
- 1 Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Cádiz, Cádiz, Spain
- 2 Laboratory of Free Radical Biology, School of Pharmacy and Biochemistry, University of Buenos Aires, Buenos Aires, Argentina
Brain senescence and neurodegeneration occur with a mitochondrial dysfunction characterized by impaired electron transfer and by oxidative damage. Brain mitochondria of old animals show decreased rates of electron transfer in complexes I and IV, decreased membrane potential, increased content of the oxidation products of phospholipids and proteins and increased size and fragility. This impairment, with complex I inactivation and oxidative damage, is named “complex I syndrome” and is recognized as characteristic of mammalian brain aging and of neurodegenerative diseases. Mitochondrial dysfunction is more marked in brain areas as rat hippocampus and frontal cortex, in human cortex in Parkinson’s disease and dementia with Lewy bodies, and in substantia nigra in Parkinson’s disease. The molecular mechanisms involved in complex I inactivation include the synergistic inactivations produced by ONOO− mediated reactions, by reactions with free radical intermediates of lipid peroxidation and by amine–aldehyde adduction reactions. The accumulation of oxidation products prompts the idea of antioxidant therapies. High doses of vitamin E produce a significant protection of complex I activity and mitochondrial function in rats and mice, and with improvement of neurological functions and increased median life span in mice. Mitochondria-targeted antioxidants, as the Skulachev cations covalently attached to vitamin E, ubiquinone and PBN and the SS tetrapeptides, are negatively charged and accumulate in mitochondria where they exert their antioxidant effects. Activation of the cellular mechanisms that regulate mitochondrial biogenesis is another potential therapeutic strategy, since the process generates organelles devoid of oxidation products and with full enzymatic activity and capacity for ATP production.
Mammalian Aging and Brain Mitochondrial Impairment in Aging and Neurodegenerative Diseases
Mammalian aging is characterized by a gradual and continuous loss, starting at full adulthood, of the quality of all physiological functions and responses involving all organs and tissues. However, the losses are more marked in the functions that depend on integrated responses of the central nervous system (Gilad and Gilad, 1995). The whole economy of the body is involved in the aging process and a decreased basal metabolic rate is recognized as one of the traits of old mammals. In the study of mammalian aging there is a continuous interplay between the concept of loosing physiological functions, adequately expressed by the fraction at a given age of the function at full adulthood, and the concept of survival or extension of life, that is expressed by the median or maximal life span.
The free radical theory of aging is based on the works of Gerschaman et al. (1954) and Harman (1956) that considered that aging is caused by the continuous inactivation of biologically essential macromolecules due to chemical modifications produced in reactions mediated by oxygen free radicals. When the free radical theory of aging, that lacked the precision of the subcellular location of the oxidative reactions mediated by free radicals, is focused in mitochondria, it emerges as the mitochondrial theory of aging (Beckman and Ames, 1998; Vina et al., 2003; Harman, 2006). Mitochondria were brought to attention in aging biology due to the central role of mitochondria in producing biochemical energy (ATP) to meet cellular requirements in aerobic cells and to the decline of basal metabolic rate and of physical performance that are characteristic of aging. Moreover, mitochondria are considered likely pacemakers of tissue aging due to their continuous production of the free radicals superoxide (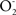 ) and nitric oxide (NO), to the mitochondrial sensitivity to free radical mediated oxidative damage and to the accumulation of phospholipid, protein, and DNA oxidation products in aged animals (Beckman and Ames, 1998; Vina et al., 2003; Harman, 2006).
) and nitric oxide (NO), to the mitochondrial sensitivity to free radical mediated oxidative damage and to the accumulation of phospholipid, protein, and DNA oxidation products in aged animals (Beckman and Ames, 1998; Vina et al., 2003; Harman, 2006).
In mammalian brain aging, more precisely in rats and mice, the accumulation of dysfunctional brain mitochondria with decreased rates of electron transfer in complexes I and IV and of ATP production is associated with the accumulation of oxidation products of phospholipids and proteins, and these characteristics appear as determining factors in brain aging. The evidence supporting this concept is the inverse relationships between accumulated oxidative damage (protein carbonyls and TBARS) and the rates of electron transfer and enzymatic activity (Figure 1A), the direct relationships between the rates of electron transfer in complexes I and IV and the enzymatic activity of mtNOS with neurological performances (neuromuscular coordination and exploratory activity) (Figure 1B) and the direct relationships between enzymatic activity of complexes I and IV and of mtNOS with the median and maximal life span in the same mice (Figure 1C) (Boveris and Navarro, 2008a). Accordingly, aged mammalian brain has a decreased capacity to produce ATP by oxidative phosphorylation and it is considered that this decreased capacity for energy production becomes limiting under physiological conditions in aged individuals. For instance, brain mitochondria isolated from mice at the time point of 50% survival (median life span) show about 50% of the activity at full adulthood of critical enzymes, such as complex I (NADH-ubiquinone reductase), complex IV (cytochrome oxidase) and mitochondrial nitric oxide synthase (mtNOS) (Navarro and Boveris, 2007a,b).
Figure 1. Statistical correlations involving: (A) mitochondrial enzyme activities (complexes I and IV and mtNOS) and mitochondrial oxidative damage (r2 = 0. 84; p < 0.01 for complexes I and IV; r2 = 0.90; p < 0.01 for mtNOS); (B) enzyme activities and neurological tests (tightrope, r2 = 0.62; p < 0.001; T-maze, r2 = 0.56; p < 0.05) and enzyme activities and median (r2 = 0.46; p < 0.05), and maximal (r2 = 0.31; p < 0.01), life spans (C) in male mice. Data from (Navarro et al., 2002, 2004, 2005a, 2007). For the neurological assays, individual mice were subjected every 2 weeks to the tightrope and the T-maze tests (Navarro et al., 2002). For the tightrope test, mice were placed hanging from their anterior legs in the middle of a 60-cm tightrope and the test was considered successful when mice reached the column at the end of the rope in less of 30 s. For the T-maze test, mice were challenged in a T-shaped maze of 50 cm arms and the test was considered successful when mice moved toward the T-intersection in less than 30 s. Modified from Boveris and Navarro (2008a,b).
The current knowledge indicates that the impairment of brain mitochondrial function in aging is mainly due to decreased electron transfer rates in complexes I and IV, among other decreased mitochondrial activities. There was a hypothesis in the field that accumulation of phospholipid hydroperoxides and other oxidation products in aging would lead to structural modifications of the bilayer with an increased permeability of the inner membrane to H+ and water. There are no reports on the effect of aging either on H+ permeability, the key property of the inner membrane in the chemiosmotic mechanism of oxidative phosphorylation, or on brain uncoupling proteins (Mattiasson and Sullivan, 2006). Increased water permeability was reported in brain mitochondria in aged rats (Navarro and Boveris, 2004). Concerning the effect of aging on brain ATP-synthase activity, it has been reported that brain aging is associated with a markedly decreased ATP-synthase activity with nitration of the Tyr269 of the β-subunit (Lam et al., 2009).
Mitochondrial dysfunction in terms of decreased rates of electron transfer in complex I and IV and decreased mitochondrial respiration with NAD-dependent substrates has been consistently observed in brain mitochondria isolated from aged rats and mice (Vitorica et al., 1985; Beckman and Ames, 1998; Navarro and Boveris, 2004, 2007b; Navarro et al., 2004, 2005a, 2007; Cocco et al., 2005). Interestingly, the phenomenon is more marked in specific rat brain areas that are understood as more affected by the aging process, such as hippocampus and cerebral cortex (Navarro et al., 2008).
Mitochondria isolated from the brain of aged rats and mice show increased contents of the oxidation products of phospholipids, proteins, and DNA, decreased membrane potential, and increased size and fragility (Beckman and Ames, 1998; Navarro and Boveris, 2004, 2007b). The oxidative damage is also more marked in hippocampus and cerebral cortex than in whole brain (Navarro et al., 2008).
Human neurodegenerative diseases are characterized by cumulative neuronal damage in specific brain areas that leads to neurological deficits when neuronal loss reaches a critical limit. For instance, Parkinson’s disease evolves for years before typical motor signs appear with a loss of about 60% of the dopaminergic neurons of substantia nigra pars compacta. There is growing evidence that mitochondrial dysfunction and impairment of the respiratory complexes is associated with the neuronal loss in neurodegenerative diseases. Decreased complex IV activity has been reported in Alzheimer’s disease (Chagnon et al., 1995) and decreased complex I activity is usually reported in the substantia nigra of postmorten samples in Parkinson’s disease (Mizuno et al., 1989; Mann et al., 1992).
