 David R. Greig
David R. Greig Amy F. W. Mikhail
Amy F. W. Mikhail Timothy J. Dallman
Timothy J. Dallman Claire Jenkins
Claire Jenkins- 1National Infection Service, Public Health England, London, United Kingdom
- 2Division of Infection and Immunity, The Royal (Dick) School of Veterinary Studies, The Roslin Institute, The University of Edinburgh, Easter Bush, United Kingdom
In December 2015, six cases of Shiga toxin (Stx)-producing Escherichia coli (STEC) O157:H7 stx2a/stx2c phage type (PT) 24 were identified by the national gastrointestinal disease surveillance system at Public Health England (PHE). Frozen grated coconut imported from India was implicated as the vehicle of infection. Short and long read sequencing data were interrogated for genomic markers to provide evidence that the outbreak strain was from an imported source. The outbreak strain belonged to a sub-lineage (IIa) rare in domestically acquired infection in the United Kingdom, and indicative of an imported strain. Phylogenetic analysis identified the most closely related isolates to the outbreak strain were from cases reporting recent travel not to India, but to Uganda. Phylo-geographical signals based on travel data may be confounded by the failure of local and/or global monitoring systems to capture the full diversity of strains in a given country. This may be due to low prevalence strains circulating in-country under the surveillance radar, or a recent importation event involving the migration of animals and/or people. Comparison of stx2a-encoding prophage harbored by the outbreak strain with publicly available stx2a-encoding prophage sequences revealed that it was most closely related to stx2a-encoding prophage acquired by STEC O157:H7 that caused the first outbreak of STEC-hemolytic uremic syndrome (HUS) in England in 1982–83. Animal and people migration events may facilitate the transfer of stx2a-encoding prophage from indigenous STEC O157:H7 to recently imported strains, or vice versa. Monitoring the global transmission of STEC O157:H7 and tracking the exchange of stx2a-encoding phage between imported and indigenous strains may provide an early warning of emerging threats to public health.
Introduction
Outbreaks of foodborne, gastrointestinal disease caused by Shiga toxin (Stx)-producing Escherichia coli (STEC) serotype O157:H7 are regarded as a significant threat to public health (Riley et al., 1983; Michino et al., 1999; Cowley et al., 2016; Launders et al., 2016a; Gobin et al., 2018). A subset of vulnerable patients at the extremes of age are at risk of developing hemolytic uremic syndrome (HUS), a condition characterized by renal failure, and cardiac and neurological complications that can be fatal (Tarr et al., 2005; Launders et al., 2016a). STEC O157:H7 is zoonotic and can be transmitted to humans via direct contact with animals and/or their environment, contaminated food or water, or person-to-person spread (Byrne et al., 2015).
There are three main lineages of STEC O157:H7 (I, II, and I/II) and eight sub-lineages (Ia, Ib, Ic, IIa, IIb, IIc, I/IIa, and I/IIb) (Dallman et al., 2015a). With respect to the clade typing scheme proposed by Iyoda et al. (2014), as described previously, lineage I corresponded to clades 1 through 6, lineage II corresponded to clade 7, and lineage I/II corresponded to clade 8 (Eppinger et al., 2011; Dallman et al., 2015a). The majority of human cases and outbreaks of STEC O157:H7 in the United Kingdom are caused by sub-lineages Ic, I/IIa, and IIc (Dallman et al., 2015a). The sub-lineages that are infrequently isolated from human cases in the United Kingdom are more likely to be associated with returning travelers or outbreaks associated with contaminated food imported to the United Kingdom from elsewhere (Gobin et al., 2018).
The defining pathogenicity factor for STEC O157:H7 is the presence of Stx-producing genes (stx). There are two types of Stx, Stx1 and 2, and at least 10 subtypes (1a–1d and 2a–2i) (Scheutz et al., 2012). The genes encoding the stx subtypes are located on mobile genetic elements (bacteriophages) that may be acquired by STEC and integrated into the genome. STEC O157:H7 harboring the stx2a-encoded phage are most commonly associated with causing HUS (Persson et al., 2007; Byrne et al., 2020).
In December 2015, six cases of STEC O157:H7 phage type (PT) 24 were identified by the national gastrointestinal disease surveillance system at Public Health England (PHE). Following the analysis of the trawling questionnaires, a specific brand of imported frozen grated coconut was implicated as the vehicle of infection. In this study, we conducted an epidemiological investigation of the outbreak, and analyzed short and long read sequencing data to look for genomic markers to provide further evidence that the outbreak strain was from a non-domestic (non-UK) source.
Materials and Methods
Microbiological and Epidemiological Data Collection
In England, all fecal specimens from hospital and community cases of gastrointestinal disease submitted to local hospital laboratories are tested for E. coli O157:H7. All isolates are submitted to the Gastrointestinal Bacterial Reference Unit (GBRU) at PHE for identification and phage typing. Since July 2015, all isolates have been whole genome sequenced for routine surveillance (National Center for Biotechnology Information Short Read Archive BioProject PRJNA315192).
The majority of isolates of STEC O157:H7 included in this study were submitted to GBRU between July 2015 (when WGS was first implemented) and December 2016 (12 months after the outbreak occurred). During this time frame, 1103 isolates were submitted to GBRU; 149 belonged to household outbreak, 283 were linked to known outbreaks, and 671 were identified as sporadic cases. Additional isolates sequenced from the archive collection submitted to GBRU between 2006 and 2016, included eight isolates belonging to the same PT as the outbreak strain, PT24, all of which reported recent (within 7 days of onset of symptoms) travel to Uganda, and all cases reporting recent travel to the Indian Sub-Continent (ISC; n = 56) (Supplementary Table S1).
Epidemiological Data Collection
In January 2009, PHE implemented the National Enhanced Surveillance System for STEC (NESSS) in England. This system has been described in detail previously (Byrne et al., 2015). Briefly, it captures standardized epidemiological on all cases of STEC reported in England through an Enhanced Surveillance Questionnaire (ESQ) including detailed demographic, clinical, and exposure data which is reconciled with microbiological data in NESSS. Following analysis of the ESQ data, each case was re-interview using a standardized trawling questionnaire.
Short Read Sequencing on the Illumina HiSeq 2500
Genomic DNA was extracted from cultures of STEC O157:H7 using the Qiagen Qiasymphony (Qiagen, Hilden, Germany). The sequencing library was prepared using the Nextera XP kit (Illumina, San Diego, CA, United States) for sequencing on the Illumina HiSeq 2500 (Illumina, San Diego, CA, United States) instrument run with the fast protocol. High quality trimmed (leading and trailing trimming at < Q30 using Timmomatic v0.27 (Bolger et al., 2014). Illumina FASTQ reads were mapped to the Sakai STEC O157 reference genome (NC 002695.1) using BWA MEM v0.7.13 (Li and Durbin, 2010) and Samtools v (Li et al., 2009). Variant positions were identified by GATK v2.6.5 UnifiedGenotyper (McKenna et al., 2010) that passed the following parameters: > 90% consensus, minimum read depth of 10, Mapping Quality (MQ) ≥ 30. Any variants called at positions that were within the known prophages in Sakai were masked from further analyses. The remaining variants were imported into SnapperDB v0.2.5 (Dallman et al., 2018).
Hierarchical single linkage clustering was performed on the pairwise single nucleotide polymorphisms (SNPs) difference between all strains at various distance thresholds (250, 100, 50, 25, 10, 5, 0). The result of the clustering is an SNP profile, or SNP address, that can be used to describe the population structure based on clonal groups (Dallman et al., 2015b, 2018). Although isolates greater than 5 SNPs apart are unlikely to be part of the same temporally linked outbreak, deeper phylogenetic relationships within the 10 or 25 SNP clusters may provide epidemiologically useful information or associations. Lineage and sub-lineage assignment were performed based on discriminatory SNPs, extracted directly from SnapperDB v0.2.5, that define the population structure, as described previously (Dallman et al., 2015b, 2018).
