 Ying Bai
Ying Bai Maria Rosales Rizzo
Maria Rosales Rizzo Rebecca J. Eisen
Rebecca J. Eisen- Bacterial Diseases Branch, Division of Vector-Borne Diseases, Centers for Disease Control and Prevention, Fort Collins, CO, United States
Rapid detection of Yersinia pestis, the causative agent of plague, is essential during field investigations to enable prompt control measures for prevention of the spread of the disease. Affordable, efficient, reliable, and simple detection assays are extremely useful, particularly in plague-endemic regions with limited resources. We developed a loop-mediated isothermal amplification (LAMP) assay that detects Y. pestis within 30 min by simply incubating at 65°C on a dry bath heater. The assay targeted the caf1A gene that is situated on the pMT1 plasmid using six specific primers. Y. pestis presence is visually detected based on the color change in the reactions. For comparison of the assay performance, a real-time LAMP with fluorescent dye detection was conducted on a real-time PCR instrument using the same six primers. Sensitivity assessment showed that the limit of detection (LOD) was 0.2 and 0.03 pg when performed on the dry bath heater and on the real-time PCR instrument, respectively. The assay was 100% specific, having no cross-reactivity with closely related Yersinia spp. and other bacterial species. We tested the LAMP assay on field-collected fleas and showed that it successfully detected Y. pestis with identical results to that of a previously published pentaplex real-time PCR assay. These findings suggest that the relatively inexpensive and simpler LAMP assay could be used to support field investigations, yielding comparable results to more expensive and complex PCR assays.
Introduction
Yersinia pestis, classified as a category “A” biothreat agent, is the bacterial causative pathogen of plague, a deadly infectious zoonotic disease. The bacterium mainly infects rodents that are its natural reservoirs and can be transmitted to humans and some other mammals through infected rodent flea bites (Stenseth et al., 2008). Plague outbreaks often occur in geographic regions that lack basic laboratory infrastructure for diagnostics (Butler, 2013; Grácio and Grácio, 2017). Due to the highly infectious nature and mortality rate of plague, accurate and rapid direct detection of Y. pestis in the early stages of epizootics is critical for containment of the plague outbreak as well as for environmental and medical management. Several surveillance and response strategies have been proposed to identify where and when plague epizootics are occurring, with the intent of directing interventions to prevent human plague cases (Gage, 1999). These strategies include monitoring plague host abundance, host mortality (die-offs or rat falls), flea loads on rodents, seropositivity of non-human hosts, or direct detection of Y. pestis in vectors or rodent hosts (Borchert et al., 2012; Moore et al., 2015; Boegler et al., 2018; Ehlers et al., 2020). The most effective and reliable strategies are dependent on accurate detection of Y. pestis (Eisen et al., 2020, 2021).
Multiple methods have been employed for plague diagnosis. Bacterial culture is the gold standard for plague diagnosis in humans, vectors, and non-human hosts; however, this procedure is time-consuming and poses biosafety risks to laboratory staff, with strict requirements of performance in biosafety level 3 laboratories (Norkina et al., 1994). Serology is mainly used in late stages of the infection for detection of plague antibodies (Splettstoesser et al., 2004; Simon et al., 2013; Hsu et al., 2018). While useful for identifying evidence of past plague activity, serology is not suitable for investigating plague in rodents due to the extremely high mortality (Enscore et al., 2020). In recent years, polymerase chain reaction (PCR) and other molecular diagnostic techniques have been developed and largely applied for Y. pestis detection (Campbell et al., 1993; Neubauer et al., 2000; Tomaso et al., 2003; Stewart et al., 2008; Bai et al., 2020). However, the requirement for a high level of technical expertise and high-precision thermal cyclers for amplification may limit this powerful technology from being widely used in areas of greatest need.
Loop-mediated isothermal amplification (LAMP) is a relatively new molecular technique that amplifies nucleic acid under isothermal conditions (Notomi et al., 2000). Using a DNA polymerase with high displacement strand activity and a set of four to six specific primers, LAMP amplifies DNA with high sensitivity, specificity, and rapidity. In contrast to PCR, LAMP does not require thermal cyclers. A simple dry/water bath that provides a constant temperature will fulfill the amplification. Furthermore, because a pH indicator dye was used in LAMP reactions, the amplified products can be visually detected based on the color change in the reaction (Pal et al., 2018). These features make LAMP an excellent application for pathogen detection, especially for use in resource-limited areas in developing countries. LAMP techniques are increasingly used in molecular diagnostics as a potential alternative to PCR-based methods (Pal et al., 2018; Bodulev and Sakharov, 2020; Mukama et al., 2020) and already have been successfully applied for detection of bacteria, fungi, parasites, and viruses (Kuboki et al., 2003; Gonçalves et al., 2014; Lass et al., 2017; Lu et al., 2020). Several LAMP assays for Y. pestis detection have been developed as well (Nunes et al., 2014; Feng et al., 2018; Randriantseheno et al., 2020; Singh et al., 2020).
During the evolutionary history of Y. pestis, the bacterium acquired two unique plasmids, pMT1 and pPCP1, in addition to pCD1 that is shared with Yersinia pseudotuberculosis and Yersinia enterocolitis (Ben-Gurion and Shafferman, 1981; Ferber and Brubaker, 1981). Nevertheless, not all three plasmids are present in all Y. pestis strains (Butler, 1972; Filippov et al., 1990; Meka-Mechenko, 2003). Previously developed Y. pestis LAMP assays have mainly used caf1 (Nunes et al., 2014; Randriantseheno et al., 2020; Singh et al., 2020), a highly specific gene that is situated in the pMT1 plasmid and plays an essential role for Y. pestis virulence by expressing the capsular antigen fraction 1 (Caf1) protein (Sebbane et al., 2009; Al-Jawdah et al., 2019). However, paired loop primers seemed difficult to generate when using the caf1 gene. There was only one loop primer included in previous assays (Nunes et al., 2014; Randriantseheno et al., 2020). In this study, we developed a new LAMP assay for rapid detection of Y. pestis. Our main goal was to apply the assay for testing fleas, rodents, and other environmental samples collected during field investigations and/or in plague-endemic regions for Y. pestis infection. When dealing with a large sample size, a simple, rapid, and efficient assay would be very helpful. Previously, Hanson et al. (2007) used a nested PCR for investigation of Y. pestis infection in large scale of fleas collected from prairie dog burrows. Although it worked, the nested PCR required a thermal cycler and took several hours to complete. Our LAMP assay targeted the caf1A gene, which is a subunit of the caf1 operon. We (1) designed a set of six caf1A-specific primers, (2) optimized reaction temperature and reaction time, (3) assessed the sensitivity and specificity, (4) evaluated the performance by checking the interference of mouse DNA and testing Y. pestis strains with different plasmid profiles, and (5) tested the applicability by screening field-collected fleas. A dry bath heater was used to perform the LAMP assay. We also performed the assay using a real-time PCR instrument to evaluate the performance of the LAMP assay under simple and less costly isothermal conditions. The terms “standard LAMP” or “real-time LAMP” were used when the assay was performed on a dry bath heater or a real-time PCR instrument, respectively.
Materials and Methods
Primer Design
Employing the Primer Explorer V4 software1 (Eiken Chemical Co., Ltd., Tokyo, Japan) and using the sequences of Y. pestis EV vaccine strain (GenBank accession no. AY450845), we first designed two outer primers (F3 and B3) and two inner primers (FIP and BIP); then two loop primers (LF and LB) were generated based on the sequences of F3/B3 and FIP/BIP. Together, a set of six LAMP primers were designed (Table 1). The screening parameters included in primer selection were the GC (guanine–cytosine) content, Tm (melting temperature), stability of the primer sequence, secondary structure formation, and distance between primers, which were assessed to determine the final primer sequences.

Table 1. Nucleotide sequences of the LAMP caf1A primers.
DNA Template of Yersinia pestis and Other Bacterial Species
Yersinia pestis strain CO96-3188 is a fully virulent North American biovar Orientalis strain (Engelthaler et al., 2000; Eisen et al., 2006). Genomic DNA extracted from this strain was used for the assay development in this work. DNA from 11 other Y. pestis strains varying in plasmid profiles was included for further evaluation of the assay (Table 2). DNAs from 10 Y. pseudotuberculosis strains, 2 Y. enterocolitica strains, and 20 non-Yersinia strains representing a wide variety of different bacterial species including Bacillus anthracis, Bartonella spp., Borrelia spp., Brucella canis, Burkholderia cenocepacia, Escherichia coli, Francisella tularensis, Leptospira interrogans, Listeria monocytogenes, Rickettsia sibirica, Salmonella enterica, Staphylococcus aureus, Toxoplasma gondii, and Trypanosoma cruzi were included for specificity testing of the assay (Table 3). Although many of these species would not be expected in flea specimens (the sample type upon which this assay was evaluated), most could be encountered if the assay is later applied to mammalian samples. DNA templates of the above-mentioned strains used in the study were obtained from BEI Resources, University of Texas Medical Branch (UTMB), and CDC Bacterial Diseases Branch (Tables 2, 3).
