
- 1 Department of Anatomy, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
- 2 Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
- 3 Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
- 4 National Neuroscience Institute, Novena, Singapore
Currently, there is still no effective therapy for neurodegenerative diseases (NDD) such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) despite intensive research and on-going clinical trials. Collectively, these diseases account for the bulk of health care burden associated with age-related neurodegenerative disorders. There is therefore an urgent need to further research into the molecular pathogenesis, histological differentiation, and clinical management of NDD. Importantly, there is also an urgency to understand the similarities and differences between these two diseases so as to identify the common or different upstream and downstream signaling pathways. In this review, the role iron play in NDD will be highlighted, as iron is key to a common underlying pathway in the production of oxidative stress. There is increasing evidence to suggest that oxidative stress predisposed cells to undergo damage to DNA, protein and lipid, and as such a common factor involved in the pathogenesis of AD and PD. The challenge then is to minimize elevated and uncontrolled oxidative stress levels while not affecting basal iron metabolism, as iron plays vital roles in sustaining cellular function. However, overload of iron results in increased oxidative stress due to the Fenton reaction. We discuss evidence to suggest that sustained exercise and diet restriction may be ways to slow the rate of neurodegeneration, by perhaps promoting neurogenesis or antioxidant-related pathways. It is also our intention to cover NDD in a broad sense, in the context of basic and clinical sciences to cater for both clinician’s and the scientist’s needs, and to highlight current research investigating exercise as a therapeutic or preventive measure.
Introduction
Neurodegenerative diseases (NDD) in on the rise, as the global population grows older, since aging is a prominent risk factor (Yankner et al., 2008). Needless to say, human’s sufferings are considerable, and the associated socioeconomic losses are enormous (Leifer, 2003). It is also reasonably clear that the different NDD (AD/PD) may share some similarities in genetic, histopathology and/or behavioral traits. This inevitably leads to problems with diagnosis, as subjective family history profiling, supported by objective genetic testing is not the optimal approach. To further compound this problem, there is a lack of suitable biomarkers for AD/PD. Even with post-mortem histopathology, there are still many gaps in knowledge that need to be addressed. This is exemplified by the positive immuno-staining of brain samples of PD and AD patients for alpha synuclein (α-syn) (Crews et al., 2009), although the findings in the latter are an exception, rather than a rule. In addition, genetic profiling without complementary evidence cannot fully clarify the above problem, as common mutations can be found for both AD and PD (Bertoli-Avella et al., 2004).
Prevailing sedentary lifestyle in this modern era has definitely contributed to the increase in age-related problems such as dementia (2009). This trend coupled with overindulgence in food consumption is worrying and may inevitably set the stage for an explosion in NDD. The effects of exercise and diet on brain function has previously been elaborated on (Gomez-Pinilla, 2008), and may be used as strategies to combat the above problem. The rationale for these shall be discussed later.
With that background, we shall provide below a review for Alzheimer’s disease (AD), and Parkinson’s disease (PD), the implications of the “Iron hypothesis” and to highlight the efficacy of exercise (mental and physical) and dietary restriction in slowing the progression of these diseases.
Types of Neurodegenerative Diseases
Alzheimer’s Disease
Alzheimer’s disease is the leading neurodegenerative disease. It is a progressive neurological disorder of broad etiology with a strong genetic influence, and with varied presentation according to age, sex and life style factors (Mattson, 2004). The disease primarily affects the aged population above the age of 65 in the USA (Leifer, 2003), although there are instances of early onset. In Singapore, the demographic profile is as follows: 0.8% amongst the 60- to 64-year-old cohort, to a staggeringly 32.2% amongst individuals above 84 years of age (Ministry of Health, Singapore, 2007). Sporadic form of AD (>90%) arises from a combination of various genetic, behavioral, dietary and environmental factors; hence there is no single magic remedy to tackle this problem. Inevitably, this presents a significant problem, in view of the extended life expectancies, and rapidly graying populations (Ministry of Health, Singapore, 2007).
In terms of clinical manifestations, AD affects the mental faculty of a patient with severe consequences inclusive of socioeconomic cost (Leifer, 2003). Typically, an AD patient loses normal cognitive function over time, including emotion, learning and memory processing skills (Mattson, 2004), ultimately leading to what is generally termed as “dementia” (Jalbert et al., 2008). In terms of pathology, there is the characteristic accumulation of inter-neuronal “Plaques” and intra-neuronal “Tangles” (Maccioni et al., 2001; Mudher and Lovestone, 2002; Mattson, 2004; Halliwell and Gutteridge, 2007). “Plaques” form when the peptide, β-Amyloid (Aβ), aggregate outside of the neuron, while the “Tangles” that build up within the cell are mainly composed of hyper-phosphorylated microtubule-associated tau protein (Mattson, 2004; Halliwell and Gutteridge, 2007). Although “Plaques and Tangles” are also found in normal aged brains, soluble Aβ is at least 5 times higher, while precipitated Aβ may be about 100 times higher in an AD patient (Halliwell and Gutteridge, 2007). Accumulation of these intra- and extra-cellular proteins coupled with poor clearance of it seems to interfere with neuronal function and induce neuronal death. However, there is still no consensus as to whether which of the two factors plays a more significant role (Mudher and Lovestone, 2002). Nonetheless, these factors are obviously the most prominent therapeutic targets (Mudher and Lovestone, 2002; Soto, 2003; Mattson, 2004; Roberson and Mucke, 2006; Bates et al., 2009).
There are known mutations in a few genes that are closely linked to AD. Some genes (APP, PSEN1, PSEN2, ApoE) (Bertram and Tanzi, 2008; Sando et al., 2008), related cholesterol and inflammatory factors have also been extensively investigated (Beffert et al., 1998; Bales et al., 2002; Lott and Head, 2005). However, it has been suggested that oxidative damage is one of the most integral neurotoxic mechanisms in both Aβ accumulation (Halliwell and Gutteridge, 2007) and tau pathologies (Ballatore et al., 2007). In that connection, we shall be reviewing some studies in relation to oxidative stress and mitochondria dysfunction in AD, to neurodegenerative disorders in general, and we shall discuss how exercise may be a promising therapy.