Long Read Sequencing Using ONT and Data Processing
Genomic DNA was extracted and purified using the Qiagen Genomic Tip, midi 100/G (Qiagen, Hilden, Germany) with minor alterations including no vigorous mixing steps (performed by inversion) and elution into 100 μl. DNA was quantified using a Qubit and the high sensitivity (HS) dsDNA Assay Kit (Thermofisher Scientific, Waltham, MA, United States) to manufacturer’s instructions. Library preparation was performed using the Native Barcoding kit (SQK-LSK108 and EXP-NBD103) (Oxford Nanopore Technologies, Oxford, United Kingdom). The prepared library was loaded on a FLO-MIN106 R9.4.1 flow cell (Oxford Nanopore Technologies, Oxford, United Kingdom) and sequenced using the MinION for 24 h.
Data produced in a raw FAST5 format were basecalled into FASTQ format and de-multiplexed using Guppy v2.3.5 (Oxford Nanopore Technologies). The reads were then de-multiplexed again using Deepbinner v0.2.0 (Wick et al., 2018) to reduce barcode contamination. Run metrics were generated using Nanoplot v1.8.1 (De Coster et al., 2018). The barcode and y-adapter from each sample’s reads were trimmed, and chimeric reads split using Porechop v0.2.4. Finally, trimmed reads were filtered using Filtlong v0.1.1 with the following parameters; min_length = 1000, keep_percent = 90, and target_bases = 275Mb, to generate approximately 50x coverage of the STEC genome with the longest and highest quality reads.
De novo Assembly, Polishing, Reorientation, and Annotation
Trimmed ONT FASTQ files were assembled using Canu v1.7.1 (Koren et al., 2017). Polishing of the assembly was performed in a three-step process first, using Nanopolish v0.11.1 (Loman et al., 2015) using both the trimmed ONT FASTQs and FAST5s for each respective sample accounting for methylation using the –methylation-aware = dam, dcm and –min-candidate-frequency = 0.1. Secondly, Pilon v1.22 (Walker et al., 2014) using Illumina FASTQ reads as the query dataset with the use of BWA v0.7.17 (Li and Durbin, 2010) and Samtools v1.7 (Li et al., 2009). Finally, Racon v1.2.1 (Vaser et al., 2017) also using BWA v0.7.17 and Samtools v1.7 (Li et al., 2009) was used with the Illumina reads to produce a final assembly for each of the samples. As the chromosome from the assembly was closed, it was re-orientated to start at the dnaA gene (NC_000913) from E. coli K-12, using the –fixstart parameter in circlator v1.5.5 (Hunt et al., 2015). Prokka v1.13 was used to annotate the draft assembly (Seemann, 2014).
Prophage Detection, Excision, and Processing
Prophages across both samples were detected and extracted using the Phage Search Tool (PHASTER) (Arndt et al., 2016). Prophage extraction from the genome occurred regardless of prophage size or quality and any detected prophages separated by less than 4 kbp were conjoined into a single phage using Propi v0.9.0 as described in Shaaban et al. (2016). From here the prophages were manually trimmed to remove any non-prophage genes and were again annotated using Prokka v 1.13 with the use of a personalized database (Amino Acid multi-FASTA) containing known STEC prophage genes was used to annotate the final assemblies. Database publicly available from https://github.com/gingerdave269/prophage_DB. The output GenBank (gbk) files were modified to color genes by function.
Mash and Phylogeny
Mash v2.2 (Ondov et al., 2016) was used to sketch (sketch length 1000, kmer length, 21) stx-encoding prophages from the outbreak isolate sequenced using ONT in this study, and the publicly available STEC genomes listed in Table 4. The pairwise Jaccard distance between the prophages was calculated and a neighbor joining tree computed and visualized using FigTree v1.4.4.
Visualization Tools
All gene diagrams were constructed using Easyfig v2.2.3 (Sullivan et al., 2011). Neighbor joining trees were visualized and annotated using FigTree v1.4.4.
Data Deposition
Illumina FASTQ files for all samples used in the study can be found under BioProject PRJNA315192. Nanopore FASTQ file is available under SRA accession: SRR10177137. The assembly can be found under the following accession: CP044350. All the above are available from BioProject: PRJNA315192.
Results and Discussion
Epidemiological Investigations
In December 2015, the Gastrointestinal Bacteria Reference Unit identified six cases of STEC O157:H7 with a rare PT, PT24. Whole genome sequencing results confirmed that the cases belong to the same 5-SNP single linkage cluster. The outbreak strain belonged to sub-lineage IIa and had the stx profile stx2a and stx2c.
An outbreak control team was convened, and trawling questionnaires were completed for each case and their symptomatic household contacts. Of the six cases, two were male and four were female, and the age range for all six cases was 1–45 years old. Dates of onset of symptoms of gastrointestinal infection fell between 17 and 25 November 2015. The cases were national distributed. Analysis of the trawling questionnaires identified consumption of frozen grated coconut belonging to the same brand was reported by four of the six cases. Of the remaining two cases, one patient recalled eating coconut yogurt, while the other reported the consumption of Indian sweets and attended a Diwali party in the days before onset of symptoms but did not recall specifically eating coconut.
Microbiological analysis of an unopened packet of the coconut product from the restaurant where two of the cases ate, detected unsatisfactory levels of E. coli (>102 cfu/g), as defined by Regulation (EC) No. 178/200211, although no STEC O157:H7 was detected in the sample.
Phylogenetic Analysis of WGS Data Linked to Patients Travel History to Determine a Domestic or Non-domestic Origin for the Outbreak Strain
Following the epidemiological investigation that provided evidence that the contaminated food vehicle may be imported, we analyzed the genome data to look for evidence that the outbreak strain had a non-domestic origin.
Of the 671 sporadic isolates of STEC O157:H7 referred to GBRU between July 2015 and December 2016, the majority belonged to sub-lineages IIc (246/671, 36.7%) and Ic (183/671, 27.2%) (Table 1). In contrast, sub-lineage IIa was less frequently isolated from sporadic cases (98/671, 14.6%), and showed the highest level of sub-lineage diversity, as measured by the number of different 250-SNP single linkage clusters within each sub-lineage (Table 1). High levels of sub-lineage diversity are representative of infrequent sampling from a geographically dispersed reservoir (Gobin et al., 2018).
