


- 1 Department of Psychology and Program Neuroscience, Stony Brook University, Stony Brook, NY, USA
- 2 Department of Psychology, Center for Developmental Neuroscience and Developmental Disabilities, College of Staten Island/City University of New York, Staten Island, NY, USA
To identify interventions for brain aging, we must first identify the processes in which we hope to intervene. Brain aging is a period of decreasing functional capacity and increasing vulnerability, which reflect a reduction in morphological organization and perhaps degeneration. Since life is ultimately dependent upon the ability to maintain cellular organization through metabolism, this review explores evidence for a decline in neural metabolic support during aging, which includes a reduction in whole brain cerebral blood flow, and cellular metabolic capacity. Capillary density may also decrease with age, although the results are less clear. Exercise may be a highly effective intervention for brain aging, because it improves the cardiovascular system as a whole, and increases regional capillary density and neuronal metabolic capacity. Although the evidence is strongest for motor regions, more work may yield additional evidence for exercise-related improvement in metabolic support in non-motor regions. The protective effects of exercise may be specific to brain region and the type of insult. For example, exercise protects striatal cells from ischemia, but it produces mixed results after hippocampal seizures. Exercise can improve metabolic support and bioenergetic capacity in adult animals, but it remains to be determined whether it has similar effects in aging animals. What is clear is that exercise can influence the multiple levels of support necessary for maintaining optimal neuronal function, which is unique among proposed interventions for aging.
Introduction
To intervene in brain aging, we must first identify the primary factors that underlie it. Brain aging is a period of decreasing functional capacity and increasing vulnerability, which reflect a reduction in morphological organization and perhaps degenerative changes. The termination of this period is death, which is defined as a point at which cell degradation is irreversible. In contrast to death, life is dependent upon metabolism for the reversal of movement toward disorder (Schrodinger, 1944). Through this lens, brain aging is a period in which neuronal metabolism may decline, leading to a reduction in function and anatomical integrity. Therefore, interventions directed at maintenance and restoration of neuronal metabolic capacity may target fundamental features of cellular aging. Here, we propose that exercise is one of the most effective interventions for aging, because it can simultaneously slow age-related decline in neuronal metabolic capacity and maintain cardiovascular support for neuronal metabolism. We will begin with a discussion of the relatively high metabolic demand of neurons and the fundamental importance of maintaining metabolic capacity for optimal neuronal function. We then review evidence for an age-related decline in both neuronal cardiovascular support and cellular metabolism, each followed by a review of evidence that exercise enhances these processes. We briefly review experiments conducted to test whether exercise can protect the brain from metabolic challenges that are more frequent during aging. We end by discussing the gaps in research, and the need for more direct tests of the effects of exercise on these systems in aging animals.
Neurons are Vulnerable to Declines in Metabolic Support and Capacity
In the brain, the demand for oxygen and glucose to drive cellular metabolism necessary for the maintenance and restoration of ionic gradients is ever present, but the function of these processes diminishes with age. And yet, the ability to continually reverse cellular entropy (movement toward disorder) is the fundamental process that maintains and prolongs the organized systems we consider as living (Schrodinger, 1944). Failure of this process causes death, first of cells, then organs, and eventually, the life of the individual. The end point is particularly notable for neurons, because neurons store information that is required over a lifetime. Paradoxically, their information processing and storage function underlie a high demand for energy that makes neurons particularly vulnerable to energy deficits and impaired metabolic support. Neurons’ high demand for energy is driven by the need to maintain ionic gradients across the large membrane surface area of dendrites. In addition to the large membrane surface area of neurons, synaptic action across the dendritic membrane continually degrades the ionic gradients. As a result, the brain uses 20% of the oxygen and 25% of glucose consumed despite making up only 2% of body weight. Oxygen and glucose are used to produce ATP through oxidative phosphorylation, a process that accounts for 95% of ATP production in the brain (Erecinska and Silver, 1989). In the anesthetized brain 40–60% of ATP is used by the sodium potassium pump to restore ionic gradients (Astrup et al., 1981; Erecinska and Silver, 1989).
There is a strong relationship between metabolic demand and the density of excitatory synapses over space and over developmental stages (Kageyama and Wong-Riley, 1982; Mjaatvedt and Wong-Riley, 1988). ATP production is coupled to the activity of cytochrome oxidase (Wong-Riley, 1989), for which a chemical reaction allows visualization of cellular energy consumption across nuclei, and across cellular compartments (Kageyama and Wong-Riley, 1982; Mjaatvedt and Wong-Riley, 1988). Within dendritic regions, which make up 78–86% of gray matter tissue volume in the hippocampus (Sousa et al., 1998; Tata et al., 2002; Tata and Anderson, 2010), the layers with small dendritic processes and higher synaptic density have higher metabolic demand than areas with large dendrites and smaller synaptic density (Kageyama and Wong-Riley, 1982; Mjaatvedt and Wong-Riley, 1988). Both have greater metabolic demand than layers with cell bodies, which tend to have low metabolic demand (Kageyama and Wong-Riley, 1982) and make up less than 20% of volume. The high metabolic demand of the dendritic layers underlies the disproportionate need for oxygen, glucose and ATP production in the brain. Depriving cells of primary afferent input causes a dramatic down-regulation of cytochrome oxidase activity, whereas restoration of afferent input up-regulates cytochrome oxidase activity (Wong-Riley, 1989). If metabolic support decreases subtly with age, dendritic retraction could effectively reduce energy demand, thereby sparing cells. So it may not be surprising that age can be associated with dendritic atrophy (Markham et al., 2005), and is not always related to cell loss (Rapp and Gallagher, 1996; Yates et al., 2008).
Despite the high and constant neural demand for ATP, it is used within minutes of being produced, leaving the brain highly vulnerable to any decline in metabolic support. ATP production is reduced when there is too little glucose or oxygen, or there is damage to cellular metabolic machinery. Aging is associated with a reduction in cytochrome oxidase reactivity (Ojaime et al., 1999), which would appear to place neurons at risk in periods of high metabolic demand. With too little ATP, the sodium potassium pumps may fail to restore resting ion gradients, resulting in excess depolarization. An increase in cell membrane depolarization allows excess calcium influx that, in turn, can cause cell death by initiating apoptosis, or necrosis. If age is related to damage to cellular metabolic machinery, then there is an increase in neuronal vulnerability to excitotoxicity.
As much as life is dependent upon metabolism, it is also dependent upon a second fundamental feature, collaboration (Dupre and O’Malley, 2009). This collaborative process is illustrated best by the dependence of cellular metabolism on the delivery of oxygen and glucose, which is carried out by the cardiovascular system. Neurons store little glucose, the fundamental source of energy, or oxygen, which is necessary for glucose metabolism. Since little ATP is stored, it must be produced within minutes of being used. Not surprisingly, neuronal activity triggers an increase in the delivery of oxygen and glucose; cerebral glucose utilization and cerebral metabolic rate for oxygen are coupled to neuronal activity (Magistretti and Pellerin, 1996). Similarly, across regions and dendritic layers, cytochrome oxidase activity correlates with glucose metabolism and capillary density (Borowsky and Collins, 1989). The dynamic and exquisite coupling between neuronal activity, cellular metabolism and oxygen and glucose delivery illustrates the fundamental role collaboration plays in supporting neuronal function, and therefore cognition.
The consequences of insufficient oxygen and glucose are illustrated by the sensory and cognitive deficits resulting from low glucose (Bie-Olsen et al., 2009; Gonder-Frederick et al., 2009) and hypoxia at high altitudes (Kramer et al., 1993) as well as mitochondrial dysfunction (Lauritzen et al., 2010). As we will see, aging can represent a period of slow decline in cellular metabolism, cardiovascular support or in the coupling between these systems, and therefore this decline may play a role in age-related cognitive deficits. As we consider targets for brain aging intervention, processes critical for metabolism must be considered. Thus, the most basic interventions in cognitive aging may require interventions in metabolic decline that span cellular to systems levels of support.
