1
Higuchi Biosciences Center, The University of Kansas, Lawrence, KS, USA
2
Department of Pharmacology and Toxicology, The University of Kansas, Lawrence, KS, USA
Oxidative stress (OS), caused by the imbalance between the generation and detoxification of reactive oxygen and nitrogen species (ROS/RNS), plays an important role in brain aging, neurodegenerative diseases, and other related adverse conditions, such as ischemia. While ROS/RNS serve as signaling molecules at physiological levels, an excessive amount of these molecules leads to oxidative modification and, therefore, dysfunction of proteins, nucleic acids, and lipids. The response of neurons to this pervasive stress, however, is not uniform in the brain. While many brain neurons can cope with a rise in OS, there are select populations of neurons in the brain that are vulnerable. Because of their selective vulnerability, these neurons are usually the first to exhibit functional decline and cell death during normal aging, or in age-associated neurodegenerative diseases, such as Alzheimer’s disease. Understanding the molecular and cellular mechanisms of selective neuronal vulnerability (SNV) to OS is important in the development of future intervention approaches to protect such vulnerable neurons from the stresses of the aging process and the pathological states that lead to neurodegeneration. In this review, the currently known molecular and cellular factors that contribute to SNV to OS are summarized. Included among the major underlying factors are high intrinsic OS, high demand for ROS/RNS-based signaling, low ATP production, mitochondrial dysfunction, and high inflammatory response in vulnerable neurons. The contribution to the selective vulnerability of neurons to OS by other intrinsic or extrinsic factors, such as deficient DNA damage repair, low calcium-buffering capacity, and glutamate excitotoxicity, are also discussed.
The approximately 100 billion neurons in the human brain orchestrate an incredibly wide range of motor and internal regulatory functions, such as body movement, balance, visual and auditory perception, pleasure, pain and thermal sensations, hormonal and metabolic regulation, as well as highly complex behaviors, such as language, memory, learning, and executive functions. Such diverse functional output is the product of molecular events occurring in neurons. Small variations in the thousands of chemical reactions occurring in neurons lead to different morphological and functional characteristics among the billions of neurons. Morphologically, central nervous system (CNS) neurons differ in size, the number and complexity of dendrites, number of synaptic connections, length of axons and distance across which synaptic connections are established, extent of axonal myelination, and other morphological characteristics. Neurons can also be classified chemically on the basis of the neurotransmitters they use for chemical transmission or neuromodulation, e.g., glutamate, GABA, acetylcholine, dopamine, adenosine, or peptide transmitters and neuromodulators. This great diversity among neuronal populations is a strong indication that although all neurons contain the same genetic code in their genome, each neuronal population has their own unique gene expression profile as to what parts of the genome are active and at what levels. While the diversity of neuronal structures and functions are well documented by the neuroscience community, what is less appreciated is the diverse response of brain neurons to stresses and adverse factors during aging or as a result of neurodegenerative diseases.
A pronounced yet underappreciated phenomenon in the response of different neuronal populations to stressful neurodegenerative conditions is the appearance of selective neuronal vulnerability (SNV). SNV refers to the differential sensitivity of neuronal populations in the CNS to stresses that cause cell injury or death and lead to neurodegeneration. For example, neurons in the entorhinal cortex, hippocampus CA1 region, frontal cortex, and amygdala are the populations of neurons most sensitive to the neurodegeneration associated with Alzheimer’s disease (AD) (Hyman et al., 1984
; Braak and Braak, 1991
; Terry et al., 1991
). In Parkinson’s disease (PD), dopaminergic neurons of the substantia nigra are the primary neurons undergoing cell death (Hirsch et al., 1988
; Damier et al., 1999
; Dauer and Przedborski, 2003
). And, amyotrophic lateral sclerosis (ALS) is characterized by the degeneration of, primarily, spinal motor neurons, but also cortical and brain stem neurons (Rowland and Shneider, 2001
). The fact that specific brain regions exhibit differential vulnerabilities to various neurodegenerative diseases is a reflection of both the specificity in the etiology of each disease and of the heterogeneity in neuronal responses to cell-damaging processes associated with each of the diseases. The appearance of SNV is not limited to cross-regional differences in the brain, as within a single brain region, such as the hippocampus or the entorhinal cortex, SNV is manifested as internal, sub-regional differences in relative sensitivities to stress and disease. For example, the hippocampal CA1 neurons are much more vulnerable than CA3 neurons to a variety of adverse conditions including global cerebral ischemia (Schmidt-Kastner and Freund, 1991
; Olsson et al., 2003
), early stages of AD (O’Banion et al., 1994
), chronic epileptic seizures (Mathern et al., 1997
), aging (Mueller et al., 2007
), and oxidative stress (OS) (Wilde et al., 1997
; Wang et al., 2005
). Similarly, transentorhinal neurons undergo neurodegeneration before entorhinal proper and CA1 hippocampus neurons (Braak and Braak, 1991
).
In the CNS, excessive production of reactive oxygen and nitrogen species (ROS/RNS) has been invoked as a mechanism for neurodegeneration associated with various insults to neurons, such as hypoxia and hypoglycemia (Halliwell et al., 1992
; Friberg et al., 2002
), as well as with the neurodegeneration seen in AD (Zhu et al., 2004b
), PD (Fahn and Cohen, 1992
; Jenner, 2003
; van Muiswinkel et al., 2004
) and ALS (Carri et al., 2003
). OS is caused by an imbalance between overproduction of ROS and/or RNS and the enzymatic or non-enzymatic detoxification of these highly reactive species. The highly reactive ROS and RNS, when over-produced or under-detoxified due to factors such as aging and disease, are detrimental to cells since they chemically modify lipids, proteins, and nucleic acids. Such oxidative modification of macromolecules may be an initiating event in the causation of neuronal injury. As mentioned above, brain neurons respond to such stresses differently. While most brain neurons can tolerate OS well, neurons in certain parts of the brain, such as those in the hippocampal CA1 region and cerebellar granule cell layer, are particularly vulnerable to OS (Wilde et al., 1997
; Wang et al., 2005
, 2007
, 2009
). The vulnerability of the cerebellar granule neurons to OS might play an important role in their significant loss in aged individuals (Renovell et al., 1996
; Andersen et al., 2003
). As OS appears to be a common underlying factor in the various adverse conditions characterized by SNV (Sayre et al., 2001
; Valko et al., 2007
), the study of SNV to OS might improve our understanding of how this particular form of cell stress causes selective neuronal losses in brain, as well as reveal potential molecular and cellular mechanisms that bring about relative resistance or sensitivity of neurons to stresses.
