringens isolates from necrotic enteritis outbreaks in broiler chicken populations. J. Clin. Microbiol. 46, 3957–3964. doi: 10.1128/JCM.01548-08 (accessed May 11, 2018).
Chalmers, G., Martin, S. W., Prescott, J. F., and Boerlin, P. (2008b). Typing of Clostridium perfringens by multiple-locus variable number of tandem repeats analysis. Vet. Microbiol. 128, 126–135. doi: 10.1016/j.vetmic.2007.09.018
Chon, J.-W., Park, J.-S., Hyeon, J.-Y., Park, C., Song, K.-Y., Hong, K.-W., et al. (2012). Development of real-time PCR for the detection of Clostridium perfringens in meats and vegetables. J. Microbiol. Biotechnol. 22, 530–534. doi: 10.4014/jmb.1107.07064
Cock, P. J., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Deguchi, A., Miyamoto, K., Kuwahara, T., Miki, Y., Kaneko, I., Li, J., et al. (2009). Genetic characterization of type A enterotoxigenic Clostridium perfringens Strains. PLoS One 4:e5598. doi: 10.1371/journal.pone.0005598
EFSA (2016). Scientific report of european food safety autority. Summary report on trends and sources of zoonoses, zoonotic agents and foodborne outbreaks. EFSA J. 14, 4634. doi: 10.2903/j.efsa.2016.4634
Felten, A., Vila Nova, M., Durimel, K., Guillier, L., Mistou, M. Y., and Radomski, N. (2017). First gene-ontology enrichment analysis based on bacterial coregenome variants: insights into adaptations of Salmonella serovars to mammalian- and avian-hosts. BMC Microbiol. 17:222. doi: 10.1186/s12866-017-1132-1
Fohler, S., Klein, G., Hoedemaker, M., Scheu, T., Seyboldt, C., Campe, A., et al. (2016). Diversity of Clostridium perfringens toxin-genotypes from dairy farms. BMC Microbiol. 16:199. doi: 10.1186/s12866-016-0812-6
Freedman, J. C., Shrestha, A., and McClane, B. A. (2016). Clostridium perfringens enterotoxin: action, genetics, and translational applications. Toxins 8:E73. doi: 10.3390/toxins8030073
Gao, Z., and McClane, B. A. (2012). Use of Clostridium perfringens enterotoxin and the enterotoxin receptor-binding domain (C-CPE) for cancer treatment: opportunities and challenges. J. Toxicol. 2012:981626. doi: 10.1155/2012/981626
Goossens, E., Valgaeren, B. R., Pardon, B., Haesebrouck, F., Ducatelle, R., Deprez, P. R., et al. (2017). Rethinking the role of alpha toxin in Clostridium perfringens-associated enteric diseases: a review on bovine necro-haemorrhagic enteritis. Vet. Res. 48:9. doi: 10.1186/s13567-017-0413-x
Grass, J. E., Gould, L. H., and Mahon, B. E. (2013). Epidemiology of foodborne disease outbreaks caused by Clostridium perfringens, United States, 1998–2010. Foodborne Pathog. Dis. 10, 131–136. doi: 10.1089/fpd.2012.1316
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hassan, K. A., Elbourne, L. D. H., Tetu, S. G., Melville, S. B., Rood, J. I., and Paulsen, I. T. (2015). Genomic analyses of Clostridium perfringens isolates from five toxinotypes. Res. Microbiol. 166, 255–263. doi: 10.1016/j.resmic.2014.10.003
Hibberd, M. C., Neumann, A. P., Rehberger, T. G., and Siragusa, G. R. (2011). Multilocus sequence typing subtypes of poultry Clostridium perfringens isolates demonstrate disease niche partitioning. J. Clin. Microbiol. 49, 1556–1567. doi: 10.1128/JCM.01884-10
Johansson, A., Aspan, A., Bagge, E., Båverud, V., Engström, B. E., and Johansson, K. E. (2006). Genetic diversity of Clostridium perfringens type A isolates from animals, food poisoning outbreaks and sludge. BMC Microbiol. 6:47. doi: 10.1186/1471-2180-6-47
Jost, B. H., Trinh, H. T., and Songer, J. G. (2006). Clonal relationships among Clostridium perfringens of porcine origin as determined by multilocus sequence typing. Vet. Microbiol. 116, 158–165. doi: 10.1016/j.vetmic.2006.03.025