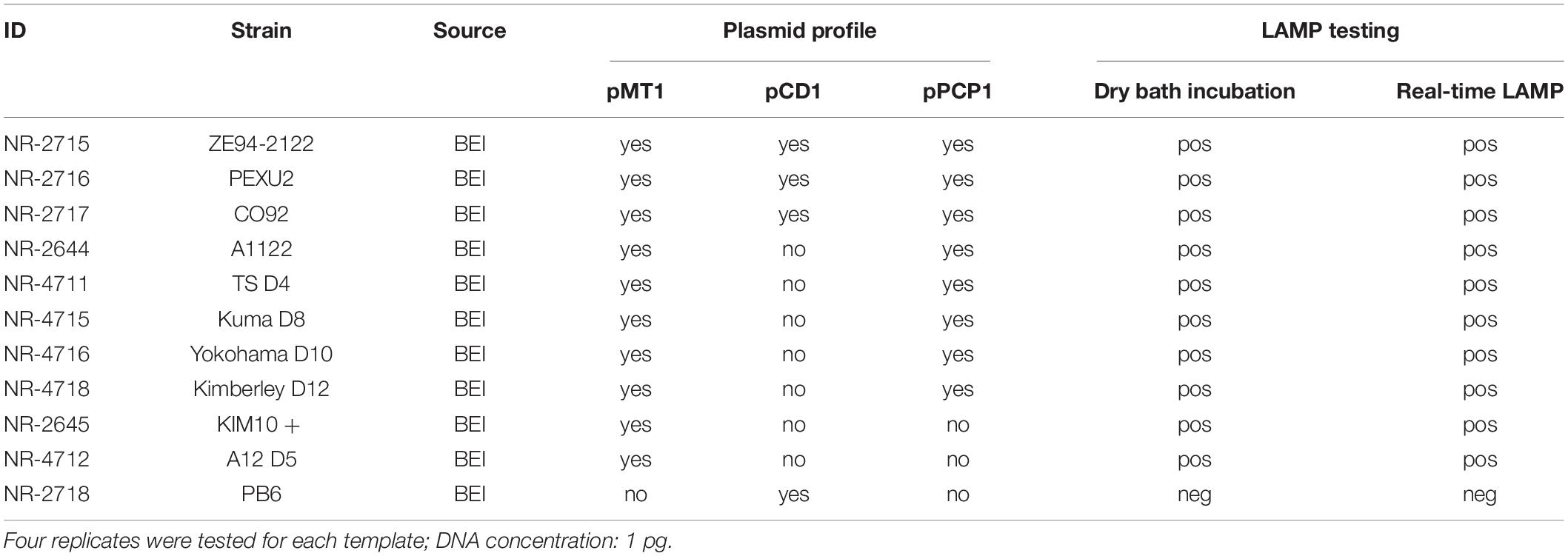
Table 2. Loop-mediated isothermal amplification testing results for Y. pestis strains with different plasmid profiles.
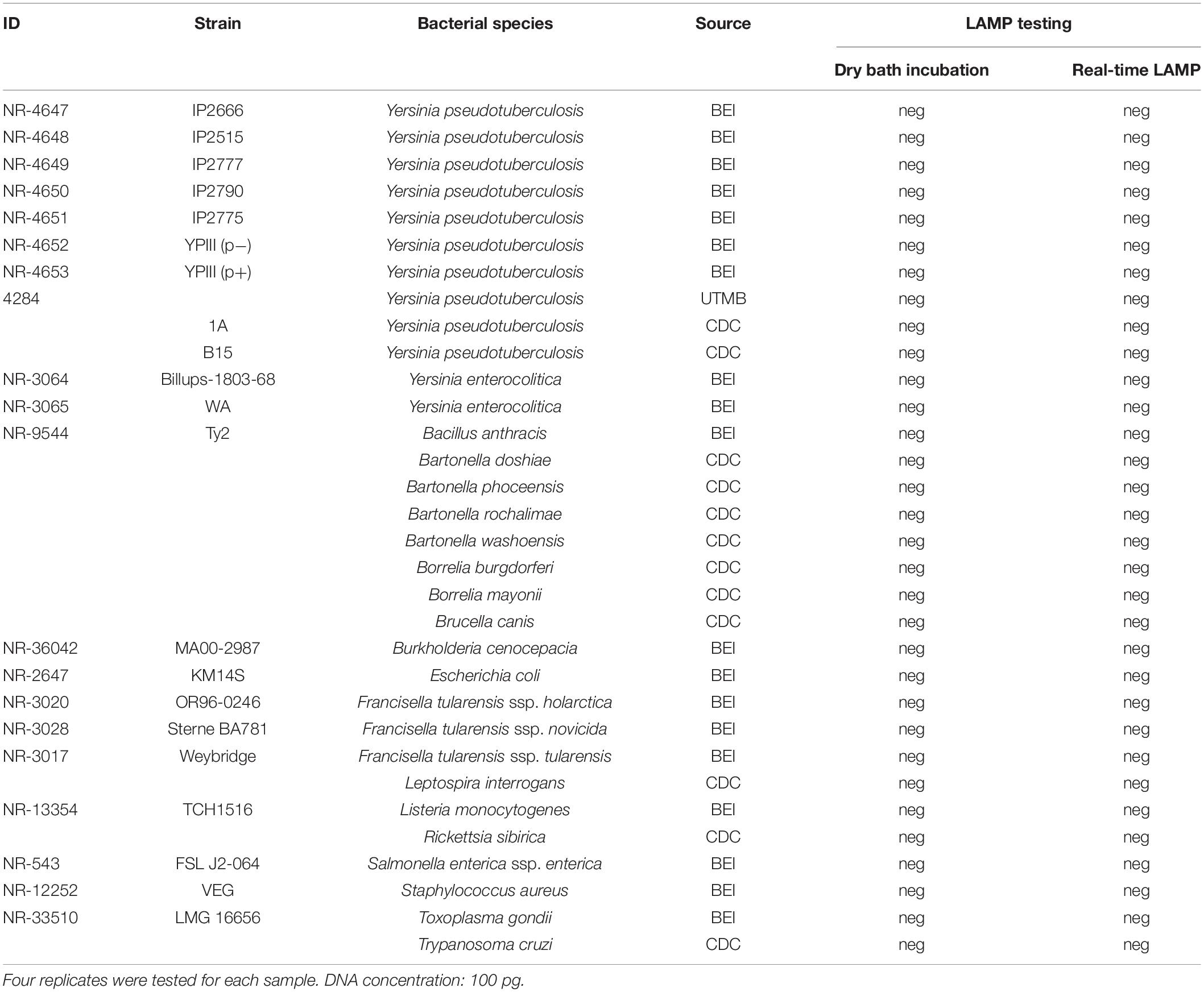
Table 3. Specificity testing of the LAMP assay.
Loop-Mediated Isothermal Amplification Reaction Setup
The caf1A primer mix (10×) was first prepared, which contained 2 μM F3 and B3 each; 16 μM FIP and BIP each; and 4 μM LF and LB each. LAMP reactions with a total volume of 25 μL were set up in two different systems depending on whether we were using the real-time PCR instrument or the dry bath heater. For the real-time LAMP, the reaction contained 12.5 μl of WarmStart LAMP 2 × Master Mix (New England Biolabs, Ipswich, MA, United States), 0.5 μl of fluorescent dye (New England Biolabs, Ipswich, MA, United States), 2.5 μl 10 × caf1A primer mix, 8.5 μl H2O, and 1 μl DNA template. The reactions were carried out on a CFX96 Real-Time Detection System (Bio-Rad, Hercules, CA, United States) at 65°C for 30 cycles with each cycle lasting 1 min. Amplification signals were detected by the fluorescent dye included in the reaction, and amplification with a sigmoidal-shaped curve was considered positive. For the standard LAMP, the reaction contained 12.5 μl of WarmStart Colorimetric LAMP 2 × Master Mix with UDG (New England Biolabs, Ipswich, MA, United States), 2.5 μl 10 × caf1A primer mix, 9 μl H2O, and 1 μl DNA template. The reactions were incubated on a 0.2 ml PCR tube-block (Thermo Fisher Scientific, Pittsburgh, PA, United States) within an enclosed Corning™ LSE™ Digital Dry Bath (Thermo Fisher Scientific, Pittsburgh, PA, United States) at 65°C for 30 min. The presence of the targeted gene was signified by visual observation with naked eyes of color change from red (cresol red as the pH indicator dye) to yellow, whereas the color remained unchanged in the absence of amplification. Positive was defined when a complete color change in a LAMP reaction was observed within 30 min of incubation at 65°C. Distilled water was always included as the negative control for both systems.