Specifically, increased oxidative damage to brain lipids, carbohydrates, proteins and DNA in AD has been reported (Markesbery, 1999; Varadarajan et al., 2000; Butterfield and Kanski, 2001; Butterfield and Lauderback, 2002; Mariani et al., 2005). Hence, the management of oxidative stress level is highly likely to play an important role, in the overall disease course. As such, there has been much interest in investigating ROS generation, and how this may be minimized. Of the various sources of oxidative stress proposed for AD: Aβ, mitochondria dysfunction, and redox-active metal ions like iron (Fe2+) and copper (Cu2+) ions play important roles. There is a current view suggesting that exercise could in fact moderate AD (Rolland et al., 2008). This is presented in Table 2.
Parkinson’s Disease
Although described about a century before AD, PD (a movement disorder) is the second most common neurodegenerative disorder worldwide. It affects approximately 1% of the aged population (≥ age of 70) in the USA. In addition to the classical resting tremor, other symptoms include bradykinesia, rigidity, and postural instability with accompanying dementia or autonomic dysfunction (Olanow and Tatton, 1999). These symptoms are clinically defined as “parkinsonism”. However, a distinction should be drawn between the latter and PD itself, as display of “parkinsonism” does not equate someone with the disease. In terms of pathology, the classical lesion occurs in the brain region called Substantia Nigra pars compacta (SNpc). Here, selective death of dopaminergic neurons occurs, thereby resulting in a reduction in the release of neurotransmitters such as dopamine. This frequently translates into impairment of gait and movements. The other cardinal pathology is the presence of Lewy bodies which are largely composed of aggregated proteins (Irvine et al., 2008), such as alpha (α) synuclein.
It is now believed that genetics influenced the etiology of this disease substantially (Hardy et al., 2006). A leap toward the understanding of PD pathogenesis took place after the discovery of mutations in α-synuclein in 1997 (Polymeropoulos et al., 1997; Kitada et al., 1998). This was followed by discoveries of mutations in other genes, including Parkin, PINK1, LRRK2 and DJ-1 (Hardy et al., 2006). In addition to the mutations in the α-synuclein gene, duplication (Chartier-Harlin et al., 2004; Ibanez et al., 2004; Eriksen et al., 2005) and triplication (Singleton et al., 2004) of the gene have also been linked to familial forms of PD. There is substantial evidence suggesting the positive role of exercise toward slowing the progression of PD (Crizzle and Newhouse, 2006; Falvo et al., 2008; Goodwin et al., 2008). This is presented in Table 2.
AD Versus PD
As mentioned earlier, PD patients exhibit much movement dysfunction, and other related issues such as balance. In that context, PD is essentially a movement disorder, primarily due to the death of dopaminergic neurons in the striatum. On the other hand, AD is a dementia disorder compounded by mood swings, depression, irritability, and learning dysfunction. Toward the terminal phase of the disease, movement disorders will also be featured. In the following section, the major differences and similarities will be discussed (Table 1).
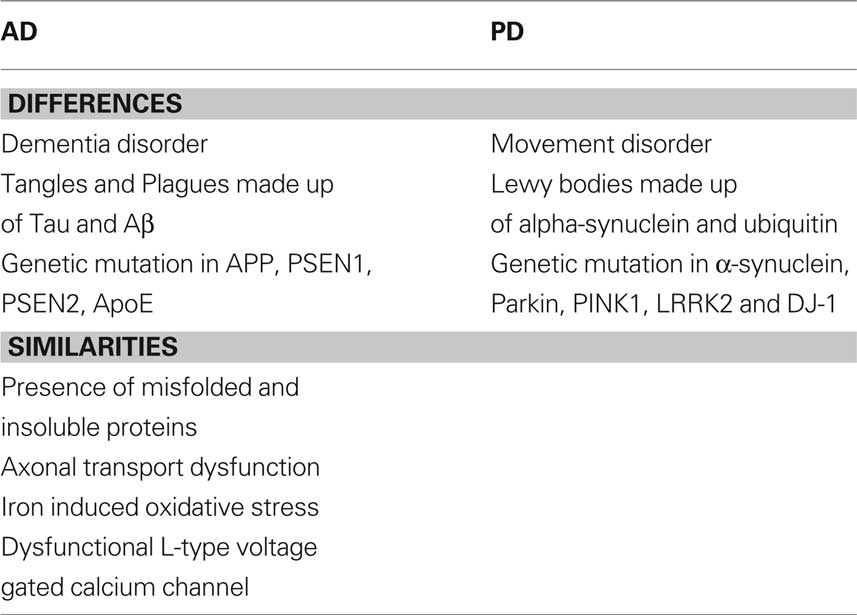
Table 1. Differences and similarities between AD and PD.