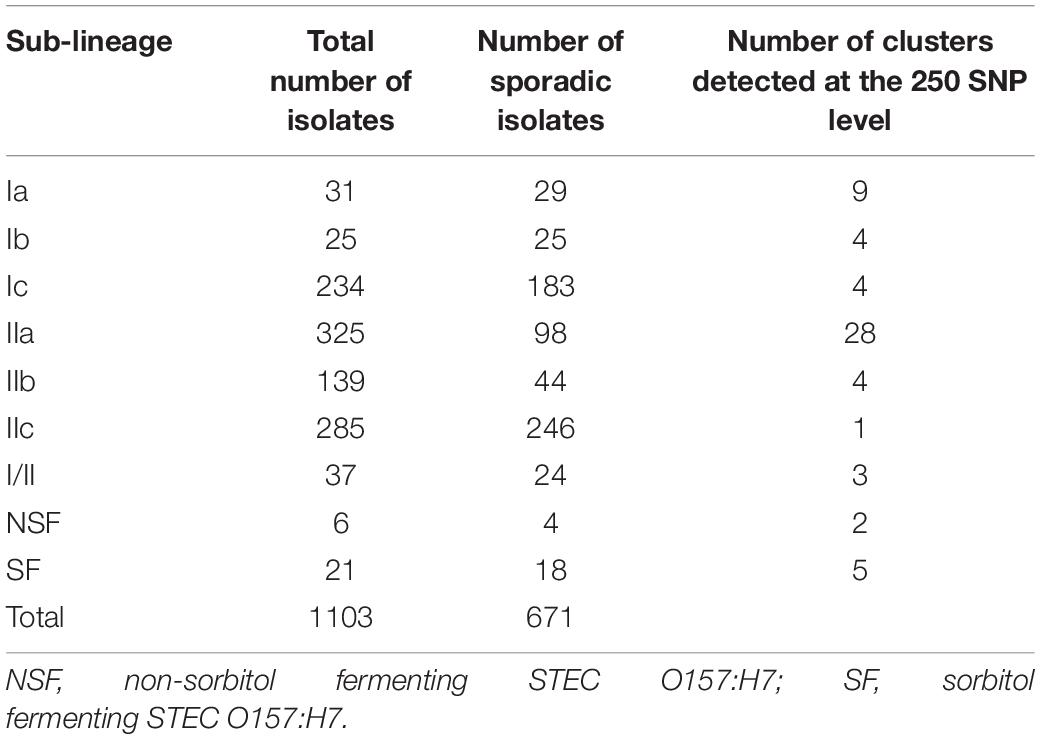
Table 1. Diversity and frequency of sampling within each sub-lineage 2015–2016.
The phylogeny of the 671 sporadic isolates included in this study were visualized using a sunburst diagram showing the distribution of isolates belonging to lineage and sub-lineage, and six descending single linkage SNP clusters at the 250 SNPs, 100 SNPs, 50 SNPs, 25 SNPs, 10 SNPs, and 5 SNPs level (Figure 1) (Dallman et al., 2018). In Figure 1, the inner circle shows the proportions of the three main lineages and the proportion of those isolates that fall outside the lineage structure. Moving outward from the inner circle, the second concentric circle shows the proportion of isolates in each sub-lineage, and the third, fourth, fifth, sixth, and seventh concentric circles from the inner circle represent the 250 SNPs, 100 SNPs, 50 SNPs, 25 SNPs, 10 SNPs, and 5 SNPs levels, respectively. The numbers represent SNP type or SNP address designation, for example, the SNP address designation for the largest 25 SNP single linkage cluster in sub-lineage IIa is t25: 5.156.490.925%.
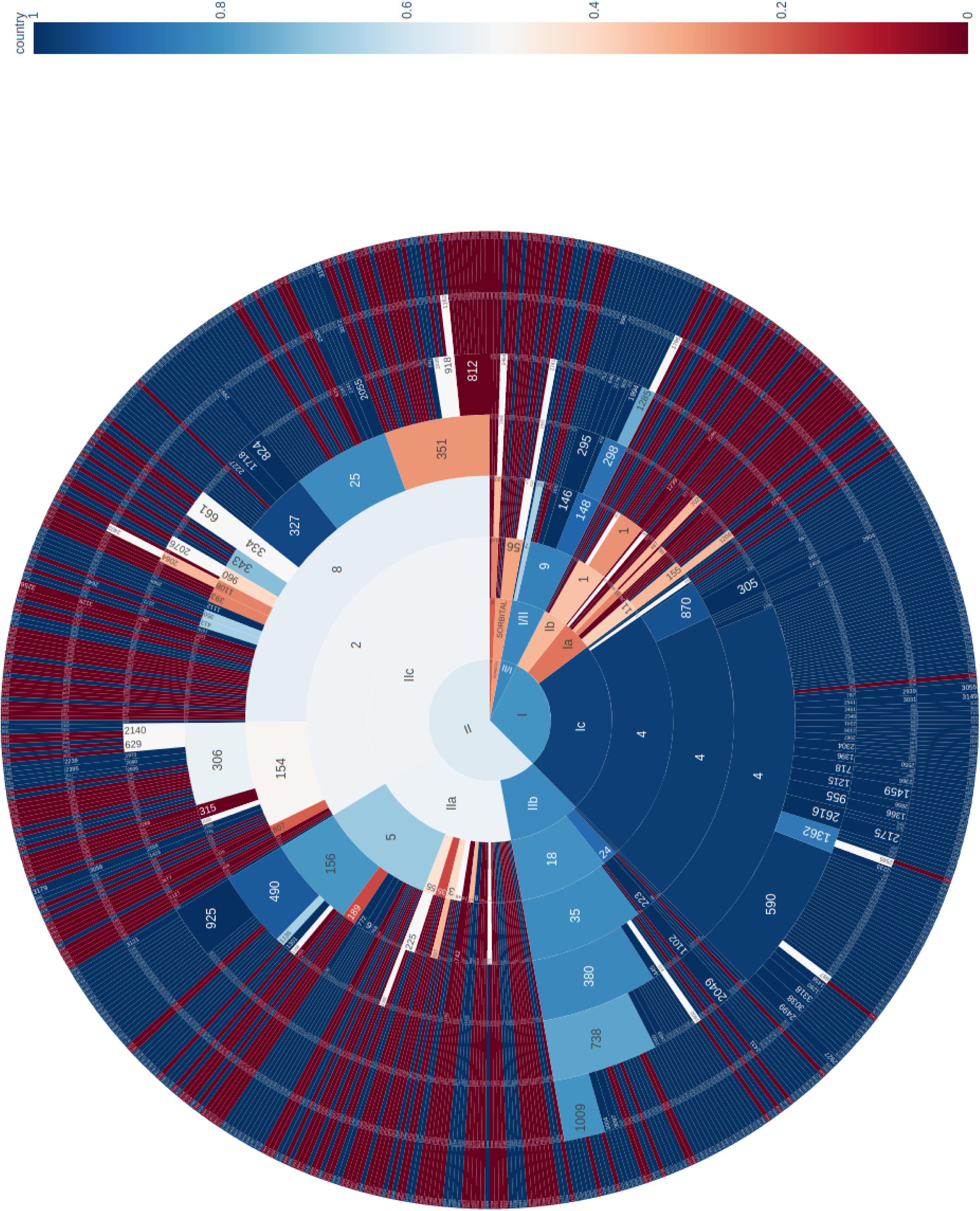
Figure 1. Sunburst diagram showing the distribution of isolates belonging to lineage and sub-lineage, and six descending concentric circles represent single linkage SNP clusters at the 250 SNP, 100 SNP, 50 SNP, 25 SNP, 10 SNP, and 5 SNP levels. The segments were colored based on the proportion of isolates from cases reporting foreign travel with 7 days of onset of symptoms, with dark blue being no cases report recent travel and dark red being all cases report recent travel outside the United Kingdom. For example, within sub-lineage IIa, with the exception of one 50-SNP level cluster (t50: 5.156.490) that was almost exclusively domestically acquired, the majority of the remaining clusters were travel associated.
The segments were colored based on the proportion of isolates from cases reporting foreign travel with 7 days of onset of symptoms, with dark blue being no cases report recent travel and dark red being all cases report recent travel outside the United Kingdom. The majority of clusters that belong to sub-lineages Ic, IIb, and I/II were domestically acquired. These data were consistent with previous studies that indicated these sub-lineages were likely to be endemic in the United Kingdom (Dallman et al., 2015a; Adams et al., 2016; Byrne et al., 2018). Sub-lineages IIa and IIc displayed a mixture of domestically acquired and travel associated clusters (Figure 1). However, within each sub-lineage, at more discriminatory SNP levels, clear cluster associations to either travel or domestic-acquisition were observed. For example, within sub-lineage IIa, with the exception of one 50-SNP level cluster (t50: 5.156.490.) that was almost exclusively domestically acquired, the majority of the remaining clusters were travel associated. The outbreak strain described in this study fell within the travel-associated clusters providing evidence that the contaminated food vehicle was most likely from an imported source.