Mitochondria and Parkinson’s Disease
There is considerable evidence for a mitochondrial involvement in sporadic Parkinson’s disease. Experimental data indicate that Parkinson’s disease is associated to two interdependent conditions of brain mitochondria: mitochondrial dysfunction and mitochondrial oxidative damage. Several studies have shown mitochondrial dysfunction and reduced activity of mitochondrial complex I in substantia nigra (Schapira et al., 1990a,b; Schapira, 2008) and in frontal cortex (Navarro et al., 2009) in Parkinson’s disease patients. Moreover, similar mitochondrial complex I dysfunctions were reported in skeletal muscle and platelets of Parkinson’s disease patients (Mann et al., 1992). This condition of complex I impairment is likely to be of pathogenic importance because intoxication of experimental animals with inhibitors of complex I (rotenone, pyridaben, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its metabolite 1-methyl-4-phenylpyridinium (MPP+) (Bougria et al., 1995; Gomez et al., 2007) reproduce the clinical symptoms of Parkinson’s disease in humans. Moreover, multiple genes in which mutations or polymorphisms increase the risk of Parkinson’s disease are linked to mitochondrial functions. Inhibition of complex I creates a biochemical environment with increased reduction of the FMN and high levels of the 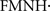 semiquinone that increase the generation of . In turn, the increased rate of generation promotes lipid peroxidation, protein oxidative damage and peroxynitrite (ONOO−) mediated protein nitration (as 3-nitrotyrosine) and nitrosylation (as –S–NO of thiol groups), in a process that lead neurons to apoptosis (Navarro and Boveris, 2009) and to aggregation of α-synuclein with the subsequent death of dopaminergic neurons (Dawson and Dawson, 2003). There are numerous reports linking α-synuclein, mitochondrial dysfunction, and oxidative stress; however, reports are neither consistent in the causality nor in the cross-effects of oxidative stress and α-synuclein expression and polymerization. It is a current hypothesis in the field that cytosolic oxidative stress (increased GSSG and H2O2 levels) favor the post-translational oxidative modification of α-synuclein and its aggregation.
semiquinone that increase the generation of . In turn, the increased rate of generation promotes lipid peroxidation, protein oxidative damage and peroxynitrite (ONOO−) mediated protein nitration (as 3-nitrotyrosine) and nitrosylation (as –S–NO of thiol groups), in a process that lead neurons to apoptosis (Navarro and Boveris, 2009) and to aggregation of α-synuclein with the subsequent death of dopaminergic neurons (Dawson and Dawson, 2003). There are numerous reports linking α-synuclein, mitochondrial dysfunction, and oxidative stress; however, reports are neither consistent in the causality nor in the cross-effects of oxidative stress and α-synuclein expression and polymerization. It is a current hypothesis in the field that cytosolic oxidative stress (increased GSSG and H2O2 levels) favor the post-translational oxidative modification of α-synuclein and its aggregation.
Mitochondrial Complex I, the “Complex I Syndrome” and Parkinson’s Disease
Mitochondrial complex I (NADH-UQ reductase) catalyzes electron transfer from NADH to ubiquinone and is the molecular pathway to connect the tricarboxylic acid cycle and NADH with the mitochondrial respiratory chain. Complex I is a supra-molecular protein complex of 850 kDa composed of about 40 polypeptide units and contains FMN and iron–sulfur clusters as redox active centers. Seven polypeptides of complex I are encoded by mitochondrial genes (ND 1, 2, 3, 4, 4L, 5, and 6). Complex I components include a 54-kDa flavoprotein, 24, 75, and 49 kDa proteins, and the proteins TYKY and PSST that are bound to the inner membrane and that transfer electrons to UQ (Walker, 1992). Two complex I-linked UQ-pools have been detected (Raha and Robinson, 2000). Non-covalent hydrophobic bonds and van der Waals’ weak bonds are important in keeping together the whole structure of complex I; low concentrations of detergents, natural and synthetic steroids (Boveris and Stoppani, 1971) and hydrophobic pesticides [rotenone and pyridaben (Gomez et al., 2007)] are effective in disrupting intra-complex I polypeptide hydrophobic bonds and in inhibiting complex I electron transfer activity. The rate of electron transfer in complex I is relatively high; NADH oxidation proceeds at 250–500 nmol/min mg protein, as compared with the rate of electron transfer of complex II (UQH2-cytochrome c reductase) of 100–150 nmol/min mg protein (Brown and Borutaite, 2004). Complex I by auto-oxidation with molecular O2 produces significant amounts of by the collisional reaction of O2 with the flavin semiquinone (Chance et al., 1979; Boveris and Cadenas, 2000; Turrens, 2003). The rate of production by complex I is increased by electron transfer inhibition with rotenone (Boveris and Chance, 1973) and by complex I dysfunction in choline deficiency (Hensley et al., 2000).
It is currently accepted that mitochondrial complex I is particularly sensitive, in terms of inactivation, to oxidants, oxygen free radicals, and reactive nitrogen species. This special characteristic is frequently referred as the “complex I syndrome”, with the symptoms of reduced mitochondrial respiration in state 3 (with ADP) and with malate–glutamate as substrate and of reduced complex I activity. This “complex I syndrome” has been observed in Parkinson’s disease and in other neurodegenerative diseases (Schapira et al., 1990a,b; Carreras et al., 2004; Schapira, 2008; Navarro et al., 2009), as well as in aging (Boveris and Navarro, 2008a) and in ischemia-reperfusion (Gonzalez-Flecha et al., 1993). The molecular mechanism involved in the inactivation of complex I is likely accounted by the sum of ONOO− mediated reactions, reactions with the free radicals intermediates of the lipid peroxidation process ( and
and 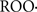 ) and amine–aldehyde adduction reactions. It is now understood that the three process above mentioned alter the native non-covalent polypeptide interactions of complex I and promote synergistically protein damage and inactivation by shifting the non-covalent bonding to covalent cross linking (Liu et al., 2003).
) and amine–aldehyde adduction reactions. It is now understood that the three process above mentioned alter the native non-covalent polypeptide interactions of complex I and promote synergistically protein damage and inactivation by shifting the non-covalent bonding to covalent cross linking (Liu et al., 2003).
Nitric Oxide and Peroxynitrite as Complex I Inhibitors
Both, NO and ONOO− have been reported as direct inhibitors of complex I. It has been claimed that NO inhibits mitochondrial complex I activity by S-nitrosylation and Fe-nitrosation (Brown and Borutaite, 2004). On the other hand, irreversible inhibition of brain mitochondrial complex I by ONOO− was reported (Riobo et al., 2001). The current ideas are that the chronic toxic effects of NO in the brain are explained by the cumulative nitrations of complex I proteins due to ONOO− increased levels in the mitochondrial matrix where it is produced in the termination reaction of the two free radicals 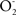 and NO. It is considered that the anion ONOO− is confined to the mitochondrial matrix, since it is charged and there is no recognized transporter. Superoxide radical is produced physiologically in mitochondria by the autoxidation reactions of
and NO. It is considered that the anion ONOO− is confined to the mitochondrial matrix, since it is charged and there is no recognized transporter. Superoxide radical is produced physiologically in mitochondria by the autoxidation reactions of 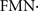 and
and 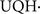 at a rate of 0.80–0.90 nmol
at a rate of 0.80–0.90 nmol 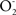 /min mg protein resulting in a steady state concentration of 0.1–0.2 nM in the mitochondrial matrix (Boveris and Cadenas, 2000; Boveris et al., 2006; Valdez et al., 2006). In the same physiologic conditions and by the enzymatic reaction of mitochondrial nitric oxide synthase (mtNOS), NO is produced at a rate of 1.0–1.4 nmol NO/min mg protein and kept at a steady state level of 200–350 nM in the mitochondrial matrix (Boveris et al., 2006; Valdez et al., 2006). Both radicals react in a diffusion controlled reaction (k = 1.9 × 1010 M−1 s−1) and generate ONOO− at a rate of 0.38 μM/s in the mitochondrial matrix or 0.92 nmol/min mg protein (Valdez et al., 2000). Peroxynitrite is normally reduced by the mitochondrial reductants NADH2, UQH2 and GSH and kept at an intramitochondrial steady state level of 2–5 nM (Valdez et al., 2000). When this level is increased in excess, up to 25–40 nM, it leads to tyrosine nitration, protein oxidation, and damage to iron sulfur centers with sustained complex I inhibition and increased generation of
/min mg protein resulting in a steady state concentration of 0.1–0.2 nM in the mitochondrial matrix (Boveris and Cadenas, 2000; Boveris et al., 2006; Valdez et al., 2006). In the same physiologic conditions and by the enzymatic reaction of mitochondrial nitric oxide synthase (mtNOS), NO is produced at a rate of 1.0–1.4 nmol NO/min mg protein and kept at a steady state level of 200–350 nM in the mitochondrial matrix (Boveris et al., 2006; Valdez et al., 2006). Both radicals react in a diffusion controlled reaction (k = 1.9 × 1010 M−1 s−1) and generate ONOO− at a rate of 0.38 μM/s in the mitochondrial matrix or 0.92 nmol/min mg protein (Valdez et al., 2000). Peroxynitrite is normally reduced by the mitochondrial reductants NADH2, UQH2 and GSH and kept at an intramitochondrial steady state level of 2–5 nM (Valdez et al., 2000). When this level is increased in excess, up to 25–40 nM, it leads to tyrosine nitration, protein oxidation, and damage to iron sulfur centers with sustained complex I inhibition and increased generation of 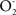 by complex I. The available evidence indicates that increased mitochondrial matrix NO levels, secondary to mtNOS over-expression or activation or to NO diffusion from cytosolic NOS, leads to ONOO−-mediated nitration, to inhibition of complex I enzymatic activity, to increased production of
by complex I. The available evidence indicates that increased mitochondrial matrix NO levels, secondary to mtNOS over-expression or activation or to NO diffusion from cytosolic NOS, leads to ONOO−-mediated nitration, to inhibition of complex I enzymatic activity, to increased production of 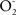 and to the turning of normal cell signaling by H2O2 and NO to ONOO−-initiated apoptotic signaling. The normal signaling role of NO and H2O2 is supported by the developmental biology observation that the 144-kDa brain mtNOS increases several folds in the pre- and post-natal periods in rats in parallel to brain and cerebellum development (Riobo et al., 2002).