Cerebral Blood Flow is Reduced in Aging Populations
At the level of the whole brain, an age-related decline in metabolic support is clearly established. Resting cerebral blood flow has been found to be lower in normal healthy elderly with a variety of methods, which include continuous arterial spin labeling (CASL) magnetic resonance (MR) imaging (Bertsch et al., 2009), PET (Frackowiak et al., 1980), and SPECT (Devous et al., 1986). Cerebral blood flow was significantly less in elderly (M = 71 years old) than in young individuals (M = 27 years old) (Stoquart-ElSankari et al., 2007). Within the elderly sample, the decline was linearly related to age. At the regional level, neurovascular coupling is impaired. In aged compared to young subjects, the BOLD signal, which is dependent upon neurovascular coupling, has a higher signal to noise ratio, and can take longer to reach half the maximum value (D’Esposito et al., 2003). It is unclear whether these reductions reflect a reduction in neural activity driving a reduction in metabolism and need for oxygen and glucose, or conversely, whether impaired metabolic support reduces the capacity for neural activity. The initiating factor may vary across individuals.
Lifestyle correlates with cardiovascular health and cerebral blood flow. Within aging populations, adults who chose to continue working at retirement age, and adults who retired but remained active maintained cerebral blood flow 3 years past retirement relative to the decline seen in adults who became physically inactive (Rogers et al., 1990). It is unclear to what extent the reduction in blood flow is a cause or reflection of age-related changes in cognition and tissue volume. Individuals with active lifestyles have tighter cerebral regulation for cerebral blood perfusion (Formes et al., 2010). Although exercise is well-known to maintain heart health, reduce blood pressure and elevate high density lipoproteins (National Institutes of Health, 1996), all effects that should support brain function, more studies need to address whether exercise can directly maintain cerebral blood flow. The importance of cardiovascular function for cognitive function in aging humans is consistent with this possibility (Kramer et al., 1993, 1999; Colcombe et al., 2004; McAuley et al., 2004).
Age Reduces Capillary Integrity and Exercise Increases Capillary Density
Total cerebral blood flow and regional cerebral blood flow are dependent upon maintenance of arteries, arterioles and capillaries. The importance of the health of the cerebral vascular network may be best illustrated by evidence that vascular damage is the primary cause of cognitive deficits reported after tumor irradiation (Brown et al., 2005, 2007). Capillary density determines the distance over which oxygen and glucose must diffuse to reach cells, and can respond to changes in cellular energy demands. Capillary density varies with energy demand across space and developmental phases (Gross et al., 1987; Borowsky and Collins, 1989; Tuor et al., 1994), and is affected by conditions such as hypoxia (Mironov et al., 1994). An uncoupling between demand and density would be expected to alter basal neuronal metabolic capacity, which in turn may affect cognition. The risk of vascular occlusion increases with age so that a low density leaves more cells vulnerable to deficient oxygen and glucose when small vessels are occluded. Recognizing the importance of cerebral capillary density, numerous studies of capillary density in aged humans and non-human animals have been conducted (Riddle et al., 2003).
In humans, there are numerous reports that capillary density is lower in autopsy tissue from aged subjects relative to young subjects (Hunziker et al., 1979; Bell and Ball, 1981, 1985, 1990; Mann et al., 1986; Abernethy et al., 1993). Capillary density has also been reported to be lower in aged animals (Buchweitz-Milton and Weiss, 1987; Jucker and Meier-Ruge, 1989; Jucker et al., 1990; Amenta et al., 1995). Although, a number of studies have also reported no age-related differences in capillary density in rats and humans (Hunziker et al., 1979; Knox and Oliveira, 1980; Meier-Ruge et al., 1980; Hughes and Lantos, 1987; Meier-Ruge and Schulz-Dazzi, 1987). The reductions reported in humans are typically 16%, and in rats as much as 30%. Nearly all of these studies labeled capillaries with alkaline phosphatase histochemistry, which, while allowing labeling of capillaries in post-mortem tissue, has disadvantages. There is often variability in staining quality within samples, requiring investigators to forego random sampling. Capillary density estimates with this method have been reported to be 30% lower than the other methods in brain (Gobel et al., 1990), and muscle tissue (Hansen-Smith et al., 1992). Since alkaline phosphatase can be inhibited by a number of factors, a reduction in labeled capillaries may represent more than just a reduction in density. Using electron microscopy, age was not associated with capillary density in white matter, but age was associated with differences in capillary integrity (Farkas et al., 2006), which has been explored for its role in Alzheimer’s disease. The consistent reports of age-related declines in cerebral blood flow, taken with the alkaline phosphatase measures of capillary density support the need for more research to test the relationship between capillary density and age. If capillary density decreases with age, an intervention that maintains capillary density or induces angiogenesis should be beneficial.
If exercise can increase capillary density, then it may spare neuronal function by maintaining adequate glucose and oxygen availability to neurons. In humans, MR angiograms offer a method for measuring arteries in live subjects, but are limited with regard to measurement of capillaries. Using this method, exercise was shown to increase the number of small vessels ranging from 0.25 to 0.6 mm radius (Bullitt et al., 2009). No studies have tested relationships between fitness levels in humans and the density of capillaries. In rats, exercise at ages that are equivalent to human adulthood and middle age has been reported to increase capillary density in motor areas of the brain. One month of voluntary or forced (1 h/day at 10 m/min) exercise in middle-aged female rats increased capillary density in the cerebellar molecular layer (Isaacs et al., 1992). Wheel running also increased capillary density and integrins in the motor cortex, as well as capillary perfusion (Kleim et al., 2002; Swain et al., 2003). In the striatum, treadmill walking for 3 weeks at 30 min/day increased capillary density in 3- and 22-month-old rats (Ding et al., 2004, 2006b). In the substantia nigra, treadmill training reversed age-related declines in vessels labeled with endothelial cell antigen (Villar-Cheda et al., 2009). Exercise in monkeys (5 days a week for 5 months) increased vascular volume fraction in the motor cortex. The effect did not persist after 3 months of sedentary conditions (Rhyu et al., 2010), suggesting that exercise must be maintained. The exercise-induced increases in vascular volume fraction were restricted to 15- to 17-month-old (mature) monkeys, and were not seen in 10- to 12-year-old (middle-aged) monkeys. This is notable because it is one of the few studies that test the effects of exercise across ages and including older subjects. In our lab, treadmill training in rats with the parameters used by Isaacs et al. (1992) did not increase capillary density or glucose transporter mRNA in the hippocampus (McCloskey and Anderson, 2005), although exercise is reported to increase endothelial cell proliferation in the hippocampus (Ekstrand et al., 2008). There is strong evidence that exercise can increase capillary density in motor regions of the brain obtained with methods that are less subject to artifacts. Unfortunately, few studies have tested for the effects of exercise in non-motor regions. Therefore more work is necessary to test whether exercise-related changes in regional vascularization are widespread, and whether these effects generalize to aging animals.
If exercise can induce capillary growth, angiogenesis, it may do so through an increase in the expression of vascular endothelial growth factor (VEGF). Exercise (1 session of treadmill running at 24 m/min, 10° incline for 1 h) in mice increases VEGF transcription, mRNA and protein in the hippocampus, but not in the cerebellum or striatum (Tang et al., 2010). In our lab, 3 weeks of voluntary exercise in adult male mice also increased hippocampal VEGF protein, measured using ELISA (unpublished observations). In the substantia nigra pars compacta, both VEGF mRNA and vascularization are reduced in aging, and these reductions are reversed by exercise (Villar-Cheda et al., 2009). The correspondence between vessel density and VEGF in this study is notable because it is the only study in which both factors are studied in the same animals. The lack of correspondence between exercise-induced VEGF expression and exercise-induced angiogenesis within other brain areas needs to be further explored by addressing both variables in the same species with the same exercise conditions, and in middle-aged and aging animals.