Optimization of Reaction Temperature by Real-Time Loop-Mediated Isothermal Amplification
Real-time LAMP was performed using the DNA of Y. pestis strain CO96-3188 (1 pg) for optimization of the reaction temperature. The reactions were carried out on a CFX96 Real-Time Detection System (Bio-Rad, Hercules, CA, United States) at a temperature gradient of 60–70°C (60, 60.7, 62, 64, 66.4, 68.4, 69.5, and 70°C) for 30 cycles, with each cycle lasting 1 min. Four replicates were tested for each temperature. Ct values were converted to TTR (time to result) for comparison purposes. A temperature with lower TTR and all four replicates amplified was considered an optimized temperature.
Cutoff of Reaction Time and Sensitivity Assessment Using a Dry Bath Heater
The DNA of Y. pestis strain CO96-3188 of 11 different concentrations (100, 50, 10, 5, 1, 0.5, 0.4, 0.3, 0.2, 0.1, and 0.05 pg) was used to optimize the cutoff time of the LAMP reaction using a dry bath heater. Sensitivity when using the dry bath heater was assessed simultaneously in this step. Limit of detection (LOD) was used for estimating sensitivity. Four replicates were tested for each concentration. LAMP reactions were loaded on a preheated 0.2 ml PCR tube-block and incubated in a Corning™ LSE™ Digital Dry Bath (Thermo Fisher Scientific, Pittsburgh, PA, United States) at 65°C based on the optimization results of reaction temperatures by the real-time LAMP. A color change of the reactions was initially checked after 20 min of incubation and monitored every additional 5 min until there was no more color change observed in the reactions containing tested DNA or until negative controls started to show some level of color change. A longer time with a complete color change in all four replicates of a lower DNA concentration or no color change in all four replicates of negative control was considered the cutoff of the reaction time, and the DNA concentration was considered the LOD using the dry bath heater.
Sensitivity Assessment by Real-Time Loop-Mediated Isothermal Amplification
The DNA of Y. pestis strain CO96-3188 at concentrations of 100, 50, 10, 5, 1, 0.5, 0.1, 0.05, 0.04, 0.03, 0.02, 0.01, and 0.005 pg was tested using real-time LAMP. Reactions were carried out on a CFX96 Real-Time Detection System (Bio-Rad, Hercules, CA, United States) at 65°C for 30 one-minute cycles based on the optimization results for reaction temperature and cutoff of reaction time from the above sections. Four replicates of each concentration were tested. The lower concentration with all four replicates amplified or no amplification in all four replicates of negative control was considered the LOD using real-time LAMP.
Specificity Assessment
Specificity of the LAMP assay was assessed by testing DNA from genetically close bacterial species within the genus of Yersinia, including 10 strains of Y. pseudotuberculosis and 2 strains of Y. enterocolitica. In addition, a wide variety of other non-Yersinia bacterial species (Table 3) was also tested. The reason to include these microorganisms is because some of them share a similar transmission route with Yersinia species, either by fleas or other blood-feeding vectors (Brook et al., 2015; Bai et al., 2017; Pawelczyk et al., 2019) or by consumption of contaminated water (Casanovas-Massana et al., 2018). High nucleic acid concentrations (100 pg) with four replicates of each sample were tested. LAMP reactions were carried out on both a dry bath heater and real-time PCR instrument at 65°C for 30 min (which were the optimization results from the above section). A negative result was defined when targeted DNA was not detected in any of the four replicates for each sample.
Evaluation of the Loop-Mediated Isothermal Amplification Assay
The liver and spleen of rodents are two tissue types commonly tested for Y. pestis infection. To evaluate the performance of the LAMP assay, DNA was extracted from the liver and spleen collected from ten 2–4-month-old outbred female CD-1 white mice (Charles River Laboratories, Wilmington, MA, United States) and was first screened for the absence of Y. pestis DNA using a pentaplex real-time PCR (Bai et al., 2020). All tissue DNAs were then spiked with the DNA of Y. pestis strain CO96-3188 with a concentration of 1 pg and tested using both real-time LAMP and dry bath incubation at 65°C for 30 min to check if any interference would occur from the mouse tissue DNA. Furthermore, one liver DNA sample and one spleen DNA sample collected from the same mouse were randomly picked for sensitivity testing and to compare with pure Y. pestis DNA. The final DNA concentrations of Y. pestis spiked in the mouse DNA matched the sensitivity testing using the pure DNA of Y. pestis strain CO96-3188. To improve quantification, real-time LAMP was conducted for this step. Four replicates of each concentration were tested.
Testing Yersinia pestis Strains With Different Plasmid Profiles
DNAs extracted from 11 Y. pestis strains were tested using both the standard LAMP and the real-time LAMP. Among them, three strains possess all three plasmids; five strains lack the pCD1 plasmid; two strains lack the pCD1 and pPCP1 plasmids; and the last one (strain PB6) lacks the pMT1 and pPCP1 plasmids (Table 1). The final DNA concentration of Y. pestis was 1 pg in the LAMP reactions, and four replicates of each template were tested.
Application of the Assay for Testing Field-Collected Fleas
To evaluate the utility of the LAMP assay, we tested 32 fleas (30 Oropsylla hirsuta and 2 Oropsylla tuberculata) that were collected from prairie dog burrows after plague activities (Enscore et al., 2021) using both the standard LAMP and the real-time LAMP. These fleas had been tested previously using a pentaplex real-time PCR, and two of the samples were reported positive for Y. pestis DNA while all others were negative (Bai et al., 2020). The results from the LAMP assay were compared with those from the pentaplex real-time PCR.
Results
Optimization of Reaction Temperature by Real-Time Loop-Mediated Isothermal Amplification
When the real-time LAMP is run at a temperature gradient of 60–70°C (60, 60.7, 62, 64, 66.4, 68.4, 69.5, and 70°C), all four replicates of Y. pestis DNA in the LAMP reactions were amplified at all temperatures except for 70°C (Figure 1 and Table 4). The TTR differed among the temperatures, ranging from 12.4 to 19.6 min (Table 4). The least TTR was at 66.4 and 64°C (12.4 and 12.8 min, respectively). At 64 and 66.4°C, the LAMP reactions not only had shorter TTR but also amplified more consistently (smaller standard deviation) compared to other temperatures (Table 4). Inconsistent amplification (larger standard deviation) was observed at lower or higher temperature; for example, at 69.5°C, one of the four replicates was amplified at about 30 min (Figure 1). Based on the results, 65°C was selected as the optimized reaction temperature and was applied for all subsequent experiments in this work.