and to the turning of normal cell signaling by H2O2 and NO to ONOO−-initiated apoptotic signaling. The normal signaling role of NO and H2O2 is supported by the developmental biology observation that the 144-kDa brain mtNOS increases several folds in the pre- and post-natal periods in rats in parallel to brain and cerebellum development (Riobo et al., 2002).
At this point, it is convenient to briefly address the current status of mtNOS, the mitochondria-located member of the NOS family. This enzyme activity was first described in rat liver mitochondria by Ghafourifar and Richter (1997) and by Giulivi et al. (1998). At the beginning there was a skeptical attitude toward this new enzyme but in a few years mtNOS activities were reported in a series of mouse and rat organs such as brain, heart, kidney, thymus, and skeletal muscle with a mean activity of about 0.90 nmol NO/min × mg protein (Boveris and Navarro, 2008b). The idea of contamination faded away because it seemed unreasonable that mitochondria of various tissues would show a similar contamination with cytosolic NOS. In 2002, Giulivi and co-workers in a transcendent contribution (Elfering et al., 2002) sequenced rat liver mtNOS and identified the enzyme as an inner membrane protein and as the transcript of nNOS, splice variant α, myristylated and phosphorylated. Decisive evidence was provided by Kanai et al. (2001) with the electrochemical determination of the Ca2+-induced NO release from a single mouse heart mitochondrion, a process that was abolished in nNOS −/− knock out mice. The current understanding is that mtNOS is the result of the specific and controlled translocation of a cytosolic transcript of genomic nNOS.
In this context, the transcriptional regulation of the two brain NOS, the cytosolic nNOS and the mitochondrial mtNOS, are particularly relevant. A bipolar and complementary distribution of NOS activities in cells has been proposed; one in mitochondria (mtNOS) and the other one (nNOS or eNOS) in the cytosol (Navarro and Boveris, 2008). There are reports of over-expression of nNOS in the brain of patients with Parkinson’s disease (Eve et al., 1998) and of increased levels of cytosolic nNOS in aging rats (Lam et al., 2009). In the latter case, proteomic analysis revealed a selective and almost specific nitration of the ATP-synthase at Tyr269, explained by the higher level of ONOO− in the mitochondrial matrix as compared with the cytosol (Lam et al., 2009).
The observed nNOS over-expression and the presence of 3-nitrotyrosine in circulating neutrophils from Parkinson’s disease patients, suggests a generalized deregulation of the nNOS gene (Gatto et al., 2000). The role of increased levels of NO in Parkinson’s disease gained significance by the finding of 3-nitrotyrosine in the core of Lewy bodies, the pathological hallmark of the disease (Good et al., 1998). The presence of 3-nitrotyrosine, the ‘‘footprint’’ of ONOO− high levels, was observed in brain mitochondria of rats treated with MTPT and its metabolite MPP+ and prevented by previous administration of 7-nitroindazole, a relatively specific nNOS inhibitor. Moreover, nNOS-gene deficient mice are more resistant to the toxic effects of MPTP than wild-type animals. In agreement, in SH-SY5Y neuroblastoma cells, MPTP and MPP+ increase the mitochondrial production of NO, suggesting an activating effect on mtNOS that is associated to Bax increase, release of cytochrome c and caspase activation (Dennis and Bennett, 2003). It was also reported that oxidative stress induced by complex I inhibition biphasically activated the pro-apoptotic factor c-Jun-N-terminal kinase (JNK) and the transcription factor NF-κB (Cassarino et al., 2000).
The action of the mentioned inhibitors (ONOO−, ROOH, 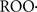 , intermediates and aldehydes from the lipid peroxidation processes), on complex I leads to denaturation of the protein structure and further increases of
, intermediates and aldehydes from the lipid peroxidation processes), on complex I leads to denaturation of the protein structure and further increases of 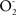 and ONOO− production at the vicinity of complex I itself, tending to the perpetuity of this metabolic abnormality which is relevant for the chronic situations of neurodegeneration.
and ONOO− production at the vicinity of complex I itself, tending to the perpetuity of this metabolic abnormality which is relevant for the chronic situations of neurodegeneration.
Complex I Syndrome in Patients with Parkinson’s Disease and with Dementia with Lewy Bodies
Recently, moderate and marked impairments of tissue respiration, state 3 mitochondrial respiration with malate–glutamate as substrate and complex I decreased activity, associated with oxidative damage were determined in frozen samples of frontal cortex (area 8) of patients with Parkinson’s disease and with dementia with Lewy bodies in comparison to age-matched healthy controls (Table 1) (Navarro et al., 2009). The observed mitochondrial impairment is properly described as a reduced tissue respiration of frontal cortex with reduced mitochondrial respiration with NAD-dependent substrates and with a marked decrease in complex I activity. The decreases in respiratory rates and in enzymatic activities were associated with oxidative damage, as determined by the increased contents of phospholipid and protein oxidation products. It is to note that cortex mitochondrial dysfunction is now added to the classical recognition of mitochondrial dysfunction in the dopaminergic neurons of substantia nigra in Parkinson’s disease which was early considered as specific (Schapira et al., 1990a).

Table 1. Oxygen uptake and complex I and mtNOS activities of human brain cortex in healthy controls and in patients with Parkinson’s disease or with dementia with Lewy bodies.
The markedly higher mtNOS activity observed in human frontal cortex (4.0–7.0 nmol NO/min mg protein (Table 1) in comparison with the mtNOS activities in mouse and rat whole brain and frontal cortex (0.64–0.67 nmol NO/min mg protein (Navarro et al., 2008) set the basis for the speculation that this level of brain mtNOS expression corresponds to an adaptive response of the highly evolved human brain, as part of the human extraordinary capacity for homeostatic maintenance and longevity. Cutler and co-workers introduced the concept that Cu,Zn-SOD and Mn-SOD activities in various organs, evolved in mammals as determinants of life span (Tolmasoff et al., 1980; Cutler, 1991). Similarly, it is reasonable to consider that human brain acquired upon evolution an adaptive resistance to brain cortex mitochondrial dysfunction and oxidative damage.
The mitochondrial adaptive response of mtNOS in neurodegenerative diseases is speculated as an increased mtNOS activity that supports an increased mitochondrial biogenesis in order to provide an increased energy supply. A 48% increase in the mitochondrial mass of human brain cortex was reported in Parkinson’s disease patients (Navarro et al., 2009) and the observation was interpreted as an adaptive response for the respiratory impairment with a presumable shortage in ATP supply. The decrease in maximal (state 3) respiratory activity is estimated in the range of 34–59%, seeming that the adaptive response in mitochondrial mass provides the same energy production than in the state of health.
The current views on Parkinson’s disease consider that this disease is not only characterized by substantia nigra dysfunction but that also involves the frontal cortex with a cognitive decline at the early stages of parkinsonism (McNamara et al., 2007). Oxidative and nitrosative damage and mitochondrial dysfunction in the human frontal cortex are to be considered factors leading to impaired cognition in Parkinson’s disease.
It is currently considered that the energy demands of brain and central nervous system are provided by very active glucose oxidation and aerobic metabolism; it is accepted than the O2 uptake of brain and central nervous system accounts for about 20% of the O2 uptake in basal metabolic conditions. It has been recently postulated that brain phospholipid oxidation products, as TBARS and organic hydroperoxides, reach systemic circulation and that the final product malonaldehyde (measured as TBARS) is increased by 21% in the plasma of patients with Parkinson’s and Alzheimer’s diseases and with vascular dementia (Serra et al., 2009). An estimation of the rate of brain lipid peroxidation in the mentioned neurodegenerative diseases indicates an about twice increased rate as compared to the normal condition.