Oxygen is essential for the survival of aerobic organisms. Because of its high redox potential, it serves as the terminal electron acceptor in the process of metabolic energy generation through a series of redox reactions. Unfortunately, its high redox potential can also be damaging to cells, if it is not completely reduced. Partial reduction of molecular oxygen is an unintended occurrence during aerobic metabolism and may lead to the generation of highly reactive species, including singlet oxygen, superoxide anion, hydrogen peroxide, hydroxyl radical, and peroxyl radical. These ROS can disrupt the redox balance inside cells if not properly neutralized. RNS, including nitric oxide, nitrogen dioxide, dinitrogen trioxide and peroxynitrite, are similarly disruptive to the redox state of cells if not detoxified. Nitric oxide is a product of the reaction catalyzed by nitric oxide synthase. Other RNS species, such as peroxynitrite, are derived from the reaction of nitric oxide with superoxide. Nitric oxide is relatively unreactive, with the exception of reaction with sulfhydryl-containing peptides and proteins (nitrosylation reactions) (Stamler et al., 1992
); on the other hand, peroxynitrite is a more highly reactive species that can nitrate proteins on select tyrosine residues and thus alter the structure and function of such proteins (Viner et al., 1996
). In cells, over-production and/or under-detoxification of ROS/RNS, or even normal demands for these reactive species due to their beneficial effects (see below), may cause oxidative/nitrosative stress.
Many of the modifications of lipids, nucleic acids, and proteins result in structural changes in the respective macromolecules and lead to either dysfunction or loss of activity of these molecules. To protect themselves from the detrimental effects of these oxidative modifications, neurons employ a variety of defensive mechanisms that include lipid turnover, protein re-folding or degradation, and DNA base excision and repair. When these mechanisms are compromised, neuronal homeostasis is disturbed and OS ensues. Among all organs in the body, the brain is particularly prone to OS-induced damage because of the high oxygen demand of this organ, the abundance of redox-active metals (iron and copper), the high levels of oxidizable polyunsaturated fatty acids, and the fact that neurons are post-mitotic cells with relatively restricted replenishment by progenitor cells during the lifespan of an organism.
Induction of OS in neurodegenerative diseases may result from changes in neuronal metabolism brought about by the accumulation of certain macromolecules. For example, the accumulation of the oligomeric form of amyloid-β peptide (Aβ), the most toxic form of Aβ, induces OS in neurons. The redox potential of different types of Aβ species (human Aβ42 > human Aβ40 >> rodent Aβ) matches the order of their toxicity to neurons (Huang et al., 1999a
), which suggests that OS plays a causative role in the development of AD. In addition, redox-active transition metal ions that are known to increase ROS formation through the Fenton reaction, such as iron and copper, bind with high affinity to Aβ (Huang et al., 1999a
,b
; Atwood et al., 2000
). The link between AD and OS is further supported by the increased levels of lipid peroxidation, protein and DNA oxidation in neurons affected byAD (Smith et al., 1997
; Gabbita et al., 1998
; Montine et al., 1998
; Butterfield and Lauderback, 2002
; Markesbery and Lovell, 2006
; Pratico, 2008
). In addition, neurons affected by AD are characterized by decreased ATP production and diminished cytochrome c oxidase content, both of which are indices of mitochondrial dysfunction and, as dysfunctional mitochondria are a major source of ROS generation, increased OS (Zhu et al., 2004a
; Onyango and Khan, 2006
; Reddy, 2007
).
In PD, neurons in the substantia nigra pars compacta region are selectively lost. The role of OS in this disease is supported by the observation of dramatically diminished levels of glutathione as one of the earliest detectable changes during the development of this disease in this region (Pearce et al., 1997
). The iron levels in this brain region are high, as is the activity of monoamine oxidase, and both of these entities contribute to the generation of ROS in these neurons. The accumulated free radicals in the substantia nigra pars compacta neurons can lead to aberrant oxidation of dopamine and the formation of 6-hydrodopamine (6-OHDA), which then undergoes auto-oxidation to the quinone form with the generation of superoxide (Heikkila and Cohen, 1973
). This cascade of events, amplified by the redox cycling of the quinone, leads to significant increases in OS and ultimately the demise of the dopaminergic neurons in this region. Several agents that lead to the formation of ROS, including MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and paraquat, can reproduce a selective neuronal death pattern similar to that observed in PD (Moratalla et al., 1992
; Brooks et al., 1999
; McCormack et al., 2002
).
The involvement of OS in brain aging and neurodegenerative conditions, such as ischemia, seizures, calcium dysfunction and glutamate-mediated excitotoxicity, are described in existing reviews (Coyle and Puttfarcken, 1993
; Sayre et al., 2001
; Floyd and Hensley, 2002
; Valko et al., 2007
) and not repeated here.
Over millions of years of evolution, cells have developed a multitude of anti-oxidant mechanisms to cope with the potential damaging effects of ROS and RNS. These mechanisms include anti-oxidant enzymes (such as superoxide dismutase, catalase, glutathione peroxidase, and the thioredoxins), as well as non- protein antioxidants (such as glutathione [GSH], α-tocopherol [vitamin E], ascorbic acid [vitamin C], bilirubin, and coenzyme Q10). Glutathione is a major non-protein antioxidant in cells and the ratio of its reduced and oxidized form (GSH/GSSG) is an indicator of cellular redox status (Schafer and Buettner, 2001
; Jones, 2002
). The combination of anti-oxidant proteins and smaller molecules offers a versatile and flexible system to control intracellular levels of ROS/RNS. The high efficiency of this system in cells, including neurons, is maintained through the diverse subcellular localization of the antioxidants, their biochemical properties, and the differential inducibility of anti-oxidant enzymes at both transcriptional and translational levels.
Although cellular antioxidants are highly efficient in maintaining redox homeostasis, such anti-oxidant defenses do not completely eradicate ROS/RNS from the intracellular environment. Nor would such eradication be desirable as cells also use ROS/RNS as signaling molecules to monitor changes in the internal and external environment. Scientific thinking with regard to OS has evolved over the years from one of viewing all ROS and RNS as “evil-doers”, to one of considering them as “double-edged swords”, because these damaging reactive species are also beneficial to cells at physiological levels (Valko et al., 2007
). The coining of the term “oxidative stress” is a reflection of the historical view of seeing ROS and RNS as purely harmful molecules. More recent scientific data support the idea that these species are essential components of a repertoire of signals that neurons depend on to respond to environmental and developmental cues (Gutierrez et al., 2006
). In the CNS, ROS and RNS are used as secondary messengers in many neurological processes. Neurons can sense, transmit and convert ROS/RNS signals into appropriate intracellular responses, including synaptic plasticity (Kamsler and Segal, 2004
, 2007
; Serrano and Klann, 2004
; Kishida and Klann, 2007
). Such redox sensing and signal transduction processes require a network of redox-sensitive metabolic and signaling pathways. These pathways contain redox-sensitive proteins that can undergo reversible oxidation/reduction and thus modulate their functions based on the cellular redox status (Kohr et al., 1994
; Suzuki et al., 1997
; Forman et al., 2002
; Lipton et al., 2002
; Hidalgo, 2005
; Janssen-Heininger et al., 2008
). The modulation of redox-sensitive proteins can be conferred by ROS/RNS either directly or indirectly through other redox-sensitive molecules such as glutathione or thioredoxins. It is currently known that many of these proteins contain redox-sensitive cysteine residues that may be oxidized to either sulfenic acid or disulfide bonds, thus producing changes in structural and functional states of the respective proteins (Denu and Tanner, 1998
; Droge, 2003
; Piotukh et al., 2007
). The reversible oxidation/reduction changes in these redox-sensing proteins lead to downstream metabolic and signaling events, for example, modulation of cellular metabolism and synaptic plasticity through activation/deactivation of a variety of signaling effectors, such as kinases, phosphatases and transcription factors (e.g., Nrf2 and NF-κB) (Arrigo, 1999
).