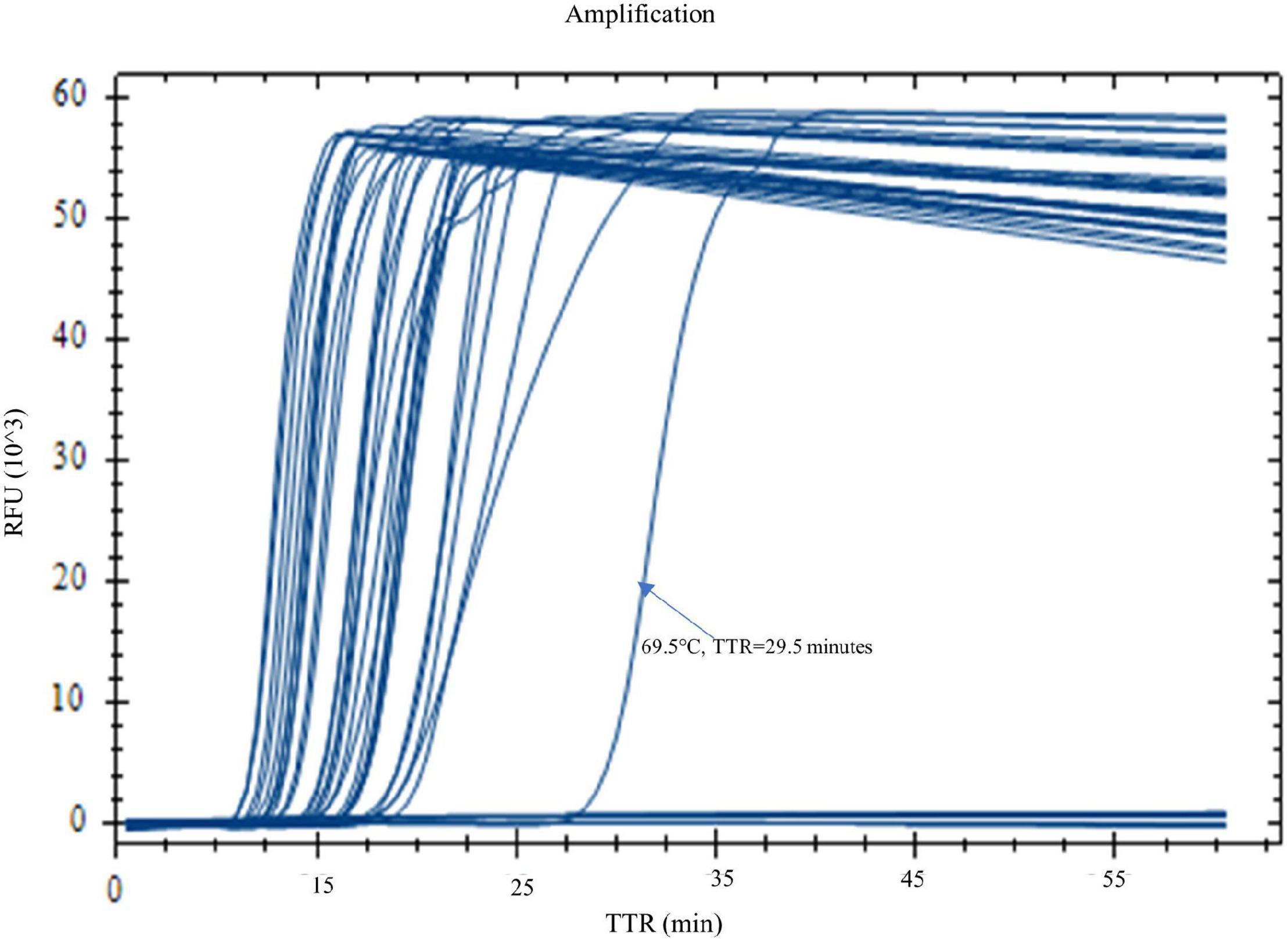
Figure 1. Optimization of reaction temperature by real-time LAMP. LAMP reactions with Yersinia pestis DNA (1 pg) were carried out on a real-time PCR instrument at a temperature gradient of 60–70°C (60, 60.7, 62, 64, 66.4, 68.4, 69.5, and 70°C). Four replicates were tested for each temperature. Four out of four replicates were amplified at all temperatures, except for 70°C. Temperatures 66.4 and 64°C generated the best results with shorter TTR and a more even amplification curve.
Table 4. Time to result (mean of the TTRs of four replicates) at different temperatures.
Cutoff of Reaction Time and Sensitivity Assessment Using the Dry Bath Heater
A color change from red (negative) to yellow (positive) was observed at different time points for the LAMP reactions with varying DNA concentrations. The higher the DNA concentration, the shorter the TTR for color change. At 20 min, a complete color change was observed in all four replicates of reactions with DNA concentrations of 100 and 50 pg while reactions with lower concentrations and negative control remained unchanged. At 25 min, a complete color change was observed in all four replicates of reactions with DNA concentrations of 10, 5, 1, and 0.5 pg while reactions with lower concentrations and negative control remained unchanged. At 30 min, a complete color change was observed in all four replicates of reactions with DNA concentrations of 0.4, 0.3, and 0.2 pg, and the negative control remained unchanged, while reactions with lower DNA concentrations showed color change in partial replicates, with 3/4 and 2/4 replicates for DNA concentrations of 0.1 and 0.05 pg, respectively (Figure 2A). The LAMP reactions with these two DNA concentrations were considered negative based on our criteria requiring that all four replicates be called positive. At 35 min, the color change for the DNA concentrations of 0.1 and 0.05 pg was similar to that at 30 min, with some incomplete color change observed (Figure 2B, tubes B2 and C4), and the two DNA concentrations still were considered negative. However, at 35 min, two of four replicates of the negative control started to show some minor color change (Figure 2B, tubes N3 and N4), suggesting that some non-specific reactivity occurred. This was an indication to stop the incubation. In the comparison of the results between 30 and 35 min, 30 min was determined as the cutoff of LAMP reaction time using the dry bath heater. Simultaneously, the LOD was identified as 0.2 pg by visual detection using the dry bath heater.
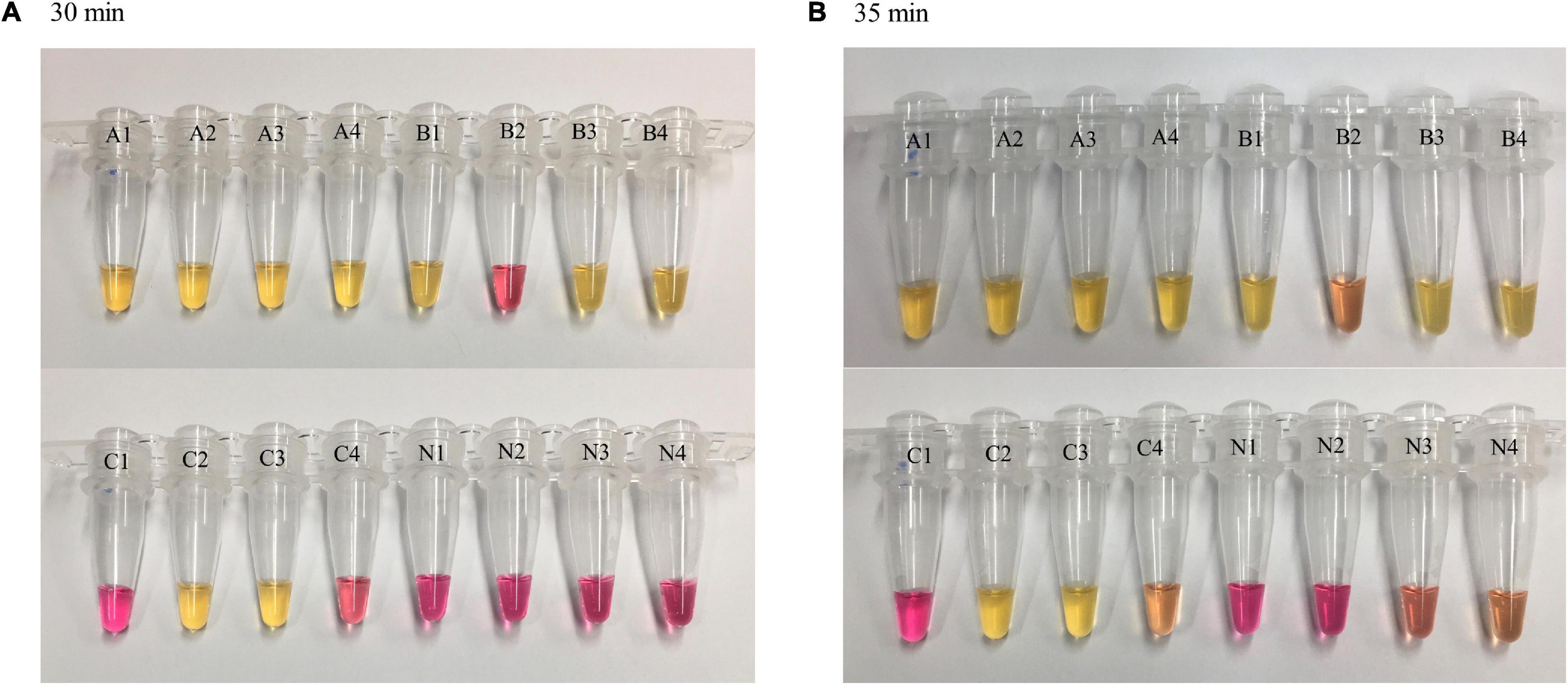
Figure 2. Loop-mediated isothermal amplification reaction time and visual observations of color change. LAMP reactions with different concentrations of Y. pestis DNA were carried out on a dry bath heater. Four replicates were tested for each concentration and negative control. At 30 min (A), color change was observed in four of four replicates of reactions with a DNA concentration of 0.2 pg (as well as higher concentration, not shown here) and in partial replicates of reactions with lower DNA concentrations (0.1 and 0.05 pg). Negative control remained unchanged; at 35 min (B), a minor color change was observed in negative control (N3 and N4). A = 0.2 pg, B = 0.1 pg, C = 0.05 pg, N = negative control.
