 Sebastián Bernales1
Sebastián Bernales1 Marisol Morales Soto1,2 and Emma McCullagh1*
Marisol Morales Soto1,2 and Emma McCullagh1*- 1Fundación Ciencia y Vida, Santiago, Chile
- 2Facultad de Ciencias Biológicas, Universidad Andrés Bello, Santiago, Chile
Protein-folding occurs in several intracellular locations including the endoplasmic reticulum and mitochondria. In normal conditions there is a balance between the levels of unfolded proteins and protein folding machinery. Disruption of homeostasis and an accumulation of unfolded proteins trigger stress responses, or unfolded protein responses (UPR), in these organelles. These pathways signal to increase the folding capacity, inhibit protein import or expression, increase protein degradation, and potentially trigger cell death. Many aging-related neurodegenerative diseases involve the accumulation of misfolded proteins in both the endoplasmic reticulum and mitochondria. The exact participation of the UPRs in the onset of neurodegeneration is unclear, but there is significant evidence for the alteration of these pathways in the endoplasmic reticulum and mitochondria. Here we will discuss the involvement of endoplasmic reticulum and mitochondrial stress and the possible contributions of the UPR in these organelles to the development of two neurodegenerative diseases, Parkinson's disease (PD) and Alzheimer's disease (AD).
Introduction
Proper protein folding is fundamental for cells to carry out normal functions and survive. The endoplasmic reticulum (ER) is a specialized organelle that folds and modifies secretory and integral membrane proteins. Mitochondria (MT) also contain machinery to fold proteins encoded by the mitochondrial genome or refold proteins imported into MT from the cytoplasm. Disruption of protein folding homeostasis in these two organelles results in the accumulation of misfolded proteins and induces stress response pathways termed the unfolded protein responses (ER-UPR and MT-UPR).
The loss of protein folding homeostasis and the intracellular deposition of aggregated proteins are pathologies common to numerous neurodegenerative diseases. Insoluble protein aggregates have been detected in PD and AD, among others. It remains unknown whether these protein aggregates are the cause of cytotoxicity and disease or the by-product of a diseased state. There is evidence, however, that unfolded protein response (UPR) pathways in the ER and in MT are either upregulated or abrogated in many of these diseases. This raises the possibility that the accumulation of unfolded proteins triggers stress-response pathways that induce the neurotoxicity and cell death exhibited in brains of afflicted patients. In this review we examine the ER- and MT-UPRs and the evidence linking unfolded protein stress in these organelles to neurodegenerative disease.
Misfolded Protein Stress in the Endoplasmic Reticulum
Protein Folding in the ER
Most secreted and plasma membrane proteins fold and mature in the ER. Polypeptides are recognized in the cytoplasm by an N-terminal signal peptide and are co-translationally imported into the ER through the translocon pore, the SEC61 (secretory 61) complex (Rapoport, 2007). The majority of proteins that pass through the ER are met on the luminal side of the ER by molecular chaperones and protein-modifying enzymes that fold polypeptides modified by disulfide bond formation and append chemical groups such as glycans to produce fully mature proteins (Braakman and Bulleid, 2011). The highly conserved ER-resident heat shock protein 70 (HSP70) family member BIP (binding immunoglobulin protein, also called 78 kDa glucose-regulated protein GRP78) is an essential protein that is thought to interact with most nascent polypeptides as they enter the ER (Schroder and Kaufman, 2005). BIP is aided in its protein folding functions by various co-chaperones including the ER-localized J-proteins (ERDJ1-5) that transfer newly imported polypeptides from the translocon to BIP. While BIP does not actively fold proteins, it is thought to maintain peptides in a folding-competent state by binding and preventing aggregation during the folding process (Gething, 1999; Schroder and Kaufman, 2005). Protein disulfide isomerase (PDI) catalyzes and aids in disulfide bond formation and chaperones such as the calcium-dependent calnexin and calreticulin promote folding of glycosylated proteins (Braakman and Bulleid, 2011).
Cells utilize several mechanisms to resolve aberrant folding or modification of peptides in the ER. Protein folding appears to be a kinetic process that is in constant competition with a degradation pathway, or ER-associated degradation (ERAD) (Trombetta and Parodi, 2003; Meusser et al., 2005; Braakman and Bulleid, 2011). Protein misfolding affects the affinity of peptides for chaperones and retards the folding process. Thus, a misfolded protein may be re-folded by ER chaperones or, if caught in a misfolded state for too long, may be degraded via ERAD. The exact mechanisms by which proteins are targeted to the ERAD pathway remain unclear, but misfolded peptides are retrotranslocated across the ER membrane by protein complexes including the Sec61 translocon. Once in the cytoplasm, ERAD targets are met by ubiquitin conjugating enzymes and are degraded by the proteasome machinery, thus linking the activity of the ER to that of the ubiquitin-proteasome system (UPS). Consistent with this, it has been found that inhibiting the UPS with general proteasome inhibitors or mutated proteins can cause the accumulation of unfolded proteins in the ER (Bush et al., 1997; Lee et al., 2003).
The ER-UPR
Protein folding in the ER is sensitive to various conditions including the redox state of the environment, calcium concentrations, and ATP levels (Figure 1) (Schroder and Kaufman, 2005). Changes in these environmental conditions or the presence of a chemical ER stressor such as tunicamycin, which inhibits protein glycosylation, cause unfolded proteins to accumulate in the ER and trigger the ER-UPR. In general, the ER-UPR is activated when there is a loss in protein folding homeostasis and the quantity of unfolded proteins outweighs the folding capacity of the ER. The ER-UPR transmits the signal of unfolded protein stress from the ER to the nucleus in an attempt to regain homeostasis by increasing levels of protein folding and degradation machinery while inhibiting general translation (Schroder and Kaufman, 2005). Under unmitigated ER stress, the ER-UPR can activate apoptosis (Tabas and Ron, 2011).
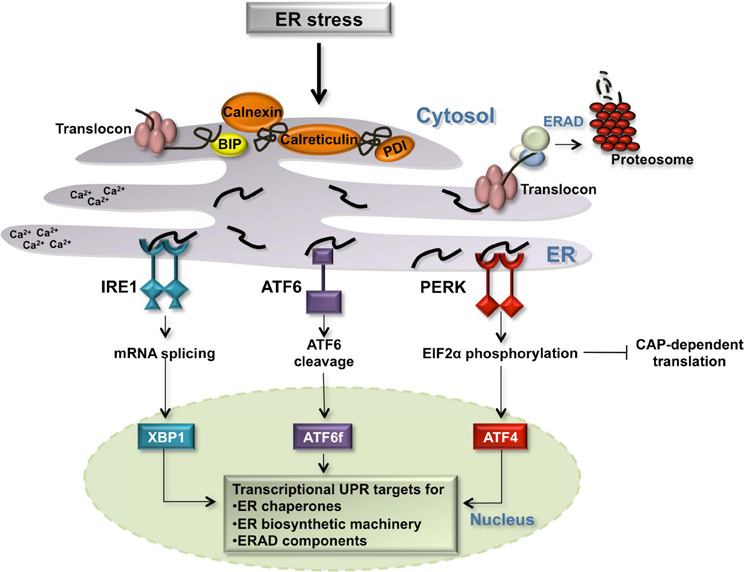
Figure 1. The Unfolded Protein Response in the ER. Several environment alterations including changes in the cellular redox state, calcium concentration, ATP concentration, and inhibition of protein glycosylation affect the protein folding capacity of the ER. Three transmembrane ER receptors, IRE1, ATF6, and PERK, sense unfolded proteins in the lumen of the ER and promote the transcription of genes whose protein products increase the folding capacity of the cell: ER chaperones [BIP, calnexin, protein disulfide isomerase (PDI), and calreticulin], ER biosynthetic machinery, and components of the ERAD degradation pathway.
