

- Centre for Neuroscience, Division of Experimental Medicine, Imperial College London, London, UK
There is significant epidemiological evidence to suggest that there are beneficial effects of treatment with non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease, although these effects have not been reproduced in clinical trials. The failure of the clinical trials may be attributed to several possible facts: (1) NSAIDS may have been delivered too late to patients, as they may only be effective in early stages of the disease and possibly counterproductive in the late stages; (2) the beneficial effect may depend on the drug, because different NSAIDs may have different molecular targets; (3) the NSAID concentration reaching the brain and the duration of the treatment could also be critical, so increasing drug penetration is important in order to improve the efficacy and avoid secondary gastro-intestinal effects of the NSAIDs. In this report we analyze these different factors, with special emphasis on the role of NSAIDs in microglia activation over time.
Many inflammatory pathways have been implicated in Alzheimer’s disease (AD), yet these pathways are not sufficiently well delineated to define those processes and targets that may be pathogenic as opposed to those that may be protective. A clearer understanding of these distinctions is critical to the design of therapeutic strategies. The finding that treatment with non-steroidal anti-inflammatory drugs (NSAIDs) is associated with a reduced risk and age of onset of AD reinforces the hypothesis that modulating inflammation could have therapeutic efficacy. The beneficial effects of NSAIDs have also been associated with reductions in Aβ generation, since experiments in vitro and in AD animal models indicate that certain NSAIDs are able to decrease Aβ levels, plaque size and tau phosphorylation ( Yoshiyama et al., 2007 ; El Khoury and Luster, 2008 ).
However, clinical trials have failed to reproduce the beneficial effects of NSAIDs in AD patients. This has led to further analysis of the previously published epidemiological data, which has revealed that the use of NSAIDs prevents cognitive decline in older adults if started in midlife (prior to age 65) rather than late in life (Hayden et al., 2007 ). In addition, it was recently shown that while NSAIDs may indeed protect those with healthier brains, they can accelerate AD pathogenesis in patients with advanced stages of the disease (Breitner et al., 2009 ). This is supported by studies in transgenic mice, in which NSAIDs can prevent the appearance of cell cycle protein markers in neurons in young mice, but not after cell cycle entry has been initiated (Varvel et al., 2009 ). Therefore, it seems that the protective effects of NSAIDs depend very much on the stage of the disease at which the medication is started as well as the duration of the treatment. Here we discuss four possible reasons why clinical trials with NSAIDs have been unsuccessful.
Temporal Profile of Microglial Activation
A potential target of NSAIDs is thought to be the microglia associated with the senile plaques. This is supported by a study by Mackenzie and Munoz (1998) showing in non-demented patients that those treated with NSAIDs had three times less activated microglia as non-treated controls. These data have been confirmed by in vivo treatment with NSAIDs in mouse models of AD, which have shown decreases in microglial activation and in inflammatory mediators such as iNOS, COX and cytokines (Lim et al., 2000 ; Heneka et al., 2005 ). Experiments carried out in cultured microglia have revealed that incubation with NSAIDs decreased the secretion of pro-inflammatory cytokines and may increase Aβ phagocytosis (Lleo et al., 2007 ). However, the reduction of activated microglia and astroglia by NSAIDs was not significant in AD patients, indicating an age or stage dependent difference in the glial response i.e. in their activation rate (Alafuzoff et al., 2000 ). Recently, these findings have been backed up by observations indicating that microglia may change from alternative to classical phenotype over time (Colton et al., 2006 ; Hickman et al., 2008 ; Jimenez et al., 2008 ; Lucin and Wyss-Coray, 2009 ), although probably there is a heterogeneous population. In addition, a paper of Meyer-Luehmann et al. (2008) using two-photon microscopy showed that microglia in the aged mouse brain is less motile and posses fewer processes. Therefore, the response of NSAIDs may differ in “young” vs “old” microglia (Figure 1 ).