Mitochondria Targeted Antioxidant Therapy
The experimental recognition of the association between dysfunctional mitochondria, inhibition of electron transfer, and accumulation of oxidation products is interpreted as the result of the continuous production of the free radicals 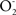 and NO by the enzymes of the inner mitochondrial membrane. Determinations of the mitochondrial rates of
and NO by the enzymes of the inner mitochondrial membrane. Determinations of the mitochondrial rates of 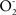 and of NO production indicate that they account for 1 % (
and of NO production indicate that they account for 1 % (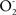 ) and 2 % (NO) of the rates of brain O2 uptake. The chain reactions initiated by
) and 2 % (NO) of the rates of brain O2 uptake. The chain reactions initiated by 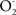 and NO establish a continuous free radical processes that entail lipid peroxidation and protein oxidation. In terms of causality it seems that the processes of oxidative damage to phospholipids, of enzyme inactivation, and of mitochondrial dysfunction are simultaneous. The mitochondrial increase in oxidation products has been interpreted as the result of a continuous process of phospholipid and protein oxidation. It is becoming clear that mitochondria continuously and simultaneously respire, produce
and NO establish a continuous free radical processes that entail lipid peroxidation and protein oxidation. In terms of causality it seems that the processes of oxidative damage to phospholipids, of enzyme inactivation, and of mitochondrial dysfunction are simultaneous. The mitochondrial increase in oxidation products has been interpreted as the result of a continuous process of phospholipid and protein oxidation. It is becoming clear that mitochondria continuously and simultaneously respire, produce 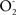 and NO, are subjected to free radical mediated processes that produce lipid peroxidation and protein oxidation and loose enzymatic activity. The loosing of enzymatic activity is also called the appearance of dysfunctional mitochondria. It is speculated that dysfunctional mitochondria, as they are defined by sub-normal or partially inactivated respiration, enzymatic activity, and ATP-production, have an extended time for mitochondrial turnover, which is associated with a higher level of mitochondrial oxidation products. It is also speculated that this high level of oxidation products is enough to trigger apoptosis, before de novo mitochondrial biogenesis takes the level of oxidized products to one half (non-oxidized phospholipids and proteins are incorporated in mitochondrial biogenesis) (Figure 2).”
and NO, are subjected to free radical mediated processes that produce lipid peroxidation and protein oxidation and loose enzymatic activity. The loosing of enzymatic activity is also called the appearance of dysfunctional mitochondria. It is speculated that dysfunctional mitochondria, as they are defined by sub-normal or partially inactivated respiration, enzymatic activity, and ATP-production, have an extended time for mitochondrial turnover, which is associated with a higher level of mitochondrial oxidation products. It is also speculated that this high level of oxidation products is enough to trigger apoptosis, before de novo mitochondrial biogenesis takes the level of oxidized products to one half (non-oxidized phospholipids and proteins are incorporated in mitochondrial biogenesis) (Figure 2).”
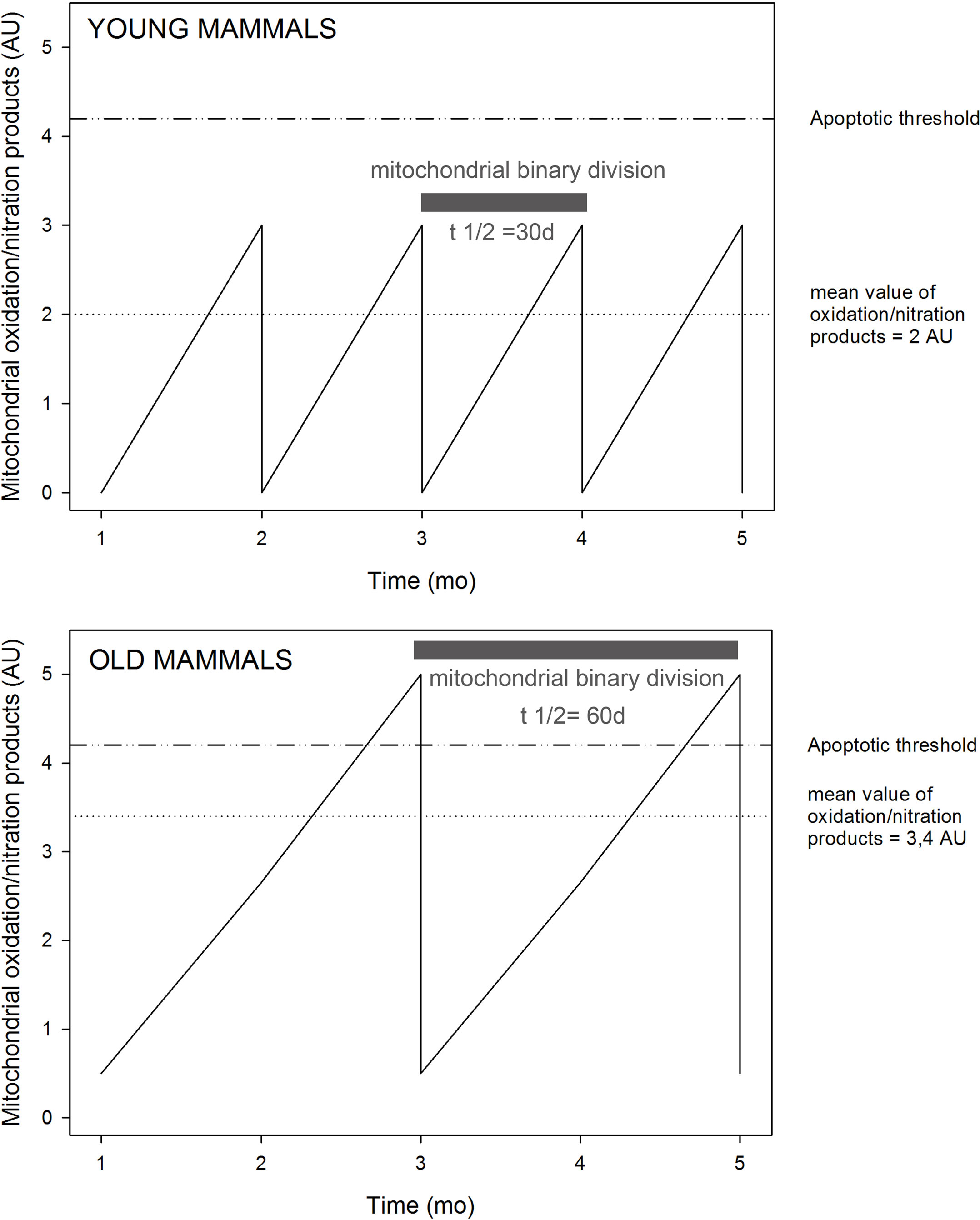
Figure 2. Scheme illustrating the hypothesis of the time course of the levels of mitochondrial oxidation and nitration products, associated with mitochondrial turnover for brain mitochondria as a function of time and age. AU, arbitrary units. Modified from Navarro and Boveris (2008).
It is expected that slowing down the processes of brain mitochondrial dysfunction that occurs upon aging will provide a decrease of the neurological deficits in aged humans. Antioxidants targeted to mitochondria are promising therapeutic agents for human neurodegenerative diseases, however a huge research has to be performed (Reddy, 2007).
A series of antioxidants that can be described as mitochondria-targeted are considered as follows.
Vitamin E
Chronic supplementation with high doses of vitamin E extended median life span by 39%, improved neurological functions by 25–28% in aging mice (Navarro et al., 2005a) and improved brain mitochondrial function in aging mice and rats. Vitamin E reduced the inhibition of brain complex I activity produced by aging from 36% to 12–14% in mice (Navarro et al., 2005a) and the inhibition of the malate-glutamate supported state 3 respiration of hippocampal and frontal cortex mitochondria from 23–29% to 4–10% (Table 2). Dietary supplementation with vitamin E increased α-tocopherol levels in mouse brain from 11.5 to 26.2 nmol/g brain (Navarro et al., 2005a). It is then clear that vitamin E (431 Da) crosses the blood-brain barrier, a process reserved to relatively small molecules (<450 Da) with lipophylic character, given to vitamin E by the chroman ring and the isoprenoid chain, and with less than 10 hydrogen bonds (Fand and McNally, 1981; Vatassery et al., 2004). The effect of vitamin E on the prevention of the aging-dependent decline in mitochondrial function was dose-dependent: 2.0 and 5.0 g of α-tocopherol acetate/kg food produced a prevention of 22% and 34% of the decline of respiration in rat hippocampal mitochondria (Table 2). These two α-tocopherol levels (2.0 and 5.0 g of α-tocopherol acetate/kg food) in the rat diet would correspond, on the basis of the ratio: mg α-tocopherol/kJ of basal metabolic rate, to 0.90 and 2.1 g vitamin E/day in humans.