Three transmembrane proteins have been identified as sensors of unfolded proteins in the ER in mammalian cells: IRE1 (inositol-requiring protein 1), ATF6 (activating transcription factor 6) and PERK (protein kinase RNA-like ER kinase) (Figure 1) (Schroder and Kaufman, 2005; Kohno, 2007, 2010). IRE1 and PERK contain highly homologous ER luminal unfolded protein-sensing domains and divergent cytosolic kinase domains. The cytosolic domain of IRE1 is comprised of an endoribonuclease domain that, when IRE1 is active, participates in the non-canonical splicing of cytosolic XBP1 (X-binding protein 1) RNA (Sidrauski and Walter, 1997; Calfon et al., 2002). Spliced XBP1 mRNA is translated into an active transcription factor (Yoshida et al., 2001). Active PERK phosphorylates the alpha subunit of eukaryotic initiation translation factor 2 (EIF2alpha) promoting an inhibition of general translation and thereby reducing the protein burden on the ER. Under these conditions, CAP-independent transcripts, including the transcription factor ATF4, are preferentially translated (Ron and Walter, 2007). Upon detection of unfolded proteins, the third sensor ATF6 travels from the ER membrane to the Golgi apparatus where it is cleaved by two proteases, S1P (site 1 protease) and S2P (site 2 protease), releasing the cytosolic ATF6 fragment, ATF6f (Figure 1). ATF6f is an active transcription factor that shuttles to the nucleus to upregulate the expression of target genes. The three transcription factors activated by the ER sensors, XBP1, ATF6f, and ATF4, individually and combinatorially stimulate the expression of ER chaperones, ER biosynthetic machinery, ERAD components, and apoptotic signals in order to resolve ER stress (Figure 1).
The ER-UPR Induces Apoptosis
In the event of chronic or unmitigated ER stress, the ER-UPR triggers apoptosis by various means (Figure 2). One mechanism involves the cleavage of the ER membrane associated caspase 12 initiating a cascade of caspase proteolysis that promotes apoptosis (Szegezdi et al., 2003; Martinez et al., 2010; Tabas and Ron, 2011). Cleavage of caspase 12 is caused by the release of calcium from the ER, which under normal conditions serves as a reservoir for cellular calcium. Signaling through the PERK branch of the UPR also promotes apoptosis. The transcription factor ATF4 induces the expression of a second transcription factor, CHOP (C/EBP-homologous protein). CHOP inhibits the expression of anti-apoptotic BCL 2 family proteins and activates the transcription of pro-apoptotic BCL 2 family members (Figure 2). While the exact mechanism is unclear, this transcriptional regulation of BCL 2 family members precipitates the post-translational activation of two ER-localized BCL 2 family members, BAK and BAX (Kim et al., 2009). Upon activation, these proteins re-localize to MT, which leads to caspase activation and the release of pro-apoptotic signals such as cytochrome c. Signaling through the IRE1 branch of the ER-UPR can induce apoptosis through the association of IRE1 with TNF-receptor-associated factor 2 (TRAF2) and apoptosis-signal-regulating kinase (ASK1) under ER stress conditions (Tabas and Ron, 2011). ASK1 enhances the pro-apoptotic activity of BCL 2 family members through JNK signaling, while TRAF2 induces the clustering and cleavage of caspase 12 (Yoneda et al., 2001).
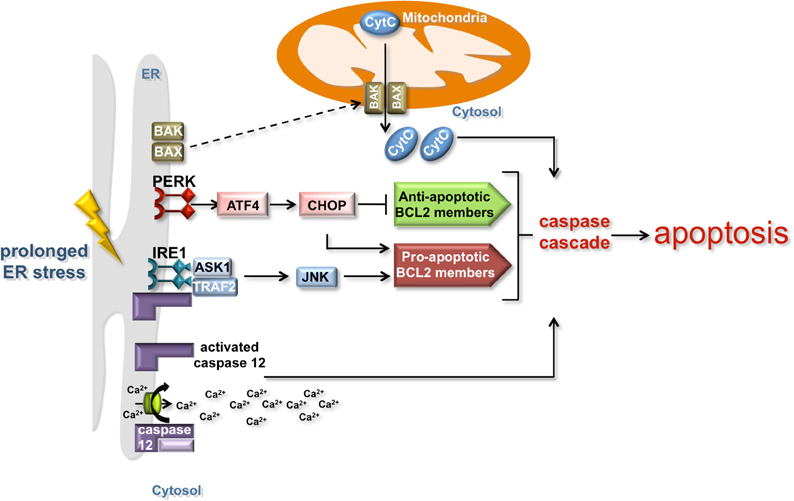
Figure 2. The induction of apoptosis by the ER and mitochondria. Prolonged ER stress triggers apoptosis by mitochondria-dependent and independent mechanisms. IRE1 and PERK signaling can induce apoptosis in a mitochondria-dependent manner by activating the expression of anti-apoptotic BCL 2 family proteins (green box) and inhibiting the expression of pro-apoptotic BCL 2 family members (red box). Although the mechanism is unclear, the regulation of BCL 2 family members precipitates the post-translational activation of the ER-localized BCL 2 family members BAK and BAX (dashed line), which re-localize to the mitochondria and lead to caspase activation and the release of pro-apoptotic signals such as cytochrome c. The release of calcium from the ER induces the cleavage of the ER membrane-associated caspase 12 and the activation of the caspase cascade that promotes apoptosis in a mitochondria-independent manner.
The ER-UPR is responsible for promoting cell survival by increasing the folding capacity of the cell or for inducing cell death depending on the type of stress. It is unclear how this balance between life and death is controlled, but the regulation of IRE1 binding partners may play an important role. The kinetics of IRE1, ATF6, and PERK activation and inactivation are also thought to affect the life and death output. IRE1 inactivation, as determined by the diminution of XBP1 splicing, occurs on a much shorter timescale than the inactivation of ATF6 or PERK, as measured by the reduction in ATF6f and the attenuation of PERK phosphorylation (Lin et al., 2007). This evokes a model in which prolonged stress induces apoptosis through ATF6 and PERK. However, understanding the totality of the ER-UPR requires simultaneous investigation of all three branches under specific stress conditions.
The ER-UPR and Disease
Due to the importance of protein folding for cellular function, it is not surprising that the UPR is an essential pathway. It is equally unsurprising the disruption or malfunction of the ER-UPR may contribute to the onset of many diseases. Pancreatic β-cells, for example, require a large folding and secretory capacity to produce hormones and, therefore, depend on the ER-UPR to maintain ER homeostasis (Lin et al., 2008). Increasing evidence suggests that chronic ER stress in these cells leads to the development of diabetes mellitus (Lin et al., 2008). Several types of cancer including multiple myeloma are thought to depend on the ER-UPR for survival (Lin et al., 2008; Michallet et al., 2011). In such cases disrupting the ER-UPR could preferentially kill cancer cells while causing little injury to normal tissue. Efforts are currently underway to develop ER-UPR inhibitors to test this hypothesis. Finally, numerous neurodegenerative diseases are thought to involve protein misfolding and misregulation of the ER-UPR. The contributions of ER stress in the pathogenesis of the two most common neurodegenerative diseases, AD and PD, will be discussed below in the section "Misfolded proteins and neurodegeneration".