Increasingly vascular pathology is being explored for its role in the etiology of AD (Farkas and Luiten, 2001), which increases in risk with age. Basement membrane thickness and fibrosis are pathological features in AD that increase in the brain with age (Farkas and Luiten, 2001; Farkas et al., 2006). Thickened microvessels may represent impairments in nutrient, glucose, and oxygen extraction as well as impaired elimination of waste. Furthermore, they are predicted to affect elasticity, and therefore the potential for a reduction in neurovascular coupling. The increase in basement membrane thickening in rats begins to appear at 20 months, but is visible by 30 months (de Jong et al., 1992). The age-related changes in humans are accelerated by risk factors such as hypertension (Farkas et al., 2000). Few studies have addressed whether exercise affects basement membrane thickness in cerebral capillaries. However, in muscle tissue, 9 months of endurance training decreased skeletal muscle capillary basement membrane thickening in older humans (M = 63 years old), producing a return to values similar to those seen in young subjects (M = 28 years old) (Williamson et al., 1996). It is possible these effects generalize to the brain since pathology in peripheral vasculature correlates with pathology in central nervous system arteries (Farkas and Luiten, 2001). If exercise can maintain capillary integrity, it may not only protect or slow age-related deficits in cognition, but also provide partial protection from vascular dementia and Alzheimer’s disease. A stronger relationship between age and capillary density needs to be established, but age clearly affects capillary integrity, which in turn may underlie some age-related neuropathologies.
Cellular Metabolic Capacity Declines With Age, and is up-Regulated by Exercise
Age-related decline in metabolism is apparent at the cellular level. At autopsy, there is a relationship between age and cytochrome c oxidase activity in the frontal cortex, temporal cortex, superior temporal gyrus, cerebellum and putamen (Ojaimi et al., 1999). Similarly, 26-month-old rats had a decrease in cytochrome oxidase subunits in the cerebral cortex (Nicoletti et al., 1995). Cytochrome oxidase increases after exercise in motor areas of the rat brain. Six months of voluntary wheel running in rats increases cytochrome oxidase reactivity in limb representations of the striatum and motor cortex, but not the hippocampus (McCloskey et al., 2001, see Figure 1). The lack of an effect in the hippocampus may suggest that the relationship between exercise and metabolic capacity is less clear beyond motor regions. For example, Molteni et al. (2002) found that 7 days of exercise produced marginal increases in gene expression for cytochrome oxidase subunits IV, V, VI, VIII, 4 of the 13 protein subunits necessary for a functional enzyme. Yet, Tong et al. (2001) found that subunits I, II, and III (3 of 13) were decreased in expression in male rats that exercised for 3 weeks. Although there is evidence that the regulation of gene expression for the 13 subunits is coordinated when there is dramatic increase or reduction in afferent input (Liang et al., 2006), it seems possible that exercise may produce a less dramatic increase in neural activity in the hippocampus, and therefore may be less capable of initiating the coordinated up-regulation necessary for a functional enzyme. Exercise has also been shown to influence anti-oxidants (Radak et al., 2001, 2008a,b), which should protect mitochondrial DNA, and in turn maintain the capacity to produce functional cytochrome oxidase enzyme. The demonstration that exercise can affect cellular metabolic capacity, at least in motor regions, is a key demonstration that any restoration in cardiovascular support can be utilized by cells.
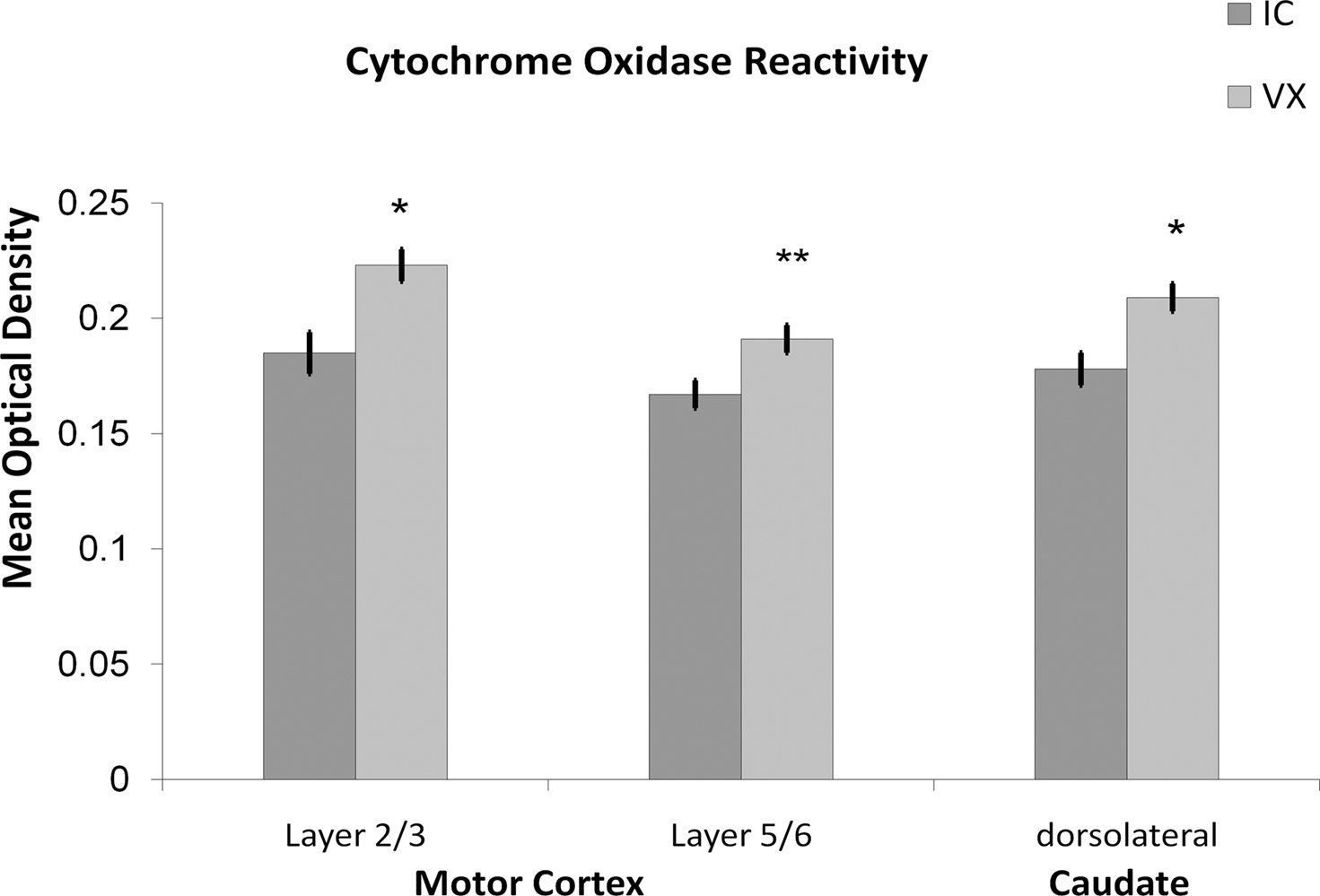
Figure 1. Exercise increases metabolic capacity in motor regions. Voluntary wheel running for 6 months increased the cytochrome oxidase reactivity in the dorsolateral striatum and limb representation areas of the motor cortex relative to inactive controls.
Exercise Protection Against Metabolic Challenges
Neuron loss may not always occur in healthy aging (Rapp and Gallagher, 1996; Yates et al., 2008), but neurons are at risk from the rising probability of an imbalance between metabolic capacity and energy demands as age increases. Age is associated with an increased risk for epilepsy (Hauser, 1992) and stroke. During ischemia, the energy demand of basal neural activity exceeds the diminished metabolic support, whereas during seizures, neural activity is high and produces metabolic demand that exceeds available supply. In both, the energy demands of neural activity exceed supply leading, in turn, to excess depolarization and potential cell death. Animal models of ischemia and seizures provide the opportunity to test for neuroprotective effects of exercise.