Keyburn, A. L., Boyce, J. D., Vaz, P., Bannam, T. L., Ford, M. E., Parker, D., et al. (2008). NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 4:e26. doi: 10.1371/journal.ppat.0040026
Kim, Y. B., Kim, J. Y., Song, H. S., Lee, C., Kwon, J., Kang, J., et al. (2017). Complete genome sequence of Clostridium perfringens CBA7123 isolated from a faecal sample from Korea. Gut Pathog. 9:32. doi: 10.1186/s13099-017-0181-1
Kiu, R., Caim, S., Alexander, S., Pachori, P., and Hall, L. J. (2017). Probing genomic aspects of the multi-host pathogen Clostridium perfringens reveals significant pangenome diversity, and a diverse array of virulence factors. Front. Microbiol. 8:2485. doi: 10.3389/fmicb.2017.02485
Kiu, R., and Hall, L. J. (2018). An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 7:141. doi: 10.1038/s41426-018-0144-8
Kosugi, S., Hirakawa, H., and Tabata, S. (2015). GMcloser: closing gaps in assemblies accurately with a likelihood-based selection of contig or long-read alignments. Bioinformatics 31, 3733–3741. doi: 10.1093/bioinformatics/btv465
Lacey, J. A., Allnutt, T. R., Vezina, B., Van, T. T. H., Stent, T., Han, X., et al. (2018). Whole genome analysis reveals the diversity and evolutionary relationships between necrotic enteritis-causing strains of Clostridium perfringens. BMC Genom. 19:379. doi: 10.1186/s12864-018-4771-1
Law, J. W.-F., Ab Mutalib, N.-S., Chan, K.-G., and Lee, L. H. (2015). Rapid methods for the detection of foodborne bacterial pathogens: principles, applications, advantages and limitations. Front. Microbiol. 5:770. doi: 10.3389/fmicb.2014.00770
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, J., Adams, V., Bannam, T. L., Miyamoto, K., Garcia, J. P., Uzal, F. A., et al. (2013). Toxin plasmids of Clostridium perfringens. Microbiol. Mol. Biol. Rev. 77, 208–233. doi: 10.1128/MMBR.00062-12
Li, J., Miyamoto, K., Sayeed, S., and McClane, B. A. (2010). Organization of the cpe locus in CPE-Positive Clostridium perfringens Type C and D isolates. PLoS One 5:e10932. doi: 10.1371/journal.pone.0010932
Li, J., Paredes-Sabja, D., Sarker, M. R., and McClane, B. A. (2016). Clostridium perfringens sporulation and sporulation-associated toxin production. Microbiol. Spectr. 4, 1–27. doi: 10.1128/microbiolspec.TBS-0022-2015
Lin, Y.-T., and Labbe, R. (2003). Enterotoxigenicity and genetic relatedness of Clostridium perfringens isolates from retail foods in the United States. Appl. Environ. Microbiol. 69, 1642–1646. doi: 10.1128/AEM.69.3.1642-1646.2003
Lindström, M., Heikinheimo, A., Lahti, P., and Korkeala, H. (2011). Novel insights into the epidemiology of Clostridium perfringens type A food poisoning. Food Microbiol. 28, 192–198. doi: 10.1016/j.fm.2010.03.020
Lukinmaa, S., Takkunen, E., and Siitonen, A. (2002). Molecular epidemiology of Clostridium perfringens related to food-borne outbreaks of disease in Finland from 1984 to 1999. Appl. Environ. Microbiol. 68, 3744–3749. doi: 10.1128/AEM.68.8.3744-3749.2002
McClane, B. A., Robertson, S. L., and Li, J. (2013). “Clostridium perfringens,” in Food Microbiology, eds M. Doyle and R. Buchanan (Washington, DC: ASM Press), 465–489. doi: 10.1128/9781555818463.ch18
Mehdizadeh Gohari, I., Kropinski, A. M., Weese, S. J., Whitehead, A. E., Parreira, V. R., Boerlin, P., et al. (2017). NetF-producing Clostridium perfringens: clonality and plasmid pathogenicity loci analysis. Infect. Genet. Evol. 49, 32–38. doi: 10.1016/j.meegid.2016.12.028