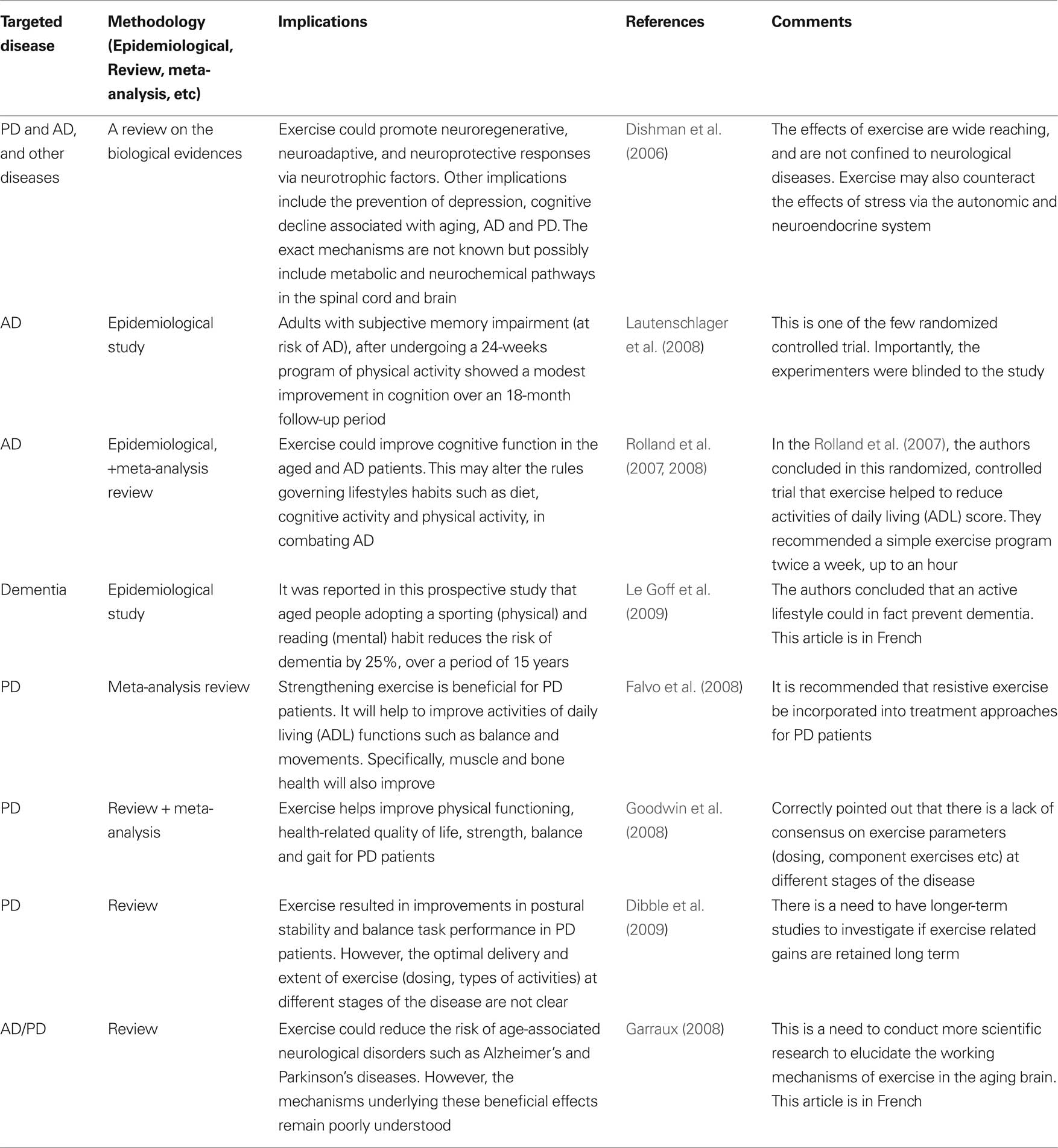
Table 2. Selected literatures on the effects of exercise on the brain (from a PubMed search: keywords – Alzheimer’s, Parkinson’s disease, exercise, brain).
Neurodegeneration Diseases – “The Iron Hypothesis” and Others
It is obvious that old age alone is a key factor in the onset of NDD (Yankner et al., 2008). A simplistic notion would be aging results in the accumulation of iron, which will increase oxidative stress, thus causing diseases like AD and PD. This occurs primarily via the Fenton reaction in producing the highly reactive hydroxyl radicals, which are involved in causing damage to DNA, lipid and protein (see box below). Aptly, “The iron hypothesis” deserves special attention as it is tightly linked to aging and the various NDD (Egana et al., 2003; Ke and Ming Qian, 2003; Zecca et al., 2004).
Fe2+ + H2O2→Fe3++ OH−+•OH
Fe3+ + O2•−→Fe2++ O2
H2O2+O2•−→ OH−+•OH + O2
Fenton reaction. This involves the conversion of iron (II)(Fe2+) to iron (III) (Fe3+) through the reaction with hydrogen peroxide (H2O2). Hydroxyl radical (•OH) is generated as a by-product. Additonally, in the presence of superoxide anion (O2•-), Fe3+ can be converted back to Fe2+, which in turn can go through another cycle of reaction with H2O2 to generate more •OH. Besides, •OH could be generated through the reaction between H2O2 and O2•
In AD, iron does not only contribute to oxidative stress (Mattson, 2004), but it is also involved in plaque pathology, and the resulting plaque-related oxidative damage (Zecca et al., 2004). This is important because it was shown that physiological iron accumulation in AD brains is independent of normal age-related increase in ferritin (Zecca et al., 2004). Specifically, iron can participate directly in Aβ plaque formation by modulating the ability of α-secretase to cleave APP (Zecca et al., 2004) or by facilitating Aβ aggregation (Maccioni et al., 2001). Oxidative stress contributed by iron or from Aβ plaque, may also further promote amyloidogenesis (Mattson, 2004). These processes are likely taking place concurrently with iron build up during the aging process. Furthermore, iron accumulation is both regional- and cell-specific (e.g., hippocampus and astrocytes) possibly accounting for the preferential oxidative damage characteristic in AD (Zecca et al., 2004). In summary, the role of iron related oxidative stress in relation to NDD is significant and have been thoroughly discussed previously by others (Emerit et al., 2004; Mariani et al., 2005; Shibata and Kobayashi, 2008).
Moving away from iron as a causative agent, one has to be mindful of the interaction between environmental (iron is considered to be an environmental influence) and genetic factors in causing diseases such as PD (Wszolek et al., 1997). These factors are often cited in an attempt to differentiate NDD into the familial and sporadic subtypes (Goldberg et al., 1993; Rossor et al., 1996; Gros-Louis et al., 2006; McNaught and Olanow, 2006).
Without dwelling into the other NDD such as ALS or HD, there are other genetic factors which play important roles in the pathogenesis of AD and PD. Such include the influences of misfolded proteins (Apolipoprotein E, α-synuclein, etc), mitochondrial dysfunction and defective myelination (Figure 1). For instance, the role of ApoE (Apolipoprotein E) has been implicated in both AD and PD (Masliah et al., 1996; Gallardo et al., 2008). Other proteins such as α-syn are also known to affect AD and PD. The importance of protein aggregation and misfolding has also been thoroughly reviewed in NDD such as AD, PD, ALS, HD (Agorogiannis et al., 2004; Ross and Poirier, 2004; Irvine et al., 2008). Similarly, there are also interesting studies suggesting the role of certain pro-inflammatory cytokines (TNF-α) in causing AD and PD (Figure 1). Indeed, there are now initiatives to find suitable ways to combat NDD via anti-inflammatory strategy (Tweedie et al., 2007). For instance, the use of non-steroidal anti-inflammatory drugs (NSAID) to reduce exceedingly high level of age-related neuroinflammation has been undertaken. Unfortunately to this date, there is still a lack of consensus regarding its efficacy (Aisen et al., 2003; de Craen et al., 2005). This was further confounded by the controversies involving C0X-2 inhibitors in recent years.