Exercise Protects Against Cell Loss From Ischemia
If exercise increases capillary density and the cellular metabolic capacity, then exercise should protect cells when they face metabolic challenges that increase in frequency with age. The risk for ischemia increases with age, but exercise pre-conditioning protects some cell populations from ischemia (Stummer et al., 1994, 1995; Sim et al., 2005). Exercise pre-conditioning reduced mortality from a 20-min occlusion of the carotid artery (Stummer et al., 1994). Gerbils that ran for 2 weeks in a running wheel prior to a 15-min occlusion of the carotid artery had only 50% cell loss in the limb representations of the striatum relative to 90% cell loss in control gerbils. Similarly, treadmill training for 30 min a day over 1–3 weeks in rats prior to middle cerebral artery occlusion reduced inflammatory injury in the striatum and reduced infarct volume (Ding et al., 2005, 2006a). It is as of yet unclear whether the greater metabolic capacity and capillary density protect against damage from capillary occlusion since these studies tested for protection after occlusion of major arteries. After MCAO occlusions, the protective effects of exercise are at least partly due to a reduction in inflammatory processes during reperfusion (Li et al., 2004). Exercise pre-conditioning increased TNF-alpha expression, which reduced NFκB expression. Reperfusion injury, mediated by TNF-alpha binding to NFκB, was reduced because of the exercise-related reduction in NFκB. Consequently, reperfusion injury, mediated by inflammatory processes, was reduced in exercising animals (Ding et al., 2006c), but angiogenesis also plays a role (Ding et al., 2004). Exercise was not protective when MCAO occlusion was permanent. Together, the data suggest that exercise protects striatal cells from metabolic challenge, in part through an increase in metabolic support.
Exercise also protects against ischemia in the hippocampus. Fifty percent of neurons in the CA3 subregion of the hippocampus of exercising gerbils survived after a 15-min occlusion of the carotid artery, whereas only 10% of neurons survived in the control gerbils (Stummer et al., 1994). However, protection was restricted to CA3, and was not seen in CA1, a subregion of the hippocampus particularly vulnerable to ischemia. Treadmill training (30 min/day for 10 days) in gerbils decreased apoptosis and caspase-3 expression in the dentate gyrus after 5 min occlusion of both carotid arteries (Sim et al., 2004). One month of treadmill training (30 min/day for four consecutive weeks) attenuated functional deficits (Sim et al., 2004, 2005). Altogether, there is substantial evidence that exercise can spare neurons from death following ischemia. However, there is less evidence that exercise can enhance metabolic support in the hippocampus, so it remains unclear whether metabolic factors account for the protective effects of exercise in the hippocampus or enhanced expression of growth factors.
Exercise Protects Against Seizures
Epilepsy and acute unprovoked seizures are predominant in patients older than 65 years and younger than 12 months of age (Hauser and Beghi, 2008). Although the onset of seizures is often the result of an antecedent event, 50% of new epilepsy cases in the elderly have no known antecedent event (Hauser, 1992; Hauser et al., 1993; Li et al., 1997; Leppik, 2006). Late onset epilepsy is correlated with a decline in vascular factors, suggesting that age-related declines increase vulnerability to epilepsy (Li et al., 1997). Seizures are events in which energy demand is high, and at times exceed the cell’s metabolic capacity. Since exercise can up-regulate cellular bioenergetic capacity and oxygen and glucose availability, it may modulate the progression of seizures and cell loss. However, exercise regulation of cell energy metabolism has not been reported in the hippocampus, which is the focus of models of temporal lobe epilepsy. In our own studies, 4–7 weeks of wheel running reduced the proportion of rats progressing to continuous generalized seizures (status epilepticus, SE) after systemic injections of kainic acid (10 mg/kg, see Figure 2). When rats ran for only 1 month, the protection from status epilepticus was restricted to rats that ran over the median number of wheel rotations (unpublished observations). High runners (above the median split) had a significant protection from SE relative to the combined group of control and low runners (Yates χ2 (1) = 9.235, p < 0.05). SE causes a dramatic loss of CA3 pyramidal cells, whereas moderate seizures cause cell loss in only small patches, if at all. As a consequence, exercise, by protecting rats from progressing to continuous generalized seizures, provides significant protection against cell death. In another study, voluntary wheel running had no effect on seizure severity (based on the Racine scale) when the dose was below 10 mg/kg, but reduced seizure severity when kainate was given at a dose above 10 mg/kg (Reiss et al., 2009). These studies suggest that exercise moderates seizure progression when kainic acid is administered systemically.
Consistent with previous studies, exercise raised the threshold for kindling induced seizures (Arida et al., 1998), although this effect was restricted to exercise in adulthood; exercise during development did not protect adult animals (Arida et al., 2007). After 21 days of voluntary wheel running, seizure ratings were lower after ventricular infusion of low, but not moderate doses of kainic acid (Reiss et al., 2009). We found that injections of 0.55 μg kainic acid into the lateral ventricles at a site most anterior to the tip of the hippocampus yielded no difference in the proportion of animals progressing to SE, despite using 6 rather than 3 weeks of exercise. Only 21% of the animals entered SE, which is consistent with the low dose.
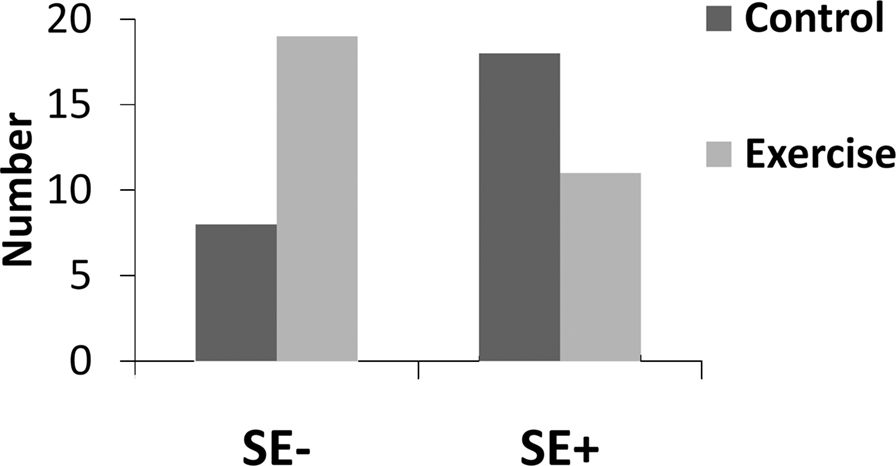
Figure 2. Exercise protected animals from status epilepticus. The number of animals that did not progress to continuous generalized seizures (SE−) was greater in the exercise group than in the control group. Conversely, the number that progressed to continuous generalized seizures (SE+) was greater after standard housing (control) than after 4–7 weeks of voluntary wheel running (Exercise). χ2 (1) = 5.96, p < 0.05.
Several investigators have also addressed the possibility that exercise protects from seizure-induced cell death. Following systemic injections of domoic acid, mice trained to run on a treadmill for 1 km/day had less cell loss in the hilus of the hippocampus (Carro et al., 2001). In contrast, when kainic acid was infused directly into the hippocampus, wheel running in female rats exacerbated, rather than protected against cell loss in the CA3 region (Ramsden et al., 2003). It is important to note that Ramsden infused kainic acid in animals anesthetized with a drug used to control seizures. We carried out a series of experiments to further test the effects of exercise on cell loss. Like Ramsden et al. (2003), we used kainic acid infusion into the lateral ventricles, but in contrast to their methods, we infused kainic acid into awake male rats. When we restrict our analysis to cell loss in animals that did not progress to SE, we find, like Ramsden et al. (2003), that exercise exacerbates damage. Together, these latter studies leave us to conclude that exercise does not protect CA3 cells from moderate seizures, but instead increases neuron vulnerability. The discrepancy in exercise effects on cell vulnerability in the hippocampus may not be surprising given that there is little evidence for exercise-related up-regulation of cellular metabolic capacity or capillary density in this region. The two reports that exercise protects the striatum from ischemic damage parallel exercise-related enhancement of metabolic support in that region. Whereas these studies support a beneficial effect of exercise in the aging striatum, future studies need to directly test that hypothesis in aging animals..