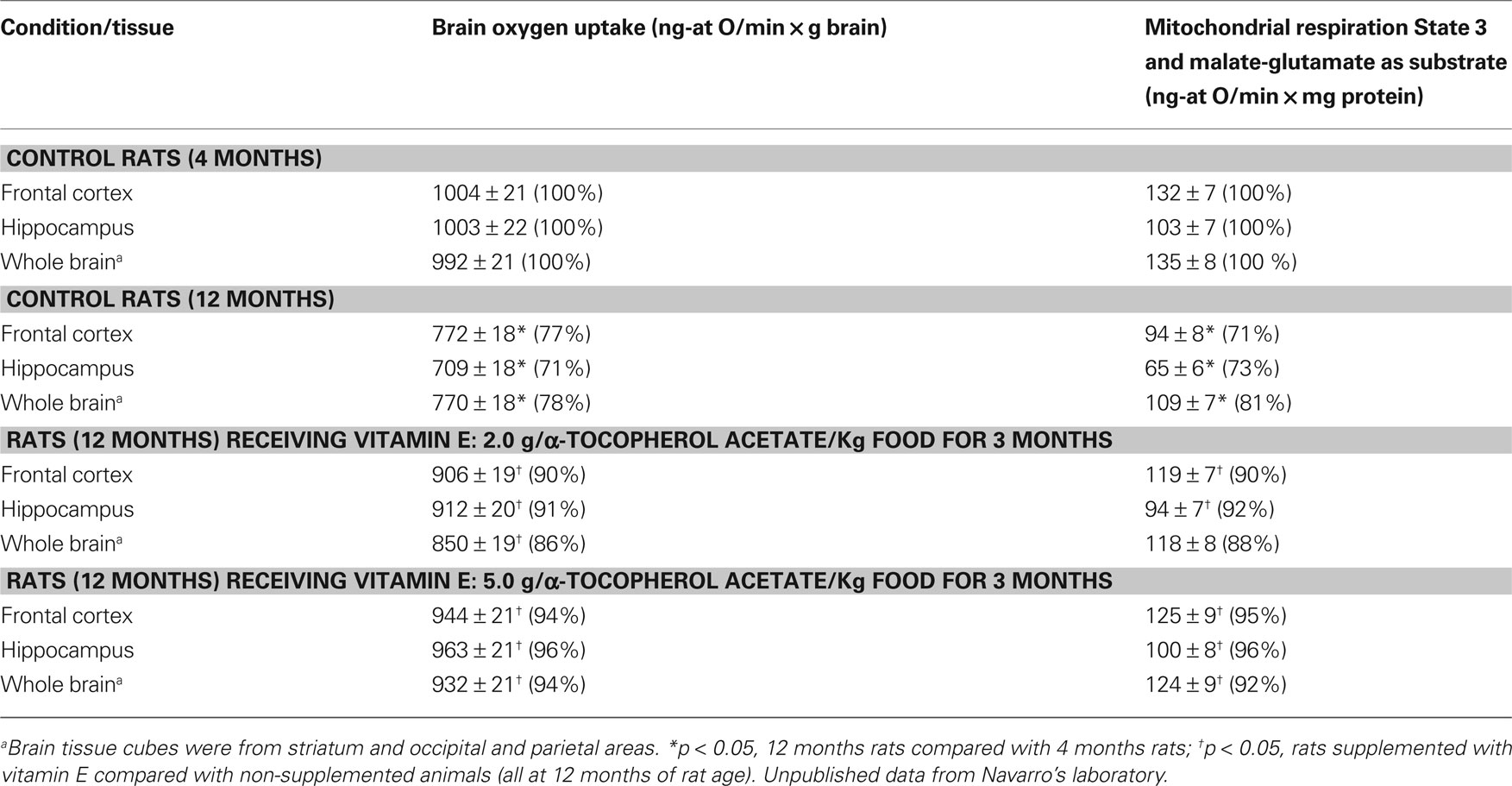
Table 2. Effect of high doses of dietary vitamin E in tissue and mitochondrial respiration in frontal cortex, hippocampus, and whole brain of aging rats.
The effect of high doses of vitamin E on mice survival is to be taken into account in the controversy on the use of vitamin E supplementation in humans. The claim that vitamin E supplementation increases human mortality, based on meta-analysis (Bjelakovic et al., 2007) is challenged by the clinical evidence that vitamin E supplements are safe at high intakes (Hathcock et al., 2005) and by the reported effects of vitamins E and C in the reduction of prevalence and incidence of Alzheimer disease in an elderly population (Zandi et al., 2004).
Other Antioxidants
Other antioxidants, such as acetylcarnitine and lipoic acid (Hagen et al., 1998; Liu et al., 2002) and flavonoid-rich vegetable extracts (Sastre et al., 1998; Bickford et al., 1999), chronically administered to mice and rats also prevented the age-associated decline in neurological functions and oxidative damage in brain mitochondria. In the case of the flavonoid rich extract Egb 761 given orally to rats, its administration was effective in preventing the appearance of enlarged mitochondria, of increased levels of 8-HO-d-guanosine and hydroperoxides, and of a decrease in membrane potential (Sastre et al., 1998). Similarly, rats treated chronically with acetylcarnitine showed a lower age-dependent decline in the mitochondrial oxidation rate of NAD-dependent substrates (Cocco et al., 2005) and in the mitochondrial gene expression of complexes I, IV, and V and of adenine nucleotide translocase (Nicoletti et al., 2005).
The Skulachev Cations Attached to Antioxidant Molecules
A new family of mitochondrial antioxidants is being developed. The new mitochondria-targeted antioxidants are covalent derivatives of vitamin E, ubiquinone, and PBN (α-phenyl-N-tert-butyl nitrone) that are covalently coupled to a triphenylphosphonium cationic group (Murphy and Smith, 2007). The phosphonium derivatives have been used for years in the determination of inner membrane potential and, following to the development and use by Russian bionergeticists, are commonly known as the “Skulachev cations.” Lipophilic triphenyl-phosphonium cations are actively taken up by mitochondria due to the inner membrane potential (160–170 mV) with the inside negative, that according to the electrochemical potential (Guggenheim equation) produces an intramitochondrial accumulation of about 700 times (Skulachev, 2005, 2007). The development and use of the phosphonium antioxidants takes advantage of the unique mitochondrial biophysical and biochemical characteristics that provide a negatively charged compartment and a reducing environment that allows regeneration of the free radical scavengers.
The molecule resulting from the coupling of triphenylphosphonium cation (TPP+) with α-tocopherol, is called MitoVitE and was developed to prevent mitochondrial oxidative damage. Mitochondria incubated with 1–20 μM MitoVitE take up the phosphonium cations in about 15–30 min with accumulation ratios of up to 1000 times (Sheu et al., 2006). Higher levels of MitoVitE (50 μM) in the incubation medium are cytotoxic for Jurkat cells (Sheu et al., 2006). It is considered that MitoVitE is inserted in the lipid bilayer of the mitochondrial inner membrane and that the chroman group becomes redox active. The semiquinone, formed after detoxifying a free radical by hydrogen donation, is reduced by electron transfer or tunneling from a reduced component of the respiratory chain. MitoVitE was reported to reduce H2O2-induced caspase activity (Hughes et al., 2005) and to prevent cell death in fibroblasts from patients with Friedrich ataxia, an inherited nervous system disease associated with decreased frataxin and increased iron-catalyzed oxidative damage (Jauslin et al., 2003), to inhibit cytochrome c release and caspase-3 activation, to inhibit complex I inactivation and to restore mitochondrial membrane potential in bovine aortic epithelial cells after oxidative stress (Reddy, 2006).
MitoQ10 is a similar TPP+ derivative with ubiquinone-50 (Q10) that is similarly accumulated in mitochondria. Internalized MitoQ10 is inmobilized by anchoring the isoprenoid chain into the lipid bilayer and becomes redox active: the oxidized form is readily reduced by complex II and is reduced by complex I. The ubiquinol molecule inserted in the mitochondrial inner membrane acts as a free radical trap and antioxidant preventing oxidative damage (Reddy, 2006). The electrical charge and the consequent superficial position of the TPP+ moiety and the solubility of Q10 in non-polar solvents indicate the isoprenoid chain of MitoQ10 concentrates in the membrane core where it quenches fluorophors deep within the membrane. In isolated cells, MitoQ10 protects from H2O2-induced apoptosis but not from the apoptosis induced by staurosporine or TNF-α (Murphy and Smith, 2007).
MitoPBN is another TPP+ derivative, in this case with phenoxy-butyl-nitrone, that was designed to prevent mitochondrial lipid peroxidation and oxidative damage based on the well known and relatively selective PBN reaction with carbon-centered radicals (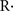 ) and peroxyl radicals (
) and peroxyl radicals (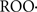 ) (Murphy et al., 2003). MitoPBN is rapidly taken up by mitochondria reaching intramitochondrial levels of 2.2–4.0 mM. MitoPBN was observed to block the oxygen-induced activation of uncoupling proteins (Murphy et al., 2003). An amphiphilic molecule derived from PBN and that is a nitronium cation (LPBNAH) shows a neuroprotective activity antagonizing oxidative damage of mitochondrial origin (Poeggeler et al., 2005).