Sensitivity and Limit of Detection Identification by Real-Time Loop-Mediated Isothermal Amplification
With real-time LAMP, the reactions with a DNA concentration of 100–0.03 pg were amplified by all four replicates. The TTR, ranging 10.1 min for 100 pg to 16.2 min for 0.03 pg, is negatively correlated with the DNA concentrations (Figure 3 and Table 5). For lower DNA concentrations, there were 3/4, 3/4, and 1/4 replicate(s) amplified for 0.02, 0.01, and 0.005 pg, respectively, which was considered a negative based on our criteria. Therefore, 0.03 pg was determined as the LOD for the LAMP assay when using the real-time PCR instrument.
Figure 3. Amplification and standard curve with DNA concentrations 100, 50, 10, 5, 1, 0.5, 0.1, 0.05, 0.04, 0.03, 0.02, 0.01, and 0.005 pg. Four replicates were tested for each concentration. Four of four replicates were amplified for DNA concentrations 100–0.03 pg. TTR was negatively correlated to the DNA concentrations.
Table 5. Time to result (mean of the TTRs of four replicates) for different concentrations of Y. pestis DNA.
Specificity Assessment
Testing a high DNA concentration (100 pg) of Y. pseudotuberculosis, Y. enterocolitica, and 20 other non-Yersinia bacterial species using the LAMP assay showed no amplification of the targeted gene in any of these microorganisms by either real-time LAMP or dry bath incubation (Table 3), showing 100% specificity of the assay.
Evaluation of the Loop-Mediated Isothermal Amplification Assay
DNA extracted from mouse spleen and liver spiked with Y. pestis DNA (1 pg) all produced amplification within 30 min with both dry bath incubation and real-time LAMP. The average TTR by real-time LAMP was very similar between the two types of Y. pestis DNA-spiked tissue with 16.7 and 17.1 min for liver DNA and spleen DNA, respectively (t = 2.02, p = 0.27), but was significantly longer than that of pure Y. pestis DNA (13.2 min, t = 1.68, p < 0.01).
The LOD was identified as 0.05 pg for both Y. pestis-spiked liver DNA and spleen DNA using real-time LAMP. This was lower than the LOD of pure Y. pestis DNA (0.03 pg). The average TTR was similar between Y. pestis-spiked liver DNA and spleen DNA, with 18.2 and 21.8 min, respectively (t = 3.18, p = 0.12), but was significantly longer than that of pure Y. pestis DNA of the same concentration (15.3 min, t = 1.94, p < 0.01). These results suggest that the mouse tissue presented some interference with the Y. pestis DNA when performing the LAMP assay.
Testing Yersinia pestis Strains With Different Plasmid Profiles
When Y. pestis strains with different plasmid profiles were tested using the LAMP assay, all strains that possess the pMT1 plasmid were detected by both real-time LAMP and dry bath incubation within 30 min, although these strains differ in other plasmids; one strain (strain PB6) was not detected with either real-time LAMP or dry bath incubation. This strain, PB6, lacks the pMT1 plasmid (Table 2), and the negative result was expected.
Application of the Assay for Testing Field-Collected Fleas
When fleas collected from prairie dog burrows were tested after plague activities using the LAMP assay, Y. pestis DNA was detected in one O. hirsuta flea (#41) and one O. tuberculata flea (#83) with both real-time LAMP and dry bath incubation (Figure 4). The two fleas had been reported Y. pestis positive previously by using a pentaplex real-time PCR assay (Bai et al., 2020). The LAMP testing results were identical to the pentaplex real-time PCR testing.
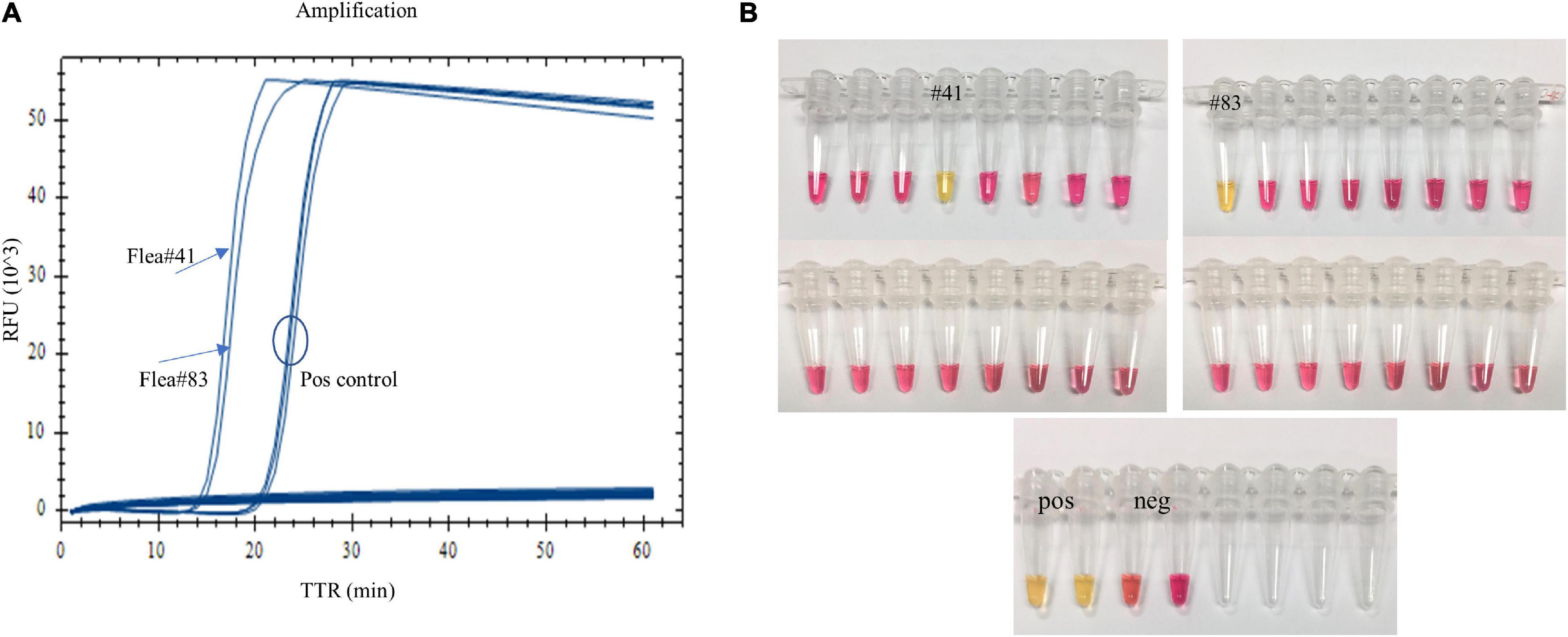
Figure 4. Testing field-collected fleas using the LAMP assay. One Oropsylla hirsuta flea (#41) and one O. tuberculata flea (#83) previously tested positive were amplified with real-time LAMP (A), and a color change was observed with dry bath incubation (B).
Discussion
In this work, we developed a LAMP assay targeting the caf1A gene for Y. pestis detection. As a subunit of the caf1 operon, caf1A plays an important role in capsular biogenesis (Galyov et al., 1990; Karlyshev et al., 1992; Runco et al., 2008; Sebbane et al., 2009; Dubnovitsky et al., 2010). Situated on the pMT1 plasmid, the highly specific caf1 has been a common target used in developing PCR assays for Y. pestis detection (Norkina et al., 1994; Tomaso et al., 2003; Stewart et al., 2008; Singh et al., 2020). Although retaining the same specificity as caf1, caf1A has not been explored much as a target for Y. pestis detection. To our knowledge, this is the first LAMP assay that targets caf1A for Y. pestis detection.
An ideal LAMP assay uses six of three pairs of primers, which include two outer primers, two inner primers, and two loop primers in the reaction. However, the LAMP primer design is very challenging due to the requirement of the selection of eight separate regions of target nucleic acid sequences, restriction on positioning, and other conditions. It is extremely difficult to design paired loop primers for Y. pestis LAMP assays when targeting caf1. In the previously reported Y. pestis LAMP assays, only one assay included two paired loop primers (Singh et al., 2020), while other assays used only one single loop primer in their assay development (Nunes et al., 2014; Randriantseheno et al., 2020), which possibly was because the primers failed to generate. Using caf1A, we were able to generate paired loop primers, which gave us a total of six caf1A-specific primers (Table 1) for our LAMP assay development. Although a LAMP assay can work without loop primers, the technology was far too slow for most practical applications in this manifestation, while the addition of loop primers in conjunction with the other primers will significantly increase the speed (Nagamine et al., 2002; Nunes et al., 2014). As a result, our LAMP assay can detect Y. pestis in less than 30 min. This faster completion time greatly expedites results compared to other assays which run between 45 and 60 min using four or five primers (Nunes et al., 2014; Feng et al., 2018; Randriantseheno et al., 2020; Singh et al., 2020).