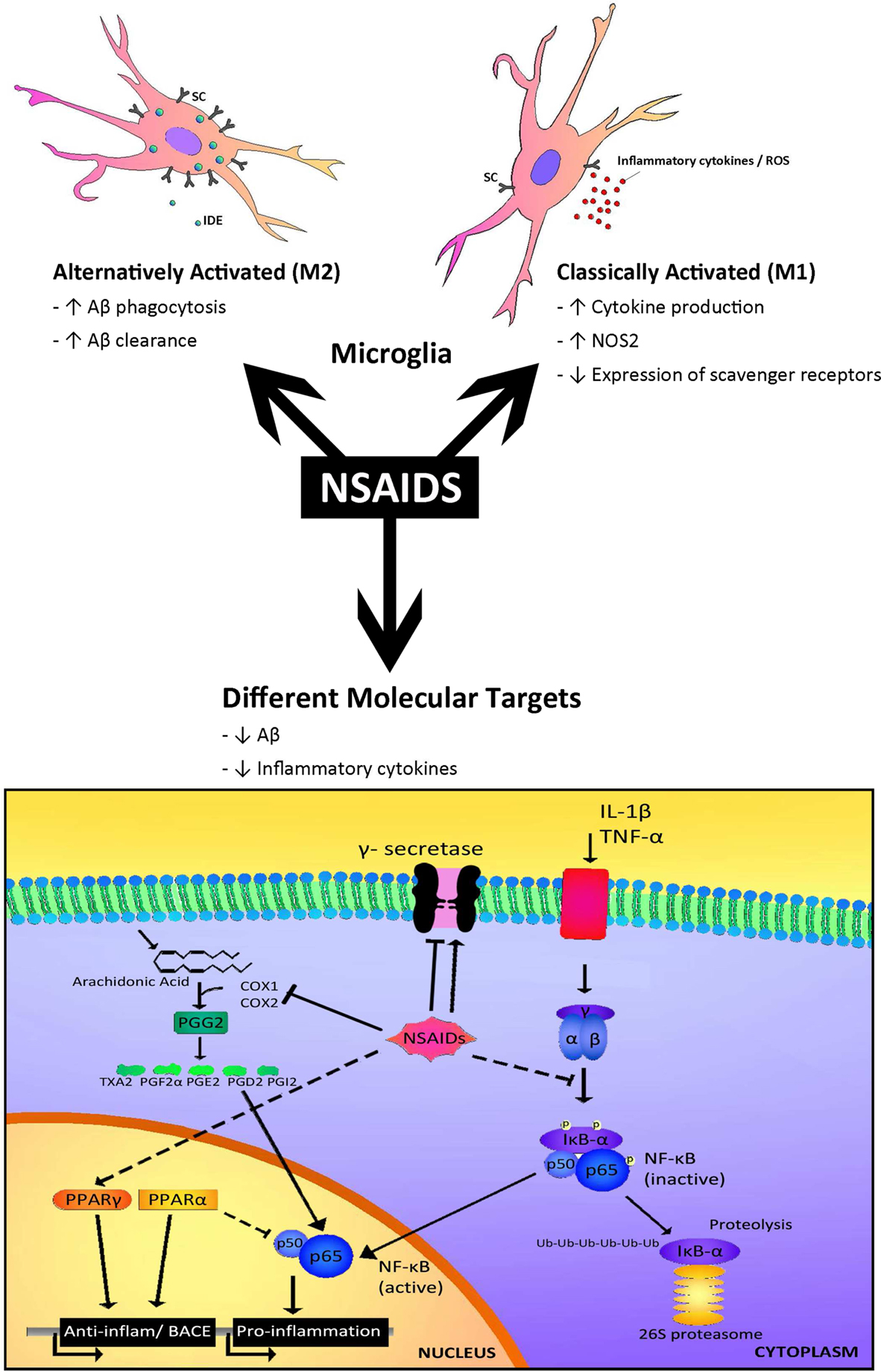
Figure 1. Different targets for NSAIDs. The response to NSAIDs may differ depending on whether they are used in early stages of disease, in which microglia present an alternatively activated phenotype compared with late stages which is associated with a classical microglia phenotype. On the other hand, different subsets of NSAIDs have different affinity for targets such as COX1, COX2, NFκB, PPARγ or γ-secretase, resulting in a range of effects including reductions in inflammatory mediators, such as cytokines and alterations in Aβ generation.