Misfolded Protein Stress in Mitochondria
Protein Import and Folding in Mitochondria
Mammalian MT contain 1000–1500 proteins, 13 of which are encoded by the endogenous genome. These 13 proteins are expressed and folded by mitochondrial machinery and function as integral subunits of the electron transport chain. The remaining mitochondrially localized proteins are nuclearly encoded and are imported from the cytoplasm (Milenkovic et al., 2007). Mitchondrially bound proteins are often recognized by import machinery by a signal sequence. After translocation through the outer membrane, polypeptides are delivered to their appropriate destination and the signal sequences are cleaved. Other mitochondrial proteins that do not contain such pre-sequences are instead recognized by other markers such as internal targeting sequences, structure, or conserved cysteine motifs and are imported by an array of protein complexes (Chacinska et al., 2009; Mokranjac and Neupert, 2009).
Translocation into MT requires that proteins fully or partially unfold prior to import (Eilers et al., 1988; Lithgow, 2000). Imported proteins are refolded by resident mitochondrial molecular chaperones such as HSP70 and HSP60 (heat shock protein 60) upon entry (Manning-Krieg et al., 1991; Hartl et al., 1992; Deocaris et al., 2006). Mitochondrial HSP70 interacts directly with incoming peptides on the matrix side of the inner membrane and drives protein import with its ATPase activity. While both mitochondrial HSP70 and HSP60 have roles in refolding newly imported proteins, they have been shown recently to play additional roles in MT quality control (Raza et al., 2002; Zhao et al., 2002; Yoneda et al., 2004).
Similarly to the ER, MT encounter stresses that can cause the accumulation of misfolded or damaged proteins leading to mitochondrial dysfunction. Such stress is reported to occur during mitochondrial biogenesis when the accelerated input of nuclear-encoded proteins may exceed the folding capacity of MT (Tsang and Lemire, 2002; Benedetti et al., 2006; Durieux et al., 2011). Misregulation of mitochondrial gene expression through the mutation or loss of mitochondrial DNA (mtDNA) can also result in the accumulation of unfolded proteins (Martinus et al., 1996; Yoneda et al., 2004). Finally, the interior environment of MT is highly oxidative and mtDNA and mitochondrially localized proteins can accrue oxidative damage over time. MT have several mechanisms to combat the stress of damaged or unfolded proteins and contain an array of molecular chaperones and proteases that refold or degrade unfolded proteins. One such mechanism that was described recently is the MT-UPR.
The MT-UPR
The MT-UPR (Figure 3) was first discovered in mammalian cells using a system in which a mutant of the mitochondrially localized enzyme ornithine transcarbamylase (OTC-Δ) was over-expressed (Zhao et al., 2002). This mutant is prone to misfold and aggregate in MT. The mechanisms by which misfolded proteins are detected in MT and how this signal is transmitted to the nucleus remain unclear but accumulation of OTC-Δ was found to elicit the transcriptional upregulation of several genes including that of the mitochondrial chaperone HSP60 and the matrix-localized AAA protease CLPP (Zhao et al., 2002). Further studies revealed that OTC-Δ overexpression results in the upregulation of double-stranded RNA activated protein kinase (PKR) expression, phosphorylation of EIF2alpha (Rath et al., 2011), and the phosphorylation of c-JUN (Horibe and Hoogenraad, 2007). Phosphorylated cJUN activates the transcription of CHOP and C/EBPβ, which in turn activate the transcription of MT-UPR responsive genes such as CLPP, HSP60, and the mitochondrial chaperone MPPβ (Zhao et al., 2002; Horibe and Hoogenraad, 2007). Interestingly, MT-UPR responsive genes also contain two other conserved elements, MURE1 and MURE2 (mitochondrial unfolded protein response elements 1 and 2), but the proteins, if any, that bind to these regions are yet to be discovered (Aldridge et al., 2007).
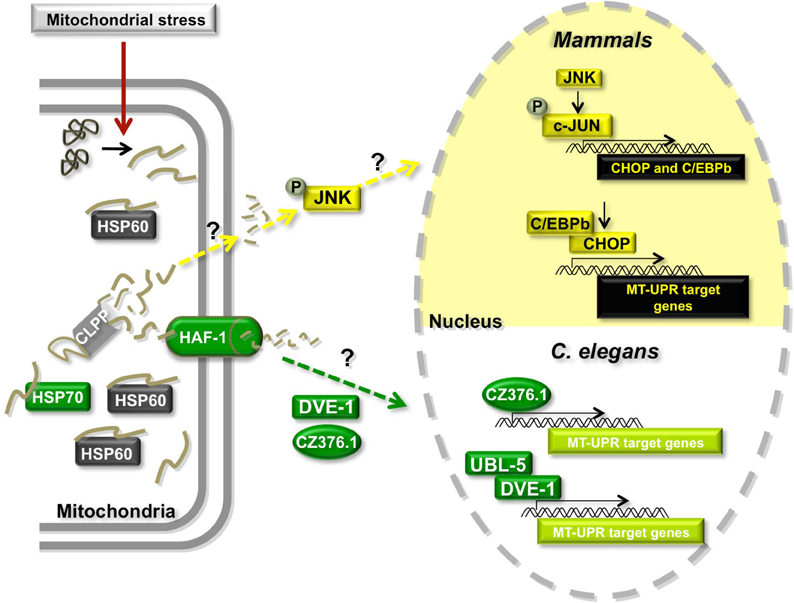
Figure 3. Model of the mitochondrial Unfolded Protein Response (MT-UPR). Misregulation of import of nuclearly encoded mitochondrial proteins, misregulation of mitochondrial gene expression or damage of mtDNA or mitochondrially localized proteins can cause the accumulation of misfolded or damaged proteins leading to mitochondrial stress. The MT-UPR pathway is thought to restore protein folding homeostasis in MT. This pathway has been described in mammalian cells and in C. elegans. Proteins shown in gray are shared between the two models. Proteins in yellow are unique to the mammalian model and proteins in green have only been described in C. elegans.
A similar pathway was shown to exist in C. elegans (Figure 3). Initial studies showed that chronic ethidium bromide treatment to remove mtDNA and induce the accumulation of unfolded proteins in MT resulted in the upregulation of mitochondrial HSP60 expression (Yoneda et al., 2004). Unlike the mammalian system, mitochondrial HSP70 (HSP-6 in worms) is also transcriptionally activated under these conditions. Using a genome-wide RNAi screening approach in worms that contain a construct of GFP driven by the MT-UPR responsive hsp-60 promoter, authors uncovered several proteins that appear to be involved in the pathway, including the worm CLPP ortholog CLPP-1 (Haynes et al., 2007). Additionally, knocking down the expression of ubiquitin-like protein 5 (UBL-5) (Benedetti et al., 2006), an ABC transporter HAF-1 (Haynes et al., 2010), the homeobox DNA-binding protein DVE-1 (Haynes et al., 2007) and a leucine zipper protein ZC376.7 (Haynes et al., 2007) increased the expression of the GFP reporter. The authors proposed a model in which the protease CLPP-1 degrades misfolded proteins to small peptides, which are transported into the cytoplasm through the HAF-1 transporter triggering the relocalization of DVE-1, UBL-5, and ZC376.7 to the nucleus (Figure 3) (Haynes and Ron, 2010). DVE-1 and UBL-5 are known to form a complex and activate the transcription of MT-UPR target genes. ZC376.7 is also a transcription factor, but it is unknown whether it binds to the DVE-1/UBL5 complex. Intriguingly, mammalian cells contain a HAF-1 ortholog (ABCB10) in the inner membrane of the MT, as well as orthologs of UBL-5 (UBL5) and DVE-1 (SATB2) that form a complex in vivo (Haynes et al., 2007). It remains to be seen whether these orthologs play similar roles in the mammalian MT-UPR.