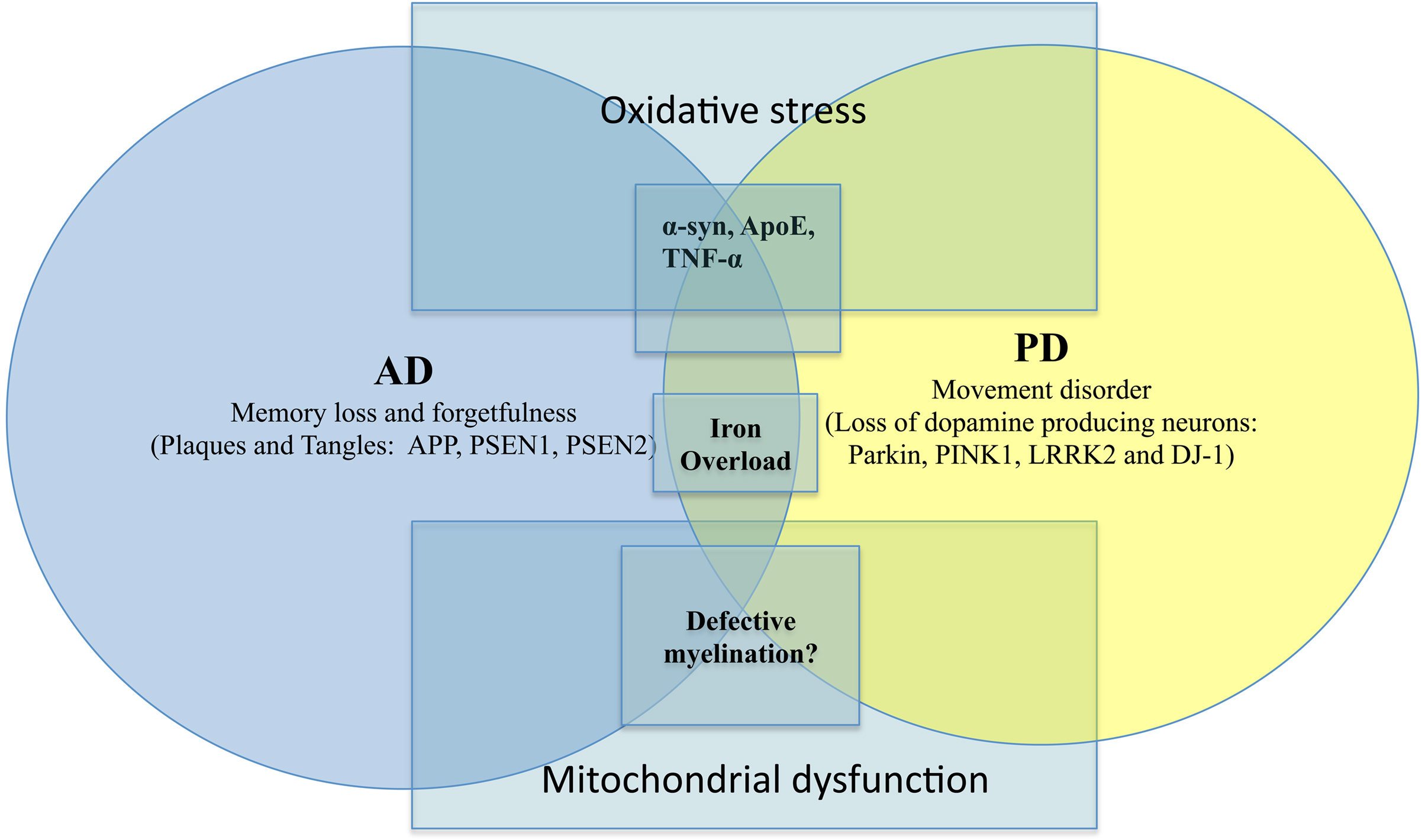
Figure 1. AD versus PD with overlapping causative factors. Salient signs of the disease are shown here together with key genetic/environmental influences. The overlaps were intentionally meant to suggest possible contribution by the same factor to both scenarios.
On another note, dysfunctional axonal transport in NDD (De Vos et al., 2008) has been reported suggesting a possibility that there may be defective myelination. However, other damaged structures such as mitochondria, kinesin and microtubules may also contribute to compromised axonal transport (De Vos et al., 2008). There are now studies alluding to the role of dysfunctional myelin in AD and other NDD (Bartzokis, 2004; Bartzokis et al., 2007) (Figure 1). Typically, in NDD there is myelin breakdown in the CNS or PNS. It is also interesting to note that myeloarchitecture may be laden with iron deposits (Fukunaga et al., 2010), again reaffirming the possible contributory role of iron dysregulation in NDD. Another recurring theme in AD/PD pathology is the involvement of mitochondrial dysfunction. This often leads to severe consequences such as impaired calcium buffering, generation of free radicals, activation of the mitochondrial permeability transition and secondary excitotoxicity (Beal, 1998). There is a chance that via the buffering of intracellular ATP and free radicals, the onset of NDD may be prevented (Beal, 1998).
Perhaps, more importantly, in considering the vast overlapping causative agents for AD and PD (Figure 1), we have to ask how NDD patients may be genetically predisposed to developing dementia (Novakovic et al., 2005). Knowing some of these factors may help in early diagnosis and treatment for these patients.
Exercising to Prevent AD/PD
The benefits of exercise to the brain and its cognitive functions are voluminous, and have been thoroughly reviewed by others (Dishman et al., 2006; Ang and Gomez-Pinilla, 2007; Winter et al., 2007). In AD patients, recent randomized controlled trials have shown that exercise could in fact help slow disease progression both directly and indirectly (Rolland et al., 2007, 2008; Lautenschlager et al., 2008). It is postulated that exercise could help to clear amyloid-beta peptide (main pathological driver) in AD patients (Bates et al., 2009). Via exercise induced nerve growth factor production, the death of cholinergic neurons could be prevented and perhaps attenuate cognitive decline (Scott and Crutcher, 1994). With aging, the basal metabolism will naturally fall, and it is speculated that with exercise, this may also help to reverse some of the risk factors (such as lower testosterone) for AD.