The duality of ROS/RNS as both signaling and stress molecules is exemplified by the effects of these reactive species on synaptic plasticity. At high concentrations, ROS and RNS, such as hydrogen peroxide and hydroxyl radical, attenuate long-term potentiation (LTP) and synaptic neurotransmission (Colton et al., 1989
; Pellmar et al., 1991
; Gahtan et al., 1998
; Avshalumov et al., 2000
). This might be partially due to the inhibition of glutamatergic N-methyl-D-aspartate (NMDA) receptors by ROS/RNS through excessive oxidation of the extracellular redox-sensitive sites of these receptors (Steullet et al., 2006
). On the other hand, at lower (“physiological”) levels, ROS such as superoxide and hydrogen peroxide enhance LTP and synaptic neurotransmission (Thiels et al., 2000
; Thiels and Klann, 2002
; Kamsler and Segal, 2003
, 2004
). Signaling in neurons based on formation and release of ROS and RNS is produced through reversible modification of redox-sensitive sites on target proteins, such as ryanodine receptors (RyRs) (Hidalgo, 2005
; Huddleston et al., 2008
). These redox modulations lead to a cascade of downstream events, such as phosphorylation and activation of ERK (extracellular signal-regulated kinase), PKC (protein kinase C), and CREB (cAMP-responsive element-binding protein), all of which are indispensable for LTP and synaptic plasticity (English and Sweatt, 1997
; Klann et al., 1998
; Knapp and Klann, 2002
; Silva, 2003
; Hidalgo, 2005
; Kemmerling et al., 2007
).
Excessive amounts of ROS and RNS cause OS in all neurons, yet, the vulnerability of neurons to OS varies from one brain region to another. Selective vulnerability of neurons to OS is one type of SNV in the brain. Since OS is a pervasive stress involved in many neurodegenerative conditions, studies on SNV to OS not only provide mechanistic insights into this particular form of SNV but may also shed light on the phenomenon of SNV, in general.
The hippocampus is an ideal brain area for the study of SNV caused by OS. The majority of neurons in this important brain area are densely packed into a single layer, which is divided into several regions, i.e., CA1 through CA4. The hippocampal regions CA1 and CA3 are adjacent to each other and are composed of morphologically similar neurons, pyramidal neurons. Despite their physical proximity and cell morphological similarity, CA1 and CA3 neurons respond to OS very differently. When exposed to OS-generating agents, the pyramidal neurons in the CA1 region suffer massive cell death while those in CA3 mostly survive (Wilde et al., 1997
; Vornov et al., 1998
; Sarnowska, 2002
; Wang et al., 2005
). This is the case regardless of the agents used to generate OS. Superoxide-producing agents, such as duroquinone or paraquat, lead to the selective destruction of CA1 neurons (Wilde et al., 1997
; Vornov et al., 1998
; Wang et al., 2005
). Ferrous sulphate (FeSO4), a hydroxyl radical generator, also produces the same effect (Wang et al., 2005
), as does another ROS molecule, hydrogen peroxide (Sarnowska, 2002
). This pattern of selective sensitivity of CA1 neurons to OS matches that of selective vulnerability of CA1 neurons to other adverse or disease-related conditions, such as hypoxia, ischemia, and neurodegeneration in AD.
Outside the hippocampus, OS-inducing agents, such as paraquat or xanthine/xanthine oxidase, cause extensive death of neurons in the cerebellar granule cell layer, but not in the cerebral cortex (layers IV-VI) (Satoh et al., 1998
; Wang et al., 2009
). Cerebellar granule neurons are so sensitive to OS that even the oxygen tension in the ambient air (∼20%) under in vitro culture conditions causes increased cell death among these neurons as compared with effects of the oxygen tension similar to that in the body (5%) (Wang et al., 2009
). In comparison, most cerebral cortical neurons in primary cultures survive either induction of OS by paraquat, or exposure to 20% oxygen tension. This differential response to OS of cerebellar granule and cerebral cortical neurons is consistent with other observations under conditions involving OS, including methyl mercury (Kaur et al., 2007
), ischemia and re-oxygenation (Scorziello et al., 2001
). The vulnerability of cerebellar granule neurons to OS is also in agreement with their significant loss in aged individuals (Renovell et al., 1996
; Andersen et al., 2003
). Selective vulnerability of cerebellar granule neurons to OS might underlie the poor motor coordination and impaired motor learning associated with the aging process.
The two major populations of dopaminergic neurons in midbrain, i.e. those in the substantia nigra pars compacta (A9) and the nearby ventral tegmental area (A10), also show differential vulnerability to OS. Although they are electrophysiologically similar, A9 neurons are susceptible to OS while A10 neurons are mostly resistant. The A9 neurons are also the ones that are selectively lost in PD. In both populations of dopaminergic neurons, the auto-oxidation of dopamine itself and its metabolites can lead to the production of ROS, which is worsened by abundant content of iron and copper in this region (Olney et al., 1990
; Jenner et al., 1992
). In addition, treating the two neuronal populations with MPTP, 6-OHDA, or paraquat, all of which induce OS, can easily differentiate these two groups of neurons based on their survival pattern (Burns et al., 1983
; Waters et al., 1987
; German et al., 1988
; Hung and Lee, 1998
; Rodriguez et al., 2001
; McCormack et al., 2005
, 2006
). Most A9 neurons die following such treatments, while A10 neurons mostly survive the same type of treatment.
Studies of SNV to OS have been focused for the most part on neurons in the CNS, yet neurons in the peripheral nervous system also display differential vulnerability to OS. For example, sympathetic neurons in the celiac and superior mesenteric ganglia (CG/SMG) are more sensitive to menadione-induced OS than those in the superior cervical ganglion (SCG) (Semra et al., 2006
). The induction of OS by menadione is through its one-electron reduction to a semiquinone radical, which then generates large amounts of superoxide via redox cycling with molecular oxygen (Thor et al., 1982
). Whereas the concentration of 1 nM menadione caused a significant loss of viability in CG/SMG neurons, the same concentration applied to SCG neurons had no significant effect. This differential vulnerability is consistent with the selective degeneration of CG/SMG neurons during aging or glucose-induced OS in diabetes-associated autonomic neuropathy (Schmidt et al., 1993
, 1997
).
SNV is a reflection of the heterogeneity of neurons in the nervous system. From the list of OS-vulnerable neurons described in the preceding sections, it is clear that different populations of neurons, in different brain regions, have different morphologies and biochemical characteristics. Due to this diversity, it is very likely that each neuronal population has a unique molecular composition that determines its level of vulnerability to OS. Thus, mechanisms underlying the vulnerability of the hippocampal CA1 neurons may be somewhat different from those of the dopaminergic neurons in the substantia nigra pars compacta. Close examination of the existing literature in the field, although still limited, may identify some common factors that are shared by many currently known OS-vulnerable neurons. The purpose of this review is to first present an overview on these common factors from existing studies on OS-induced neuronal vulnerability, and second to raise the awareness in the neuroscience field on this particular form of SNV and on the general phenomenon of SNV.