Analysis of WGS Data of Isolates From UK Residents Reporting Recent Travel to the Indian Sub-Continent
A closer look at the phylogenetic context of the outbreak strain revealed the most closely related strains were isolates from cases reporting recent travel to Uganda (Figure 2). As the epidemiological investigation indicated that the country of origin of the outbreak strains was India, we included sequences of all the isolates of STEC O157:H7 in the PHE database isolated from travelers recently returned to the United Kingdom from the ISC (Figure 2).

Figure 2. Maximum likelihood phylogenetic tree of strains belonging to sub-lineage IIa, most closely related to the outbreak cluster, showing SRA accession, stx type profile, and travel history where available. *denotes Oxford Nanopore Technology sequenced sample 194195.
Interrogation of the PHE database of all isolates submitted to GBRU between 2006 and 2016 (n = 11,339), identified 56 isolates from UK travelers recently returned from the ISC, the majority reporting recent travel to India (n = 32) or Pakistan (n = 16) (Table 2). The majority of isolates from the ISC belonged to sub-lineage IIa (53/56), and all had stx2c only (n = 56). Evidence from studies testing samples from humans, food, and animals in India indicates that the prevalence of STEC O157:H7 is low in this country (Sehgal et al., 2008; Lanjewar et al., 2010; Shrivastava et al., 2017).
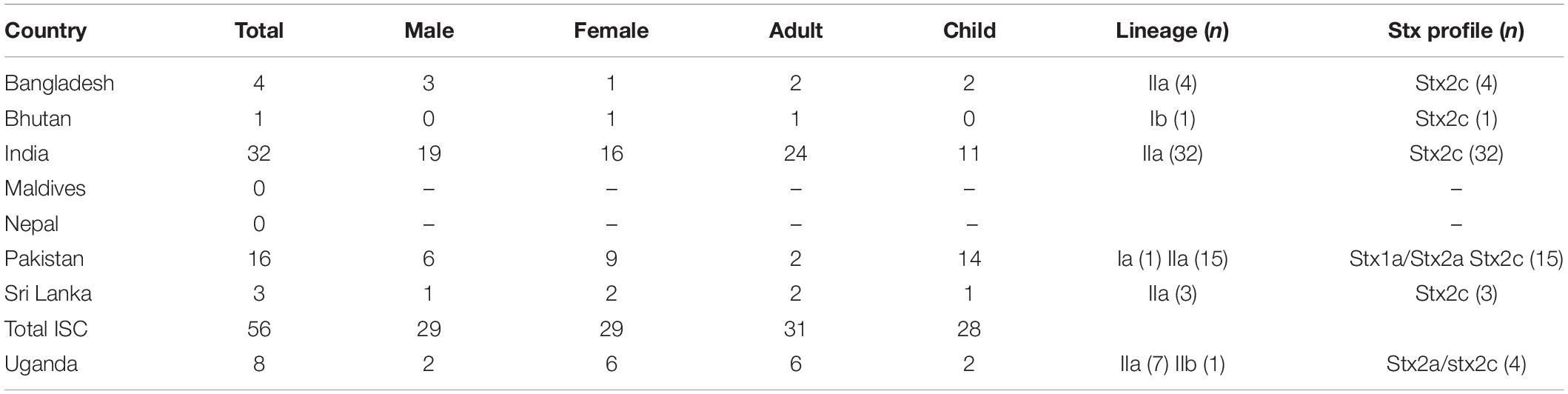
Table 2. Age and sex ratio and characteristics of isolates from UK travelers returning from the ISC and Uganda 2006–2016 (number in parentheses).
There were eight isolates from UK travelers recently returned from Uganda, all but one belonged to sub-lineage IIa (7/8). Of these, 4/8 had the same stx profile (stx2a/stx2c) as the outbreak strain, and all belonged to a unique clade that had acquired a stx2a-encoding bacteriophage. Evidence for the prevalence of STEC O157:H7 in East Africa from human clinical specimens is sparse although STEC O157:H7 has been detected in a number of studies sampling the animal reservoir in this region (Kaddu-Mulindw et al., 2001; Grace et al., 2008; Dulo et al., 2015; Beyi et al., 2017).
Phylo-geographical signals have proved useful in providing evidence for the likely country of origin of STEC and Salmonella causing outbreaks of foodborne gastrointestinal disease (Allard et al., 2016; Cowley et al., 2016; Hoffmann et al., 2016; Gobin et al., 2018; Pijnacker et al., 2019). However, there are factors that may confound this signal. Strains of STEC O157:H7 isolated from returning travelers only provide a snapshot of strains endemic to the country visited and may not reflect the full diversity of strains circulating in a given region. Although the outbreak strain may be associated with travel to Uganda, it may also be circulating at low prevalence in India, perhaps due to a recent importation event.
Analysis of the Outbreak Strain Using Oxford Nanopore Technology
We analyzed long read sequencing data of the outbreak strain, primarily to determine the sequence and genomic structure of the stx2-encoding bacteriophage. The outbreak isolate assembled into a chromosome of 5,753,376 bp and a single plasmid, pO157 (111,625 bp, IncFIB). The genome of the outbreak isolate comprised 19 prophages (Table 3 and Figure 3).
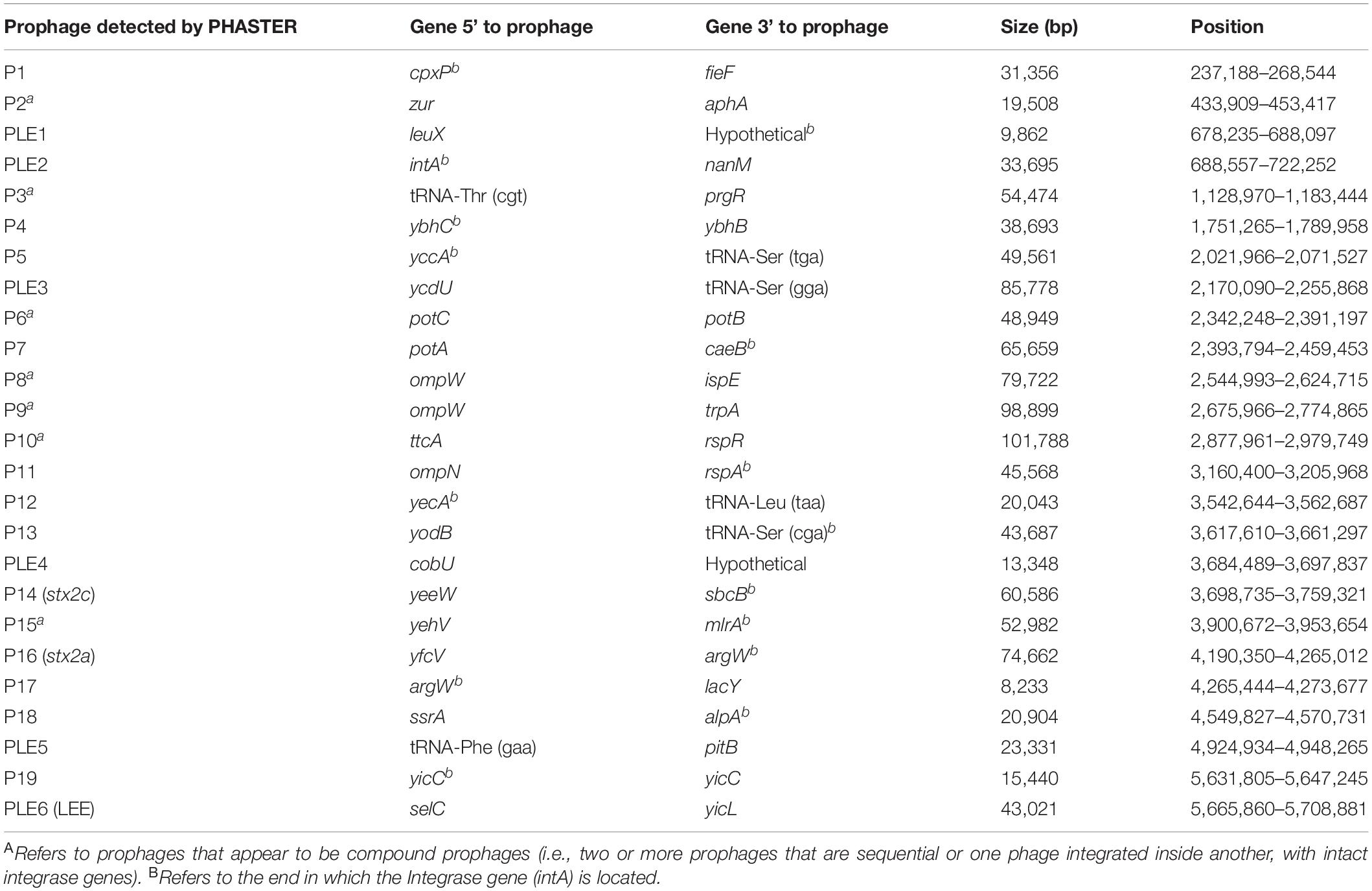
Table 3. Size, position, and integration sites of all 19 prophages (P) and six prophage-like elements (PLE) within sample 194195.