In PD patients, exercise could also alter the course of the disease (Crizzle and Newhouse, 2006; Dishman et al., 2006; Falvo et al., 2008; Goodwin et al., 2008). Exercise could help to improve postural stability, balance (Dibble et al., 2009), and tremor (Schalow et al., 2005) in this group of patients. It could also help to overcome musculoskeletal deficiencies (Falvo et al., 2008). At the basic level, it has been found that exercise could reduce the production of reactive oxygen and nitrogen species in this group of patients (Bloomer et al., 2008). Exercise could also possibly alleviate depression or negative mood (Peluso and Guerra de Andrade, 2005) often associated with AD and PD. It may also maintain cerebral blood flow in the retirees (Rogers et al., 1990) thereby preventing cognitive decline. It is also possible that exercise induced growth hormone changes may play a role in promoting neuroregeneration (by increasing neural stem cells) (Blackmore et al., 2009).
From animal studies, the benefits of exercise are even more startling. It has been reported that exercise could help with learning and memory (Winter et al., 2007). There are also evidences to suggest that exercise could in fact enhance neurogenesis (Aberg et al., 2008; Naylor et al., 2008; Van Praag, 2008; Wu et al., 2008). Furthermore, sustained exercise could in fact facilitate axonal transport of certain proteins such as acetylcholinesterase (Jasmin et al., 1988). Pertinent to this review, there is some evidence from animal studies that exercise could help to alter the antioxidant status in the brain under certain circumstances (Ozkaya et al., 2002). Collectively, this could well alleviate symptoms or even delay the progression of AD/PD. However, even with all these scientific knowledge, there is still no agreement concerning the optimal exercise intensity, and prescription strategy for patients suffering from AD/PD.
In summary, numerous epidemiological studies have pointed out that exercise is efficacious in reducing the risk of most age-related diseases such as AD and PD. However, there are still many gaps to be filled between biological findings and translating that understanding to situations in the clinic and community (Garraux, 2008). Furthermore, there is still no standardization for optimal outcomes, such as duration of exercise, intensity, point of intervention, and whether exercise could reverse the course of the disease after onset. These are issues, which undoubtedly will need further investigations and research. The following section presents a non-exhaustive review of selected literatures on the effects of exercise (mental and physical) on AD and PD.
Diet and Brain Function
It has been reported recently that caloric restriction and exercise could result in a myriad of systemic effects, including anti-inflammatory, reduction of oxidative stress; promotion of synaptic plasticity, and induction of neuroprotective factors in the brain (Gillette-Guyonnet and Vellas, 2008; Hofer et al., 2008). This is indeed promising, as oxidative stress has long been an underlying factor to trigger the onset of NDD (Linseman, 2009). In terms of mechanisms, since the mitochondria is known as the major source of ROS in the cell, dietary restriction would directly reduce the generation of it by minimizing proton leakage (Hagopian et al., 2005; Civitarese et al., 2007). Conversely, a high fat diet is capable of inducing the activation and expression of certain transcription factors like NF-kappa B and hence inflammation (Liao et al., 1993), which might lead to increased cellular stress. Free radical damage also underlies age-related diseases (Egana et al., 2003; Stankiewicz and Brass, 2009). The damage due to exposure to free radical throughout life, especially ROS generated by the mitochondria (Bayir and Kagan, 2008), is ameliorated by a repertoire of antioxidant systems acting at the cellular level to protect the integrity of biomolecules from oxidative damage. Emerging evidences, both in in vitro and in vitro studies have shown that increased energy expenditure (ATP usage) and dietary (calorie) restriction could exert beneficial effects on bodily systems, working primarily through improved antioxidant systems, upregulated growth factors (BDNF and NGF) as well as cytoprotective proteins (Lane et al., 1996; Hofer et al., 2008). As a consequence, these molecular mechanisms work to enhance the efficiency of free radical neutralization so as to reduce the risk factor of age-related oxidative load and diseases.
On another note, caloric restriction could also promote adult neurogenesis, and hence prevent AD (Levenson and Rich, 2007) although the neurobiological mechanism is still unknown. Selecting diet enriched with Omega 3 fatty acids could also help brain function (Sinclair et al., 2007; Tassoni et al., 2008). In summary, hormesis is perhaps taking place while protecting the body against aging and neurodegeneration (Gomez-Pinilla, 2008; Mattson, 2008a,b).
Perspectives for Future Research
It may be worthwhile if one could design a blood test to screen for certain biomarkers which could predict the chances of developing AD and PD, and indeed there are now growing interest in such endeavors (Rachakonda et al., 2004). In conclusion, we propose here that there is much overlap in the mechanisms leading to AD/PD and the possible strategies to combat it (Figure 2). We propose in this review that exercise coupled with diet/caloric restriction may be the way forward.
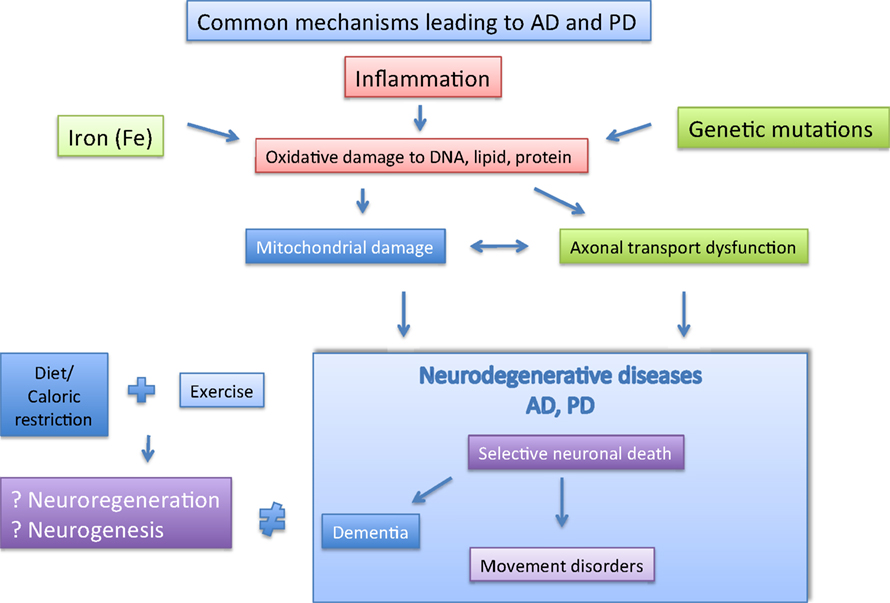
Figure 2. The major player implicated in AD and PD are depicted here. The possible pathological factor (boxed), and likely interactions between them are shown. Separately, Caloric restriction and exercise may help to prevent AD and PD via neuroregeneration and neurogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are funded by a Singapore BMRC research grant (04/1/21/19/335). We would also like to thank Dr Karthik Harve (Department of Anatomy, NUS) for proof reading this manuscript.