High Intrinsic OS in Vulnerable Neurons
It might sound overly simplistic to explain the sensitivity of vulnerable neurons as being the result of pre-existing high OS in these neurons. The reasoning behind this hypothesis is that when further OS increase is encountered, due to either endogenous or exogenous factors, the vulnerable neurons will be overwhelmed and consequently suffer cell death. High baseline OS condition is indeed an intrinsic characteristic of a number of vulnerable neuronal populations. In the case of hippocampal CA1 and CA3 neurons, a measurement of superoxide formation using the dihydroethidine method showed that the vulnerable CA1 neurons contain significantly higher levels of superoxide anion than the resistant CA3 neurons (Wang et al., 2005
). Another line of evidence for a higher level of endogenous OS in CA1 neurons comes from measurement of ROS production from purified mitochondria using H2DCFDA (2’,7’-dichlorodihydrofluorescein-diacetate). Mitochondria isolated from the CA1 release more ROS than those from the CA3 region (Mattiasson et al., 2003a
).
Transcriptomic studies also show that neurons from brain regions vulnerable to OS express higher levels of genes related to OS response than those in resistant regions, which is a further indication of an endogenous higher OS status in neurons within vulnerable regions. A mapping of basal condition CA1/CA3 microarray data to an OS response pathway that consists of induced antioxidant and repressed ROS-producing genes (Morel and Barouki, 1999
), showed that CA1 neurons express both anti-oxidant and ROS-producing genes at significantly higher levels than those in CA3 (Wang et al., 2005
). One of these genes, Nqo1, encodes the enzyme NAD(P)H:quinone oxidoreductase 1, an important anti-oxidant in the maintenance of cellular redox homeostasis. Due to its importance in maintaining the redox status in cells, Nqo1 is often employed as an indicator of cellular redox status (Raina et al., 1999
; Wang et al., 2000
; SantaCruz et al., 2004
; van Muiswinkel et al., 2004
). The significantly higher expression of this gene in CA1 than in CA3 is an additional indication of endogenously higher levels of OS status in the CA1 region. Nqo1 is one of the target genes of the key anti-oxidant transcription factor Nrf2, which is also significantly more active in CA1 neurons (Wang et al., 2005
). Nrf2 activates the transcription of a multitude of downstream anti-oxidant genes via binding to the cis-acting anti-oxidant response element (ARE) of these genes (Nguyen et al., 2004
). The concerted higher expression of Nqo1, Nrf2 and other anti-oxidant genes in CA1, in comparison to CA3, is further evidence for the presence of high OS in neurons of this vulnerable region under basal conditions. After treatment with ROS-generating agents, such as duroquinone, stress-response genes in CA1 are consistently expressed at higher levels than their counterparts in CA3, indicating that the stress level stays higher in vulnerable neurons before reaching the point of neuronal death (Wang et al., 2007
).
The same differential OS profile is characteristic of the contrasting case of vulnerable cerebellar granule neurons vs. the resistant cerebral cortical neurons, with the vulnerable cerebellar granule neurons expressing higher levels of genes related to OS than the resistant cortical neurons (Wang et al., 2009
). In addition, the A9/A10 dopaminergic neurons in midbrain showed a similar transcriptomic pattern to CA1 or cerebellar granule neurons that is characterized by higher expression of anti-oxidant genes in the vulnerable A9 than in the resistant A10 neurons (Chung et al., 2005
). Included among the genes of higher expression in A9 neurons were Glrx3 (or Txnl2, glutaredoxin 3), Gpx4 (glutathione peroxidase 4), Oxr1 (oxidation resistance 1), Prdx2 (peroxiredoxin 2), and Sod2 (superoxide dismutase 2). Because this pattern of gene expression is suggestive of a high intrinsic OS status in vulnerable neurons, despite their location in different regions of the brain, it appears that a commonality among these various types of neurons is an endogenously high level of ROS/RNS formation. Logical questions to ask then are: why do vulnerable neurons contain more ROS/RNS and why do they have higher intrinsic OS than resistant neurons? The answer may lie in the differential requirements of vulnerable and resistant neurons for ROS and RNS as signaling molecules as detailed next.
Differential Signaling Requirements for ROS and RNS
As mentioned earlier, while ROS and RNS are neurotoxic at high concentrations, they are also indispensable as signaling molecules at physiological levels for crucial neuronal functions, including enhancing synaptic plasticity, LTP and memory formation. In connection with the phenomenon of SNV, the enhancement of synaptic plasticity and LTP by ROS/RNS seems to be neuron-type-specific. For example, CA1 neurons, but not CA3 neurons, require superoxide for LTP, and the use of scavengers of superoxide leads to impaired LTP in CA1 but not in CA3 (Klann, 1998
; Thiels et al., 2000
; Knapp and Klann, 2002
; Huddleston et al., 2008
). The ROS-induced form of LTP requires the activation of RyR3, a subtype of the RyRs (Huddleston et al., 2008
). The modulation of RyR3 activity by ROS leads to the activation of downstream factors, including ERK and CREB. Intriguingly, this specific subtype of ryanodine receptor is particularly enriched in CA1 neurons but not in CA3 (Mori et al., 2000
). The differential signaling requirements for ROS/RNS in different neuronal populations might explain why vulnerable neurons maintain higher levels of these reactive species and exhibit the signs of higher levels of intrinsic OS.
Besides LTP and synaptic plasticity, ROS/RNS also serve as signaling molecules for other bio-functions such as apoptosis. To connect to SNV, some neurons are found to be more responsive to these signaling molecules than others. For example, in a recent study on ROS as signaling intermediates for apoptosis in the substantia nigra pars compacta, the vulnerable dopaminergic neurons were found to be much more sensitive than the resistant non- dopaminergic neurons in this area to the ROS-mediated apoptosis through a pathway involving receptor-mediated Ca2+ influx (Agrawal et al., 2010
). The authors postulated several possible underlying factors for the differential effects of ROS on the dopaminergic and non-dopaminergic neurons, including differences in receptor density and distribution.
Low ATP Levels and Mitochondrial Dysfunction
Neurons require high levels of energy in order to operate. Therefore, either exceedingly high demand for ATP or diminished production of ATP can affect normal neuronal function and the response of neurons to heightened stress encountered during aging and in the development of neurodegenerative diseases. The neurons that are vulnerable to OS may have a high demand for ATP to counteract the high intrinsic OS status of these neurons; such neuronal efforts as synthesizing anti-oxidant proteins, repairing and degrading oxidatively modified macromolecules (proteins, lipids, and nucleic acids) may require additional energy. In addition, the production of ATP in these neurons may be reduced due to mitochondrial dysfunction (to be detailed next). Currently available data, although still limited, support the negative effects of low ATP levels on vulnerable neurons. Measurement of ATP levels in OS- vulnerable cerebellar granule neurons and in OS-resistant cerebral cortical neurons showed that the level of ATP in the cerebellar granule neurons was 25% lower than that in cortical neurons under basal conditions. Following exposure to exogenous OS, in order to maintain intracellular redox balance, ATP levels in both neuronal populations dropped, but this drop was more precipitous in cerebellar granule neurons (Wang et al., 2009
). It is expected that the higher energy demand and low ATP levels in vulnerable neurons can lead to energy crises in case of increased stress, which can seriously affect their ability to mount effective defenses against the stress increase.