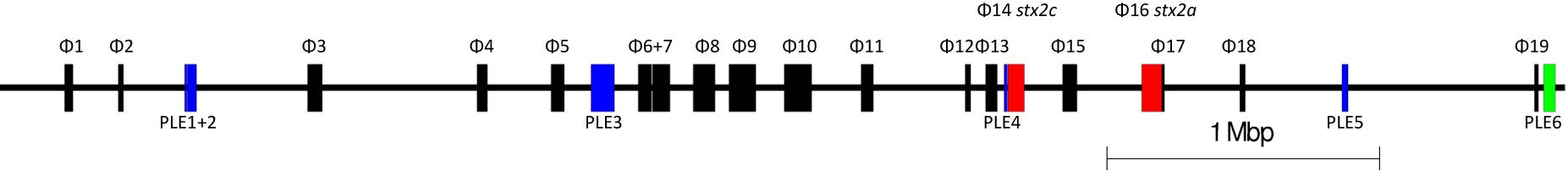
Figure 3. The relative positions of the 19 prophages and six prophage-like elements within the chromosome of 194195. Red prophages indicate stx-encoding prophages. Blue indicates prophage like elements. Green indicates the locus of enterocyte effacement (LEE).
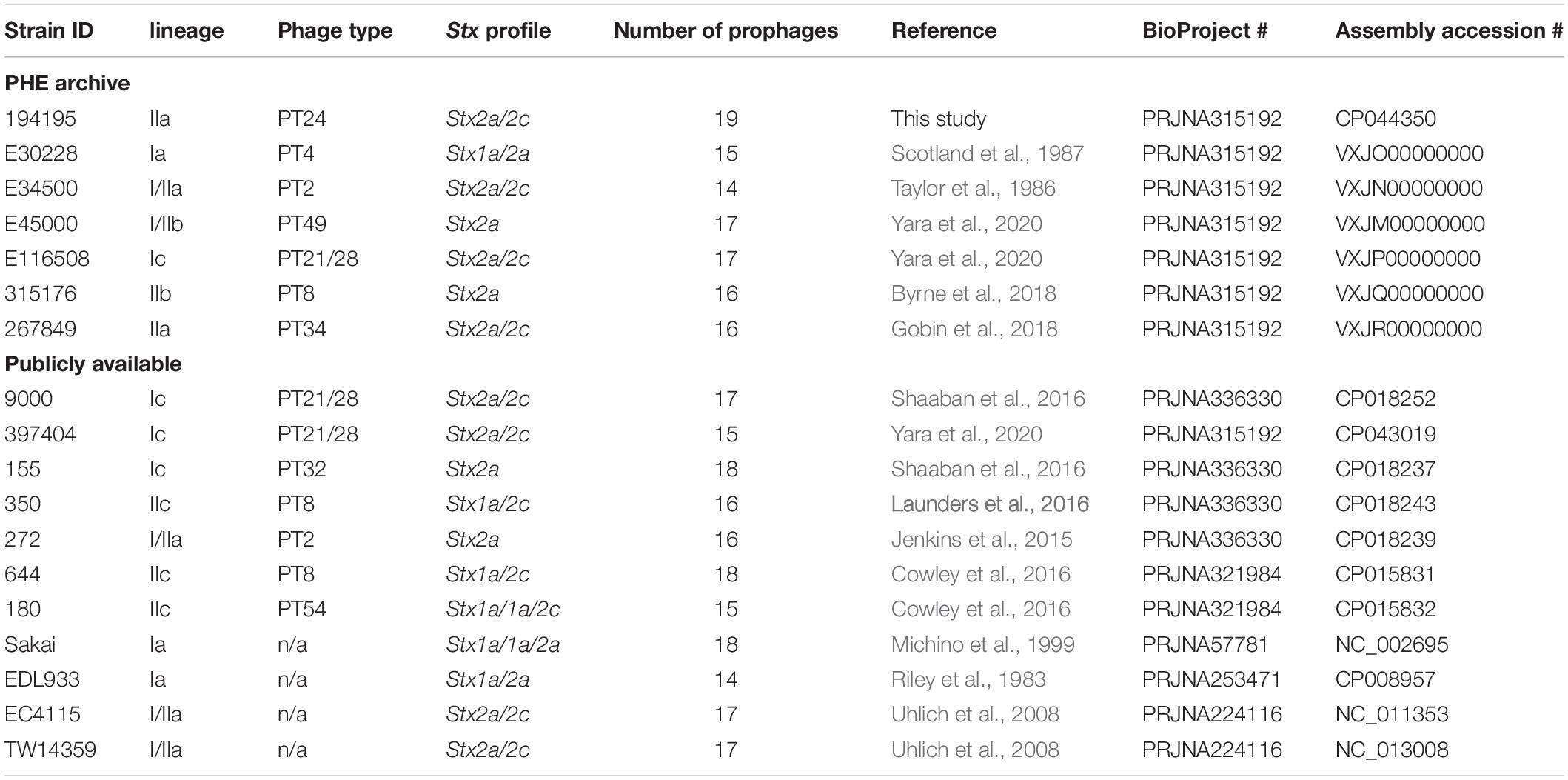
Table 4. Summary of the PHE archived and publicly available strains used within this study, their Strain ID, lineage, phage type, stx profile, assembly accession numbers, and NCBI BioProject.
Based on Mash distance, the stx2c-encoding prophage located at the stx-encoding bacteriophage insertion (SBI) site sbcB, clustered on the same branch as stx2c prophage from strains from different time frames, geographical regions, and sub-lineages, most closely related to other stx2c-encoding prophage from sub-lineage IIa (Figure 4). The stx2c-encoding prophage from the outbreak strains aligned across the length of other stx2c-encoding prophage with few structural variations (Figure 5). The conserved nature of the stx2c-encoding bacteriophage was consistent with the hypothesis that the stx2c-encoding bacteriophage was acquired prior to the global dissemination and regional expansions of STEC O157:H7 (Dallman et al., 2015a).