Conclusions
It is important to note that the majority of evidence for the ability of exercise to influence regional vasculature and cellular metabolic machinery come from motor systems like the striatum and motor cortex. Exercise also affects the hippocampus, but those effects may be through growth factor expression rather than through an increase in capillary density or metabolic capacity (Kesslak et al., 1998; Gomez-Pinilla, 2008). For example, VEGF, which is increased in the hippocampus by exercise, can decrease seizure-like activity there (McCloskey et al., 2005), and protect hippocampal neurons from seizures (Nicoletti et al., 2008), independent of its ability to increase vasculature. This may explain why the evidence for cell protection is mixed in the hippocampus.
Many of the studies reviewed here utilized animal models, which allow greater control over exercise conditions as well as the application of methods that can directly test the biological mechanisms of aging and exercise. Exercise manipulations vary in these animal studies. The use of voluntary exercise is common because activity levels can be increased without inducing stress. When voluntary exercise is used, animals can run in wheels any time over the 24-h day, although running has a circadian rhythm. Voluntary exercise in rats may be analogous to humans with physically active lives, and jobs with manual labor. In contrast, forced exercise on a treadmill usually constitutes forced running for 1 h a day for 5–7 days a week, while rats are sedentary for the remaining 23 h a day. This may be analogous to gym workouts by individuals with desk jobs. Therefore the two types of exercise used in animal studies serve as models for very different physical activity patterns in humans. Nevertheless, both wheel running and treadmill training increase capillary density in motor regions. Wheel running has been shown to increase cellular metabolic capacity in these regions, and treadmill training has not been tested.
Here, we propose that the functional deficits that occur with age are at least partially related to the decline in cellular function resulting from a decline in metabolism. This decline involves cardiovascular function at the level of the organism and brain, in cerebral blood flow, in cytochrome oxidase activity, and possibility capillary density. There is substantial evidence that exercise can influence metabolic support at a number of levels, including cardiovascular function, regional capillary density, and cellular bioenergetic capacity. We hypothesize that exercise can counter the age-related decline in metabolic support (see Figure 3). Unfortunately, there is little evidence to support this hypothesis, because evidence that exercise can influence vasculature and cellular metabolism is primarily limited to adult and middle-aged animals. If exercise can induce similar effects in aging animals, then it may be a powerful intervention through its influence on the multiple levels of support necessary for cellular metabolism. It is important to note that most of the studies demonstrating exercise effects on human cognition have studied aging rather than young or adult samples. Accordingly, the evidence that exercise improves cognition in elderly humans provides incentive for understanding the neurobiological mechanisms, and supports the possibility that effects established in adult animals may generalize to aging animals. To bridge this gap, studies of exercise effects on regional vasculature and cellular metabolism should be extended to aging animals, and should include analyses of both motor and non-motor structures.
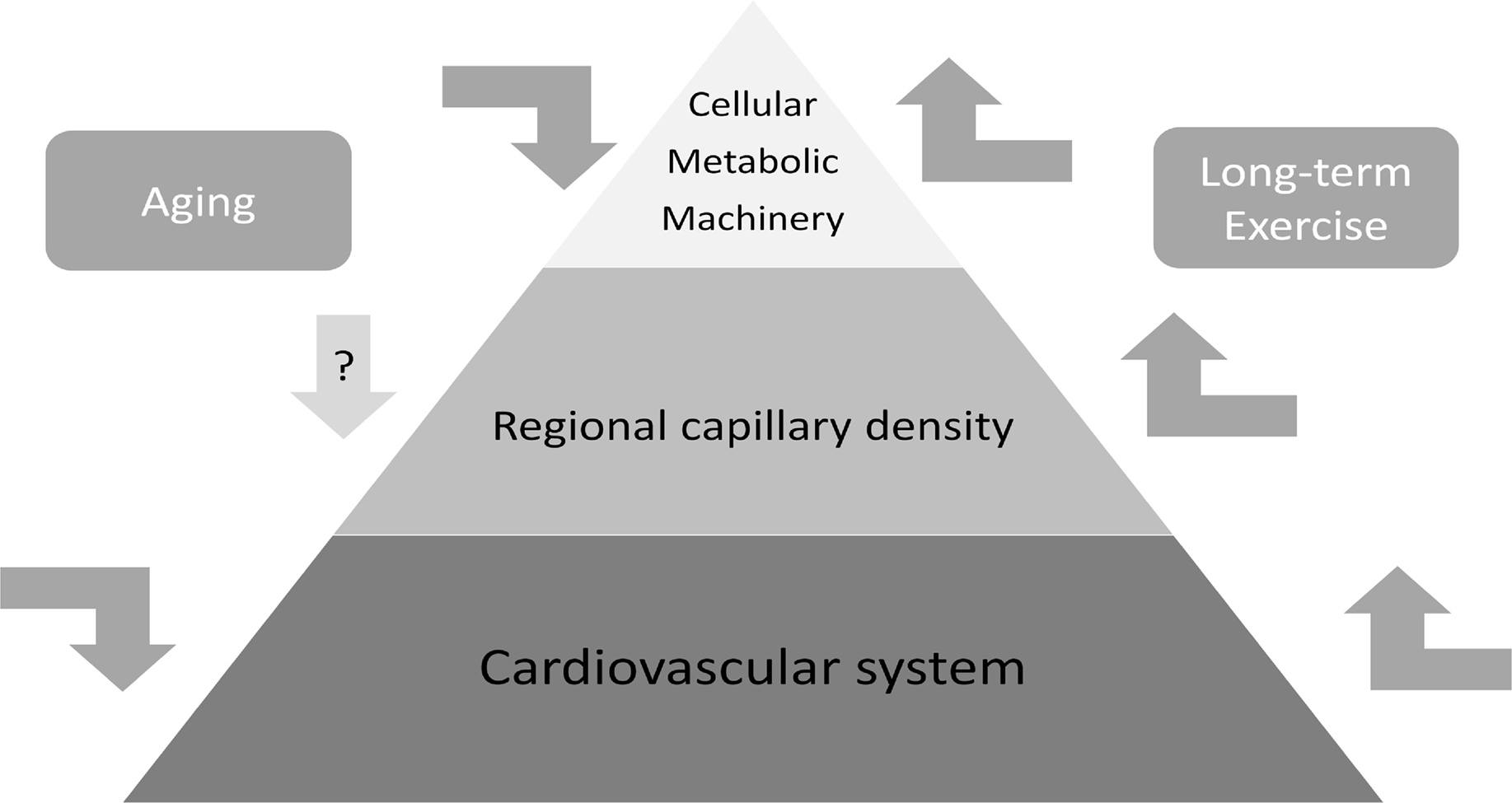
Figure 3. Exercise has potential as a powerful intervention in brain aging because it can enhance cellular metabolic capacity, while simultaneously enhancing cardiovascular support at the level of the organism and individual brain region. If these beneficial effects of exercise extend to aging, then exercise could be used as a multi-level strategy for maintaining or reversing the age-related reductions in cardiovascular support, cellular metabolic capacity, and possibly capillary density.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
W. Burghardt Turner Fellowship and the NSF Bridges to the Doctorate Fellowship and Turner Summer Research Fellowship to Shayri J. Greenwood. We thank Sara Mellers for helpful suggestions on manuscript.