Transcriptomic data on the vulnerable (CA1 and cerebellar granule neurons) and resistant (CA3 and cerebral cortical) neuronal populations also support the concept of lower ATP and energy reserves in vulnerable as compared with resistant neurons. Transcriptomic analyses show that genes related to “DNA damage and repair”, “RNA damage and repair”, “Response to unfolded protein”, and “Lipid metabolism” were significantly more active in the vulnerable neurons (Wang et al., 2007
, 2009
). While the expression of enzymes involved in these repair processes was up-regulated, the expression of nuclear genes involved in energy generation was lower in vulnerable as compared with resistant neurons (Brooks et al., 2007
; Liang et al., 2008
; Wang et al., 2009
). The reason for such under-expression of genes related to energy generation is not known but might be due to oxidative modifications in the promoter region of these genes, or due to mitochondrial loss. In support of the idea that there might be loss of mitochondria in vulnerable neurons is the observation that neurons of the substantia nigra pars compacta contain less mitochondrial mass than those of the ventral tegmental area (Liang et al., 2007
).
Mitochondria are not only the powerhouses for ATP generation in cells, they are also the major site of free radical generation and initiation of OS. The electron transport chain localized in the inner membrane of mitochondria is a major source of superoxide in cells due to side reactions of electron carriers along the chain (especially Complexes I and III) with molecular oxygen. The outer mitochondrial membrane, on the other hand, produces a sizable amount of hydrogen peroxide from the membrane enzyme monoamine oxidase during the catalysis of oxidative deamination of monoamines (such as the catecholamines). This represents one of the major sources for hydrogen peroxide generation in both the mitochondrial matrix and cytosol. As indicated previously, the superoxide and hydrogen peroxide generated by the mitochondria can react further to produce other ROS, such as hydroxyl radical and peroxynitrite, thus accounting for the close relationship between mitochondrial activity and cellular OS (Cadenas and Davies, 2000
; Balaban et al., 2005
).
Mitochondria, however, may also be damaged by the free radicals they produce. The mitochondrial DNA molecules, being close to the site of free radical generation, accumulate mutations more quickly than nuclear DNA does. Such damage to mitochondrial DNA tends to be more extensive than nuclear DNA also because of the limited DNA repair capacity in mitochondria. In addition to mitochondrial DNA, metabolic enzymes in these organelles are frequently modified by the highly reactive ROS or RNS formed. Therefore, the high intrinsic OS status in vulnerable neurons may have direct detrimental effects on mitochondrial function, including reduced ATP production. Dysfunctional mitochondria in vulnerable neurons can release more ROS and thus maintain a vicious cycle of oxidative modification of mitochondrial proteins leading to further OS. Currently available evidence shows that mitochondria isolated from CA1 neurons release more ROS than those from CA3 neurons (Mattiasson et al., 2003a
). The link among ATP decline, dysfunctional mitochondria, and SNV is further supported by the use of some mitochondrial toxicants (such as MPTP and rotenone) to induce ATP depletion and produce very similar patterns of selective vulnerability of dopaminergic neurons as that seen in PD (Hasegawa et al., 1990
; Betarbet et al., 2000
).
The mitochondrial permeability transition (MPT) pore leads to mitochondrial swelling and ultimately cell death. Differential activation of the MPT pore in vulnerable vs. resistant neurons provides another link between mitochondria and SNV. In the hippocampus, the MPT pore can be more readily activated upon calcium induction in CA1 than in CA3 (Mattiasson et al., 2003a
). In a study of selective dopaminergic vulnerability in PD, the MPT pore was activated by a potent dopamine metabolite much more readily from energetically compromised mitochondria than fully energized mitochondria, and this MPT pore activation was suggested to be a mechanism for the selective vulnerability of neurons to dopamine in PD (Kristal et al., 2001
). Finally, the differential activation of the MPT pore in several brain regions was found to be correlated with the differential susceptibility of neurons to ischemia-induced injury (Friberg et al., 1999
).
Uncoupling protein 2 (UCP2) provides a further potential connection between mitochondria and SNV. This mitochondrial inner membrane protein can protect neurons from OS by reducing ROS production through the uncoupling of oxidative phosphorylation and ATP synthesis (Horvath et al., 2003
). As such, after ischemia and traumatic brain injury, both involving OS, high expression of Ucp2 is neuroprotective through activation of neuronal redox signaling or prevention of apoptosis (Mattiasson et al., 2003b
). Consistent with the higher ROS production and selective vulnerability of hippocampal CA1, neurons in this region exhibit lower expression of Ucp2 compared with those in CA3 (Richard et al., 2001
).
Glia and Chronic Inflammatory Response
There is emerging evidence that suggests neuron-glial crosstalk may play a role in SNV. Although often overlooked, glial cells, such as astrocytes and microglia, play important roles in maintaining overall CNS homeostasis, providing trophic support to neurons, clearing synapses of the released neurotransmitters, re-establishing ionic gradients near the synapse, mediating immune responses in the brain, and reducing OS (Dringen et al., 2000
; Park et al., 2001
). When CNS regional homeostasis is disturbed because of redox shifts or other neurodegenerative conditions, astrocytes and microglia release various cytokines in an effort to re- establish regional integrity and repair damaged cells. While these glial responses are beneficial to neurons, the continuous or repeated activation of astrocytes and microglia under conditions of chronic inflammatory stresses can lead to the increased production of ROS/RNS and other neurotoxic mediators, which can lead to severe neuronal damage (Banati et al., 1993
; Banati and Graeber, 1994
; Aloisi, 2001
; Streit, 2002
). Because properly functioning glia are essential for neuronal health, glial dysfunction can increase the vulnerability of neurons to neurotoxic conditions such as OS. In the substantia nigra pars compacta of individuals suffering from PD, glial dysfunction has been shown to underlie the selective degeneration of dopaminergic neurons (McNaught and Jenner, 1999
; Marchetti et al., 2005
). In the hippocampus, astrocytes in the CA1 region under ischemia, display selective loss of glutamate transport activity, increased mitochondrial ROS generation, and reduced mitochondrial membrane potential. This selective dysfunction of CA1 astrocytes has been suggested as being important in determining the selective loss of CA1 neurons under ischemia and other OS-related adverse conditions (Ouyang et al., 2007
). In a different study focused on the relationship between neuron-glial interactions and SNV during ischemia, it was shown that microglia are specifically activated in areas containing susceptible neurons (Bernaudin et al., 1998
). In trying to reproduce in neuronal cultures the pattern of SNV observed in vivo, it became apparent that the interactions between glial cells and neurons are essential to the appearance of SNV among neuronal populations (Bernaudin et al., 1998
).