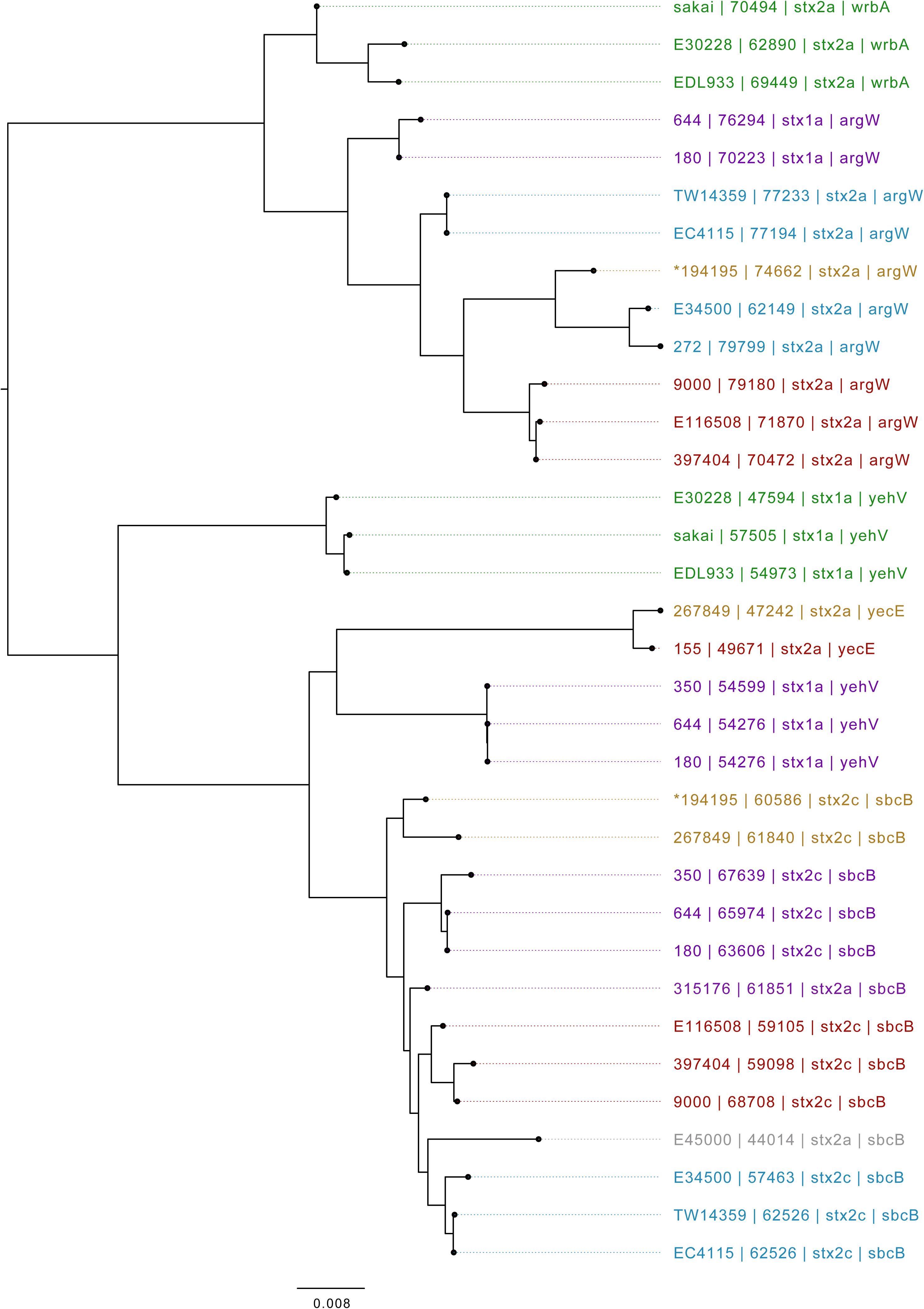
Figure 4. Mid-rooted neighbor joining tree of Shiga toxin-encoding prophages based on Jaccard distance produced from Mash. Strains are annotated as Strain ID, length, and stx type profile. Strains are colored by lineage—green: Ia, red: Ic, blue: I/IIa, gray: I/IIb, orange: IIa, purple: IIb. *denotes Oxford Nanopore Technology sequenced sample 194195.

Figure 5. Easyfig plots comparing the stx2c-encoding prophages from 194195, 267849, 180, 397404, E34500, and TW14359 in descending order. Arrows indicate gene directions. stx genes are shown in red; recombination/replication genes are shown in light blue; regulation-associated genes are shown in dark blue; effector genes are shown in pink; structure- and lysis-associated genes are shown in light and dark green, respectively; tRNAs are shown as purple lines; finally, hypothetical genes are shown in gray.
The stx2a-encoding prophage acquired by the outbreak strain was located at SBI site argW. Compared to the stx2c-encoding prophage, the stx2a-encoding prophage exhibits greater diversity, based on Mash distance and whole prophage alignment (Figure 4). In certain countries, the regional expansion of specific sub-lineages, or clades within sub-lineages, has involved acquisition of a stx2a-encoding prophage (Dallman et al., 2015a; Yara et al., 2020).
The stx2a-encoding prophage from the outbreak strain was most closely related to stx2a-encoding prophage acquired by a strain belonging to lineage I/II that caused the first outbreak of STEC-HUS in the West Midlands in England in the early 1980s (Figures 4, 6) (Taylor et al., 1986; Yara et al., 2020). Previous studies have shown that this lineage I/II strain of STEC O157:H7 harboring stx2c-encoding prophage had been indigenous in UK domestic cattle population for decades, and that it emerged as a threat to public health in 1983, following the acquisition of a stx2a-encoding prophage at some point during the previous decade (Dallman et al., 2015a; Yara et al., 2020).
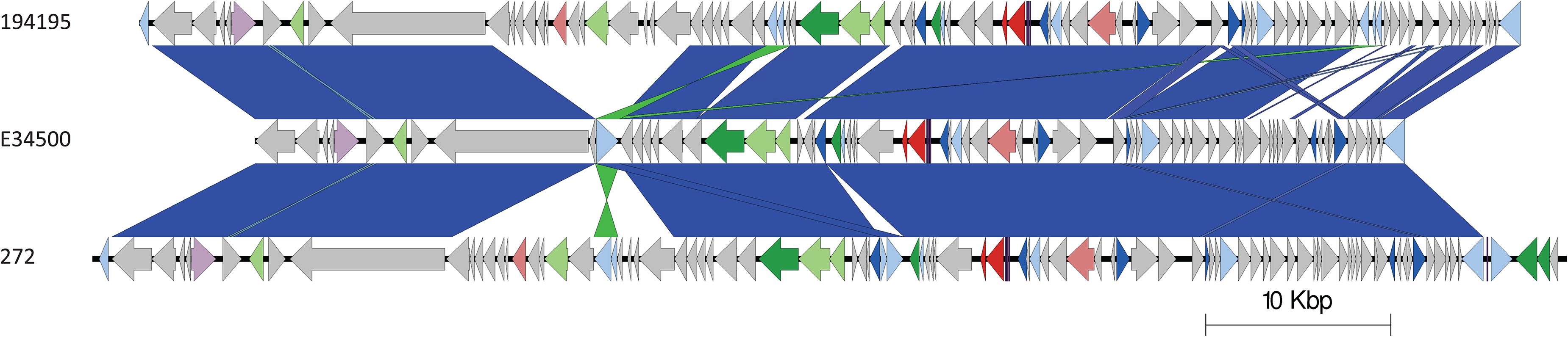
Figure 6. Easyfig plots comparing the stx2a-encoding prophages from 194195 with E34500 and 272, in descending order. Arrows indicate gene directions. stx genes are shown in red; recombination/replication genes are shown in light blue; regulation-associated genes are shown in dark blue; effector genes are shown in pink; structure- and lysis-associated genes are shown in light and dark green, respectively; tRNAs are shown as purple lines; finally, hypothetical genes are shown in gray.