References
Aberg, E., Perlmann, T., Olson, L., and Brene, S. (2008). Running increases neurogenesis without retinoic acid receptor activation in the adult mouse dentate gyrus. Hippocampus 18, 785–792.
Agorogiannis, E. I., Agorogiannis, G. I., Papadimitriou, A., and Hadjigeorgiou, G. M. (2004). Protein misfolding in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 30, 215–224.
Aisen, P. S., Schafer, K. A., Grundman, M., Pfeiffer, E., Sano, M., Davis, K. L., Farlow, M. R., Jin, S., Thomas, R. G., and Thal, L. J. (2003). Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826.
Ang, E. T., and Gomez-Pinilla, F. (2007). Potential therapeutic effects of exercise to the brain. Curr. Med. Chem. 14, 2564–2571.
Bales, K. R., Dodart, J. C., Demattos, R. B., Holtzman, D. M., and Paul, S. M. (2002). Apolipoprotein E, amyloid, and Alzheimer disease. Mol. Interv. 2, 363–375, 339.
Ballatore, C., Lee, V. M., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8, 663–672.
Bartzokis, G. (2004). Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol. Aging 25, 5–18; author reply 49–62.
Bartzokis, G., Lu, P. H., and Mintz, J. (2007). Human brain myelination and amyloid beta deposition in Alzheimer’s disease. Alzheimers Dement. 3, 122–125.
Bates, K. A., Verdile, G., Li, Q. X., Ames, D., Hudson, P., Masters, C. L., and Martins, R. N. (2009). Clearance mechanisms of Alzheimer’s amyloid-beta peptide: implications for therapeutic design and diagnostic tests. Mol. Psychiatry.14, 469–486.
Bayir, H., and Kagan, V. E. (2008). Bench-to-bedside review: Mitochondrial injury, oxidative stress and apoptosis–there is nothing more practical than a good theory. Crit. Care 12, 206.
Beal, M. F. (1998). Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta 1366, 211–223.
Beffert, U., Danik, M., Krzywkowski, P., Ramassamy, C., Berrada, F., and Poirier, J. (1998). The neurobiology of apolipoproteins and their receptors in the Cns and Alzheimer’s disease. Brain Res. Brain Res. Rev. 27, 119–142.
Bertoli-Avella, A. M., Oostra, B. A., and Heutink, P. (2004). Chasing genes in Alzheimer’s and Parkinson’s disease. Hum. Genet. 114, 413–438.
Bertram, L., and Tanzi, R. E. (2008). Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat. Rev. Neurosci. 9, 768–778.
Blackmore, D. G., Golmohammadi, M. G., Large, B., Waters, M. J., and Rietze, R. L. (2009). Exercise increases neural stem cell number in a growth hormone-dependent manner, augmenting the regenerative response in aged mice. Stem Cells 27, 2044–2052.
Bloomer, R. J., Schilling, B. K., Karlage, R. E., Ledoux, M. S., Pfeiffer, R. F., and Callegari, J. (2008). Effect of resistance training on blood oxidative stress in Parkinson disease. Med. Sci. Sports Exerc. 40, 1385–1389.
Butterfield, D. A., and Kanski, J. (2001). Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 122, 945–962.
Butterfield, D. A., and Lauderback, C. M. (2002). Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 32, 1050–1060.
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., Levecque, C., Larvor, L., Andrieux, J., Hulihan, M., Waucquier, N., Defebvre, L., Amouyel, P., Farrer, M., and Destee, A. (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169.
Civitarese, A. E., Carling, S., Heilbronn, L. K., Hulver, M. H., Ukropcova, B., Deutsch, W. A., Smith, S. R., and Ravussin, E. (2007). Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 4, e76. doi:10.1371/journal.pmed.0040076.
Crews, L., Tsigelny, I., Hashimoto, M., and Masliah, E. (2009). Role of synucleins in Alzheimer’s disease. Neurotox. Res. 16, 306–317.
Crizzle, A. M., and Newhouse, I. J. (2006). Is physical exercise beneficial for persons with Parkinson’s disease? Clin. J. Sport Med. 16, 422–425.
de Craen, A. J., Gussekloo, J., Vrijsen, B., and Westendorp, R. G. (2005). Meta-analysis of nonsteroidal antiinflammatory drug use and risk of dementia. Am. J. Epidemiol. 161, 114–120.
De Vos, K. J., Grierson, A. J., Ackerley, S., and Miller, C. C. (2008). Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151–173.
Dibble, L. E., Addison, O., and Papa, E. (2009). The effects of exercise on balance in persons with Parkinson’s disease: a systematic review across the disability spectrum. J. Neurol. Phys. Ther. 33, 14–26.
Dishman, R. K., Berthoud, H. R., Booth, F. W., Cotman, C. W., Edgerton, V. R., Fleshner, M. R., Gandevia, S. C., Gomez-Pinilla, F., Greenwood, B. N., Hillman, C. H., Kramer, A. F., Levin, B. E., Moran, T. H., Russo-Neustadt, A. A., Salamone, J. D., Van Hoomissen, J. D., Wade, C. E., York, D. A., and Zigmond, M. J. (2006). Neurobiology of exercise. Obesity (Silver Spring) 14, 345–356.
Egana, J. T., Zambrano, C., Nunez, M. T., Gonzalez-Billault, C., and Maccioni, R. B. (2003). Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. Biometals 16, 215–223.
Emerit, J., Edeas, M., and Bricaire, F. (2004). Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 58, 39–46.
Eriksen, J. L., Przedborski, S., and Petrucelli, L. (2005). Gene dosage and pathogenesis of Parkinson’s disease. Trends. Mol. Med. 11, 91–96.
Falvo, M. J., Schilling, B. K., and Earhart, G. M. (2008). Parkinson’s disease and resistive exercise: rationale, review, and recommendations. Mov. Disord. 23, 1–11.