Sensitivity assessment showed that the LOD of our LAMP assay was 0.2 pg of DNA when performed on the dry bath heater where positive results are visually detected by a color change. In the previous caf1 LAMP assays, Singh et al. (2020) reported the LOD at 0.5 pg; the other two assays reported a much higher LOD, 10 and 3.79 pg, respectively (Nunes et al., 2014; Randriantseheno et al., 2020). Noticeably, Singh et al. included one pair of loop primers in their assay while the other two included one single loop primer. Although different reagents and/or equipment used in each laboratory may have some impacts on the results, the comparisons suggest that inclusion of one pair of loop primers can significantly increase the sensitivity of a LAMP assay. Our LAMP assay is even more sensitive when performed on a real-time PCR instrument with an LOD of 0.03 pg. This was lower than that found by a pentaplex real-time PCR assay that had an LOD of 0.05 pg for caf1 (Bai et al., 2020). These results may have suggested that caf1A is more sensitive than caf1 in Y. pestis detection. Although the LOD of the LAMP assay was low, it still needs further validation for its biological relevance. Nevertheless, because of the extremely high sensitivity, a LAMP assay may risk generating some false amplification if overamplified (Chou et al., 2011). Using the dry bath heater, we observed some minor color change in the negative control when incubation time reached 35 min (Figure 2B). This suggested that the LAMP reaction must be stopped at an earlier time point, and we have set 30 min as the cutoff at which there was no color change for any replicates of the negative control (Figure 2A). One should take extreme caution when using a LAMP assay and make sure that a stringent reaction time cutoff is maintained during testing. Surprisingly, a cutoff time for the LAMP reaction was not determined in many other studies, which may have risked yielding false-positive results. Interestingly, the reaction time seems to have had less of an impact when performing real-time LAMP, from which we did not observe amplifications for the negative control with longer time. On the other hand, if an incomplete color change was observed within 30 min of incubation, intermediate might be considered. Such a result may suggest the targeted DNA is at a very low concentration. An electrophoresis gel may be run to check the presence of the amplicons. Alternatively, visualizing the LAMP products under UV lights may get clearer results as previous LAMP assays did (Nunes et al., 2014; Singh et al., 2020). Although some minor color change in the negative control was observed at 35 min when using the dry bath heater, testing of Y. pseudotuberculosis, Y. enterocolitica, and a variety of other bacterial species using both real-time PCR instrument and the dry bath heater has demonstrated that the LAMP assay has a 100% specificity. This again suggested having a cutoff time for the LAMP reaction is critical.
Amplification time was negatively related to the DNA concentration in the LAMP reactions. In the setup with visual detection, if the DNA concentration is high enough, the assay may take only a few minutes before color change is observed. A LAMP assay can be stopped at any time if the targeted DNA is detected before the assay completes. This is another advantage of LAMP assay over conventional PCR. Nevertheless, a full 30 min incubation should be accomplished when testing field samples.
Inhibition is often a problem when applying a molecular method for pathogen detection, which sometimes causes a false-negative detection (Schrader et al., 2012). The inhibiting substances may present in the analyzed samples and may affect the sensitivity of the assay (Kaneko et al., 2007; Nkouawa et al., 2010). A previous study reported inhibition from mouse liver, spleen, and lung DNA when testing for the presence of Y. pestis using LAMP assay and PCR (Feng et al., 2018). In the current study, we also observed some interference from mouse tissue, which would decrease the sensitivity and increase the time needed for amplification. A strategy to remove the inhibiting effects may be adapted for better consequences (Schrader et al., 2012). This highlights the need to evaluate assay performance when testing alternative samples types.
Using the LAMP assay, we tested 10 Y. pestis strains presenting different plasmid profiles. Strains that possess the pMT1 plasmid were all detected. Other plasmids, present or absent, do not impact the detection of the assay. One strain lacking the pMT1 plasmid was not detected as expected. Over time, Y. pestis strains lacking certain plasmids have been reported infrequently (Butler, 1972; Meka-Mechenko, 2003). To successfully detect different variants and to elucidate the plasmid profile in a particular strain, multi-target LAMP assay(s) would be required.
We applied the assay to test field-collected fleas. The DNA from these fleas had been tested previously using a pentaplex PCR assay (Bai et al., 2020). The results from the LAMP assay were identical to those using the pentaplex real-time PCR, suggesting the utility of the assay for field investigations.
The quality of DNA differs largely when using different extraction methods. DNA samples used in the current work were prepared using commercial kits, e.g., Qiagen kits, but these might not be available in resource-poor settings. Instead, a low-tech solution, such as boiling preparation, may have to be used. One should be aware that the performance of the LAMP assay may be affected by the DNA quality.
In conclusion, we have developed a highly sensitive, specific, and rapid LAMP assay targeting caf1A for rapid detection of Y. pestis. The assay can be performed by simply using a dry bath heater and completes within 30 min by incubation at 65°C. The results can be detected by a visual observation-based color change of the reaction. This feature would make the assay extremely useful in regions with limited resources or during field investigations where immediate access to an equipped facility may not be available. The rapidity of the assay is extremely important for medical management, control, and surveillance of plague. The assay also can be performed using a real-time PCR instrument with higher sensitivity and is recommended when quantitative analysis is favored.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Animal Care and Use Committee at the Division of Vector-Borne Diseases, Centers for Disease Control and Prevention.
Author Contributions
YB conceived, designed, and performed the experiment, analyzed the data, and wrote the manuscript. MR, CP, and SM performed the experiment and reviewed the manuscript. RE supervised, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Author Disclaimer
The conclusion, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the United States Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors’ affiliated institutions.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The following reagents were obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH: genomic DNA from Yersinia pestis, strain A1122, NR-2644; genomic DNA from Yersinia pestis, strain KIM10+, NR-2645; genomic DNA from Yersinia pestis, strain ZE94-2122, NR-2715; genomic DNA from Yersinia pestis, strain PEXU2, NR-2716; genomic DNA from Yersinia pestis, strain CO92, NR-2717; genomic DNA from Yersinia pestis, strain PB, NR-2718; genomic DNA from Yersinia pestis, strain TS D4, NR-4711; genomic DNA from Yersinia pestis, strain A12 D5, NR-4712; genomic DNA from Yersinia pestis, strain Kuma D8, NR-4715; genomic DNA from Yersinia pestis, strain Yokohama D10, NR-4716; genomic DNA from Yersinia pestis, strain Kimberley Derivative 12 (D12), NR-4718; genomic DNA from Yersinia pseudotuberculosis, strain IP2666, NR-4647; genomic DNA from Yersinia pseudotuberculosis, strain IP2515, NR-4648; genomic DNA from Yersinia pseudotuberculosis, strain IP2777, NR-4649; genomic DNA from Yersinia pseudotuberculosis, strain IP2790, NR-4650; genomic DNA from Yersinia pseudotuberculosis, strain IP2775, NR-4651; genomic DNA from Yersinia pseudotuberculosis, strain YPIII (p−), NR-4652; genomic DNA from Yersinia pseudotuberculosis, strain YPIII(p+), NR-4653; genomic DNA from Yersinia enterocolitica, strain Billups-1803-68, NR-3064; genomic DNA from Yersinia enterocolitica, strain Billups-1803-68, NR-3064; genomic DNA from Burkholderia cenocepacia, strain MA00-2987, NR-36042; genomic DNA from Escherichia coli, strain KM14S, NR-2647; genomic DNA from Francisella tularensis ssp. holarctica, strain OR96-0246, NR-3020; genomic DNA from Francisella tularensis ssp. novicida, strain Sterne BA781, NR-3028; genomic DNA from Francisella tularensis ssp. tularensis, strain Weybridge, NR-3017; genomic DNA from Listeria monocytogenes, strain TCH1516, NR-13354; genomic DNA from Salmonella enterica ssp. enterica, strain FSL J2-064, NR-543; genomic DNA from Staphylococcus aureus, strain VEG, NR-12252; and genomic DNA from Toxoplasma gondii, strain LMG 16656, NR-33510. We thank Scott Bearden from the Bacterial Disease Branch, Division of Vector-Borne Diseases, Centers for Disease Control and Prevention for providing genomic DNA from Yersinia pestis, strain CO96-3166, and genomic DNA from Yersinia pseudotuberculosis, strain 1A and strain B15. We thank Vladimir Motin from The University of Texas Medical Branch (UTMB) for providing genomic DNA from Yersinia pseudotuberculosis with ID 4284. We thank Shelby Ford from the Bacterial Diseases Branch, Division of Vector-Borne Diseases, Centers for Disease Control and Prevention for helping with the animal experiment setup. We also thank Erik Foster and Lynn Osikowicz for providing fleas collected from fields.