References
Abernethy, W. B., Bell, M. A., Morris, M., and Moody, D. M. (1993). Microvascular density of the human paraventricular nucleus decreases with aging but not hypertension. Exp. Neurol. 121, 270–274.
Amenta, F., Ferrante, F., Mancini, M., Sabbatini, M., Vega, J. A., and Zaccheo, D. (1995). Effect of long-term treatment with the dihydropyridine-type calcium channel blocker darodipine (PY 108-068) on the cerebral capillary network in aged rats. Mech. Ageing Dev. 78, 27–37.
Arida, R. M., de Jesus Vieira, A., and Cavalheiro, E. A. (1998). Effect of physical exercise on kindling development. Epilepsy Res. 30, 127–132.
Arida, R. M., Scorza, F. A., de Lacerda, A. F., da Silva, S. G., and Cavalheiro, E. A. (2007). Physical training in developing rats does not influence the kindling development in the adult life. Physiol. Behav. 90, 629–633.
Astrup, J., Sorensen, P. M., and Sorensen, H. R. (1981). Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke 12, 726–730.
Bell, M. A., and Ball, M. J. (1981). Morphometric comparison of hippocampal microvasculature in ageing and demented people: diameters and densities. Acta Neuropathol. 53, 299–318.
Bell, M. A., and Ball, M. J. (1985). Laminar variation in the microvascular architecture of normal human visual cortex (area 17). Brain Res. 335, 139–143.
Bell, M. A., and Ball, M. J. (1990). Neuritic plaques and vessels of visual cortex in aging and Alzheimer’s dementia. Neurobiol. Aging 11, 359–370.
Bertsch, K., Hagemann, D., Hermes, M., Walter, C., Khan, R., and Naumann, E. (2009). Resting cerebral blood flow, attention, and aging. Brain Res. 1267, 77–88.
Bie-Olsen, L. G., Kjaer, T. W., Pedersen-Bjergaard, U., Lonsdale, M. N., Holst, J. J., Law, I., and Thorsteinsson, B. (2009). Changes of cognition and regional cerebral activity during acute hypoglycemia in normal subjects: A H2 15O positron emission tomographic study. J. Neurosci. Res. 87, 1922–1928.
Borowsky, I. W., and Collins, R. C. (1989). Metabolic anatomy of brain: a comparison of regional capillary density, glucose metabolism, and enzyme activities. J. Comp. Neurol. 288, 401–413.
Brown, W. R., Blair, R. M., Moody, D. M., Thore, C. R., Ahmed, S., Robbins, M. E., and Wheeler, K. T. (2007). Capillary loss precedes the cognitive impairment induced by fractionated whole-brain irradiation: a potential rat model of vascular dementia. J. Neurol. Sci. 257, 67–71.
Brown, W. R., Thore, C. R., Moody, D. M., Robbins, M. E., and Wheeler, K. T. (2005). Vascular damage after fractionated whole-brain irradiation in rats. Radiat. Res. 164, 662–668.
Buchweitz-Milton, E., and Weiss, H. R. (1987). Perfused capillary morphometry in the senescent brain. Neurobiol. Aging 8, 271–276.
Bullitt, E., Rahman, F. N., Smith, J. K., Kim, E., Zeng, D., Katz, L. M., and Marks, B. L. (2009). The effect of exercise on the cerebral vasculature of healthy aged subjects as visualized by MR angiography. AJNR Am. J. Neuroradiol. 30, 1857–1863.
Carro, E., Trejo, J. L., Busiguina, S., and Torres-Aleman, I. (2001). Circulating insulin-like growth factor I mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J. Neurosci. 21, 5678–5684.
Colcombe, S. J., Kramer, A. F., McAuley, E., Erickson, K. I., and Scalf, P. (2004). Neurocognitive aging and cardiovascular fitness: recent findings and future directions. J. Mol. Neurosci. 24, 9–14.
D’Esposito, M., Deouell, L. Y., and Gazzaley, A. (2003). Alterations in the BOLD fMRI signal with ageing and disease: a challenge for neuroimaging. Nat. Rev. Neurosci. 4, 863–872.
de Jong, G. I., Jansen, A. S., Horvath, E., Gispen, W. H., and Luiten, P. G. (1992). Nimodipine effects on cerebral microvessels and sciatic nerve in aging rats. Neurobiol. Aging 13, 73–81.
Devous, M. D. Sr., Stokely, E. M., Chehabi, H. H., and Bonte, F. J. (1986). Normal distribution of regional cerebral blood flow measured by dynamic single-photon emission tomography. J. Cereb. Blood Flow Metab. 6, 95–104.
Ding, Q., Vaynman, S., Souda, P., Whitelegge, J. P., and Gomez-Pinilla, F. (2006a). Exercise affects energy metabolism and neural plasticity-related proteins in the hippocampus as revealed by proteomic analysis. Eur. J. Neurosci. 24, 1265–1276.
Ding, Y. H., Li, J., Yao, W. X., Rafols, J. A., Clark, J. C., and Ding, Y. (2006b). Exercise preconditioning upregulates cerebral integrins and enhances cerebrovascular integrity in ischemic rats. Acta Neuropathol. 112, 74–84.
Ding, Y. H., Mrizek, M., Lai, Q., Wu, Y., Reyes, R., Jr., Li, J., Davis, W. W., and Ding, Y. (2006c). Exercise preconditioning reduces brain damage and inhibits TNF-alpha receptor expression after hypoxia/reoxygenation: an in vivo and in vitro study. Curr. Neurovasc. Res. 3, 263–271.
Ding, Y., Li, J., Luan, X., Ding, Y. H., Lai, Q., Rafols, J. A., Phillis, J. W., Clark, J. C., and Diaz, F. G. (2004). Exercise pre-conditioning reduces brain damage in ischemic rats that may be associated with regional angiogenesis and cellular overexpression of neurotrophin. Neuroscience 124, 583–591.
Ding, Y. H., Young, C. N., Luan, X., Li, J., Rafols, J. A., Clark, J. C., McAllister, J. P., 2nd,, and Ding, Y. (2005). Exercise preconditioning ameliorates inflammatory injury in ischemic rats during reperfusion. Acta Neuropathol. (Berl) 109, 237–246.
Dupre, J., and O’Malley, M. A. (2009). Varieties of living things: life at the intersection of lineage and metabolism. Philos. Theory Biol. 1, 1–22.
Ekstrand, J., Hellsten, J., and Tingstrom, A. (2008). Environmental enrichment, exercise and corticosterone affect endothelial cell proliferation in adult rat hippocampus and prefrontal cortex. Neurosci. Lett. 442, 203–207.
Erecinska, M., and Silver, I. A. (1989). ATP and brain function. J. Cereb. Blood Flow Metab. 9, 2–19.
Farkas, E., de Vos, R. A., Donka, G., Jansen Steur, E. N., Mihaly, A., and Luiten, P. G. (2006). Age-related microvascular degeneration in the human cerebral periventricular white matter. Acta Neuropathol. 111, 150–157.
Farkas, E., De Vos, R. A., Jansen Steur, E. N., and Luiten, P. G. (2000). Are Alzheimer’s disease, hypertension, and cerebrocapillary damage related? Neurobiol. Aging 21, 235–243.
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 64, 575–611.
Formes, K., Zhang, P., Tierney, N., Schaller, F., and Shi, X. (2010). Chronic physical activity mitigates cerebral hypoperfusion during central hypovolemia in elderly humans. Am. J. Physiol. Heart Circ. Physiol. 298, H1029–H1037.
Frackowiak, R. S., Lenzi, G. L., Jones, T., and Heather, J. D. (1980). Quantitative measurement of regional cerebral blood flow and oxygen metabolism in man using 15O and positron emission tomography: theory, procedure, and normal values. J. Comput. Assist. Tomogr. 4, 727–736.