The intermembrane space (IMS)-localized mitochondrial protease HTRA2/OMI has also been reported to be involved in clearing unfolded proteins from the IMS of MT in mammalian cells (Radke et al., 2008; Moisoi et al., 2009; Papa and Germain, 2011). Knocking out HTRA2/OMI in mice leads to the aggregation of misfolded electron transport chain proteins in the mitochondrial membrane (Moisoi et al., 2009). HTRA2/OMI was also found to target misfolded endonuclease-G (endoG) in the IMS (Radke et al., 2008; Papa and Germain, 2011). The absence of HTRA2/OMI results in increased CHOP expression and mitochondrial dysfunction suggesting that it is required for cell survival (Moisoi et al., 2009).
Although many components participating in the MT-UPR in mammalian cells and C. elegans have been identified, crucial details about the pathway remain unknown. The mechanisms by which unfolded proteins in MT are detected and recruit proteases for degradation have not been determined. It is also unclear how mitochondrial peptides in the cytoplasm are sensed and induce the nuclear relocalization of transcription factors. Finally, although MT are critical for regulating apoptosis and controlling cell life and death decisions, it is not well understood whether the MT-UPR can induce apoptosis like the ER-UPR. However, several lines of evidence suggest a connection. First, the accumulation of unfolded proteins in MT upregulates the expression of CHOP, which is important for inducing apoptosis by the ER-UPR. It is possible that CHOP plays a similar role in the MT-UPR. Second, mice containing a proofreading-deficient mtDNA polymerase g, which results in increased levels of mtDNA mutations and is thought to induce the MT-UPR, express high levels of apoptotic markers (Kujoth et al., 2005). Third, recent studies of human tumor cells indicate a relationship between folding environment of the MT and apoptosis. In these studies, a mitochondrially targeted small molecule heat shock protein 90 (HSP90) inhibitor was used to disrupt protein folding specifically in MT (Kang et al., 2009; Siegelin et al., 2011). Treatment with this compound led to the induction of classic apoptotic markers and reduced cell viability (Siegelin et al., 2011). While these data suggest an intriguing link between protein folding in MT and the induction of apoptosis, further investigation is required to determine the exact involvement of the MT-UPR.
Misfolded Proteins and Neurodegeneration
Protein misfolding has long been implicated in neurodegeneration and the accumulation of protein aggregates has been described in the pathology of numerous neurodegenerative diseases. For example, insoluble deposits of [alpha]-synuclein (α-syn) are found in neurons of patients with Parkinson's disease (PD). Extracellular and intracellular plaques of amyloid beta (Aβ) protein are archetypal indicators of Alzheimer's disease (AD). The presence of protein aggregates is also associated with Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), and others. While the contribution of misfolded proteins to these diseases remains controversial, studies of familial disease cases have revealed that mutations in proteins found in aggregates, as well as those involved in clearing unfolded proteins from the ER and MT, are linked to disease incidence (Lindholm et al., 2006; Reddy, 2009; Morais and de Strooper, 2010; Matus et al., 2011). This suggests that protein misfolding contributes significantly to the onset of some neurodegenerative diseases. Here we will review data linking unfolded protein stress in the ER and in MT to the onset or progression of PD and AD.
Parkinson's Disease
PD is characterized by the selective degeneration of dopaminergic neurons in the substantia nigra pars compacta region of the brainstem (Olanow and Tatton, 1999; Schapira, 2006) and the disease is accompanied by the accumulation within neurons of protein aggregates termed Lewy bodies. These insoluble inclusions are comprised of several proteins including α-syn (Spillantini et al., 1997; Baba et al., 1998). The wild type function of α-syn is not well understood, but it is thought to participate in presynaptic vesicle formation (Chandra et al., 2005). Genetic studies of PD have uncovered mutations in α-syn itself that result in the over-expression or misfolding of this major Lewy body component (Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003; Zarranz et al., 2004; Simon-Sanchez et al., 2009). While the toxicity of Lewy bodies and their contribution to the onset and progression of PD is controversial, data from brains of PD patients as well as animal and cellular models suggest that these aggregates induce cellular stress responses and can induce apoptosis (Dawson and Dawson, 2003).
PD and ER stress
Studies of PD reveal evidence that the ER is subject to unfolded protein stress and suggest the involvement of ER-UPR signaling in neuronal death (Figure 4). Aggregates of α-syn have been localized to several cellular compartments including the ER and have been found to induce ER stress (Bellucci et al., 2011). Interestingly, over-expression of α-syn in yeast was found to block vesicle trafficking from the ER to the Golgi (Cooper et al., 2006). In the same study, co-overexpression of α-syn with proteins involved in ER-to-Golgi vesicle fusion such as Rab guanosine triphosphatases (GTPase), was found to suppress the trafficking defect. Similar results were found when the mammalian homolog of one of these GTPases, Rab1, was expressed in an α-syn over-expression model of PD in mammalian DA neurons (Cooper et al., 2006). This study suggests that the aggregation of α-syn in the ER inhibits a step of ER-to-Golgi trafficking likely leading to the accumulation of proteins in the ER and ER stress. Interestingly α-syn misfolding has also been reported to inhibit the UPS thereby leading to unfolded protein stress in the ER (Cook and Petrucelli, 2009).
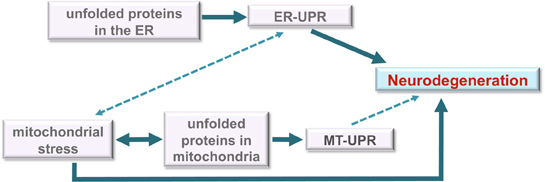
Figure 4. Model for roles of unfolded protein stress in neurodegeneration. Under unmitigated stress, as may be the case in many neurodegenerative diseases, the ER-UPR can induce cell death. The disruption or malfunction of the ER-UPR may also contribute to the development of important pathologies like neurodegenerative diseases. Although the data is unclear, the ER-UPR may induce mitochondrial stress (dashed line) through calcium signaling or other means. Mitochondrial stress is well known to induce apoptosis and contribute to neurodegeneration. We propose a model in which the MT-UPR stimulated by protein misfolding that is characteristic of neurodegenerative diseases may also play a role in cell death (dashed line).
Studies of familial PD lend further support to the presence of unfolded protein stress in the ER of affected cells. The cytosolic protein parkin, a RING-finger containing E3 ubiquitin ligase, has been proposed to function in the ERAD pathway by targeting recently retrotranslocated misfolded ER proteins to the proteasome (Imai et al., 2000; Meacham et al., 2001). Like many ERAD components, parkin is transcriptionally regulated by the ER-UPR, specifically by the transcription factor ATF4 (Bouman et al., 2011). Mutations in the two RING domains of this protein are associated with the majority of cases of recessive familial PD (Matsumine et al., 1997; Kitada et al., 1998). Many of these mutations abolish the E3 ligase activity of parkin leading to the accumulation of parkin targets in the ER and cytoplasm, suggesting that this accumulation may be responsible for ER stress and neuronal degeneration. Interestingly, parkin is thought to bind and ubiquitinate α-syn in neurons (Shimura et al., 2001). Other ERAD/UPS components may play roles in PD. Mutations in ubiquitin carboxy-terminal hydrolase 1 (UCHL1) were reported to be associated with the incidence of PD in one family, but this remains controversial (Dauer and Przedborski, 2003; Schapira, 2006).