) (Murphy et al., 2003). MitoPBN is rapidly taken up by mitochondria reaching intramitochondrial levels of 2.2–4.0 mM. MitoPBN was observed to block the oxygen-induced activation of uncoupling proteins (Murphy et al., 2003). An amphiphilic molecule derived from PBN and that is a nitronium cation (LPBNAH) shows a neuroprotective activity antagonizing oxidative damage of mitochondrial origin (Poeggeler et al., 2005).
Currently, other antioxidants of the phosphonium type are developed as effective mitochondria-targeted antioxidants (Kelso et al., 2001; James et al., 2007). For instance, the derivative with the selenium-containing ebselen shows hydroperoxide peroxidase activity (Mugesh et al., 2001).
Tetrapeptide Antioxidants
The series of the “SS tetrapeptides” is constituted by aromatic-cationic peptides that have the structural motif of alternating aromatic and basic amino acids with 2, 6-dimethyltyrosine residues (Sheu et al., 2006). These tetrapeptides, that were originally prepared to act as opioid analgesics, are taken up by isolated cells and mitochondria due to their positive charge at physiological pH and show the antioxidant properties of dimethyltyrosine, a classic phenolic antioxidant (Zhao et al., 2003a,b). Four main SS tetrapeptides have been developed: SS-02, SS-19, SS-20, and SS-31 (Sheu et al., 2006). Peptides SS-02 and SS-19 are actively taken up by mouse liver mitochondria and human CaCo-2 cells, with an intracellular localization in mitochondria and with a 100-fold accumulation in mitochondria. The mitochondrial uptake of SS-19 was decreased by the uncoupler FCCP, indicating a potential-dependent accumulation (Zhao et al., 2004).
Three structure–property relationships illustrate how this antioxidants work: SS-20 that lacks the dimethyltyrosine of SS-02 also lacks antioxidant properties; SS-31 contains the same amino acids that SS-02, but in a sequence with better antioxidant properties; and SS-19 is more lipophylic and fluorescent due to the anthranyl group. Concerning antioxidant properties, SS-02 showed antioxidant properties in a cell-free system and inhibited the H2O2-promoted oxidation of linoleic acid and of low-density lipoproteins (Zhao et al., 2003b) and SS-31 protected against oxidant-induced mitochondrial dysfunction and apoptosis in isolated neurons of the N2a and SH-SY5Y cell lines (Zhao et al., 2004). Treatment of the neurons with t-butyl-hydroperoxide resulted in increased lipid peroxidation, phosphatidylserine translocation, mitochondrial depolarization, increased caspase activity, nuclear condensation, and cell death by apoptosis and SS-31 was able to partially prevent the oxidative damage produced by t-butyl-hydroperoxide. The remarkable potency of SS-31 (effects are reported at 1 nM) is explained by extensive cellular and mitochondrial accumulation (about 5000-fold in the mitochondrial pellet (Zhao et al., 2005). It was reported that SS-19 decreases mitochondrial 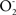 production and cellular lipid peroxidation and improves the contractile force and myocardial stunning in perfused hearts subjected to ischemia-reperfusion (Zhao et al., 2005).
production and cellular lipid peroxidation and improves the contractile force and myocardial stunning in perfused hearts subjected to ischemia-reperfusion (Zhao et al., 2005).
Mitochondrial Biogenesis, Cellular no, and Mitochondrial no Production
Nitric oxide signaling for mitochondrial biogenesis and turnover is a recent concept in cell biology. The NO-dependent pathway of mitochondrial biogenesis includes activation of guanylate cyclase, increased levels of cGMP, and activation of a series of transcription factors, such as PPAR-GC-1α, nuclear respiratory factors (NRF-1 and NRF-2) and mitochondrial transcription factor A. This mechanism is supported by observations in brain, kidney, liver, heart, and muscle (Nisoli et al., 2004) and ovary (Navarro et al., 2005b). However, it has to kept in mind that NO effects are expected to be finely tuned up according to NO levels and to the cell redox environment and that in such scenario, both neurotrophic and neurotoxic effects of NO are likely. Moderate exercise has been recognized effective in increasing brain mtNOS activity; mice subjected to moderate exercise during 12 days increases 2.5 times mtNOS activity in mouse brain (Boveris and Navarro, 2008b). The effect was explained as the activation of a muscle-initiated neural pathway that activates mtNOS synthesis in the brain (as it occurs in endorphin synthesis). Pyrroloquinone has been found to increase mitochondrial biosynthesis in cell cultures and opens a perspective for diseases associated with mitochondrial dysfunction (Chowanadisai et al., 2010).
Concluding Remarks
The long standing concept of linking aging and an impaired mitochondrial energy supply is now receiving experimental support. It has been recognized that aging is associated with a decreased mitochondrial function, considered primarily as electron transfer and respiration, in a series of organs as brain, heart, liver, and kidney. Brain mitochondria are more affected by the aging process than the mitochondria of other organs. This observation agrees with the general concept that the physiological functions that depend on the integrated response of the central nervous system are the most affected by aging.
The complex series of factors contributing to brain senescence and neurodegeneration in experimental animals, mice and rats, and humans converge to show a condition of brain mitochondrial dysfunction with two simultaneous conditions: impaired electron transfer and enzymatic activity and mitochondrial oxidative damage. Brain dysfunctional mitochondria accumulate in aged rodents and show marked decreases in the rates of active (+ADP) respiration, especially with malate–glutamate as substrate, marked decreases in the rates of electron transfer in complexes I and IV, decreased membrane potential, increased content of the oxidation products of phospholipids and proteins, and increased size and fragility. In humans, data is starting to accumulate and shows about the same general picture dominated by complex I inactivation, with the corresponding impairment of mitochondrial respiration in the presence of NAD-linked substrates and the always present increased content of the oxidation products of phospholipids and proteins. Interestingly, complex I and IV activities are consistently reported decreased in aged rats and mice. However in human neurodegenerative diseases the observations are consistent with complex I inactivation but not with complex IV inactivation. Both complexes are large molecules that are composed by a series of polypeptides and that are embedded in the phospholipid bilayer of the mitochondrial inner membrane. In both cases, phospholipids are necessary for enzyme activity; this can be easily understood as due to the hydrophobic bonding that holds together enzyme polypeptides and membrane phospholipids. Subtle differences in biological species phospholipid composition may account for the difference in complex IV sensitivity to aging in rats and mice and the complex IV insensitivity to neurodegenerative diseases in humans.
The oxidative mitochondrial partial inactivation of complex I accompanied by oxidative damage is a phenomenon, in short named “complex I syndrome,” that is being recognized as characteristic of mammalian brain aging and of human neurodegenerative diseases. Mitochondrial dysfunction has been recognized as a more marked phenomenon, in terms of complex I syndrome, in brain areas as hippocampus and frontal cortex in rats, human cortex in Parkinson’s disease and dementia with Lewy bodies, and substantia nigra in Parkinson disease. The molecular mechanism involved in complex I inactivation is likely accounted by the synergistic effects of ONOO−, by reactions with the free radical intermediates of the lipid peroxidation process, and by amine–aldehyde adduction reactions.
The accumulation of mitochondrial oxidation products in rodent aging and in neurodegenerative diseases prompted the idea of antioxidant therapies. High doses of vitamin E showed a highly significant protection of complex I activity and mitochondrial function, with improvement of neurological functions and with an increased median life span in mice. Other antioxidants showed similar effects. Interestingly a whole family of mitochondria-targeted antioxidants, as the Skulachev cations (derivatives of the phosphonium cation that covalently attached to either vitamin E, or ubiquinone or PBN) and the SS tetrapeptides, are under development and testing. These molecules are negatively charged and accumulate in mitochondria where they exert their antioxidant effects.
Activation of the cellular mechanisms that regulate mitochondrial biogenesis is another potential therapeutic strategy, considering that de novo generated mitochondria are devoid of oxidation products and show full enzymatic activity and full capacity for ATP production. Then, stimulation of mitochondrial biogenesis is a novel approach with a potential of beneficial effects in neurodegenerative diseases that are associated with mitochondrial dysfunction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by grants of Ministerio de Ciencia e Innovación (SAF2008-03690), by Plan Andaluz de Investigación 2007-2008 (CTS-194) and by Ministerio de Asuntos Exteriores y Cooperación AECI (A/010977/07) in Spain and by grants UBACYT B027, CONICET PIP 6320 and ANPCYT PICT 38326 in Argentina.
References
Beckman, K. B., and Ames, B. N. (1998). The free radical theory of aging matures. Physiol. Rev. 78, 547–581.
Bickford, P. C., Shukitt-Hale, B., and Joseph, J. (1999). Effects of aging on cerebellar noradrenergic function and motor learning: nutritional interventions. Mech. Ageing Dev. 111, 141–154.
Bjelakovic, G., Nikolova, D., Gluud, L. L., Simonetti, R. G., and Gluud, C. (2007). Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA 297, 842–857.