Gobel, U., Theilen, H., and Kuschinsky, W. (1990). Congruence of total and perfused capillary network in rat brains. Circ. Res. 66, 271–281.
Gomez-Pinilla, F. (2008). The influences of diet and exercise on mental health through hormesis. Ageing Res. Rev. 7, 49–62.
Gonder-Frederick, L. A., Zrebiec, J. F., Bauchowitz, A. U., Ritterband, L. M., Magee, J. C., Cox, D. J., and Clarke, W. L. (2009). Cognitive function is disrupted by both hypo- and hyperglycemia in school-aged children with type 1 diabetes: a field study. Diabetes Care 32, 1001–1006.
Gross, P. M., Sposito, N. M., Pettersen, S. E., Panton, D. G., and Fenstermacher, J. D. (1987). Topography of capillary density, glucose metabolism, and microvascular function within the rat inferior colliculus. J. Cereb. Blood Flow Metab. 7, 154–160.
Hansen-Smith, F. M., Blackwell, L. H., and Joswiak, G. R. (1992). Expression of muscle capillary alkaline phosphatase is affected by hypoxia. J. Appl. Physiol. 73, 776–780.
Hauser, W. A., Annegers, J. F., and Kurland, L. T. (1993). Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 34, 453–468.
Hauser, W. A., and Beghi, E. (2008). First seizure definitions and worldwide incidence and mortality. Epilepsia 49(Suppl. 1), 8–12.
Hughes, C. C., and Lantos, P. L. (1987). A morphometric study of blood vessel, neuron and glial cell distribution in young and old rat brain. J. Neurol. Sci. 79, 101–110.
Hunziker, O., Abdel’Al, S., and Schulz, U. (1979). The aging human cerebral cortex: a stereological characterization of changes in the capillary net. J. Gerontol. 34, 345–350.
Isaacs, K. R., Anderson, B. J., Alcantara, A. A., Black, J. E., and Greenough, W. T. (1992). Exercise and the brain: angiogenesis in the adult rat cerebellum after vigorous physical activity and motor skill learning. J. Cereb. Blood Flow Metab. 12, 110–119.
Jucker, M., Battig, K., and Meier-Ruge, W. (1990). Effects of aging and vincamine derivatives on pericapillary microenvironment: stereological characterization of the cerebral capillary network. Neurobiol. Aging 11, 39–46.
Jucker, M., and Meier-Ruge, W. (1989). Effects of brovincamine on stereological capillary parameters in adult and old Fischer-344 rats. Microvasc. Res. 37, 298–307.
Kageyama, G. H., and Wong-Riley, M. T. (1982). Histochemical localization of cytochrome oxidase in the hippocampus: correlation with specific neuronal types and afferent pathways. Neuroscience 7, 2337–2361.
Kesslak, J. P., So, V., Choi, J., Cotman, C. W., and Gomez-Pinilla, F. (1998). Learning upregulates brain-derived neurotrophic factor messenger ribonucleic acid: a mechanism to facilitate encoding and circuit maintenance? Behav. Neurosci. 112, 1012–1019.
Kleim, J. A., Cooper, N. R., and VandenBerg, P. M. (2002). Exercise induces angiogenesis but does not alter movement representations within rat motor cortex. Brain Res. 934, 1–6.
Knox, C. A., and Oliveira, A. (1980). Brain aging in normotensive and hypertensive strains of rats. III. A quantitative study of cerebrovasculature. Acta Neuropathol. 52, 17–25.
Kramer, A. F., Coyne, J. T., and Strayer, D. L. (1993). Cognitive function at high altitude. Hum. Factors 35, 329–344.
Kramer, A. F., Hahn, S., Cohen, N. J., Banich, M. T., McAuley, E., Harrison, C. R., Chason, J., Vakil, E., Bardell, L., Boileau, R. A., and Colcombe, A. (1999). Ageing, fitness and neurocognitive function. Nature 400, 418–419.
Lauritzen, K. H., Moldestad, O., Eide, L., Carlsen, H., Nesse, G., Storm, J. F., Mansuy, I. M., Bergersen, L. H., and Klungland, A. (2010). Mitochondrial DNA toxicity in forebrain neurons causes apoptosis, neurodegeneration, and impaired behavior. Mol. Cell. Biol. 30, 1357–1367.
Li, J., Luan, X., Clark, J. C., Rafols, J. A., and Ding, Y. (2004). Neuroprotection against transient cerebral ischemia by exercise pre-conditioning in rats. Neurol. Res. 26, 404–408.
Li, X., Breteler, M. M., de Bruyne, M. C., Meinardi, H., Hauser, W. A., and Hofman, A. (1997). Vascular determinants of epilepsy: the Rotterdam Study. Epilepsia 38, 1216–1220.
Liang, H. L., Ongwijitwat, S., and Wong-Riley, M. T. (2006). Bigenomic functional regulation of all 13 cytochrome c oxidase subunit transcripts in rat neurons in vitro and in vivo. Neuroscience 140, 177–190.
Magistretti, P. J., and Pellerin, L. (1996). Cellular bases of brain energy metabolism and their relevance to functional brain imaging: evidence for a prominent role of astrocytes. Cereb. Cortex 6, 50–61.
Mann, D. M., Eaves, N. R., Marcyniuk, B., and Yates, P. O. (1986). Quantitative changes in cerebral cortical microvasculature in ageing and dementia. Neurobiol. Aging 7, 321–330.
Markham, J. A., McKian, K. P., Stroup, T. S., and Juraska, J. M. (2005). Sexually dimorphic aging of dendritic morphology in CA1 of hippocampus. Hippocampus 15, 97–103.
McAuley, E., Kramer, A. F., and Colcombe, S. J. (2004). Cardiovascular fitness and neurocognitive function in older adults: a brief review. Brain Behav. Immun. 18, 214–220.
McCloskey, D. P., Adamo, D. S., and Anderson, B. J. (2001). Exercise increases metabolic capacity in the motor cortex and striatum, but not in the hippocampus. Brain Res. 891, 168–175.
McCloskey, D. P., and Anderson, B. J. (2005). “Running away from epilepsy: exercise-regulated gene expression,” in Growth Factors and Epilepsy, eds D. K. Binder and H. E. Scharfman (New York: Nova Sciences), 207–225.
McCloskey, D. P., Croll, S. D., and Scharfman, H. E. (2005). Depression of synaptic transmission by vascular endothelial growth factor in adult rat hippocampus and evidence for increased efficacy after chronic seizures. J. Neurosci. 25, 8889–8897.
Meier-Ruge, W., Hunziker, O., Schulz, U., Tobler, H. J., and Schweizer, A. (1980). Stereological changes in the capillary network and nerve cells of the aging human brain. Mech. Ageing Dev. 14, 233–243.
Meier-Ruge, W., and Schulz-Dazzi, U. (1987). Effects of brovincamine on the stereological parameters of corticocerebral capillaries. Life Sci. 40, 943–949.
Mironov, V., Hritz, M. A., LaManna, J. C., Hudetz, A. G., and Harik, S. I. (1994). Architectural alterations in rat cerebral microvessels after hypobaric hypoxia. Brain Res. 660, 73–80.
Mjaatvedt, A. E., and Wong-Riley, M. T. (1988). Relationship between synaptogenesis and cytochrome oxidase activity in Purkinje cells of the developing rat cerebellum. J. Comp. Neurol. 277, 155–182.
Molteni, R., Ying, Z., and Gomez-Pinilla, F. (2002). Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur. J. Neurosci. 16, 1107–1116.
National Institutes of Health, N. C. D. P. o. P. A. a. C. (1996). Physical activity and cardiovascular health. NIH Consensus Development Panel on Physical Activity and Cardiovascular Health. JAMA 276, 241–246.