Footnotes
References
Al-Jawdah, A. D., Ivanova, I. G., Waller, H., Perkins, N. D., Lakey, J. H., and Peters, D. T. (2019). Induction of the immunoprotective coat of Yersinia pestis at body temperature is mediated by the Caf1R transcription factor. BMC Microbiol. 19:68. doi: 10.1186/s12866-019-1444-4
Bai, Y., Motin, V., Enscore, R. E., Osikowicz, L., Rizzo, M. R., Hojgaard, A., et al. (2020). Pentaplex real-time PCR for differential detection of Yersinia pestis and Y. pseudotuberculosis and application for testing fleas collected during plague epizootics. Microbiologyopen 9:e1105. doi: 10.1002/mbo3.1105
Bai, Y., Osikowicz, L. M., Kosoy, M. Y., Eisen, R. J., Atiku, L. A., Mpanga, J. T., et al. (2017). Comparison of zoonotic bacterial agents in fleas collected from small mammals or host-seeking fleas from a Ugandan region where plague is endemic. mSphere 2:e00402-17. doi: 10.1128/mSphere.00402-17
Ben-Gurion, R., and Shafferman, A. (1981). Essential virulence determinants of different Yersinia species are carried on a common plasmid. Plasmid 5, 183–187. doi: 10.1016/0147-619x(81)90019-6
Bodulev, O. L., and Sakharov, I. Y. (2020). Isothermal nucleic acid amplification techniques and their use in bioanalysis. Biochemistry 85, 147–166. doi: 10.1134/S0006297920020030
Boegler, K. A., Atiku, L. A., Enscore, R. E., Apangu, T., Mpanga, J. T., Acayo, S., et al. (2018). Rat fall surveillance coupled with vector control and community education as a plague prevention strategy in the West Nile Region, Uganda. Am. J. Trop. Med. Hyg. 98, 238–247. doi: 10.4269/ajtmh.17-0502
Borchert, J. N., Eisen, R. J., Atiku, L. A., Delorey, M. J., Mpanga, J. T., Babi, N., et al. (2012). Efficacy of indoor residual spraying using lambda-cyhalothrin for controlling nontarget vector fleas (Siphonaptera) on commensal rats in a plague endemic region of northwestern Uganda. J. Med. Entomol. 49, 1027–1034. doi: 10.1603/me11230
Brook, C. E., Bai, Y., Dobson, A. P., Osikowicz, L. M., Ranaivoson, H. C., Zhu, Q., et al. (2015). Bartonella spp. in fruit bats and blood-feeding Ectoparasites in Madagascar. PLoS Negl. Trop. Dis. 9:e0003532. doi: 10.1371/journal.pntd.0003532
Butler, T. (1972). A clinical study of bubonic plague. Observations of the 1970 Vietnam epidemic with emphasis on coagulation studies, skin histology and electrocardiograms. Am. J. Med. 53, 268–276. doi: 10.1016/0002-9343(72)90168-4
Butler, T. (2013). Plague gives surprises in the first decade of the 21st century in the United States and worldwide. Am. J. Trop. Med. Hyg. 89, 788–793. doi: 10.4269/ajtmh.13-0191
Campbell, J., Lowe, J., Walz, S., and Ezzell, J. (1993). Rapid and specific identification of Yersinia pestis by using a nested polymerase chain reaction procedure. J. Clin. Microbiol. 31, 758–759. doi: 10.1128/jcm.31.3.758-759.1993
Casanovas-Massana, A., Pedra, G. G., Wunder, E. A. Jr., Diggle, P. J., Begon, M., and Ko, A. I. (2018). Quantification of leptospira interrogans survival in soil and water microcosms. Appl. Environ. Microbiol. 84:e00507-18. doi: 10.1128/AEM.00507-18
Chou, P. H., Lin, Y. C., Teng, P. H., Chen, C. L., and Lee, P. Y. (2011). Real-time target-specific detection of loop-mediated isothermal amplification for white spot syndrome virus using fluorescence energy transfer-based probes. J. Virol. Methods 173, 67–74. doi: 10.1016/j.jviromet.2011.01.009
Dubnovitsky, A. P., Duck, Z., Kersley, J. E., Härd, T., MacIntyre, S., and Knight, S. D. (2010). Conserved hydrophobic clusters on the surface of the Caf1A usher C-terminal domain are important for F1 antigen assembly. J. Mol. Biol. 403, 243–259. doi: 10.1016/j.jmb.2010.08.034
Ehlers, J., Krüger, A., Rakotondranary, S. J., Ratovonamana, R. Y., Poppert, S., Ganzhorn, J. U., et al. (2020). Molecular detection of Rickettsia spp., Borrelia spp., Bartonella spp. and Yersinia pestis in ectoparasites of endemic and domestic animals in southwest Madagascar. Acta Trop. 205:105339. doi: 10.1016/j.actatropica.2020.105339
Eisen, R. J., Atiku, L. A., Enscore, R. E., Mpanga, J. T., Acayo, S., Mead, P. S., et al. (2021). Epidemiology, ecology and prevention of plague in the West Nile Region of Uganda: the value of long-term field studies. Am. J. Trop. Med. Hyg. 105, 18–23. doi: 10.4269/ajtmh.20-1381
Eisen, R. J., Atiku, L. A., Mpanga, J. T., Enscore, R. E., Acayo, S., Kaggwa, J., et al. (2020). An evaluation of the flea index as a predictor of plague epizootics in the West Nile Region of Uganda. J. Med. Entomol. 57, 893–900. doi: 10.1093/jme/tjz248
Eisen, R. J., Bearden, S. W., Wilder, A. P., Montenieri, J. A., Antolin, M. F., and Gage, K. L. (2006). Early-phase transmission of Yersinia pestis by unblocked fleas as a mechanism explaining rapidly spreading plague epizootics. Proc. Natl. Acad. Sci. U.S.A. 103, 15380–15385. doi: 10.1073/pnas.0606831103
Engelthaler, D. M., Hinnebusch, B. J., Rittner, C. M., and Gage, K. L. (2000). Quantitative competitive PCR as a technique for exploring flea-Yersina pestis dynamics. Am. J. Trop. Med. Hyg. 62, 552–560. doi: 10.4269/ajtmh.2000.62.552
Enscore, R. E., Babi, N., Amatre, G., Atiku, L., Eisen, R. J., Pepin, K. M., et al. (2020). The changing triad of plague in Uganda: invasive black rats (Rattus rattus), indigenous small mammals, and their fleas. J. Vector Ecol. 45, 333–355. doi: 10.1111/jvec.12404
Enscore, R. E., Bai, Y., Osikowicz, L., Sexton, C., and O’Leary, D. R. (2021). Evaluation of a liquid carbaryl formulation to control burrow fleas following a die-off of black-tailed prairie dogs (Cynomys ludovicianus) caused by plague (Yersinia pestis) in Converse County, Wyoming. J. Vector Ecol. 46, 230–232. doi: 10.52707/1081-1710-46.2.230
Feng, N., Zhou, Y., Fan, Y., Bi, Y., Yang, R., Zhou, Y., et al. (2018). Yersinia pestis detection by loop-mediated isothermal amplification combined with magnetic bead capture of DNA. Braz. J. Microbiol. 49, 128–137. doi: 10.1016/j.bjm.2017.03.014
Ferber, D. M., and Brubaker, R. R. (1981). Plasmids in Yersinia pestis. Infect. Immun. 31, 839–841. doi: 10.1128/iai.31.2.839-841.1981
Filippov, A. A., Solodovnikov, N. S., and Protsenko, O. A. (1990). Plasmid content in Yersinia pestis strains of different origin. FEMS Microbiol. Lett. 67, 45–48. doi: 10.1111/j.1574-6968.1990.tb13833.x
Gage, K. L. (1999). Plague Manual: Epidemiology, Distribution, Surveillance and Control. Geneva: World Health Organization, 135–165.