PD and the ER-UPR
The presence of unfolded protein stress in the ER suggests that the ER-UPR should be activated in cells affected by PD. Indeed, components of this pathway have been found to be activated in post-mortem brain samples and experimental models of PD. Levels of phosphorylated PERK (generally thought to be the activated form) and phosphorylated EIF2alpha were increased in the substantia nigra region of PD patients (Hoozemans et al., 2007). Recent data shows that BIP directly binds to α-syn in several cell-based α-syn over-expression models of PD (Bellucci et al., 2011). Additionally, the levels of BIP, as well as downstream ER-UPR signaling molecules such as ATF4 and phosphorylated EIF2alpha, are increased in such models (Smith et al., 2005; Bellucci et al., 2011). Chemically induced cell and animal models of PD also reveal activation of ER-UPR proteins such as the phosphorylation of PERK and IRE1 and the upregulation of BIP, ATF4, and CHOP (Holtz and O'Malley, 2003; Ryu et al., 2005; Silva et al., 2005).
While it is apparent that the ER-UPR is affected in PD, the connection between the ER-UPR and neuronal cell death in PD is not clear. The opposing functions of this pathway in maintaining cell survival and inducing cell death mean its effects in disease are difficult to parse. The chronic activation of the ER-UPR could induce cell death in affected neurons in PD patients or this pathway could be upregulated in an attempt to alleviate unfolded protein stress in the ER and promote cell survival. However, significant evidence lends support for the direct involvement of the ER-UPR in neuronal degeneration in PD. Chemical mimetic induction of PD in cell or animal models induces not only the ER-UPR but also apoptosis. Under these conditions, release of cytochrome c and activation of caspases have been found to coincide with the activation of CHOP, a key regulator of ER-UPR-induced apoptosis (Figure 2) (Holtz and O'Malley, 2003; Ryu et al., 2005; Silva et al., 2005). Recent data reveals that the activation of the ER-UPR and the initiation of apoptosis coincide in an α-syn over-expression model of PD (Bellucci et al., 2011). Finally, CHOP expression levels are increased in post-mortem brains of some PD patients (Moisoi et al., 2009). Together these data suggest that the ER-UPR could play an important role in the degeneration of neurons during disease progression (Figure 4).
PD and mitochondrial stress
Mitochondrial stress and damage are classic symptoms of PD and it is widely accepted that they contribute to the pathogenesis of this disease (Figure 4). Complex I of the electron transport chain in the membrane of MT has been found to be dysfunctional exclusively in neurons of the substantia nigra in post-mortem brains of PD patients (Mann et al., 1994; Keeney et al., 2006). Chemicals such as rotenone and MPP+, the active metabolite of MPTP, which directly bind to and inhibit the activity of complex I (Ramsay et al., 1991; Mao et al., 2012), mimic many PD symptoms in animal and cellular models of this disease. Reflecting the damage to MT and complex I, reactive oxygen species (ROS) are over-produced and oxidative stress has been detected in samples from PD patients. Interestingly, while mitochondrial damaging agents and gene mutations should affect most cells in the body, only neurons in the substantia nigra and muscle cells are affected in PD. Both neurons and muscle cells are significant energy consumers and, therefore, depend heavily on MT for survival (Chen and Chan, 2006). This could partially account for the sensitivity of these particular cells to mitochondrial insults.
Studies of familial PD reveal further evidence of mitochondrial stress playing an important role in disease progression. Mutations in the mitochondrial serine/threonine kinase PTEN-induced putative kinase 1 (PINK1) correlate with disease incidence (Valente et al., 2004a). Many of these mutations cluster in the kinase domain of PINK1 and result in a non-functional protein (Hatano et al., 2004; Rohe et al., 2004; Valente et al., 2004a, b; Sha et al., 2010). Deficiencies of pink1 in Drosophila lead to changes in mitochondrial morphology, increased sensitivity to oxidative stress and reduced viability (Clark et al., 2006). Additionally, over-expression of this kinase decreases the sensitivity of cells to apoptotic stresses (Shavali et al., 2008) suggesting that PINK1 has anti-apoptotic/pro-survival functions. PINK1 has been shown to interact with parkin, which localizes to MT as well as the cytoplasm. Although the mechanism has not been fully elucidated, these proteins are thought to participate together in regulating mitochondrial dynamics and quality control (Pridgeon et al., 2007; Vives-Bauza et al., 2010).
PD and the MT-UPR
Increasing evidence of the involvement of mitochondrial stress, mitochondrial protein misfolding, and mitochondrial chaperones in PD indicate a role of the MT-UPR in this disease. Misfolded proteins such as α-syn have been demonstrated to localize to and accumulate in MT both in cell models of PD and in post-mortem brains of PD patients (Devi et al., 2008; Shavali et al., 2008). It is unclear whether this localization reflects a wild-type function of α-syn or if it is aberrant and indicates cellular damage. The association of α-syn with MT triggers Ca2+ uptake by this organelle and oxidative damage (Parihar et al., 2008), suggesting that the accumulation of this protein can induce mitochondrial dysfunction and apoptosis. PINK1 has been shown to interact with several proteins involved with the MT-UPR and mitochondrial quality control including TRAP1 (Pridgeon et al., 2007), HSP60 (Rakovic et al., 2011) and HTRA2/OMI (Plun-Favreau et al., 2007) suggesting that misregulation of PINK1 could generally affect protein folding in MT. Indeed, it was shown recently that levels of misfolded mitochondrial respiratory components were increased in post-mortem brains of PD patients carrying mutant alleles of PINK1 (Pimenta de Castro et al., 2012). Correspondingly, the levels of mitochondrial HSP60 were also upregulated in these samples.
The mitochondrial protease OMI/HTRA2 also links the MT-UPR to PD incidence and neurodegeneration. OMI/HTRA2 degrades misfolded proteins in the IMS and its activity is regulated by phosphorylation by PINK1 (Plun-Favreau et al., 2007). OMI/HTRA2 deficient mice display increased levels of aggregated mitochondrial respiratory proteins (Moisoi et al., 2009) and parkinsonian-like symptoms including heightened sensitivity to mitochondrial stress, increased ROS and cell death (Martins et al., 2004). The lack of OMI/HTRA2 also resulted in a brain-specific upregulation in the apoptosis regulator CHOP (Moisoi et al., 2009). Finally, loss-of-function mutations in OMI/HTRA2 have been associated with PD incidence in a human population (Strauss et al., 2005). Interestingly mutations in the mitochondrial folding chaperone HSP70 also associate with PD and expression of these mutant alleles affect mitochondrial function in cell models (Burbulla et al., 2010).
It is difficult to prove whether the MT-UPR directly causes neurodegeneration in PD. However, the significance of MT in regulating apoptosis and the data described above suggest a crucial link (Figure 4). Further investigation of the involvement of other MT-UPR components could aid to dissect the exact involvement of the MT-UPR in neurodegeneration and PD. It would be interesting to determine whether CLPP and other MT-UPR regulated genes are misexpressed during PD progression. Finally, further genetic studies could shed light on whether mutations in other MT-UPR genes correlate with PD incidence.
Alzheimer's Disease
AD is characterized by the extracellular and intracellular deposition of protein aggregates and neuronal death. These pathologies are accompanied by loss of memory and dementia. Several hypotheses have been proposed to explain the prominent neuronal degeneration in AD. One hypothesis suggests that the hallmark extracellular plaques of Aβ protein seen in brains of AD patients are toxic and cause neuronal degeneration. However, Aβ is constitutively secreted from neurons in brains of seemingly healthy patients (Haass et al., 1992; Seubert et al., 1992; Shoji et al., 1992). Other theories propose that intracellular aggregates of Aβ or the microtubule-associated tau protein are the toxic species that induce cell death. While the underlying causes of neurodegeneration are unclear, neurons of AD patients display oxidative damage, misregulation of calcium signaling, and mitochondrial dysfunction in addition to protein aggregation, suggesting that ER and mitochondrial stress may be involved in the cell death (Figure 4) (Selkoe, 2011).