Existing functional genomics studies provide further insights into the role of glia in SNV, especially with regard to the inflammatory responses that follow exposure of cells in a brain region to OS. Compared with OS-resistant neurons, vulnerable neurons in the hippocampal CA1 region, cerebellar granule cell layer, and substantia nigra pars compacta, have a higher transcriptional activity of genes related to cytokine and chemokine formation and inflammatory response (Grunblatt et al., 2004
; Duke et al., 2007
; Wang et al., 2007
, 2009
; Simunovic et al., 2009
). These increases in gene expression are indicative of the existence of chronic inflammatory stress in vulnerable neurons. While inflammatory responses may protect neurons by removing injured cells or neuronal processes from the vulnerable regions, there is evidence showing that chronic inflammation can be a causative factor for SNV (Herrera et al., 2005
; Whitton, 2007
). For example, microglia, the principal mediator of inflammation in the CNS, release a large amount of ROS/RNS upon activation (Colton and Gilbert, 1987
; Moss and Bates, 2001
; Liu et al., 2002
; Block et al., 2007
), leading to OS increases and further neuronal damage.
Other Factors Underlying the Selective Vulnerability of Neurons to OS
Deficient DNA damage repair
One consequence of high OS in neurons is DNA oxidation. Repair of DNA in cells following oxidative modification requires a sizeable amount of energy. Energy production in vulnerable neurons, however, is less than in resistant neurons, therefore, there is an unfavorable environment in vulnerable neurons for the repair of modified DNA. It is well known that when repair of DNA damage is insufficient, then damaged DNA accumulates, especially, in the promoter regions of protein-coding genes, and this can lead to transcriptional disruption of active genes, followed by cellular dysfunction and, ultimately, apoptosis (Hanawalt, 1994
; Lu et al., 2004
; Roos and Kaina, 2006
). Existing evidence suggests that selective OS vulnerability of some neurons is partially underlied by deficient DNA damage repair. As described earlier, MPTP elicits OS and selectively affects dopaminergic neurons in the substantia nigra pars compacta. In a study of time- and region-dependent changes in the activity of OGG1 (8- oxoguanine DNA glycosylase 1), a key enzyme for base excision repair, it was shown that in a region that is vulnerable to OS, the repair of DNA damage was increased initially but did not last beyond 48 h. On the other hand, the activity of OGG1 in regions resistant to OS, was maintained up to 72 h (Sava et al., 2006
). The brain region-specific capacity for DNA repair following induction of OS may be responsible for the selective vulnerability of specific neurons to OS (Cardozo-Pelaez et al., 2002
). The relevance of these observations is attested by the fact that deficient DNA repair systems have been linked to AD, ALS, and PD, all of which are connected to SNV (Robbins et al., 1985
; Mazzarello et al., 1992
; Lovell et al., 2000
).
Calcium dysregulation and glutamate hyperactivity
Calcium signaling plays an important role in regulating and maintaining normal neuronal function, including neurotransmitter release, excitability, neurite outgrowth, synaptic plasticity, gene transcription, and cell survival (Berridge, 1998
; Yuste et al., 2000
; Burnashev and Rozov, 2005
). Intracellular Ca2+ concentrations ([Ca2+]i) represent a powerful activating stimulus for many signal transduction cascades and abnormal elevations of intracellular [Ca2+]i may lead to cell dysfunction and death. The abnormal increases in [Ca2+]i in cells may result from diminished transport of cytosolic Ca2+ to the extracellular environment (Michaelis et al., 1992
, 1996
; Zaidi et al., 1998
), decreased sequestration into mitochondria and binding to intracellular Ca2+-binding proteins (Vitorica and Satrustegui, 1986
; Iacopino and Christakos, 1990
; Martinez-Serrano et al., 1992
; de Jong et al., 1996
; Satrustegui et al., 1996
), or enhanced influx through voltage-gated Ca2+ channels (Landfield and Pitler, 1984
; Moyer and Disterhoft, 1994
; Thibault and Landfield, 1996
; Porter et al., 1997
). The elevations in [Ca2+]i can lead to further release of Ca2+ from the endoplasmic reticulum, and the activation of the caspase-dependent apoptosis pathway through changes in mitochondrial membrane permeability (Rizzuto et al., 2003
). Also, elevations in [Ca2+]i can activate mitochondrial dehydrogenases, inhibit complex I, and cause the formation of ROS and, therefore, increase OS (Beal, 1992
; Starkov et al., 2004
). Since progressive, age-associated deterioration of [Ca2+]i control processes gradually lead to cellular deterioration and organ malfunction (Streicher, 1958
; Gibson and Peterson, 1987
; Verkhratsky and Toescu, 1998
), Ca2+ dysregulation has been regarded as an important contributor to the aging process (i.e., the calcium hypothesis of aging) (Foster, 2007
; Toescu and Vreugdenhil, 2009
).
During OS, ROS/RNS usually activate Ca2+ channels and repress Ca2+ pumps (Ermak and Davies, 2002
), resulting in elevation of [Ca2+]i and initiation of downstream events mentioned above. Because of the intricate relationship between OS and Ca2+ dysfunction, Ca2+ dysfunction is another mechanistic factor that may underlie the selective vulnerability of CNS neurons to OS (Surmeier, 2007
; Surmeier et al., 2010
). Currently available literature shows that Ca2+-buffering proteins such as calbindin D-28K and parvalbumin, which buffer intracellular Ca2+ and attenuate the damaging effects of rapidly rising [Ca2+]i in cells, provide a direct link between Ca2+ dysregulation and the phenomenon of SNV. In human PD brain, neurons containing calbindin D-28K are less vulnerable than those without this protein (Yamada et al., 1990
; German et al., 1992
). In several animal models of PD, while the majority of A10 neurons (mostly resistant) contain Ca2+- binding proteins, many fewer neurons in A9 (the vulnerable region) contain such Ca2+-buffering proteins (Gerfen et al., 1985
; Liang et al., 1996a
,b
). Consistent with this regional distribution, gene expression studies also show that calbindin D-28K mRNA levels are significantly lower in A9 neurons compared with those in A10 (Chung et al., 2005
). In addition, the lack of calbindin D-28K and parvalbumin is also related to the selective vulnerability of motor neurons in ALS (Shaw and Eggett, 2000
). The inverse correlation between the levels of Ca2+-buffering proteins in neurons and neuronal vulnerability to OS also holds true for neurons within the vulnerable and resistant hippocampal subregions (Mattson et al., 1991
; Rami et al., 1992
).