It has been hypothesized that early microglial activation in AD delays disease progression by promoting clearance of Aβ before formation of senile plaques. It is conceivable that glial activation is protective through mechanisms such as phagocytosis and clearance of Aβ deposits (through release of insulin degrading enzyme, IDE), forming a protective barrier between Aβ and neurons and secretion of growth factors, early in the disease (Wyss-Coray et al., 2003 ; Maragakis et al., 2006 ; Wyss-Coray, 2006 ). In later stages, with persistent production of pro-inflammatory cytokines, microglia lose their protective effect (Hickman et al., 2008 ; Jimenez et al., 2008 ) and may become detrimental through the release of cytokines and chemokines including IL-1β, IL-6, TNFα, IL-8 and MIP-1α (Hickman et al., 2008 ). These inflammatory mediators modulate immune and inflammatory function and may also alter neuronal function. In addition, microglia from old transgenic mice have a decrease in the expression of the Aβ-binding scavenger receptors A (SRA), CD36 and RAGE, and the Aβ degrading enzymes IDE, neprilysin and MMP9, compared with wild-type controls (Hickman et al., 2008 ) (Figure 1 ). On the other hand, in aged human brain many microglia are dystrophic showing morphological features indicative of senescence, such as fragmented cytoplasmic processes (Streit et al., 2004 ). Therefore, overactivated and dysregulated microglia could cause uncontrolled inflammation that may drive the chronic progression of AD (Mrak and Griffin, 2005 ; Gao and Hong, 2008 ).
Microglia can be primed or desensitized by a stimulus (such as Aβ), which prepares the cells for an enhanced or decreased response to a second challenge (Gao and Hong, 2008 ). Microglia in aged or diseased brains are primed and usually behave differently to those in younger individuals (Gao and Hong, 2008 ). Thus, it is likely that microglia do not respond equally to anti-inflammatory therapy in old age and therefore, treatment of patients with NSAIDs in advanced stages of the disease may not produce any benefit.
One of the most controversial points is to establish whether microglia are “oversaturated” at a certain age or whether there is a loss of function. The debate on microglia function in AD progression has been intensified by a recent report showing that AD animal models with nearly complete ablation of microglia did not display differences in plaque formation (Grathwohl et al., 2009 ), raising questions as to whether inflammation may have an effect on neurodegeneration and cognitive decline rather than a direct role on Aβ deposition. Furthermore, a study using two-photon microscopy in the intact brain of living AD mice has revealed an involvement of microglia in neuron elimination, indicated by locally increased number and migration velocity of microglia around lost neurons (Fuhrmann et al., 2010 ). Therefore, evidence has started to accumulate that the function of microglia is neuroprotection in young individuals (by secretion of neurotrophic factors and anti-inflammatory cytokines) and that “senescent” microglia contribute to the onset of sporadic AD (Streit et al., 2004 , 2009 ).
Similarly, there are other factors that may change the risk for AD depending on the age, such as obesity or body mass index (BMI), which is an important predictor for late life dementia. However, in late life, low and declining BMI is associated with increased AD risk. Therefore, there is time frame for the beneficial effect of certain factors, and since AD is a disease with a long preclinical period, trials of short duration in severe cases of AD do not provide reliable information regarding development of AD (Bennett and Whitmer, 2009 ).
Multiple Molecular Targets for NSAIDs
The type of NSAID appears to affect the outcome of the clinical trial (Figure 1 ). The inhibition of the canonical targets of NSAIDs, cyclooxygenase-1 and -2 (COX-1 and COX-2), do not seem to be responsible for the protective effect of NSAIDs in AD (see review Lleo et al., 2007 ). On the contrary, some COX-2 inhibitors may raise Aβ1-42 secretion (Kukar et al., 2005 ).