AD and ER stress
The presence of intracellular Aβ and tau deposits in neurons of AD patients raises the possibility that ER stress is a key factor in the progression of this disease. Aβ is a 37–43 amino acid proteolytic product of amyloid precursor protein (APP) (Reddy et al., 2010; Haass et al., 2012), a transmembrane protein that is folded and processed in the ER and is thought to participate in synapse formation in neurons (Priller et al., 2006). APP is proteolized by β- and γ-secretases to release Aβ fragments, but the functions of these proteolytic products are unknown. Studies of Aβ localization in neurons of AD patients and in mouse and cellular models of AD reveal that in addition to the extracellular deposits, Aβ oligomers localize to intracellular compartments including the ER (Gouras et al., 2000; Fernandez-Vizarra et al., 2004). It has been suggested that these aggregates may cause ER stress and induce cell death. Consistent with this, studies of familial AD have revealed disease causing mutations in APP that localize to the β and γ cleavage sites (Selkoe, 2011). These mutations affect APP processing and cause increased production of Aβ (primarily Aβ40− and Aβ42 peptides) in the ER (Mao and Reddy, 2011; Selkoe, 2011; Umeda et al., 2011).
AD and the ER-UPR
The involvement of the ER-UPR in the onset or progression of AD appears to be complex. However, evidence suggests that this pathway could be important for inducing neurotoxicity and cell death (Figure 4). Consistent with Aβ aggregation in the ER, levels of UPR components are generally upregulated in post-mortem brain tissue from AD patients (Hoozemans et al., 2005). Specifically, levels of PERK, phosphorylated PERK, and BIP were found to be increased (Hoozemans et al., 2005). In a cell model of AD, over-expression of mutant APP that produces large amounts of Aβ sensitized cells to ER stress and ER stress-mediated toxicity (Chafekar et al., 2008). However, IRE1 activity and BIP expression have been found to be reduced in models of AD employing mutant alleles of presenilin 1 (PS1) and presenilin 2 (PS2) that are associated with familial AD (Katayama et al., 1999; Terro et al., 2002). PS1 is a transmembrane protein located in the ER and is a component of the γ-secretase complex that cleaves APP to produce Aβ. In cell models expressing mutant alleles of PS1, IRE1 induction, as measured through BIP levels, has slower kinetics and is weaker (Katayama et al., 2001) while levels of Aβ are increased (Duff et al., 1996). BIP is known to bind APP in the ER of healthy cells as this transmembrane protein passes through the secretory pathway on its way to neuronal synapses (Yang et al., 1998). Interestingly, the binding of BIP to APP limits the production of the toxic Aβ, perhaps explaining the excess of Aβ in cells with lower BIP expression. Finally, neurons expressing mutant PS1 protein are more sensitive to ER stress and undergo apoptosis at lower doses of ER-stress agents than do wild type neurons (Guo et al., 1997). This could be due to the decreased activity of IRE1 or the reduced levels of BIP and suggests that the ER-UPR could be responsible for neurotoxicity in AD. In agreement with this idea, over-expression of Aβ is less toxic in mice lacking caspase 12 (Nakagawa et al., 2000), one of the caspases activated during ER-UPR induced apoptosis (Figure 2).
The misregulation of Ca2+ signaling is another hallmark of AD that suggests that the involvement of the ER-UPR in this disease. Evidence from patients and experimental models indicate that Ca2+ regulation is altered in early stages of AD. Aβ oligomers have been found to trigger the release of Ca2+ from the ER (Mattson et al., 1992; Ferreiro et al., 2004). Mutations in PS1 have also been shown to cause misregulated calcium signaling. PC-1 cells carrying a mutant allele of PS1 associated with AD released significantly more calcium into the cytoplasm than did control cells when they were treated with agonists that induce Ca2+ release (Keller et al., 1998). Furthermore, cells expressing mutant PS1 were hypersensitive to apoptosis, which was blocked by the addition of calcium chelators suggesting that apoptosis was induced by high cytosolic calcium levels (Keller et al., 1998). The majority of cellular Ca2+ is stored in the ER and its release activates the ER-UPR, in part because some ER folding chaperones are calcium-dependent. Fluctuations in levels of cytosolic Ca2+ trigger its uptake by MT and induce apoptosis. While it is unclear whether Ca2+ misregulation is responsible for neurotoxicity seen in AD, it suggests a potential link between the ER, MT, and induction of apoptosis.
AD and mitochondrial stress
Mitochondrial damage as detected by the dysfunction of complex I and increased oxidative stress are well-known characteristics of AD. However, the underlying causes of this damage and its affects on neurodegeneration are not fully understood. Aggregates of Aβ have been localized to MT (Caspersen et al., 2005; Manczak et al., 2006; Mao and Reddy, 2011; Umeda et al., 2011), but it is unclear if this is the toxic agent because it has been suggested that oxidative damage precedes this accumulation during AD onset (Yao et al., 2009; Chang et al., 2011). On the other hand, over-expression of Aβ in cell and mouse models of AD causes oxidative damage, inhibits the activity of complex I and induces cell death (Keil et al., 2004; Caspersen et al., 2005; Manczak et al., 2006; Reddy et al., 2010). A recent study uncovered evidence suggesting a direct link between Aβ accumulation in MT and apoptosis (Lustbader et al., 2004). Aβ-binding alcohol dyhydrogenase (ABAD) is a mitochondrial matrix protein that binds and whose function is inhibited by Aβ. Expression of ABAD has been found to be increased in damaged neurons in brains of patients with AD. In this study, authors detected the binding of Aβ to ABAD in a mice over-expressing APP, the Aβ precursor and in brain tissue of AD patients (Lustbader et al., 2004). Inhibition of this binding ameliorated the oxidative damage, mitochondrial toxicity and cell death in neurons expressing Aβ peptide (Lustbader et al., 2004) and in a mouse model (Yao et al., 2011). Furthermore, similar results were obtained when ABAD was over-expressed in mice that also over-express APP. These data provide a connection between the accumulation of Aβ aggregates in MT and cell death and suggest a mode of cytotoxicity in AD.
AD and the MT-UPR
Despite the evidence that mitochondrial stress plays a role in precipitating neurotoxicity in AD, little is known about the involvement of the MT-UPR in this process (Figure 4). It was recently shown that the expression of the mitochondrial protease OMI/HTRA2 is decreased in post-mortem brains of AD patients (Westerlund et al., 2011). The specific activity of this enzyme was found to be increased in the same samples. The same study uncovered a weak association between a mutation in OMI/HTRA2 (A141S) and AD. It will be interesting to determine if this mutation is found in other populations and whether the association strengthens with the inclusion of more samples. OMI/HTRA2 interacts with Aβ (Park et al., 2004) and was shown to retard Aβ aggregation in vitro (Kooistra et al., 2009) suggesting that mutations in OMI/HTRA2 could play a role AD. We suggest that other components of the MT-UPR such as mitochondrial chaperones and proteases could be misregulated or be involved in the progression of this disease. It would be intriguing to determine, for example, whether over-expression of mitochondrial HSP60 affects the deposition of Aβ in MT and cell toxicity. Such studies could provide novel therapeutic targets for neurodegenerative diseases.