An important pathway for Ca2+ accumulation in neurons is through activation of glutamatergic synapses. The entry of Ca2+ through glutamate receptor-ion channels, e.g., NMDA receptors, or through voltage-gated Ca2+ channels, is critical to the appearance of LTP or long-term depression (Bear and Abraham, 1996
; Schiller et al., 1998
; Malenka and Nicoll, 1999
; Rammsayer, 2001
). With age there is an increase of glutamate levels in the extracellular space of the CNS, which can lead to excessive activation of glutamate receptors and excitotoxicity (Michaelis, 1998
). And, many of the lost synapses during aging are glutamatergic, including those of large neurons that are most vulnerable to age-associated degeneration (Terry et al., 1987
; Masliah et al., 1993
). Mechanistically, OS plays an important role in the selective loss of neurons or synapses from glutamate excitotoxicity (Michaelis, 1998
; Dong et al., 2009
). To directly link glutamate excitotoxicity to SNV, the effects of exogenous glutamate treatment on in vitro neuronal cultures established from different hippocampal regions were conducted in a previous study, in which neurons from CA1 were the most susceptible to excitotoxicity (Mattson and Kater, 1989
). This glutamate-induced selective vulnerability is very similar to the pattern of selective cell loss observed in organotypic slice cultures exposed to OS (Wilde et al., 1997
; Wang et al., 2005
). In another study, real-time monitoring of glutamate release from ischemic hippocampal slices also showed more glutamate being released from CA1 than CA3 neurons, with CA1 neurons being more susceptible to ischemia (Uchino et al., 2001
). The fact that MK-801, an NMDA receptor blocker, can prevent the ischemia-induced selective neuronal death in CA1 (Newell et al., 1990
) also points to an intimate relationship between glutamate and the selective sensitivity of certain neurons, such as the CA1 pyramidal cells, to OS.
Other potential factors
Among other factors that may be linked to SNV associated with OS, or SNV in general, is the physical size of neurons. Besides the molecular and cellular mechanisms described above, the physical size of neurons may, to some degree, determine neuronal vulnerability to stress. In general, vulnerable neurons are large in size, with axons projecting over long distances to their targets. Neuronal populations characterized as being relatively large in size and thus particularly susceptible to degeneration associated with diseases such as ALS and PD include the upper and lower motor neurons, and dopaminergic neurons in the substantia nigra (Shaw and Eggett, 2000
; Cleveland and Rothstein, 2001
; Rodriguez et al., 2001
). The possible reasons for the susceptibility of large neurons include the high demand for energy and mitochondrial activity, dependence on long-distance axonal transport, high content of neurofilaments which tend to form aggregates, and a relatively large surface area for increased exposure to toxicants in the extracellular environment.
Brain aging, as well as associated neurodegenerative conditions such as AD and PD, involves a number of neuron-damaging factors, including calcium dysfunction, glutamate-induced excitotoxicity, mitochondrial dysfunction, protein aggregation, genomic instability, and accumulation of OS. As components of the neurobiological system, these factors are inter-connected and cannot be easily separated from each other. For example, as outlined above, aberrant glutamate neurotransmission can lead to calcium dysregulation, altered metabolic states, abnormal generation of ROS/RNS and therefore OS. Therefore, as one characteristic of brain aging and associated neurodegenerative conditions, SNV is not caused by a single factor alone. However, to start understanding the underlying mechanisms of SNV it is essential to take a reductionist approach to pinpoint those factors that play a causal role in the phenomenon of SNV. This review focuses on OS, a stressful condition that may serve as the converging point for many of the above factors, and provides an overview on what characteristics differentiate OS-vulnerable and -resistant neurons and how OS causes SNV in the brain. From this review, it is clear that this SNV originates from the intrinsic characteristics of vulnerable neurons (Figure 1
).
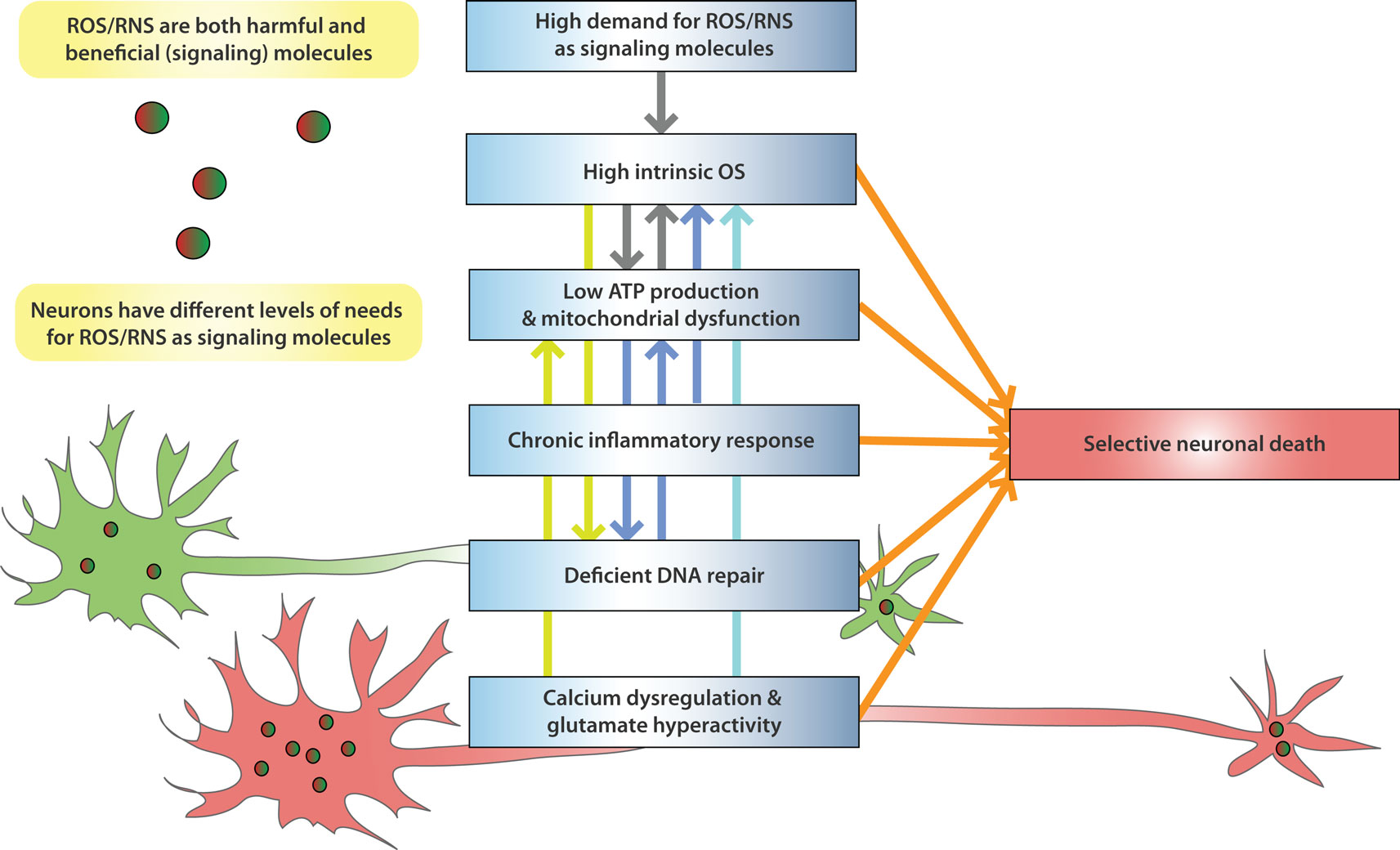
Figure 1. Molecular and cellular factors that contribute to the selective vulnerability of neurons to oxidative stress. ROS/RNS can serve as signaling molecules while they cause damages to bio-molecules at increased levels. Neurons in different parts of the brain have differential needs for ROS/RNS as signaling molecules, with some neurons (such as those in the hippocampal CA1 region) having higher demand than others. However, due to the duality of ROS/RNS, the high demand for these highly reactive species may lead to intrinsically high OS in some neurons, which can make them selectively vulnerable when facing increased stress. Vulnerable neurons are also characterized by low ATP production and mitochondrial dysfunction, possibly because of the high OS state in these neurons, and other factors such as calcium dysregulation. Low ATP production can affect DNA repair, which, when combined with high DNA oxidation, can cause change of genomic activity and decreased metabolic activity in mitochondria. Functional genomics studies also suggest existence of chronic inflammatory response in vulnerable neurons, which can further elevate OS within them. Calcium dysregulation and glutamate hyperactivity are closely connected to OS generation and underlie many adverse conditions that are characterized by SNV. There is emerging evidence that directly connects these factors, such as low calcium-buffering capacity and glutamate-mediated selective neurodegeneration, to the selective vulnerability of neurons. The colored arrows that link these mechanistic factors in the figure denote direct relationships between them. In addition, vulnerable neurons tend to be large in size, with long projecting axons to their targets.