Interestingly, some NSAIDs and other small organic molecules have been found to modulate γ-secretase and to selectively reduce Aβ1-42 levels without affecting Notch cleavage. A subset of NSAIDs including ibuprofen, sulidac sulfide, and indomethacin have been shown to decrease the levels of secreted Aβ42 in cells as well as in animal models of AD (Weggen et al., 2001 ; Eriksen et al., 2003 ). Importantly, the generation of Aβ40 was largely unaltered by these compounds, indicating that certain NSAIDs modulate rather than inhibit γ-secretase activity (Czirr and Weggen, 2006 ). In fact, NSAIDs seem to bind APP, instead of the γ-secretase (Kukar et al., 2008 ). Some positive effects on cognitive performance of AD patients have been observed with indomethacin and the (R)-enantiomer of flurbiprofen in phase-2 trials, while other NSAIDs without Aβ42 reducing activity did not show beneficial effects. However, recent clinical trials in mild AD cases revealed that R-flurbiprofen (tarenflurbil) does not slow cognitive decline or the loss of activities of daily living (Green et al., 2009 ). In addition, epidemiological studies report that the protective effect of NSAIDs seems to be independent of the Aβ42 reducing activity of the NSAID (Szekeli et al., 2008a ; Vlad et al., 2008 ).
Besides targeting molecules such as COX and γ-secretase, some NSAIDs such as ibuprofen, naproxen and indomethacin can activate PPARγ (Jaradat et al., 2001 ). PPARγ inhibition regulates the transcription of pro-inflammatory genes, such as IL1β; therefore activation of PPARγ consequently inhibits the inflammatory response. In addition, we found that PPARγ activators are able to decrease total Aβ levels under inflammatory conditions by affecting BACE1 transcription (Sastre et al., 2006b , 2008 ). Recently it was shown that ibuprofen is able to suppress RhoA activity in neuronal cells through PPARγ activation, promoting neurite elongation (Dill et al., 2010 ). Therefore, PPARγ activation could be beneficial in AD at several levels. A recent prospective randomized, open-controlled study with pioglitazone (a typical PPARγ agonist) has shown that at 6 months the WMS-R logical memory-I scores significantly increased in the pioglitazone group, but not in the control group (Hanyu et al., 2009 ). Another PPARγ agonist, rosiglitazone has been trialed with inconsistent results. In contrast to pioglitazone, rosiglitazone cannot cross the blood brain barrier (BBB) (Festuccia et al., 2008 ) and it was suggested that the protective effects are mediated through its effects on insulin and glucocorticoids that are able to penetrate into the brain.
Certain NSAIDs, such as flurbiprofen and indomethacin, inhibit the nuclear translocation of the transcription factor NF-kB. In addition, it was recently shown that R-flurbiprofen can interfere with the interaction between RXRα and 9-cis-retinoid acid, and that 9-cis-retinoid acid decreases (R)-flurbiprofen’s reduction of Aβ secretion (You et al., 2009 ). R-flurbiprofen has been shown to upregulate NGF and BDNF in vitro, which could potentially offer neuroprotection (Zhao et al., 2008 ). However, as mentioned above, R-flurbiprofen trials have not been successful, perhaps because of its low permeability into the brain and its weak pharmacological activity (Imbimbo, 2009 ).
Duration and Dose
The duration of the treatment could also influence the magnitude of the effect. A meta-analysis of nine studies revealed that the benefit of the NSAID treatment was greater in long-term users than in intermediate users (Etminan et al., 2003 ). Moreover, it has been suggested that at least 2 years of exposure are necessary to obtain full benefit, so the benefits may be greater the longer NSAIDs are used (Sastre et al., 2006a ). The effect of NSAIDs in mice has been reviewed extensively by Imbimbo (2009) . Short term studies (3- to 7-days treatment) in transgenic mice revealed decreases in Aβ levels, particularly in the Aβ42 isoform and activated microglia, although some studies have shown no changes, depending on the drug and the animal model (Eriksen et al., 2003 ; Heneka et al., 2005 ; Lanz et al., 2005 ). Long-term administration has demonstrated protective effects predominantly using ibuprofen and indomethacin treatments, in both types of Aβ, as well as brain plaque load and inflammation (see Imbimbo, 2009 ). However, chronic R-flurbiprofen produced weak effects on Aβ deposition, and was more effective as a preventive rather than a therapeutic treatment (Kukar et al., 2007 ). This is in line with the results obtained in clinical trials using flurbiprofen.