Fukunaga, M., Li, T. Q., Van Gelderen, P., De Zwart, J. A., Shmueli, K., Yao, B., Lee, J., Maric, D., Aronova, M. A., Zhang, G., Leapman, R. D., Schenck, J. F., Merkle, H., and Duyn, J. H. (2010). Layer-specific variation of iron content in cerebral cortex as a source of Mri contrast. Proc. Natl. Acad. Sci. U.S.A. 107, 3834–3839.
Gallardo, G., Schluter, O. M., and Sudhof, T. C. (2008). A molecular pathway of neurodegeneration linking alpha- synuclein to ApoE and Abeta peptides. Nat. Neurosci. 11, 301–308.
Gillette-Guyonnet, S., and Vellas, B. (2008). Caloric restriction and brain function. Curr. Opin. Clin. Nutr. Metab. Care 11, 686–692.
Goldberg, Y. P., Andrew, S. E., Theilmann, J., Kremer, B., Squitieri, F., Telenius, H., Brown, J. D., and Hayden, M. R. (1993). Familial predisposition to recurrent mutations causing Huntington’s disease: genetic risk to sibs of sporadic cases. J. Med. Genet. 30, 987–990.
Gomez-Pinilla, F. (2008). The influences of diet and exercise on mental health through hormesis. Ageing Res. Rev. 7, 49–62.
Goodwin, V. A., Richards, S. H., Taylor, R. S., Taylor, A. H., and Campbell, J. L. (2008). The effectiveness of exercise interventions for people with Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord. 23, 631–640.
Gros-Louis, F., Gaspar, C., and Rouleau, G. A. (2006). Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 956–972.
Hagopian, K., Harper, M. E., Ram, J. J., Humble, S. J., Weindruch, R., and Ramsey, J. J. (2005). Long-term calorie restriction reduces proton leak and hydrogen peroxide production in liver mitochondria. Am. J. Physiol. Endocrinol. Metab. 288, E674–E684.
Hardy, J., Cai, H., Cookson, M. R., Gwinn-Hardy, K., and Singleton, A. (2006). Genetics of Parkinson’s disease and parkinsonism. Ann. Neurol. 60, 389–398.
Halliwell, B., and Gutteridge, J. M. C. (2007). Free Radicals in Biology and Medicine. New York: Oxford University Press.
Hofer, T., Fontana, L., Anton, S. D., Weiss, E. P., Villareal, D., Malayappan, B., and Leeuwenburgh, C. (2008). Long-term effects of caloric restriction or exercise on Dna and Rna oxidation levels in white blood cells and urine in humans. Rejuvenation Res. 11, 793–799.
Ibanez, P., Bonnet, A. M., Debarges, B., Lohmann, E., Tison, F., Pollak, P., Agid, Y., Durr, A., and Brice, A. (2004). Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171.
Irvine, G. B., El-Agnaf, O. M., Shankar, G. M., and Walsh, D. M. (2008). Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 14, 451–464.
Jalbert, J. J., Daiello, L. A., and Lapane, K. L. (2008). Dementia of the Alzheimer type. Epidemiol. Rev. 30, 15–34.
Jasmin, B. J., Lavoie, P. A., and Gardiner, P. F. (1988). Fast axonal transport of labeled proteins in motoneurons of exercise-trained rats. Am. J. Physiol. 255, C731–C736.
Ke, Y., and Ming Qian, Z. (2003). Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol. 2, 246–253.
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y., and Shimizu, N. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608.
Lane, M. A., Baer, D. J., Rumpler, W. V., Weindruch, R., Ingram, D. K., Tilmont, E. M., Cutler, R. G., and Roth, G. S. (1996). Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc. Natl. Acad. Sci. U.S.A. 93, 4159–4164.
Lautenschlager, N. T., Cox, K. L., Flicker, L., Foster, J. K., Van Bockxmeer, F. M., Xiao, J., Greenop, K. R., and Almeida, O. P. (2008). Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA 300, 1027–1037.
Le Goff, M., Helmer, C., Foubert-Samier, A., Cowppli-Bony, P., Berr, C., and Dartigues, J. F. (2009). Activities in retired people and the risk of dementia. C. R. Biol. 332, 378–384.
Leifer, B. P. (2003). Early diagnosis of Alzheimer’s disease: clinical and economic benefits. J. Am. Geriatr. Soc. 51, S281–S288.
Levenson, C. W., and Rich, N. J. (2007). Eat less, live longer? New insights into the role of caloric restriction in the brain. Nutr. Rev. 65, 412–415.
Liao, F., Andalibi, A., Debeer, F. C., Fogelman, A. M., and Lusis, A. J. (1993). Genetic control of inflammatory gene induction and Nf-kappa B-like transcription factor activation in response to an atherogenic diet in mice. J. Clin. Invest. 91, 2572–2579.
Linseman, D. A. (2009). Targeting oxidative stress for neuroprotection. Antioxid. Redox Signal. 11, 421–424.
Lott, I. T., and Head, E. (2005). Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol. Aging 26, 383–389.
Maccioni, R. B., Munoz, J. P., and Barbeito, L. (2001). The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch. Med. Res. 32, 367–381.
Mariani, E., Polidori, M. C., Cherubini, A., and Mecocci, P. (2005). Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 827, 65–75.
Markesbery, W. R. (1999). The role of oxidative stress in Alzheimer disease. Arch. Neurol. 56, 1449–1452.
Masliah, E., Mallory, M., Veinbergs, I., Miller, A., and Samuel, W. (1996). Alterations in apolipoprotein E expression during aging and neurodegeneration. Prog. Neurobiol. 50, 493–503.
McNaught, K. S., and Olanow, C. W. (2006). Protein aggregation in the pathogenesis of familial and sporadic Parkinson’s disease. Neurobiol. Aging 27, 530–545.
Ministry of Health, Singapore (2007). Dementia, MOH Clinical Practice Guidelines 3/2007. Available at: http://www.moh.gov.sg/cpg.
Mudher, A., and Lovestone, S. (2002). Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 25, 22–26.
Naylor, A. S., Bull, C., Nilsson, M. K., Zhu, C., Bjork-Eriksson, T., Eriksson, P. S., Blomgren, K., and Kuhn, H. G. (2008). Voluntary running rescues adult hippocampal neurogenesis after irradiation of the young mouse brain. Proc. Natl. Acad. Sci. U.S.A. 105, 14632–14637.