Conclusion
The use of WGS for surveillance of gastrointestinal pathogens enabled us to identify a small, geographically dispersed outbreak of foodborne disease. The epidemiological analysis provided evidence that the outbreak strains originated from India, while the phylogenetic analysis of the sequencing data indicated the strain was most closely related to isolates from Uganda, and the stx2a-encoding phage was most closely related to stx2a-encoding bacteriophage harbored by the strains of STEC O157:H7 that emerged in the United Kingdom, as the most common cause of STEC-HUS in early 1980s.
These analyses described in this study are open to interpretation in a number of different ways. Microbiological investigation of the grated coconut samples did not detect STEC O157:H7, and the contaminated food vehicle may have been an imported product from elsewhere. Alternatively, the epidemiological evidence indicating India as the country of origin of the outbreak strain may have been correct, but the phylo-geographical signal was obscured by the low prevalence of the outbreak strain in that region. Strains of STEC O157:H7 that have a low prevalence in a specific region may not be captured by either local or global monitoring systems, and it is likely the full diversity of strains in a given region may circulate under the surveillance radar. Moreover, non-indigenous strains of STEC O157:H7 may be introduced into a new region by the migration of animals (including migratory birds) and people, thus further confounding the phylo-geographical signal. These animal and people migration events may facilitate the transfer of mobile genetic elements, such as the stx2a-encoding prophage, from indigenous strains of STEC O157:H7 to recently imported strains, or vice versa. Monitoring the transmission of strains of STEC O157:H7 on a global scale, and tracking the exchange of stx2a-encoding phage between imported and indigenous strains, may provide an early warning of emerging threats to public health.
Data Availability Statement
Illumina FASTQ files for all samples used in the study can be found under BioProject PRJNA315192. Nanopore FASTQ file is available under SRA accession: SRR10177137. The assembly can be found under the following accession: CP044350. All the above are available from BioProject: PRJNA315192.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements. The authors declare that there is no requirement for ethical approval for this submission. This work was undertaken to inform the delivery of patient care and to prevent the spread of infection, defined as USUAL PRACTICE in public health and health protection.
Author Contributions
AM performed epidemiological investigations. DG performed DNA extraction, library preparation, Nanopore sequencing, data processing, genome assembly, correction, and annotation, created Easyfig diagrams, and performed the prophage comparison using Mash with associated scripts designed by TD. TD created sunburst plots. CJ and DG wrote the original manuscript. CJ, DG, and TD reviewed the manuscript. CJ and TD supervised DG. All authors contributed to the article and approved the submitted version.
Funding
This research was part funded by the National Institute for Health Research (NIHR) Health Protection Research Unit in Gastrointestinal Infections at the University of Liverpool (United Kingdom), in partnership with PHE, in collaboration with the University of East Anglia (United Kingdom), the University of Oxford (United Kingdom), and the Quadram Institute (United Kingdom). CJ, TD, and DG are based at PHE. The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR, the Department of Health nor PHE.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.577658/full#supplementary-material
Footnotes
- ^ https://www.gov.uk/government/publications/ready-to-eat-foods-microbiological-safety-assessment-guidelines
References
Adams, N. L., Byrne, L., Smith, G. A., Elson, R., Harris, J. P., Salmon, R., et al. (2016). Shiga toxin-producing Escherichia coli O157, England and Wales, 1983–2012. Emerg. Infect. Dis. 22, 590–597. doi: 10.3201/eid2204.151485
Allard, M. W., Strain, E., Melka, D., Bunning, K., Musser, S. M., Brown, E. W., et al. (2016). Practical value of food pathogen traceability through building a whole-genome sequencing network and database. J. Clin. Microbiol. 54, 1975–1983. doi: 10.1128/JCM.00081-16
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acid Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Beyi, A. F., Fite, A. T., Tora, E., Tafese, A., Genu, T., Kaba, T., et al. (2017). Prevalence and antimicrobial susceptibility of Escherichia coli O157 in beef at butcher shops and restaurants in central Ethiopia. BMC Microbiol. 17:49. doi: 10.1186/s12866-017-0964-z
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Byrne, L., Adams, N., and Jenkins, C. (2020). Association between Shiga toxin-producing Escherichia coli O157:H7 stx gene subtype and disease severity, England, 2009–2019. Emerg. Infect. Dis. 26, 2394–2400. doi: 10.3201/eid2610.200319
Byrne, L., Dallman, T. J., Adams, N., Mikhail, A. F. W., McCarthy, N., and Jenkins, C. (2018). Highly pathogenic clone of shiga toxin-producing Escherichia coli O157:H7, England and Wales. Emerg. Infect. Dis. 24, 2303–2308. doi: 10.3201/eid2412.180409
Byrne, L., Jenkins, C., Launders, N., Elson, R., and Adak, G. K. (2015). The epidemiology, microbiology and clinical impact of Shiga toxin-producing Escherichia coli in England, 2009-2012. Epidemiol. Infect. 143, 3475–3487. doi: 10.1017/S0950268815000746
Cowley, L. A., Dallman, T. J., Fitzgerald, S., Irvine, N., Rooney, P. J., McAteer, S. P., et al. (2016). Short-term evolution of Shiga toxin- producing Escherichia coli O157:H7 between two food-borne outbreaks. Microb. Genom. 2:e000084. doi: 10.1099/mgen.0.000084
Dallman, T. J., Ashton, P., Schafer, U., Jironkin, A., Painset, A., Shaaban, S., et al. (2018). SnapperDB: a database solution for routine sequencing analysis of bacterial isolates. Bioinformatics 34, 3028–3029. doi: 10.1093/bioinformatics/bty212
Dallman, T. J., Ashton, P. M., Byrne, L., Perry, N. T., Petrovska, L., and Ellis, R. (2015a). Applying phylogenomics to understand the emergence of Shiga- toxin-producing Escherichia coli O157:H7 strains causing severe human disease in the UK. Microb. Genom. 1:e000029. doi: 10.1099/mgen.0.000029
Dallman, T. J., Byrne, L., Ashton, P. M., Cowley, L. A., Perry, N. T., and Adak, G. (2015b). Whole- genome sequencing for national surveillance of Shiga toxin- producing Escherichia coli O157. Clin. Infect. Dis. 61, 305–312. doi: 10.1093/cid/civ318
De Coster, W., D’Hert, S., Scheutz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Dulo, F., Feleke, A., Szonyi, B., Fries, R., Baumann, M. P., and Grace, D. (2015). Isolation of multidrug-resistant Escherichia coli O157 from goats in the somali region of ethiopia: a cross-sectional, abattoir-based study. PLoS One 10:e0142905. doi: 10.1371/journal.pone.0142905
Eppinger, M., Mammel, M. K., Leclerc, J. E., Ravel, J., and Cebula, T. A. (2011). Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc Natl. Acad. Sci. U S A 108, 20142–20147. doi: 10.1073/pnas.1107176108