Bougria, M., Vitorica, J., Cano, J., and Machado, A. (1995). Implication of dopamine transporter system on 1-methyl-4-phenylpyridinium and rotenone effect in striatal synaptosomes. Eur. J. Pharmacol. 291, 407–415.
Boveris, A., and Cadenas, E. (2000). Mitochondrial production of hydrogen peroxide regulation by nitric oxide and the role of ubisemiquinone. IUBMB Life 50, 245–250.
Boveris, A., and Chance, B. (1973). The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 134, 707–716.
Boveris, A., and Navarro, A. (2008a). Brain mitochondrial dysfunction in aging. IUBMB Life 60, 308–314.
Boveris, A., and Navarro, A. (2008b). Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free Radic. Biol. Med. 44, 224–229.
Boveris, A., and Stoppani, A. O. (1971). Inhibition of mitochondrial swelling by 19-nor-ethynyltestosterone acetate and other steroids. Arch. Biochem. Biophys. 142, 150–156.
Boveris, A., Valdez, L. B., Zaobornyj, T., and Bustamante, J. (2006). Mitochondrial metabolic states regulate nitric oxide and hydrogen peroxide diffusion to the cytosol. Biochim. Biophys. Acta 1757, 535–542.
Brown, G. C., and Borutaite, V. (2004). Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 1658, 44–49.
Carreras, M. C., Franco, M. C., Peralta, J. G., and Poderoso, J. J. (2004). Nitric oxide, complex I, and the modulation of mitochondrial reactive species in biology and disease. Mol. Aspects Med. 25, 125–139.
Cassarino, D. S., Halvorsen, E. M., Swerdlow, R. H., Abramova, N. N., Parker, W. D. Jr., Sturgill, T. W., and Bennett, J. P. Jr. (2000). Interaction among mitochondria, mitogen-activated protein kinases, and nuclear factor-kappaB in cellular models of Parkinson’s disease. J. Neurochem. 74, 1384–1392.
Chagnon, P., Betard, C., Robitaille, Y., Cholette, A., and Gauvreau, D. (1995). Distribution of brain cytochrome oxidase activity in various neurodegenerative diseases. Neuroreport 6, 711–715.
Chance, B., Sies, H., and Boveris, A. (1979). Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 59, 527–605.
Chowanadisai, W., Bauerly, K. A., Tchaparian, E., Wong, A., Cortopassi, G. A., and Rucker, R. B. (2010). Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J. Biol. Chem. 285, 142–152.
Cocco, T., Sgobbo, P., Clemente, M., Lopriore, B., Grattagliano, I., Di, P. M., and Villani, G. (2005). Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic. Biol. Med. 38, 796–805.
Dawson, T. M., and Dawson, V. L. (2003). Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302, 819–822.
Dennis, J., and Bennett, J. P. Jr. (2003). Interactions among nitric oxide and Bcl-family proteins after MPP+ exposure of SH-SY5Y neural cells I: MPP+ increases mitochondrial NO and Bax protein. J. Neurosci. Res. 72, 76–88.
Elfering, S. L., Sarkela, T. M., and Giulivi, C. (2002). Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem. 277, 38079–38086.
Eve, D. J., Nisbet, A. P., Kingsbury, A. E., Hewson, E. L., Daniel, S. E., Lees, A. J., Marsden, C. D., and Foster, O. J. (1998). Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 63, 62–71.
Fand, I., and McNally, W. P. (1981). Whole-body localization of 14C-tocopheryl acetate in the rat following oral administration. Arch. Int. Pharmacodyn. Ther. 250, 4–17.
Gatto, E. M., Riobo, N. A., Carreras, M. C., Chernavsky, A., Rubio, A., Satz, M. L., and Poderoso, J. J. (2000). Overexpression of neutrophil neuronal nitric oxide synthase in Parkinson’s disease. Nitric Oxide 4, 534–539.
Gerschman, R., Gilbert, D. L., Nye, S. W., Dwyer, P., and Fenn, W. O. (1954). Oxygen poisoning and x-irradiation: a mechanism in common. Science 119, 623–626.
Ghafourifar, P., and Richter, C. (1997). Nitric oxide synthase activity in mitochondria. FEBS Lett. 418, 291–296.
Gilad, G. M., and Gilad, V. H. (1995). Strain, stress, neurodegeneration and longevity. Mech. Ageing Dev. 78, 75–83.
Giulivi, C., Poderoso, J. J., and Boveris, A. (1998). Production of nitric oxide by mitochondria. J. Biol. Chem. 273, 11038–11043.
Gomez, C., Bandez, M. J., and Navarro, A. (2007). Pesticides and impairment of mitochondrial function in relation with the parkinsonian syndrome. Front. Biosci. 12, 1079–1093.
Gonzalez-Flecha, B., Cutrin, J. C., and Boveris, A. (1993). Time course and mechanism of oxidative stress and tissue damage in rat liver subjected to in vivo ischemia-reperfusion. J. Clin. Invest. 91, 456–464.
Good, P. F., Hsu, A., Werner, P., Perl, D. P., and Olanow, C. W. (1998). Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 57, 338–342.
Hagen, T. M., Ingersoll, R. T., Wehr, C. M., Lykkesfeldt, J., Vinarsky, V., Bartholomew, J. C., Song, M. H. and Ames, B. N. (1998). Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc. Natl. Acad. Sci. U.S.A. 95, 9562–9566.
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300.
Harman, D. (2006). Free radical theory of aging: an update: increasing the functional life span. Ann. N. Y. Acad. Sci. 1067, 10–21.
Hathcock, J. N., Azzi, A., Blumberg, J., Bray, T., Dickinson, A., Frei, B., Jialal, I., Johnston, C. S., Kelly, F. J., Kraemer, K., Packer, L., Parthasarathy, S., Sies, H. and Traber, M.G. (2005). Vitamins E and C are safe across a broad range of intakes. Am. J. Clin. Nutr. 81, 736–745.
Hensley, K., Kotake, Y., Sang, H., Pye, Q. N., Wallis, G. L., Kolker, L. M., Tabatabaie, T., Stewart, C. A., Konishi, Y., Nakae, D. and Floyd, R. A. (2000). Dietary choline restriction causes complex I dysfunction and increased H(2)O(2) generation in liver mitochondria. Carcinogenesis 21, 983–989.
Hughes, G., Murphy, M. P., and Ledgerwood, E. C. (2005). Mitochondrial reactive oxygen species regulate the temporal activation of nuclear factor kappaB to modulate tumour necrosis factor-induced apoptosis: evidence from mitochondria-targeted antioxidants. Biochem. J. 389, 83–89.
James, A. M., Sharpley, M. S., Manas, A. R., Frerman, F. E., Hirst, J., Smith, R. A., and Murphy, M. P. (2007). Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 282, 14708–14718.
Jauslin, M. L., Meier, T., Smith, R. A., and Murphy, M. P. (2003). Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 17, 1972–1974.
Kanai, A. J., Pearce, L. L., Clemens, P. R., Birder, L. A., VanBibber, M. M., Choi, S. Y., de Groat, W. C. and Peterson, J. (2001). Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. U.S.A. 98, 14126–14131.
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., Smith, R. A. and Murphy, M. P. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596.
Lam, P. Y., Yin, F., Hamilton, R. T., Boveris, A., and Cadenas, E. (2009). Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radic. Res. 43, 431–439.
Liu, J., Head, E., Gharib, A. M., Yuan, W., Ingersoll, R. T., Hagen, T. M., Cotman, C. W. and Ames, B. N. (2002). Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc. Natl. Acad. Sci. U.S.A. 99, 2356–2361.
Liu, Q., Raina, A. K., Smith, M. A., Sayre, L. M., and Perry, G. (2003). Hydroxynonenal, toxic carbonyls, and Alzheimer disease. Mol. Aspects Med. 24, 305–313.
Mann, V. M., Cooper, J. M., Krige, D., Daniel, S. E., Schapira, A. H., and Marsden, C. D. (1992). Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain 115 (Pt 2), 333–342.
Mattiasson, G., and Sullivan, P. G. (2006). The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid. Redox Signal. 8, 1–38.
McNamara, P., Durso, R., and Harris, E. (2007). “Machiavellianism” and frontal dysfunction: evidence from Parkinson’s disease. Cogn. Neuropsychiatry 12, 285–300.
Mizuno, Y., Ohta, S., Tanaka, M., Takamiya, S., Suzuki, K., Sato, T., Oya, H., Ozawa, T. and Kagawa, Y. (1989). Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 163, 1450–1455.
Mugesh, G., du Mont, W. W., and Sies, H. (2001). Chemistry of biologically important synthetic organoselenium compounds. Chem. Rev. 101, 2125–2179.
Murphy, M. P., Echtay, K. S., Blaikie, F. H., Asin-Cayuela, J., Cocheme, H. M., Green, K., Buckingham, J. A, Taylor, E. R., Hurrell, F., Hughes, G., Miwa, S., Cooper, C. E., Svistunenko, D. A., Smith, R. A., and Brand, M. D. (2003). Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: studies using a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone. J. Biol. Chem. 278, 48534–48545.