The dose and permeability of the drug could be relevant, although some epidemiological studies suggest that the daily dose is not important. In’t Veld et al. (2001) noted that low doses had effects equal to those of the higher doses typically prescribed for osteoarthritis and other inflammatory conditions. However, the failure of some clinical trials has been associated with the low permeability of certain drugs such as R-flurbiprofen and rosiglitazone. In addition, there is some conflict between the potency of certain NSAIDs to decrease Aβ42 in vitro (reaching 300 �M) and the effective drug concentration in the brain. Active concentrations of NSAIDs found in human plasma and cerebrospinal fluid are in the lower �M range (Bannwarth et al., 1990 , 1995 ), which could not account for the effects on γ-secretase cleavage. Unfortunately, the clinical dose is limited because of adverse effects such as irritation and ulceration of the gastro-intestinal (GI) mucosa. There is growing interest in improving absorption and BBB permeability of certain NSAIDs in order to allow the administration of lower doses of these drugs. Another point that has to be borne in mind is that the barrier may be compromised in neurodegenerative diseases, permitting the diffusion of molecules that usually have no access to the brain parenchyma, therefore the dose reaching the brain can be different in AD patients compared with healthy controls (Nguyen et al., 2002 ).
ApoE Genotype
Another possible confounding factor in NSAID trials is the genotype of the patients being treated, most notably in relation to ApoE. The majority of prospective studies appear to show greater benefits in those with an ApoE ε4 allele (Hayden et al., 2007 ; Szekely et al., 2008b ). In a recent trial, the ApoE ε4 carriers treated with ibuprofen were the only group without cognitive decline (Pasqualetti et al., 2009 ). Similar positive effects in ApoE4 carriers have also been seen when the NSAIDs have been taken in conjunction with Vit E (Fotuhia et al., 2008 ). In addition, the influence of NSAID use in microglia activation has been noted in all ApoE genotypes however the trend of lower counts of glial cells with regular NSAID use was more marked in patients carrying the ApoEε4/4 alleles (Alafuzoff et al., 2000 ).
By contrast, results from clinical trials with rosiglitazone suggest that those without an APOE ε4 allele exhibit cognitive and functional improvement in response to rosiglitazone, while those that do carry the allele carriers showed no improvement and some decline was noted (Risner et al., 2006 ). ApoE4 and C-terminal-truncated fragments of apoE4 [apoE4(1–272), lacking the C-terminal 27 aa] impair cytoskeletal structure and mitochondrial function and its seems that rosiglitazone reverses this effect.
Conclusions
There is still controversy regarding the reasons why clinical trials with NSAIDs have provided such disappointing results. Here we suggest that the short duration of the trials, the use of drugs targeting the wrong molecule, the wrong timing of the treatment (patients too old or too severely ill), the low levels of the drugs reaching the CNS and the genetic variability of the patients may all have contributed to the failures.
It is still unclear whether NSAIDs are beneficial because of their effects on reducing Aβ or whether it is because of their anti-inflammatory action or the interaction between both (reducing inflammation may decrease Aβ generation and the other way around). It would be helpful in the future to determine whether patients involved in trials experience changes in biomarkers in blood or CSF (such as Aβ levels, tau or inflammatory markers) and whether those correlate with cognitive performance.
Taking all the currently available evidence together, NSAIDs should be used as preventive treatment rather than a therapeutic option and this would make more sense in a disease with a long preclinical period, with evidence that microglial/cytokine events take place years or even decades before plaques or tangles are detected (Griffin et al., 1989 ; Cagnin et al., 2001 ).
Conflict of Interest Statement
The authors declare that the report was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Jonathan Cheung for his help in the design of the figure and the Alzheimer’s Research Trust for their support (grants ART/PG2009/5 and ART/PPG2009B/10).
References
Alafuzoff, I., Overmyer, M., Helisalmi, S., and Soininen, H. (2000). Lower counts of astroglia and activated microglia in patients with Alzheimer’s disease with regular use of nonsteroidal anti-inflammatory drugs. J. Alzheimers Dis. 2, 37–46.
Bannwarth, B., Netter, P., Lapicque, F., Pere, P., Thomas, P., and Gaucher, A. (1990). Plasma and cerebrospinal fluid concentrations of indomethacin in humans. Relationship to analgesic activity. Eur. J. Clin. Pharmacol. 38, 343–346.