Summary
Protein misfolding and aggregation as well as the upregulation of the ER-UPR and mitochondrial dysfunction are classic etiologies of many neurodegenerative diseases suggesting a general role for UPRs in inducing cell death in these diseases. This notion is supported by studies of hereditary spastic paraplegia (SPG), a degenerative disease of the upper motor neurons involved in voluntary movement. The incidence of this disease is associated with mutations in the mitochondrial HSP60 and protease SPG7 (Haynes and Ron, 2010). The expression of CLPP has been found to be reduced in some cases of SPG (Hansen et al., 2008). The ER-UPR is also reported to be upregulated in cases of ALS (Doyle et al., 2011), a fatal neurodegenerative disease that affects primarily motor neurons and is associated with the accumulation of aggregates of ubiquitin, a mutant form of superoxide dismutase 1 (mSOD1) and the ER chaperone, PDI. Despite the evidence of the involvement of unfolded protein stress response pathways in the ER and MT in neurodegeneration, it remains unclear whether these pathways are responsible for the cell death associated with these diseases or are upregulated as a consequence of other toxic effects.
The involvement of the ER-UPR in life-death decisions has been well documented. It is thought that early activation of the ER-UPR triggers pro-survival signals such as folding chaperones to increase the folding capacity of the cell. Chronic activation of this pathway, however, is thought to generate pro-apoptotic signals through the PERK branch and induce cell death. It is not well understood how these two opposing forces are balanced. Unlike the ER-UPR, the connection between the MT-UPR and induction of apoptosis is not known and further investigation is needed to determine whether saturation of the MT-UPR can induce cell death in a manner similar to that of the ER-UPR. We propose, however, that evidence suggests that protein misfolding in MT in neurodegenerative diseases could participate in inducing neurodegeneration. Establishing a firm connection between the neurotoxicity typical of neurodegenerative diseases such as AD and PD and the activation of the cellular unfolded protein stress responses including the MT-UPR could prove invaluable for developing novel therapeutics to treat these diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mónica Rodrigo-Brenni, Ph.D., Iván Alfaro, Ph.D., and Marc Shuman, M.D. for critically reading the manuscript and Gonzalo Ureta for his judicious and artistic review of the figures. This review was supported by FONDECYT 1080449 and Conicyt Programa PFB-16.
References
Aldridge, J. E., Horibe, T., and Hoogenraad, N. J. (2007). Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2:e874. doi: 10.1371/journal.pone.0000874
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., Trojanowski, J. Q., and Iwatsubo, T. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Bellucci, A., Navarria, L., Zaltieri, M., Falarti, E., Bodei, S., Sigala, S., Battistin, L., Spillantini, M., Missale, C., and Spano, P. (2011). Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson's disease. J. Neurochem. 116, 588–605.
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P., and Ron, D. (2006). Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229–239.
Bouman, L., Schlierf, A., Lutz, A. K., Shan, J., Deinlein, A., Kast, J., Galehdar, Z., Palmisano, V., Patenge, N., Berg, D., Gasser, T., Augustin, R., Trumbach, D., Irrcher, I., Park, D. S., Wurst, W., Kilberg, M. S., Tatzelt, J., and Winklhofer, K. F. (2011). Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 18, 769–782.
Braakman, I., and Bulleid, N. J. (2011). Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99.
Burbulla, L. F., Schelling, C., Kato, H., Rapaport, D., Woitalla, D., Schiesling, C., Schulte, C., Sharma, M., Illig, T., Bauer, P., Jung, S., Nordheim, A., Schols, L., Riess, O., and Kruger, R. (2010). Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease: functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 19, 4437–4452.
Bush, K. T., Goldberg, A. L., and Nigam, S. K. (1997). Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J. Biol. Chem. 272, 9086–9092.
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., Clark, S. G., and Ron, D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96.
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., Xu, H. W., Stern, D., Mckhann, G., and Yan, S. D. (2005). Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 19, 2040–2041.
Chacinska, A., Koehler, C. M., Milenkovic, D., Lithgow, T., and Pfanner, N. (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644.
Chafekar, S. M., Zwart, R., Veerhuis, R., Vanderstichele, H., Baas, F., and Scheper, W. (2008). Increased Abeta1-42 production sensitizes neuroblastoma cells for ER stress toxicity. Curr. Alzheimer Res. 5, 469–474.
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O. M., and Sudhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396.
Chang, J., Siedlak, S., Moreira, P., Nunomura, A., Castellani, R. J., Smith, M. A., Zhu, X., Perry, G., Casadesus, G., Basu, S., and Wiklund, L. (2011). “Oxidative stress in Alzheimer's disease: a critical appraisal of the causes and the consequences”, in Studies on Experimental Models, eds S. Basu, and L. Wiklund (New York, Dordrecht, Heidelberg, London: Humana Press), 211–220.
Chen, H., and Chan, D. C. (2006). Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Biol. 18, 453–459.
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., Yoo, S. J., Hay, B. A., and Guo, M. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166.
Cook, C., and Petrucelli, L. (2009). A critical evaluation of the ubiquitin-proteasome system in Parkinson's disease. Biochim. Biophys. Acta 1792, 664–675.
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., Liu, K., Xu, K., Strathearn, K. E., Liu, F., Cao, S., Caldwell, K. A., Caldwell, G. A., Marsischky, G., Kolodner, R. D., Labaer, J., Rochet, J. C., Bonini, N. M., and Lindquist, S. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328.
Dauer, W., and Przedborski, S. (2003). Parkinson's disease: mechanisms and models. Neuron 39, 889–909.
Dawson, T. M., and Dawson, V. L. (2003). Molecular pathways of neurodegeneration in Parkinson's disease. Science 302, 819–822.
Deocaris, C. C., Kaul, S. C., and Wadhwa, R. (2006). On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones 11, 116–128.
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100.
Doyle, K. M., Kennedy, D., Gorman, A. M., Gupta, S., Healy, S. J., and Samali, A. (2011). Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 15, 2025–2039.
Duff, K., Eckman, C., Zehr, C., Yu, X., Prada, C. M., Perez-Tur, J., Hutton, M., Buee, L., Harigaya, Y., Yager, D., Morgan, D., Gordon, M. N., Holcomb, L., Refolo, L., Zenk, B., Hardy, J., and Younkin, S. (1996). Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713.
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91.
Eilers, M., Hwang, S., and Schatz, G. (1988). Unfolding and refolding of a purified precursor protein during import into isolated mitochondria. EMBO J. 7, 1139–1145.
Fernandez-Vizarra, P., Fernandez, A. P., Castro-Blanco, S., Serrano, J., Bentura, M. L., Martinez-Murillo, R., Martinez, A., and Rodrigo, J. (2004). Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer's disease. Histol. Histopathol. 19, 823–844.
Ferreiro, E., Oliveira, C. R., and Pereira, C. (2004). Involvement of endoplasmic reticulum Ca2C release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J. Neurosci. Res. 76– 872–880.
Gething, M. J. (1999). Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 10, 465–472.
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., Greenfield, J. P., Haroutunian, V., Buxbaum, J. D., Xu, H., Greengard, P., and Relkin, N. R. (2000). Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 156, 15–20.
Guo, Q., Sopher, B. L., Furukawa, K., Pham, D. G., Robinson, N., Martin, G. M., and Mattson, M. P. (1997). Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J. Neurosci. 17, 4212–4222.
Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. [Epub ahead of print].
Haass, C., Schlossmacher, M. G., Hung, A. Y., Vigo-Pelfrey, C., Mellon, A., Ostaszewski, B. L., Lieberburg, I., Koo, E. H., Schenk, D., Teplow, D. B., and Selkoe, D. J. (1992). Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325.