The hypothesis advanced in this review is that the selective vulnerability of neurons to OS is the result of high intrinsic OS, high demand for ROS/RNS as signaling molecules, chronic inflammatory response, low energy generation and mitochondrial dysfunction, as well as deficient DNA damage repair, calcium dysregulation and glutamate hyperactivity, in vulnerable neurons (Figure 1
). The duality of ROS/RNS seems to be one of the root causes of OS-induced SNV. On one hand, these highly reactive species are damaging to cellular molecules and can wreak havoc in neurons when overly produced; on the other hand, they serve as signal molecules and are beneficial to neurons. The delicate balance maintained in vulnerable neurons can be more readily disrupted than in resistant neurons, when challenged by excessive levels of OS produced in some neurodegenerative diseases and during aging, and therefore more prone to synaptic destruction, dendrite and axonal pathology, and eventual neuronal death. Mechanistically, the production of such excessive levels of OS under these adverse conditions is most likely mediated by increases in intracellular Ca2+, glutamate-induced changes in neuronal excitability, and altered mitochondrial function.
Because OS-vulnerable and OS-resistant neurons are located in different regions of the brain and have their own unique characteristics, in order to identify key mechanistic factors that determine SNV, it is essential to study multiple populations of vulnerable neurons and focus on their commonalities in comparison with multiple populations of resistant neurons. As part of this endeavor, more neuronal populations that are vulnerable to OS need to be identified and added to the list of vulnerable neurons. Technologically, to have an unbiased understanding of the molecular mechanisms of SNV, it is essential that further genomic, proteomic, and metabolomic studies be performed on vulnerable neurons and compared to resistant neurons. To a large degree, the functional status of each neuronal population is reflected by its genomic, proteomic and metabolomic activities. As demonstrated in this review, this high-throughput approach has already begun to provide some insights into the causation of SNV. Of course, findings from these high-throughput technologies need to be experimentally validated and further characterized.
It is a step forward in neuroscience research from studying the commonalities of different brain regions or neuronal populations to realizing the individuality of each region and population. The experimental evidence summarized herein of SNV reminds us of the heterogeneity of brain neurons. Even inside a seemingly homogeneous brain structure, such as the hippocampus or the substantia nigra, there are many types of intrinsically different neurons. OS is only an adverse condition under which the differences among these neurons are manifested.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank the U.S. National Institute on Aging (NIA) for grants AG12993 and AG025350, the U.S. Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) for HD02528, the U.S. National Center for Research Resources (NCRR) for RR-P20 17708, the Kansas Technology Enterprise Corporation, and the Miller-Hadwiger Fund for financial support.
Atwood, C. S., Scarpa, R. C., Huang, X., Moir, R. D., Jones, W. D., Fairlie, D. P., Tanzi, R. E., and Bush, A. I. (2000). Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J. Neurochem. 75, 1219–1233.
Burns, R. S., Chiueh, C. C., Markey, S. P., Ebert, M. H., Jacobowitz, D. M., and Kopin, I. J. (1983). A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6- tetrahydropyridine. Proc. Natl. Acad. Sci. U.S.A. 80, 4546–4550.
Grunblatt, E., Mandel, S., Jacob-Hirsch, J., Zeligson, S., Amariglo, N., Rechavi, G., Li, J., Ravid, R., Roggendorf, W., Riederer, P., and Youdim, M. B. (2004). Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural Transm. 111, 1543–1573.
Huang, X., Atwood, C. S., Hartshorn, M. A., Multhaup, G., Goldstein, L. E., Scarpa, R. C., Cuajungco, M. P., Gray, D. N., Lim, J., Moir, R. D., Tanzi, R. E., and Bush, A. I. (1999a). The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry Mosc. 38, 7609–7616.
Huang, X., Cuajungco, M. P., Atwood, C. S., Hartshorn, M. A., Tyndall, J. D., Hanson, G. R., Stokes, K. C., Leopold, M., Multhaup, G., Goldstein, L. E., Scarpa, R. C., Saunders, A. J., Lim, J., Moir, R. D., Glabe, C., Bowden, E. F., Masters, C. L., Fairlie, D. P., Tanzi, R. E., and Bush, A. I. (1999b). Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274, 37111–37116.
Liang, W. S., Reiman, E. M., Valla, J., Dunckley, T., Beach, T. G., Grover, A., Niedzielko, T. L., Schneider, L. E., Mastroeni, D., Caselli, R., Kukull, W., Morris, J. C., Hulette, C. M., Schmechel, D., Rogers, J., and Stephan, D. A. (2008). Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 4441–4446.
Marchetti, B., Serra, P. A., L’Episcopo, F., Tirolo, C., Caniglia, S., Testa, N., Cioni, S., Gennuso, F., Rocchitta, G., Desole, M. S., Mazzarino, M. C., Miele, E., and Morale, M. C. (2005). Hormones are key actors in gene x environment interactions programming the vulnerability to Parkinson’s disease: glia as a common final pathway. Ann. N. Y. Acad. Sci. 1057, 296–318.
Mattiasson, G., Shamloo, M., Gido, G., Mathi, K., Tomasevic, G., Yi, S., Warden, C. H., Castilho, R. F., Melcher, T., Gonzalez-Zulueta, M., Nikolich, K., and Wieloch, T. (2003b). Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 9, 1062–1068.
McCormack, A. L., Thiruchelvam, M., Manning-Bog, A. B., Thiffault, C., Langston, J. W., Cory-Slechta, D. A., and Di Monte, D. A. (2002). Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 10, 119–127.
Moratalla, R., Quinn, B., DeLanney, L. E., Irwin, I., Langston, J. W., and Graybiel, A. M. (1992). Differential vulnerability of primate caudate-putamen and striosome-matrix dopamine systems to the neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc. Natl. Acad. Sci. U.S.A. 89, 3859–3863.
Scorziello, A., Pellegrini, C., Forte, L., Tortiglione, A., Gioielli, A., Iossa, S., Amoroso, S., Tufano, R., Di Renzo, G., and Annunziato, L. (2001). Differential vulnerability of cortical and cerebellar neurons in primary culture to oxygen glucose deprivation followed by reoxygenation. J. Neurosci. Res. 63, 20–26.