Bannwarth, B., Lapicque, F., Pehourcq, F., Gillet, P., Schaeverbeke, T., Laborde, C., Dehais, J., Gaucher, A., and Netter, P. (1995). Stereoselective disposition of ibuprofen enantiomers in human cerebrospinal fluid. Br. J. Clin. Pharmacol. 40, 266–269.
Bennett, D. A., and Whitmer, R. A. (2009). NSAID exposure and risk of Alzheimer disease: is timing everything? Neurology 72, 1884–1885.
Breitner, J. C., Haneuse, S. J., Walker, R., Dublin, S., Crane, P. K., Gray, S. L., and Larson, E. B. (2009). Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 72, 1899–1905.
Cagnin, A., Brooks, D. J., Kennedy, A. M., Gunn, R. N., Myers, R., Turkheimer, F. E., Jones, T., and Banati, R. B. (2001). In-vivo measurement of activated microglia in dementia. Lancet 358, 461–467.
Colton, C. A., Mott, R. T., Sharpe, H., Xu, Q., Van Nostrand, W. E., and Vitek, M. P. (2006). Expression profiles for macrophage alternative activation genes in AD and mouse models of AD. J. Neuroinflammation 3, 27.
Czirr, E., and Weggen,. S. (2006). Gamma-secretase modulation with Abeta42-lowering nonsteroidal anti-inflammatory drugs and derived compounds. Neurodegener. Dis. 3, 298–304.
Dill, J., Patel, A. R., Yang, X. L., Bachoo, R., Powell, C. M., and Li, S. (2010). A molecular mechanism for ibuprofen-mediated RhoA inhibition in neurons. J. Neurosci. 30, 963–972.
El Khoury, J., and Luster, A. D. (2008). Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol. Sci. 29, 626–632.
Eriksen, J. L., Sagi, S. A., Smith, T. E., Weggen, S., Das, P., McLendon, D. C., Ozols, V. V., Jessing, K. W., Zavitz, K. H., Koo, E. H., and Golde, T. E. (2003). NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ 42 in vivo. J. Clin. Invest. 112, 440–449.
Etminan, M., Gill, S., and Samii, A. (2003). Effect of nonsteroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ 327, 128.
Festuccia, W. T., Oztezcan, S., Laplante, M., Berthiaume, M., Michel, C., Dohgu, S., Denis, R. G., Brito, M. N., Brito, N. A., Miller, D. S., Banks, W. A., Bartness, T. J., Richard, D., and Deshaies, Y. (2008). Peroxisome proliferator-activated receptor–mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology 149, 2121–2130.
Fotuhia, M., Zandi, P. P., Hayden, K. M., Khachaturian, A. S., Szekely, C. A., Wengreen, H., Munger, R. G., Norton, M. C., Tschanz, J. T., Lyketsos, C. G., Breitner, J. C., and Welsh-Bohmer, K. (2008). Better cognitive performance in elderly taking antioxidant vitamins E and C supplements in combination with nonsteroidal anti-inflammatory drugs: the Cache County Study. Alzheimers Dement. 4, 223–227.
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., Haass, C., Laferla, F. M., Kretzschmar, H., and Herms, J. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413.
Gao, H. M., and Hong, J. S. (2008). Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365.
Grathwohl, S. A., Kälin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., Odenthal, J., Radde, R., Eldh, T., Gandy, S., Aguzzi, A., Staufenbiel, M., Mathews, P. M., Wolburg, H., Heppner, F. L., and Jucker, M. (2009). Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363.
Green, R. C., Schneider, L. S., Amato, D. A., Beelen, A. P., Wilcock, G., Swabb, E. A., and Zavitz, K. H.; Tarenflurbil Phase 3 Study Group. (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564.
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., White, C. L. III, and Araoz, C. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615.
Hanyu, H., Sato, T., Kiuchi, A., Sakurai, H., and Iwamoto, T. (2009). Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J. Am. Geriatr. Soc. 57, 177–179.