Novakovic, K. E., Villemagne, V. L., Rowe, C. C., and Masters, C. L. (2005). Rare genetically defined causes of dementia. Int. Psychogeriatr. 17(Suppl. 1), S149–S194.
Olanow, C. W., and Tatton, W. G. (1999). Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 22, 123–144.
Ozkaya, Y. G., Agar, A., Yargicoglu, P., Hacioglu, G., Bilmen-Sarikcioglu, S., Ozen, I., and Aliciguzel, Y. (2002). The effect of exercise on brain antioxidant status of diabetic rats. Diabetes Metab. 28, 377–384.
Peluso, M. A., and Guerra De Andrade, L. H. (2005). Physical activity and mental health: the association between exercise and mood. Clinics 60, 61–70.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C., Di Iorio, G., Golbe, L. I., and Nussbaum, R. L. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047.
Rachakonda, V., Pan, T. H., and Le, W. D. (2004). Biomarkers of neurodegenerative disorders: how good are they? Cell Res. 14, 347–358.
Roberson, E. D., and Mucke, L. (2006). 100 years and counting: prospects for defeating Alzheimer’s disease. Science 314, 781–784.
Rogers, R. L., Meyer, J. S., and Mortel, K. F. (1990). After reaching retirement age physical activity sustains cerebral perfusion and cognition. J. Am. Geriatr. Soc. 38, 123–128.
Rolland, Y., Abellan Van Kan, G., and Vellas, B. (2008). Physical activity and Alzheimer’s disease: from prevention to therapeutic perspectives. J. Am. Med. Dir. Assoc. 9, 390–405.
Rolland, Y., Pillard, F., Klapouszczak, A., Reynish, E., Thomas, D., Andrieu, S., Riviere, D., and Vellas, B. (2007). Exercise program for nursing home residents with Alzheimer’s disease: a 1-year randomized, controlled trial. J. Am. Geriatr. Soc. 55, 158–165.
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl.), S10–S17.
Rossor, M. N., Fox, N. C., Freeborough, P. A., and Harvey, R. J. (1996). Clinical features of sporadic and familial Alzheimer’s disease. Neurodegeneration 5, 393–397.
Sando, S. B., Melquist, S., Cannon, A., Hutton, M. L., Sletvold, O., Saltvedt, I., White, L. R., Lydersen, S., and Aasly, J. O. (2008). Apoe epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 8, 9.
Schalow, G., Paasuke, M., and Jaigma, P. (2005). Integrative re-organization mechanism for reducing tremor in Parkinson’s disease patients. Electromyogr. Clin. Neurophysiol. 45, 407–415.
Scott, S. A., and Crutcher, K. A. (1994). Nerve growth factor and Alzheimer’s disease. Rev. Neurosci. 5, 179–211.
Shibata, N., and Kobayashi, M. (2008). The role for oxidative stress in neurodegenerative diseases. Brain Nerve 60, 157–170.
Sinclair, A. J., Begg, D., Mathai, M., and Weisinger, R. S. (2007). Omega 3 fatty acids and the brain: review of studies in depression. Asia Pac. J. Clin. Nutr. 16(Suppl. 1), 391–397.
Singleton, A., Gwinn-Hardy, K., Sharabi, Y., Li, S. T., Holmes, C., Dendi, R., Hardy, J., Singleton, A., Crawley, A., and Goldstein, D. S. (2004). Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain 127, 768–772.
Soto, C. (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60.
Stankiewicz, J. M., and Brass, S. D. (2009). Role of iron in neurotoxicity: a cause for concern in the elderly? Curr. Opin. Clin. Nutr. Metab. Care 12, 22–29.
Tassoni, D., Kaur, G., Weisinger, R. S., and Sinclair, A. J. (2008). The role of eicosanoids in the brain. Asia Pac. J. Clin. Nutr. 17(Suppl. 1), 220–228.
Tweedie, D., Sambamurti, K., and Greig, N. H. (2007). Tnf-alpha inhibition as a treatment strategy for neurodegenerative disorders: new drug candidates and targets. Curr. Alzheimer Res. 4, 378–385.
Van Praag, H. (2008). Neurogenesis and exercise: past and future directions. Neuromolecular Med. 10, 128–140.
Varadarajan, S., Yatin, S., Aksenova, M., and Butterfield, D. A. (2000). Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 130, 184–208.
Winter, B., Breitenstein, C., Mooren, F. C., Voelker, K., Fobker, M., Lechtermann, A., Krueger, K., Fromme, A., Korsukewitz, C., Floel, A., and Knecht, S. (2007). High impact running improves learning. Neurobiol. Learn. Mem. 87, 597–609.
Wszolek, Z. K., Vieregge, P., Uitti, R. J., Gasser, T., Yasuhara, O., Mcgeer, P., Berry, K., Calne, D. B., Vingerhoets, F. J., Klein, C., and Pfeiffer, R. F. (1997). German-Canadian family (family A) with parkinsonism, amyotrophy, and dementia – Longitudinal observations. Parkinsonism Relat. Disord. 3, 125–139.
Wu, C. W., Chang, Y. T., Yu, L., Chen, H. I., Jen, C. J., Wu, S. Y., Lo, C. P., and Kuo, Y. M. (2008). Exercise enhances the proliferation of neural stem cells and neurite growth and survival of neuronal progenitor cells in dentate gyrus of middle-aged mice. J. Appl. Physiol. 105, 1585–1594.
Keywords: Alzheimer’s, Parkinson’s, iron, oxidative stress, exercise
Citation: Ang E-T, Tai Y-K, Lo S-Q, Seet R and Soong T-W (2010) Neurodegenerative diseases: exercising toward neurogenesis and neuroregeneration. Front. Ag. Neurosci. 2:25. doi: 10.3389/fnagi.2010.00025
Received: 26 November 2009;
Paper pending published: 17 March 2010;
Accepted: 14 June 2010;
Published online: 21 July 2010
Edited by:
Massimo Tabaton, University of Genoa, ItalyReviewed by:
Gianluigi Zanusso, University of Verona, ItalyFabrizio Tagliavini, Istituto Neurologico C. Besta, Italy
Copyright: © 2010 Ang, Tai, Lo, Seet and Soong. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Tuck-Wah Soong, Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, MD9, 2 Medical Drive, Singapore 117597, Singapore. e-mail:cGhzc3R3QC5udXMuZWR1LnNn