Galyov, E. E., Smirnov, O., Karlishev, A. V., Volkovoy, K. L., Denesyuk, A. I., Nazimov, I. V., et al. (1990). Nucleotide sequence of the Yersinia pestis gene encoding F1 antigen and the primary structure of the protein. Putative T and B cell epitopes. FEBS Lett. 277, 230–232. doi: 10.1016/0014-5793(90)80852-a
Gonçalves, D. S., Cassimiro, A. P. A., de Oliveira, C. D., Rodrigues, N. B., and Moreira, L. A. (2014). Wolbachia detection in insects through LAMP: loop mediated isothermal amplification. Parasit. Vectors 7:228. doi: 10.1186/1756-3305-7-228
Grácio, A. J. D. S., and Grácio, M. A. A. (2017). Plague: a millenary infectious disease reemerging in the XXI century. Biomed. Res. Int. 17:5696542. doi: 10.1155/2017/5696542
Hanson, D. A., Britten, H. B., Restani, M., and Washburn, L. R. (2007). High prevalence of Yersinia pestis in black-tailed prairie dog colonies during an apparent enzootic phase of sylvatic plague. Conserv. Genet. 8, 789–795. doi: 10.1007/s10592-006-9226-6
Hsu, C. C., Chuang, C. C., Liang, C. C., Chiao, D. J., Wu, H. L., Wu, Y. P., et al. (2018). Rapid and sensitive detection of Yersinia pestis by lateral-flow assay in simulated clinical samples. BMC Infect. Dis. 18:402. doi: 10.1186/s12879-018-3315-2
Kaneko, H., Kawana, T., Fukushima, E., and Suzutani, T. (2007). Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J. Biochem. Biophys. Methods 70, 499–501. doi: 10.1016/j.jbbm.2006.08.008
Karlyshev, A. V., Galyov, E. E., Smirnov, O., Guzayev, A. P., Abramov, V. M., and Zav’yalov, V. P. (1992). A new gene of the f1 operon of Y. pestis involved in the capsule biogenesis. FEBS Lett. 297, 77–80. doi: 10.1016/0014-5793(92)80331-a
Kuboki, N., Inoue, N., Sakurai, T., Cello, F. D., Grab, D. J., Suzuk, H., et al. (2003). Loop-mediated isothermal amplification for detection of African trypanosomes. J. Clin. Microbiol. 41, 5517–5524. doi: 10.1128/JCM.41.12.5517-5524.2003
Lass, A., Szostakowska, B., Korzeniewski, K., and Karanis, P. (2017). The first detection of Toxoplasma gondii DNA in environmental air samples using gelatine filters, real-time PCR and loop-mediated isothermal (LAMP) assays: qualitative and quantitative analysis. Parasitology 144, 1791–1801. doi: 10.1017/S0031182017001172
Lu, R., Wu, X., Wan, Z., Li, Y., Jin, X., and Zhang, C. (2020). A novel reverse transcription loop-mediated isothermal amplification method for rapid detection of SARS-CoV-2. Int. J. Mol. Sci. 21:2826. doi: 10.3390/ijms21082826
Meka-Mechenko, T. V. (2003). F1-negative natural Y. pestis strains. Adv. Exp. Med. Biol. 529, 379–381. doi: 10.1007/0-306-48416-1_76
Moore, S. M., Monaghan, A., Borchert, J. N., Mpanga, J. T., Atiku, L. A., Boegler, K. A., et al. (2015). Seasonal fluctuations of small mammal and flea communities in a Ugandan plague focus: evidence to implicate Arvicanthis niloticus and Crocidura spp. as key hosts in Yersinia pestis transmission. Parasit. Vectors 8:11. doi: 10.1186/s13071-014-0616-1
Mukama, C., Nie, J. D., Habimana, X., Meng, Y., Ting, F., Songwe, A., et al. (2020). Synergetic performance of isothermal amplification techniques and lateral flow approach for nucleic acid diagnostics. Anal. Biochem. 600:113762. doi: 10.1016/j.ab.2020.113762
Nagamine, K., Hase, T., and Notomi, T. (2002). Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 16, 223–229. doi: 10.1006/mcpr.2002.0415
Neubauer, H., Meyer, H., Prior, J., Aleksic, S., Hensel, A., and Splettstosser, W. (2000). A combination of different polymerase chain reaction (PCR) assays for the presumptive identification of Yersinia pestis. J. Vet. Med. B Infect. Dis. Vet. Public Health 47, 573–580. doi: 10.1046/j.1439-0450.2000.00384.x
Nkouawa, A., Sako, Y., Li, T., Chen, X., Wandra, T., Swastika, I. K., et al. (2010). Evaluation of a loop-mediated isothermal amplification method using fecal specimens for differential detection of Taenia species from humans. J. Clin. Microbiol. 48, 3350–3352. doi: 10.1128/JCM.00697-10
Norkina, O. V., Kulichenko, A. N., Gintsburg, A. L., Tuchkov, I. V., Popov, Y., Aksenov, M. U., et al. (1994). Development of a diagnostic test for Yersinia pestis by the polymerase chain reaction. J. Appl. Bacteriol. 76, 240–245.
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:E63.
Nunes, M. L., Mendes-Marques, C. L., Almeida, A. M. P., and Leal, N. C. (2014). The development of a loop-mediated isothermal amplification (LAMP) procedure for plague diagnostic. Am. J. Analyt. Chem. 5, 1069–1077. doi: 10.4236/ajac.2014.516114
Pal, V., Saxena, A., Singh, S., Goel, A. K., Kumar, J. S., Parida, M. M., et al. (2018). Development of a real-time loop-mediated isothermal amplification assay for detection of Burkholderia mallei. Transbound. Emerg. Dis. 65, e32–e39. doi: 10.1111/tbed.12665
Pawelczyk, O., Asman, M., and Solarz, K. (2019). The molecular detection of Anaplasma phagocytophilum and Rickettsia spp. in cat and dog fleas collected from companion animals. Folia Parasitol. 66:020. doi: 10.14411/fp.2019.020
Randriantseheno, L. N., Rahantamalala, A., Randrianierenana, A. L., Rajerison, M., and Andrianaivoarimanana, V. (2020). Development and evaluation of loop-mediated isothermal amplification for detection of Yersinia pestis in plague biological samples. PLoS One 15:e0237655. doi: 10.1371/journal.pone.0237655
Runco, L. M., Myrczek, S., Bliska, J. B., and Thanassi, D. G. (2008). Biogenesis of the fraction 1 capsule and analysis of the ultrastructure of Yersinia pestis. J. Bacteriol. 190, 3381–3385. doi: 10.1128/JB.01840-07
Schrader, C., Schielke, A., Ellerbroek, L., and Johne, R. (2012). PCR inhibitors - occurrence, properties and removal. J. Appl. Microbiol. 113, 1014–1026. doi: 10.1111/j.1365-2672.2012.05384.x
Sebbane, F., Jarrett, C., Gardner, D., Long, D., Hinnebusch, B. J., and Sebbane, F. (2009). The Yersinia pestis caf1M1A1 fimbrial capsule operon promotes transmission by flea bite in a mouse model of bubonic plague. Infect. Immun. 77, 1222–1229. doi: 10.1128/IAI.00950-08
Simon, S., Demeure, C., Lamourette, P., Filali, S., Plaisance, M., Creminon, C., et al. (2013). Fast and simple detection of Yersinia pestis applicable to field investigation of plague foci. PLoS One 8:e54947. doi: 10.1371/journal.pone.0054947
Singh, R., Pal, V., Tripathi, N. K., and Goel, A. K. (2020). Development of a pair of real-time loop mediated isothermal amplification assays for detection of Yersinia pestis, the causative agent of plague. Mol. Cell. Probes 54:101670. doi: 10.1016/j.mcp.2020.101670
Splettstoesser, W. D., Rahalison, L., Grunow, R., Neubauer, H., and Chanteau, S. (2004). Evaluation of a standardized F1 capsular antigen capture ELISA test kit for the rapid diagnosis of plague. FEMS Immunol. Med. Microbiol. 41, 149–155. doi: 10.1016/j.femsim.2004.02.005
Stenseth, N. C., Atshabar, B. B., Begon, M., Belmain, S., Bertherat, E., Carniel, E., et al. (2008). Plague: past, present, and future. PLoS Med. 5:e3. doi: 10.1371/journal.pmed.0050003
Stewart, A., Satterfield, B., Cohen, M., O’Neill, K., and Robison, R. (2008). A quadruplex real-time PCR assay for the detection of Yersinia pestis and its plasmids. J. Med. Microbiol. 57(Pt 3), 324–331. doi: 10.1099/jmm.0.47485-0
Keywords: assay development, LAMP, dry bath heater, real-time LAMP, plague, Yersinia pestis
Citation: Bai Y, Rizzo MR, Parise C, Maes S and Eisen RJ (2022) A Novel Loop-Mediated Isothermal Amplification Assay for Rapid Detection of Yersinia pestis. Front. Microbiol. 13:863142. doi: 10.3389/fmicb.2022.863142
Received: 26 January 2022; Accepted: 23 February 2022;
Published: 07 April 2022.
Edited by:
Jens Andre Hammerl, Bundesinstitut für Risikobewertung, GermanyReviewed by:
Jason Sahl, Northern Arizona University, United StatesHugh Britten, University of South Dakota, United States
Copyright © 2022 Bai, Rizzo, Parise, Maes and Eisen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Bai, bby5@cdc.gov