Hayden, K. M., Zandi, P. P., Khachaturian, A. S., Szekely, C. A., Fotuhi, M., Norton, M. C., Tschanz, J. T., Pieper, C. F., Corcoran, C., Lyketsos, C. G., Breitner, J. C., and Welsh-Bohmer, K. A.; Cache County Investigators. (2007). Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology 69, 275–282.
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Kreutz, A., Dewachter, I., Kuiperi, C., Klockgether, T., Van Leuven, F., and Landreth, G. (2005). The PPARγ agonist pioglitazone reduces inflammation and Aβ1-42 levels in APP V717I transgenic mice. Brain 128, 1442–1453.
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360.
Imbimbo, B. P. (2009). An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer’s disease. Expert Opin. Investig. Drugs 18, 1147–1168.
In’t Veld, B. A., Ruitenberg, A., Hofman, A., Launer, L. J., van Duijn, C. M., Stijnen, T., Breteler, M. M. B., and Stricker, B. H. C. (2001). Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 345, 1515–1521.
Jaradat, M. S., Wongsud, B., Phornchirasilp, S., Rangwala, S. M., Shams, G., Sutton, M., Romstedt, K. J., Noonan, D. J., and Feller, D. R. (2001). Activation of peroxisome proliferator activated receptor isoforms and inhibition of prostaglandin H(2) synthases by ibuprofen, naproxen, and indomethacin. Biochem. Pharmacol. 62, 1587–1595.
Jimenez, S., Baglietto-Vargas, D., Caballero, C., Moreno-Gonzalez, I., Torres, M., Sanchez-Varo, R., Ruano, D., Vizuete, M., Gutierrez, A., and Vitorica, J. (2008). Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 28, 11650–11661.
Kukar, T., Murphy, M. P., Eriksen, J. L., Sagi, S. A., Weggen, S., Smith, T. E., Ladd, T., Khan, M. A., Kache, R., Beard, J., Dodson, M., Merit, S., Ozols, V. V., Anastasiadis, P. Z., Das, P., Fauq, A., Koo, E. H., and Golde, T. E. (2005). Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Aβ42 production. Nat. Med. 11, 545–550.
Kukar, T., Prescott, S., Eriksen, J. L., Holloway, V., Murphy, M. P., Koo, E. H., Golde, T. E., and Nicolle, M. M. (2007). Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 8, 54.
Kukar, T. L., Ladd, T. B., Bann, M. A., Fraering, P. C., Narlawar, R., Maharvi, G. M., Healy, B., Chapman, R., Welzel, A. T., Price, R. W., Moore, B., Rangachari, V., Cusack, B., Eriksen, J., Jansen-West, K., Verbeeck, C., Yager, D., Eckman, C., Ye, W., Sagi, S., Cottrell, B. A., Torpey, J., Rosenberry, T. L., Fauq, A., Wolfe, M. S., Schmidt, B., Walsh, D. M., Koo, E. H., and Golde, T. E. (2008). Substrate-targeting gamma-secretase modulators. Nature 453, 925–929.
Lanz, T. A., Fici, G. J., and Merchant, K. M. (2005). Lack of specific amyloid-beta(1-42) suppression by nonsteroidal anti-inflammatory drugs in young, plaque-free Tg2576 mice and in guinea pig neuronal cultures. J. Pharmacol. Exp. Ther. 312, 399–406.
Lim, G. P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., Tran, T., Ubeda, O., Ashe, K. H., Frautschy, S. A., and Cole, G. M. (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 20, 5709–5714.
Lleo, A., Galea, E., and Sastre, M. (2007). Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell. Mol. Life Sci. 64, 1403–1418.
Lucin, K. M., and Wyss-Coray, T. (2009). Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64, 110–122.
Mackenzie, I. R., and Munoz, D. G. (1998). Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 50, 986–990.
Maragakis, N. J., and Rothstein, J. D. (2006). Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2, 679–689.
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., Koenigsknecht-Talboo, J., Holtzman, D. M., Bacskai, B. J., and Hyman, B. T. (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724.
Mrak, R. E., and Griffin, W. S. T. (2005). Glia and their cytokines in progression of neurodegeneration. Neurobiol. Aging 26, 349–354
Nguyen, M. D., Julien, J. P., and Rivest, S. (2002). Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 3, 216–227.