Mehdizadeh Gohari, I., Parreira, V. R., Nowell, V. J., Nicholson, V. M., Oliphant, K., and Prescott, J. F. (2015). A Novel pore-forming toxin in type A Clostridium perfringens is associated with both fatal canine hemorrhagic gastroenteritis and fatal foal necrotizing enterocolitis. PLoS One 10:e0122684. doi: 10.1371/journal.pone.0122684
Miyamoto, K., Chakrabarti, G., Morino, Y., and McClane, B. A. (2002). Organization of the plasmid cpe Locus in Clostridium perfringens type A isolates. Infect. Immun. 70, 4261–4272. doi: 10.1128/IAI.70.8.4261-4272.2002
Myers, G. S. A., Rasko, D. A., Cheung, J. K., Ravel, J., Seshadri, R., DeBoy, R. T., et al. (2006). Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 16, 1031–1040. doi: 10.1101/gr.5238106
Myers, L., and Sirois, M. J. (2004). Spearman correlation coefficients, differences between. Encycl. Stat. Sci. 2, 1–11. doi: 10.1002/0471667196.ess5050.pub2
Obana, N., and Nakamura, K. (2011). A novel toxin regulator, the CPE1446-CPE1447 protein heteromeric complex, controls toxin genes in Clostridium perfringens. J. Bacteriol. 193, 4417–4424. doi: 10.1128/JB.00262-11
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Pattengale, N. D., Alipour, M., Bininda-Emonds, O. R., Moret, B. M., and Stamatakis, A. (2010). How many bootstrap replicates are necessary. J. Comput. Biol. 17, 337–354. doi: 10.1089/cmb.2009.0179
Revitt-Mills, S. A., Rood, J. I., and Adams, V. (2015). Clostridium perfringens extracellular toxins and enzymes: 20 and counting. Microbiol. Aust. 36, 114–117. doi: 10.1071/MA15039
Rey, D., and Neuhäuser, M. (2011). Wilcoxon-Signed-Rank Test, in: International Encyclopedia of Statistical Science. Berlin: Springer, 1658–1659. doi: 10.1007/978-3-642-04898-2_61
Rood, J. I., Adams, V., Lacey, J., Lyras, D., McClane, B. A., Melville, S. B., et al. (2018). Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 53, 5–10. doi: 10.1016/j.anaerobe.2018.04.011
Sandmann, S., De Graaf, A. O., Karimi, M., Van Der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating variant calling tools for non-matched next-generation sequencing data. Sci. Rep. 7:43169. doi: 10.1038/srep43169
Santé Publique France (2016). Surveillance des Toxi-Infections Alimentaires Collectives. Données de la Déclaration Obligatoire. Saint-Maurice: Santé Publique France, 11.
Sarker, M. R., Carman, R. J., and McClane, B. A. (2000a). Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 35, 249–249. doi: 10.1046/j.1365-2958.1999.01534.x
Sarker, M. R., Shivers, R. P., Sparks, S. G., Juneja, V. K., and McClane, B. A. (2000b). Comparative experiments to examine the effects of heating on vegetative cells and spores of Clostridium perfringens isolates carrying plasmid genes versus chromosomal enterotoxin genes. Appl. Environ. Microbiol. 66, 3234–3240. doi: 10.1128/AEM.66.8.3234-3240.2000
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M.-A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.P11101
Shimizu, T., Ohtani, K., Hirakawa, H., Ohshima, K., Yamashita, A., Shiba, T., et al. (2002). Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. U.S.A. 99, 996–1001. doi: 10.1073/pnas.022493799
Stamatakis, A. (2015). Using RAxML to infer phylogenies. Curr. Protoc. Bioinform. 51, 6.14.1–6.14.14. doi: 10.1002/0471250953.bi0614s51
Talukdar, P. K., Udompijitkul, P., Hossain, A., and Sarker, M. R. (2016). Inactivation strategies for Clostridium perfringens spores and vegetative cells. Appl. Environ. Microbiol. 83, e02731-16. doi: 10.1128/AEM.02731-16