Murphy, M. P., and Smith, R. A. (2007). Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 47, 629–656.
Navarro, A., and Boveris, A. (2004). Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1244–R1249.
Navarro, A., and Boveris, A. (2007a). Brain mitochondrial dysfunction in aging: conditions that improve survival, neurological performance and mitochondrial function. Front. Biosci. 12, 1154–1163.
Navarro, A., and Boveris, A. (2007b). The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 292, C670–C686.
Navarro, A., and Boveris, A. (2008). Mitochondrial nitric oxide synthase, mitochondrial brain dysfunction in aging, and mitochondria-targeted antioxidants. Adv. Drug Deliv. Rev. 60, 1534–1544.
Navarro, A., and Boveris, A. (2009). Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J. Bioenerg. Biomembr. 41, 517–521.
Navarro, A., Boveris, A., Bandez, M. J., Sanchez-Pino, M. J., Gomez, C., Muntane, G., et al. (2009). Human brain cortex: mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radic. Biol. Med. 46, 1574–1580.
Navarro, A., Gomez, C., Lopez-Cepero, J. M., and Boveris, A. (2004). Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R505–R511.
Navarro, A., Gomez, C., Sanchez-Pino, M. J., Gonzalez, H., Bandez, M. J., Boveris, A. D., and Boveris, A. (2005a). Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1392–R1399.
Navarro, A., Torrejon, R., Bandez, M. J., Lopez-Cepero, J. M., and Boveris, A. (2005b). Mitochondrial function and mitochondria-induced apoptosis in an overstimulated rat ovarian cycle. Am. J. Physiol. Endocrinol. Metab. 289, E1101–E1109.
Navarro, A., Lopez-Cepero, J. M., Bandez, M. J., Sanchez-Pino, M. J., Gomez, C., Cadenas, E., Boveris, A. (2008). Hippocampal mitochondrial dysfunction in rat aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R501–R509.
Navarro, A., Sanchez Del Pino, M. J., Gomez, C., Peralta, J. L., and Boveris, A. (2002). Behavioral dysfunction, brain oxidative stress, and impaired mitochondrial electron transfer in aging mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R985–R992.
Navarro, A., Sanchez-Pino, M. J., Gomez, C., Bandez, M. J., Cadenas, E., and Boveris, A. (2007). Dietary thioproline decreases spontaneous food intake and increases survival and neurological function in mice. Antioxid. Redox Signal. 9, 131–141.
Nicoletti, V. G., Marino, V. M., Cuppari, C., Licciardello, D., Patti, D., Purrello, V. S., and Stella, A. M. (2005). Effect of antioxidant diets on mitochondrial gene expression in rat brain during aging. Neurochem. Res. 30, 737–752.
Nisoli, E., Falcone, S., Tonello, C., Cozzi, V., Palomba, L., Fiorani, M., Pisconti, A., Brunelli, S., Cardile, A., Francolini, M., Cantoni, O., Carruba, M.O., Moncada, S. and Clementi, E. (2004). Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A. 101, 16507–16512.
Poeggeler, B., Durand, G., Polidori, A., Pappolla, M. A., Vega-Naredo, I., Coto-Montes, A., Boker, J., Hardeland, R. and Pucci, B. (2005). Mitochondrial medicine: neuroprotection and life extension by the new amphiphilic nitrone LPBNAH acting as a highly potent antioxidant agent. J. Neurochem. 95, 962–973.
Raha, S., and Robinson, B. H. (2000). Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 25, 502–508.
Reddy, P. H. (2006). Mitochondrial oxidative damage in aging and Alzheimer’s disease: implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 1–13.
Reddy, P. H. (2007). Mitochondrial dysfunction in aging and Alzheimer’s disease: strategies to protect neurons. Antioxid. Redox Signal. 9, 1647–1658.
Riobo, N. A., Clementi, E., Melani, M., Boveris, A., Cadenas, E., Moncada, S., and Poderoso, J. J. (2001). Nitric oxide inhibits mitochondrial NADH: ubiquinone reductase activity through peroxynitrite formation. Biochem. J. 359, 139–145.
Riobo, N. A., Schopfer, F. J., Boveris, A. D., Cadenas, E., and Poderoso, J. J. (2002). The reaction of nitric oxide with 6-hydroxydopamine: implications for Parkinson’s disease. Free Radic. Biol. Med. 32, 115–121.
Sastre, J., Millan, A., Garcia-de-la-Asuncion, J., Pla, R., Juan, G., Pallardo, F. O'Connor, E., Martin, J. A., Droy-Lefaix, M. T. and Vina, J. (1998). A Ginkgo biloba extract (EGb 761) prevents mitochondrial aging by protecting against oxidative stress. Free Radic. Biol. Med. 24, 298–304.
Schapira, A. H. (2008). Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 7, 97–109.
Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990a). Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827.
Schapira, A. H., Mann, V. M., Cooper, J. M., Dexter, D., Daniel, S. E., Jenner, P., Clark, J. B. and Marsden, C. D. (1990b). Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 55, 2142–2145.
Serra, J. A., Dominguez, R. O., Marschoff, E. R., Guareschi, E. M., Famulari, A. L., and Boveris, A. (2009). Systemic oxidative stress associated with the neurological diseases of aging. Neurochem. Res. 34, 2122–2132.
Sheu, S. S., Nauduri, D., and Anders, M. W. (2006). Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim. Biophys. Acta 1762, 256–265.
Skulachev, V. P. (2005). How to clean the dirtiest place in the cell: cationic antioxidants as intramitochondrial ROS scavengers. IUBMB Life 57, 305–310.
Skulachev, V. P. (2007). A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry (Mosc) 72, 1385–1396.
Tolmasoff, J. M., Ono, T., and Cutler, R. G. (1980). Superoxide dismutase: correlation with life-span and specific metabolic rate in primate species. Proc. Natl. Acad. Sci. U.S.A. 77, 2777–2781.
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344.
Valdez, L. B., Alvarez, S., Lores-Arnaiz, S., Schopfer, F., and Boveris, A. (2000). Intramitochondrial reactions of peroxynitrite. Free Radic. Biol. Med. 29, 349–356.
Valdez, L. B., Zaobornyj, T., and Boveris, A. (2006). Mitochondrial metabolic states and membrane potential modulate mtNOS activity. Biochim. Biophys. Acta 1757, 166–172.
Vatassery, G. T., Adityanjee, A., Quach, H. T., Smith, W. E., Kuskowski, M. A., and Melnyk, D. (2004). Alpha and gamma tocopherols in cerebrospinal fluid and serum from older, male, human subjects. J. Am. Coll. Nutr. 23, 233–238.
Vina, J., Sastre, J., Pallardo, F., and Borras, C. (2003). Mitochondrial theory of aging: importance to explain why females live longer than males. Antioxid. Redox Signal. 5, 549–556.
Vitorica, J., Clark, A., Machado, A., and Satrustegui, J. (1985). Impairment of glutamate uptake and absence of alterations in the energy-transducing ability of old rat brain mitochondria. Mech. Ageing Dev. 29, 255–266.
Walker, J. E. (1992). The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q. Rev. Biophys. 25, 253–324.
Zandi, P. P., Anthony, J. C., Khachaturian, A. S., Stone, S. V., Gustafson, D., Tschanz, J. T., Norton, M. C., Welsh-Bohmer, K. A. and Breitner, J. C. (2004). Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch. Neurol. 61, 82–88.
Zhao, G. M., Qian, X., Schiller, P. W., and Szeto, H. H. (2003a). Comparison of [Dmt1]DALDA and DAMGO in binding and G protein activation at mu, delta, and kappa opioid receptors. J. Pharmacol. Exp. Ther. 307, 947–954.
Zhao, K., Luo, G., Zhao, G. M., Schiller, P. W., and Szeto, H. H. (2003b). Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J. Pharmacol. Exp. Ther. 304, 425–432.
Zhao, K., Luo, G., Giannelli, S., and Szeto, H. H. (2005). Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem. Pharmacol. 70, 1796–1806.
Keywords: complex I syndrome, vitamin E, antioxidant therapy, mitochondria-targeted antioxidants
Citation: Navarro A and Boveris A (2010) Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front. Ag. Neurosci. 2:34. doi: 10.3389/fnagi.2010.00034
Received: 04 January 2010;
Paper pending published: 23 January 2010;
Accepted: 14 July 2010;
Published online: 01 September 2010.
Edited by:
Paula I. Moreira, University of Coimbra, PortugalReviewed by:
Enrique Cadenas, University of Southern California, USAAna Rego, University of Coimbra, Portugal
James Bennett, Virginia Commonwealth University, USA
Copyright: © 2010 Navarro and Boveris. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ana Navarro, Departamento de Bioquímica y Biología Molecular, Facultad de Medicina, University of Cádiz, Plaza Fragela 9, 11003 Cádiz, Spain. e-mail:YW5hLm5hdmFycm9AdWNhLmVz