Pasqualetti, P., Bonomini, C., Dal Forno, G., Paulon, L., Sinforiani, E., Marra, C., Zanetti, O., and Rossini, P. M. (2009). A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin. Exp. Res. 21, 102–110.
Risner, M. E., Saunders, A. M., Altman, J. F., Ormandy, G. C., Craft, S., Foley, I. M., Zvartau-Hind, M. E., Hosford, D. A., and Roses, A. D. (2006). Rosiglitazone in Alzheimer’s Disease Study Group. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254.
Sastre, M., Klockgether, T., and Heneka, M. T. (2006a). Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int. J. Dev. Neurosci. 24, 167–176.
Sastre, M., Roßner, S., Bogdanovic, N., Rosen, E., Dewachter, I., Thal, D., Evert, B., Klockgether, T., van Leuven, F., and Heneka, M. T. (2006b). Non-steroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448.
Sastre, M., Walter, J., and Gentleman, S. M. (2008). Interactions between APP secretases and inflammatory mediators. J. Neuroinflammation 5, 25.
Streit, W., Braak, H., Xue, Q. S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485.
Streit, W., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212.
Szekely, C. A., Green, R. C., Breitner, J. C., Ostbye, T., Beiser, A. S., Corrada, M. M., Dodge, H. H., Ganguli, M., Kawas, C. H., Kuller, L. H., Psaty, B. M., Resnick, S. M., Wolf, P. A., Zonderman, A. B., Welsh-Bohmer, K. A., and Zandi, P. P. (2008a). No advantage of A{beta}42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology 70, 2291–2298.
Szekely, C. A., Breitner, J. C. S., Fitzpatrick, A. L., Rea, T. D., Psaty, B. M., Kuller, L. H,, and Nandi, P. P. (2008b). NSAID use and dementia risk in the Cardiovascular Health Study*: role of APOE and NSAID type. Neurology 70, 17–24.
Varvel, N. H., Bhaskar, K., Kounnas, M. Z., Wagner, S. L., Yang, Y., Lamb, B. T., and Herrup, K. (2009). NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J. Clin. Invest. 119, 3692–3702.
Vlad, S. C., Miller, D. R., Kowall, N. W., and Felson, D. T. (2008). Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677.
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., Findlay, K. A., Smith, T. E., Murphy, M. P., Bulter, T., Kang, D. E., Marquez-Sterling, N., Golde, T. E., and Koo, E. H. (2001). A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414, 212–216.
Wyss-Coray, T. (2006). Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015.
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anankov, R., Yan, F., Silverstein, S. C., and Husemann, J. (2003). Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 9, 453–457.
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., Maeda, J., Suhara, T., Trojanowski, J. Q., and Lee, V. M. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351.
You, X., Zhang, Y. W., Chen, Y., Huang, X., Xu, R., Cao, X., Chen, J., Liu, Y., Zhang, X., and Xu, H. (2009). Retinoid X receptor-alpha mediates (R)-flurbiprofen’s effect on the levels of Alzheimer’s beta-amyloid. J. Neurochem. 111, 142–149.
Keywords: NSAIDs, Alzheimer’s disease, microglia, PPARγ, amyloid
Citation: Sastre M and Gentleman SM (2010) NSAIDs: how they work and their prospects as therapeutics in Alzheimer’s disease. Front. Ag. Neurosci. 2:20. doi: 10.3389/fnagi.2010.00020
Received:01 March 2010;
Paper pending published:22 March 2010;
Accepted:24 April 2010;
Published online:18 May 2010
Edited by:
Elena Galea, Universitat Autònoma de Barcelona, SpainReviewed by:
Sue T. Griffin, University of Arkansas for Medical Sciences, USAKerry O'Banion, University of Rochester, USA
Elena Galea, Universitat Autònoma de Barcelona, Spain
Copyright: © 2010 Sastre and Gentleman. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Magdalena Sastre, Centre for Neuroscience, Division of Experimental Medicine, Imperial College London, Hammersmith Hospital, Du Cane Road, London, W12 0NN, UK. e-mail:bS5zYXN0cmVAaW1wZXJpYWwuYWMudWs=