Tang, P., Croxen, M. A., Hasan, M. R., Hsiao, W. W. L., and Hoang, L. M. (2017). Infection control in the new age of genomic epidemiology. Am. J. Infect. Control 45, 170–179. doi: 10.1016/j.ajic.2016.05.015
Tewari, A., and Abdullah, S. (2015). Bacillus cereus food poisoning: international and Indian perspective. J. Food Sci. Technol. 52, 2500–2511. doi: 10.1007/s13197-014-1344-4
Uzal, F. A., Freedman, J. C., Shrestha, A., Theoret, J. R., Garcia, J., Awad, M. M., et al. (2014). Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 9, 361–377. doi: 10.2217/fmb.13.168
Uzal, F. A., McClane, B. A., Cheung, J. K., Theoret, J., Garcia, J. P., Moore, R. J., et al. (2015). Animal models to study the pathogenesis of human and animal Clostridium perfringens infections. Vet. Microbiol. 179, 23–33. doi: 10.1016/j.vetmic.2015.02.013
Vaillant, V., Valk, H. D., Baron, E., Ancelle, T., Colin, P., Delmas, M. C., et al. (2005). Foodborne infections in France. Foodbourne Pathog. Dis. 2, 221–232. doi: 10.1089/fpd.2005.2.221
Veshnyakova, A., Piontek, J., Protze, J., Waziri, N., Heise, I., and Krause, G. (2012). Mechanism of Clostridium perfringens enterotoxin interaction with claudin-3/-4 protein suggests structural modifications of the toxin to target specific claudins. J. Biol. Chem. 287, 1698–1708. doi: 10.1074/jbc.M111.312165
Wen, Q., and McClane, B. A. (2004). Detection of enterotoxigenic Clostridium perfringens type A isolates in American retail foods. Appl. Environ. Microbiol. 70, 2685–2691. doi: 10.1128/AEM.70.5.2685-2691.2004
Xiao, Y. (2014). Clostridium perfringens Sporulation, Germination and Outgrowth in Food: A functional Genomics Approach. Ph. D. Thesis. Wageningen: Wageningen University.
Xiao, Y., Wagendorp, A., Moezelaar, R., Abee, T., and Wells-Bennik, M. H. (2012). A wide variety of Clostridium perfringens type A food-borne isolates that carry a chromosomal cpe gene belong to one multilocus sequence typing cluster. Appl. Environ. Microbiol. 78, 7060–7068. doi: 10.1128/AEM.01486-12
Xu, S., Ackerman, M. S., Long, H., Bright, L., Spitze, K., Ramsdell, J. S., et al. (2015). A male-specific genetic map of the Microcrustacean Daphnia pulex based on single-sperm whole-genome sequencing. Genetics 201, 31–38. doi: 10.1534/genetics.115.179028
Yasugi, M., Sugahara, Y., Hoshi, H., Kondo, K., Talukdar, P. K., Sarker, M. R., et al. (2015). In vitro cytotoxicity induced by Clostridium perfringens isolate carrying a chromosomal cpe gene is exclusively dependent on sporulation and enterotoxin production. Microb. Pathog. 85, 1–10. doi: 10.1016/j.micpath.2015.04.003
Keywords: foodborne outbreak, Clostridium perfringens enterotoxin, virulence gene profiles, virulence factors, real-time PCR, whole genome sequencing, coregenome SNP
Citation: Mahamat Abdelrahim A, Radomski N, Delannoy S, Djellal S, Le Négrate M, Hadjab K, Fach P, Hennekinne J-A, Mistou M-Y and Firmesse O (2019) Large-Scale Genomic Analyses and Toxinotyping of Clostridium perfringens Implicated in Foodborne Outbreaks in France. Front. Microbiol. 10:777. doi: 10.3389/fmicb.2019.00777
Received: 30 July 2018; Accepted: 26 March 2019;
Published: 17 April 2019.
Edited by:
Panagiotis Skandamis, Agricultural University of Athens, GreeceReviewed by:
Jihong Li, University of Pittsburgh, United StatesGeorges Daube, University of Liège, Belgium
Copyright © 2019 Mahamat Abdelrahim, Radomski, Delannoy, Djellal, Le Négrate, Hadjab, Fach, Hennekinne, Mistou and Firmesse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Firmesse, olivier.firmesse@anses.fr