Nicoletti, J. N., Shah, S. K., McCloskey, D. P., Goodman, J. H., Elkady, A., Atassi, H., Hylton, D., Rudge, J. S., Scharfman, H. E., and Croll, S. D. (2008). Vascular endothelial growth factor is up-regulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience 151, 232–241.
Nicoletti, V. G., Tendi, E. A., Lalicata, C., Reale, S., Costa, A., Villa, R. F., Ragusa, N., and Giuffrida Stella, A. M. (1995). Changes of mitochondrial cytochrome c oxidase and FoF1 ATP synthase subunits in rat cerebral cortex during aging. Neurochem. Res. 20, 1465–1470.
Ojaimi, J., Masters, C. L., Opeskin, K., McKelvie, P., and Byrne, E. (1999). Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 111, 39–47.
Radak, Z., Chung, H. Y., and Goto, S. (2008a). Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 44, 153–159.
Radak, Z., Chung, H. Y., Koltai, E., Taylor, A. W., and Goto, S. (2008b). Exercise, oxidative stress and hormesis. Ageing Res. Rev. 7, 34–42.
Radak, Z., Kaneko, T., Tahara, S., Nakamoto, H., Pucsok, J., Sasvari, M., Nyakas, C., and Goto, S. (2001). Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem. Int. 38, 17–23.
Ramsden, M., Berchtold, N. C., Patrick Kesslak, J., Cotman, C. W., and Pike, C. J. (2003). Exercise increases the vulnerability of rat hippocampal neurons to kainate lesion. Brain Res. 971, 239–244.
Rapp, P. R., and Gallagher, M. (1996). Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc. Natl. Acad. Sci. U.S.A. 93, 9926–9930.
Reiss, J. I., Dishman, R. K., Boyd, H. E., Robinson, J. K., and Holmes, P. V. (2009). Chronic activity wheel running reduces the severity of kainic acid-induced seizures in the rat: possible role of galanin. Brain Res. 1266, 54–63.
Rhyu, I. J., Bytheway, J. A., Kohler, S. J., Lange, H., Lee, K. J., Boklewski, J., McCormick, K., Williams, N. I., Stanton, G. B., Greenough, W. T., and Cameron, J. L. (2010). Effects of aerobic exercise training on cognitive function and cortical vascularity in monkeys. Neuroscience 167, 1239–1248.
Riddle, D. R., Sonntag, W. E., and Lichtenwalner, R. J. (2003). Microvascular plasticity in aging. Ageing Res. Rev. 2, 149–168.
Rogers, R. L., Meyer, J. S., and Mortel, K. F. (1990). After reaching retirement age physical activity sustains cerebral perfusion and cognition. J. Am. Geriatr. Soc. 38, 123–128.
Schrodinger, E. (1944). Is Life Based on the Laws of Physics? In What is Life? with Minda and Matter and Autobiographical Sketches. Cambridge: Cambridge University Press, 76–85.
Sim, Y. J., Kim, H., Kim, J. Y., Yoon, S. J., Kim, S. S., Chang, H. K., Lee, T. H., Lee, H. H., Shin, M. C., Shin, M. S., and Kim, C. J. (2005). Long-term treadmill exercise overcomes ischemia-induced apoptotic neuronal cell death in gerbils. Physiol. Behav. 84, 733–738.
Sim, Y. J., Kim, S. S., Kim, J. Y., Shin, M. S., and Kim, C. J. (2004). Treadmill exercise improves short-term memory by suppressing ischemia-induced apoptosis of neuronal cells in gerbils. Neurosci. Lett. 372, 256–261.
Sousa, N., Madeira, M. D., and Paula-Barbosa, M. M. (1998). Effects of corticosterone treatment and rehabilitation on the hippocampal formation of neonatal and adult rats. An unbiased stereological study. Brain Res. 794, 199–210.
Stoquart-ElSankari, S., Baledent, O., Gondry-Jouet, C., Makki, M., Godefroy, O., and Meyer, M. E. (2007). Aging effects on cerebral blood and cerebrospinal fluid flows. J. Cereb. Blood Flow Metab. 27, 1563–1572.
Stummer, W., Baethmann, A., Murr, R., Schurer, L., and Kempski, O. S. (1995). Cerebral protection against ischemia by locomotor activity in gerbils. Underlying mechanisms. Stroke 26, 1423–1429; discussion 1430.
Stummer, W., Weber, K., Tranmer, B., Baethmann, A., and Kempski, O. (1994). Reduced mortality and brain damage after locomotor activity in gerbil forebrain ischemia. Stroke 25, 1862–1869.
Swain, R. A., Harris, A. B., Wiener, E. C., Dutka, M. V., Morris, H. D., Theien, B. E., Konda, S., Engberg, K., Lauterbur, P. C., and Greenough, W. T. (2003). Prolonged exercise induces angiogenesis and increases cerebral blood volume in primary motor cortex of the rat. Neuroscience 117, 1037–1046.
Tang, K., Xia, F. C., Wagner, P. D., and Breen, E. C. (2010). Exercise-induced VEGF transcriptional activation in brain, lung and skeletal muscle. Respir. Physiol. Neurobiol. 170, 16–22.
Tata, D. A., and Anderson, B. J. (2010). The effects of chronic glucocorticoid exposure on dendritic length, synapse numbers and glial volume in animal models: implications for hippocampal volume reductions in depression. Physiol. Behav. 99, 186–193.
Tata D. A., Coburn-Litvak, P. S., McCloskey, D. P., Pothakos, K., and Anderson, B. J. (2002). Two-Month Administration of a High Dose of Corticosterone Decreases Neuropil Volume of Middle Hippocampal Ca3 and Suppresses Activity in Rats. Washington, DC: Society for Neuroscience.
Tong, L., Shen, H., Perreau, V. M., Balazs, R., and Cotman, C. W. (2001). Effects of exercise on gene-expression profile in the rat hippocampus. Neurobiol. Dis. 8, 1046–1056.
Tuor, U. I., Kurpita, G., and Simone, C. (1994). Correlation of local changes in cerebral blood flow, capillary density, and cytochrome oxidase during development. J. Comp. Neurol. 342, 439–448.
Villar-Cheda, B., Sousa-Ribeiro, D., Rodriguez-Pallares, J., Rodriguez-Perez, A. I., Guerra, M. J., and Labandeira-Garcia, J. L. (2009). Aging and sedentarism decrease vascularization and VEGF levels in the rat substantia nigra. Implications for Parkinson’s disease. J. Cereb. Blood Flow Metab. 29, 230–234.
Williamson, J. R., Hoffmann, P. L., Kohrt, W. M., Spina, R. J., Coggan, A. R., and Holloszy, O. (1996). Endurance exercise training decreases capillary basement membrane width in older nondiabetic and diabetic adults. J. Appl. Physiol. 80, 747–753.
Wong-Riley, M. T. (1989). Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 12, 94–101.
Keywords: physical activity, metabolism, capillary density, cytochrome oxidase, seizures, motor cortex, striatum, exercise
Citation: Anderson BJ, Greenwood SJ and McCloskey D (2010) Exercise as an intervention for the age-related decline in neural metabolic support. Front. Ag. Neurosci. 4:30. doi: 10.3389/fnagi.2010.00030
Received: 02 April 2010;
Paper pending published: 15 April 2010;
Accepted: 27 June 2010;
Published online: 13 August 2010
Edited by:
Arthur F. Kramer, University of Illinois at Urbana-Champaign, USAReviewed by:
Phil Holmes, Princeton University, USAJustin Rhodes, University of Illinois at Urbana-Champaign, USA
Margaret Wong-Riley, Medical College of Wisconsin, USA
Copyright: © 2010 Anderson, Greenwood and McCloskey. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Brenda J. Anderson, Department of Psychology, Stony Brook University, Stony Brook, NY 11794-2500, USA. e-mail:YmFuZGVyc29uQG5vdGVzLmNjLnN1bnlzYi5lZHU=