Gobin, M., Hawker, J., Cleary, P., Inns, T., Gardiner, D., and Mikhail, A. (2018). National outbreak of Shiga toxin-producing Escherichia coli O157:H7 linked to mixed salad leaves, United Kingdom, 2016. Euro Surveill. 23:e0017–197. doi: 10.2807/1560-7917.ES.2018.23.18.17-00197
Grace, D., Omore, A., Randolph, T., Kang’ethe, E., Nasinyama, G. W., and Mohammed, H. O. (2008). Risk assessment for Escherichia coli O157:H7 in marketed unpasteurized milk in selected East African countries. J. Food Prot. 71, 257–263. doi: 10.4315/0362-028x-71.2.257
Hoffmann, M., Luo, Y., Monday, S. R., Gonzalez-Escalona, N., Ottesen, A. R., and Muruvanda, T. (2016). Tracing origins of the Salmonella bareilly strain causing a food-borne outbreak in the United States. J. Infect. Dis. 213, 502–508. doi: 10.1093/infdis/jiv297
Hunt, M., Silva, N. D., Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Iyoda, S., Manning, S. D., Seto, K., Kimata, K., Isobe, J., Etoh, Y., et al. (2014). Phylogenetic clades 6 and 8 of enterohemorrhagic Escherichia coli O157:H7 with particular stx subtypes are more frequently found in isolates from hemolytic uremic syndrome patients than from asymptomatic carriers. Open Forum Infect. Dis. 1:ofu061. doi: 10.1093/ofid/ofu061
Jenkins, C., Dallman, T. J., Launders, N., Willis, C., Byrne, L., Jorgensen, F., et al. (2015). Public health investigation of two outbreaks of Shiga toxin-producing Escherichia coli O157 associated with consumption of watercress. Appl. Environ. Microbiol. 81, 3946–3952. doi: 10.1128/AEM.04188-14
Kaddu-Mulindw, D. H., Aisu, T., Gleier, K., Zimmermann, S., and Beutin, L. (2001). Occurrence of Shiga toxin-producing Escherichia coli in fecal samples from children with diarrhea and from healthy zebu cattle in Uganda. Int. J. Food Microbiol. 66, 95–101. doi: 10.1016/s0168-1605(00)00493-1
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Lanjewar, M., De, A. S., Mathru, M., and Diarrheagenic, E. (2010). Coli in hospitalized patients: special reference to shiga-like toxin producing Escherichia Coli. Indian J. Pathol. Microbiol. 53, 75–78. doi: 10.4103/0377-4929.59188
Launders, N., Byrne, L., Jenkins, C., Harker, K., Charlett, A., and Adak, G. K. (2016a). Disease severity of Shiga toxin-producing E. coli O157 and factors influencing the development of typical haemolytic uraemic syndrome: a retrospective cohort study, 2009-2012. BMJ Open 6:e009933. doi: 10.1136/bmjopen-2015-009933
Launders, N., Locking, M. E., Hanson, M., Willshaw, G., Charlett, A., Salmon, R., et al. (2016b). A large Great Britain-wide outbreak of STEC O157 phage type 8 linked to handling of raw leeks and potatoes. Epidemiol. Infect. 144, 171–181. doi: 10.1017/S0950268815001016
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). 1000 genome project data processing subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Michino, H., Araki, K., Minami, S., Takaya, S., Sakai, N., Miyazaki, M., et al. (1999). Massive outbreak of Escherichia coli O157:H7 infection in schoolchildren in Sakai City, Japan, associated with consumption of white radish sprouts. Am. J. Epidemiol. 150, 787–796. doi: 10.1093/oxfordjournals.aje.a010082
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Persson, S., Olsen, K. E. P., Ethelberg, S., and Scheutz, F. (2007). Subtyping method for Escherichia Coli shiga toxin (verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 45, 2020–2024. doi: 10.1128/jcm.02591-06
Pijnacker, R., Dallman, T. J., Tijsma, A. S. L., Hawkins, G., Larkin, L., and Kotila, S. M. (2019). International Outbreak Investigation Team. An international outbreak of Salmonella enterica serotype Enteritidis linked to eggs from Poland: a microbiological and epidemiological study. Lancet Infect. Dis. 19, 778–786. doi: 10.1016/S1473-3099(19)30047-7
Riley, L. W., Remis, R. S., Helgerson, S. D., McGee, H. B., Wells, J. G., Davis, B. R., et al. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308, 681–685. doi: 10.1056/NEJM198303243081203
Scheutz, F., Teel, L. D., Beutin, L., Piérard, D., Buvens, G., and Karch, H. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/jcm.00860-12
Scotland, S. M., Willshaw, G. A., Smith, H. R., and Rowe, B. (1987). Properties of strains of Escherichia coli belonging to serogroup O 157 with special reference to production of Vero cytotoxins VTl and VT2. Epidemiol. Infect. 99, 613–624. doi: 10.1017/s0950268800066462
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sehgal, R., Kumar, Y., and Kumar, S. (2008). Prevalence and geographical distribution of Escherichia coli O157 in India: a 10-year survey. Trans. R. Soc. Trop. Med. Hyg. 102, 380–383. doi: 10.1016/j.trstmh.2008.01.015
Shaaban, S., Cowley, L., McAteer, S. P., Jenkins, C., Dallman, T. J., Bono, J. L., et al. (2016). Evolution of a zoonotic pathogen: investigating prophage diversity in enterohaemorrhagic Escherichia coli O157 by long-read sequencing. Microb. Gen. 2:e000096. doi: 10.1099/mgen.0.000096
Shrivastava, A. K., Kumar, S., Mohakud, N. K., Suar, M., and Sahu, P. S. (2017). Multiple etiologies of infectious diarrhea and concurrent infections in a pediatric outpatient-based screening study in Odisha, India. Gut. Pathog. 9:16. doi: 10.1186/s13099-017-0166-0
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Tarr, P. I., Gordon, C. A., and Chandler, W. L. (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086. doi: 10.1016/s0140-6736(05)71144-2
Taylor, C. M., White, R. H., Winterborn, M. H., and Rowe, B. (1986). Haemolytic-uraemic syndrome: clinical experience of an outbreak in the West Midlands. BMJ 292, 1513–1516. doi: 10.1136/bmj.292.6534.1513
Uhlich, G. A., Sinclair, J. R., Warren, N. G., Chmielecki, W. A., and Fratamico, P. (2008). Characterization of Shiga toxin-producing Escherichia coli isolates associated with two multistate food-borne outbreaks that occurred in 2006. Appl. Environ. Microbiol. 74, 1268–1272. doi: 10.1128/AEM.01618-07
Vaser, R., Soviæ, I., Nagarajan, N., and Šikiæ, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wick, R. R., Judd, L. M., and Holt, K. E. (2018). Deepbinner: demultiplexing barcoded Oxford Nanopore reads with deep convolutional neural networks. PLoS Comput. Biol. 14:e1006583. doi: 10.1371/journal.pcbi.1006583
Keywords: Shiga toxin-producing E. coli, outbreak, genomics, public health, epidemiology, coconut
Citation: Greig DR, Mikhail AFW, Dallman TJ and Jenkins C (2020) Analysis Shiga Toxin-Encoding Bacteriophage in a Rare Strain of Shiga Toxin-Producing Escherichia coli O157:H7 stx2a/stx2c. Front. Microbiol. 11:577658. doi: 10.3389/fmicb.2020.577658
Received: 29 June 2020; Accepted: 09 September 2020;
Published: 21 October 2020.
Edited by:
Grzegorz Wegrzyn, University of Gdańsk, PolandReviewed by:
Edward G. Dudley, Pennsylvania State University (PSU), United StatesJames L. Bono, United States Department of Agriculture, United States
Angelika Fruth, Robert Koch Institute (RKI), Germany
Copyright © 2020 Greig, Mikhail, Dallman and Jenkins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire Jenkins, claire.jenkins1@phe.gov.uk