Hansen, J., Corydon, T. J., Palmfeldt, J., Durr, A., Fontaine, B., Nielsen, M. N., Christensen, J. H., Gregersen, N., and Bross, P. (2008). Decreased expression of the mitochondrial matrix proteases Lon and ClpP in cells from a patient with hereditary spastic paraplegia (SPG13). Neuroscience 153, 474–482.
Hartl, F. U., Martin, J., and Neupert, W. (1992). Protein folding in the cell: the role of molecular chaperones Hsp70 and Hsp60. Annu. Rev. Biophys. Biomol. Struct. 21, 293–322.
Hatano, Y., Li, Y., Sato, K., Asakawa, S., Yamamura, Y., Tomiyama, H., Yoshino, H., Asahina, M., Kobayashi, S., Hassin-Baer, S., Lu, C. S., Ng, A. R., Rosales, R. L., Shimizu, N., Toda, T., Mizuno, Y., and Hattori, N. (2004). Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol. 56, 424–427.
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480.
Haynes, C. M., and Ron, D. (2010). The mitochondrial UPR – protecting organelle protein homeostasis. J. Cell Sci. 123, 3849–3855.
Haynes, C. M., Yang, Y., Blais, S. P., Neubert, T. A., and Ron, D. (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529–540.
Holtz, W. A., and O'Malley, K. L. (2003). Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J. Biol. Chem. 278, 19367–19377.
Hoozemans, J. J., van Haastert, E. S., Eikelenboom, P., De Vos, R. A., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson's disease. Biochem. Biophys. Res. Commun. 354, 707–711.
Hoozemans, J. J., Veerhuis, R., van Haastert, E. S., Rozemuller, J. M., Baas, F., Eikelenboom, P., and Scheper, W. (2005). The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol. 110, 165–172.
Horibe, T., and Hoogenraad, N. J. (2007). The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One 2:e835. doi: 10.1371/journal.pone.0000835
Imai, Y., Soda, M., and Takahashi, R. (2000). Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 275, 35661–35664.
Kang, B. H., Plescia, J., Song, H. Y., Meli, M., Colombo, G., Beebe, K., Scroggins, B., Neckers, L., and Altieri, D. C. (2009). Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Invest. 119, 454–464.
Katayama, T., Imaizumi, K., Honda, A., Yoneda, T., Kudo, T., Takeda, M., Mori, K., Rozmahel, R., Fraser, P., George-Hyslop, P. S., and Tohyama, M. (2001). Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer's disease-linked presenilin-1 mutations. J. Biol. Chem. 276, 43446–43454.
Katayama, T., Imaizumi, K., Sato, N., Miyoshi, K., Kudo, T., Hitomi, J., Morihara, T., Yoneda, T., Gomi, F., Mori, Y., Nakano, Y., Takeda, J., Tsuda, T., Itoyama, Y., Murayama, O., Takashima, A., St George-Hyslop, P., Takeda, M., and Tohyama, M. (1999). Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1, 479–485.
Keeney, P. M., Xie, J., Capaldi, R. A., and Bennett, J. P. Jr. (2006). Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 26, 5256–5264.
Keil, U., Bonert, A., Marques, C. A., Scherping, I., Weyermann, J., Strosznajder, J. B., Muller-Spahn, F., Haass, C., Czech, C., Pradier, L., Muller, W. E., and Eckert, A. (2004). Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J. Biol. Chem. 279, 50310–50320.
Keller, J. N., Guo, Q., Holtsberg, F. W., Bruce-Keller, A. J., and Mattson, M. P. (1998). Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J. Neurosci. 18, 4439–4450.
Kim, H., Tu, H. C., Ren, D., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. (2009). Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499.
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y., and Shimizu, N. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608.
Kohno, K. (2007). How transmembrane proteins sense endoplasmic reticulum stress. Antioxid. Redox Signal. 9, 2295–2303.
Kohno, K. (2010). Stress-sensing mechanisms in the unfolded protein response: similarities and differences between yeast and mammals. J. Biochem. 147, 27–33.
Kooistra, J., Milojevic, J., Melacini, G., and Ortega, J. (2009). A new function of human HtrA2 as an amyloid-beta oligomerization inhibitor. J. Alzheimers Dis. 17, 281–294.
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J. T., Schols, L., and Riess, O. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108.
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., Hofer, T., Seo, A. Y., Sullivan, R., Jobling, W. A., Morrow, J. D., van Remmen, H., Sedivy, J. M., Yamasoba, T., Tanokura, M., Weindruch, R., Leeuwenburgh, C., and Prolla, T. A. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484.
Lee, A. H., Iwakoshi, N. N., Anderson, K. C., and Glimcher, L. H. (2003). Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. U.S.A. 100, 9946–9951.
Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., Shokat, K. M., Lavail, M. M., and Walter, P. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949.
Lin, J. H., Walter, P., and Yen, T. S. (2008). Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 3, 399–425.
Lindholm, D., Wootz, H., and Korhonen, L. (2006). ER stress and neurodegenerative diseases. Cell Death Differ. 13, 385–392.
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., Caspersen, C., Chen, X., Pollak, S., Chaney, M., Trinchese, F., Liu, S., Gunn-Moore, F., Lue, L. F., Walker, D. G., Kuppusamy, P., Zewier, Z. L., Arancio, O., Stern, D., Yan, S. S., and Wu, H. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304, 448–452.
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Abeta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449.
Mann, V. M., Cooper, J. M., Daniel, S. E., Srai, K., Jenner, P., Marsden, C. D., and Schapira, A. H. (1994). Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann. Neurol. 36, 876–881.
Manning-Krieg, U. C., Scherer, P. E., and Schatz, G. (1991). Sequential action of mitochondrial chaperones in protein import into the matrix. EMBO J. 10, 3273–3280.
Mao, P., Meshul, C. K., Thuillier, P., Goldberg, N. R., and Reddy, P. H. (2012). CART peptide is a potential endogenous antioxidant and preferentially localized in mitochondria. PLoS One 7:e29343. doi: 10.1371/journal.pone.0029343
Mao, P., and Reddy, P. H. (2011). Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer's disease: implications for early intervention and therapeutics. Biochim. Biophys. Acta 1812, 1359–1370.
Martinez, J. A., Zhang, Z., Svetlov, S. I., Hayes, R. L., Wang, K. K., and Larner, S. F. (2010). Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis 15, 1480–1493.
Martins, L. M., Morrison, A., Klupsch, K., Fedele, V., Moisoi, N., Teismann, P., Abuin, A., Grau, E., Geppert, M., Livi, G. P., Creasy, C. L., Martin, A., Hargreaves, I., Heales, S. J., Okada, H., Brandner, S., Schulz, J. B., Mak, T., and Downward, J. (2004). Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 24, 9848–9862.
Martinus, R. D., Garth, G. P., Webster, T. L., Cartwright, P., Naylor, D. J., Hoj, P. B., and Hoogenraad, N. J. (1996). Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 240, 98–103.
Matsumine, H., Saito, M., Shimoda-Matsubayashi, S., Tanaka, H., Ishikawa, A., Nakagawa-Hattori, Y., Yokochi, M., Kobayashi, T., Igarashi, S., Takano, H., Sanpei, K., Koike, R., Mori, H., Kondo, T., Mizutani, Y., Schaffer, A. A., Yamamura, Y., Nakamura, S., Kuzuhara, S., Tsuji, S., and Mizuno, Y. (1997). Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 60, 588–596.
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., and Rydel, R. E. (1992). beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12, 376–389.
Matus, S., Glimcher, L. H., and Hetz, C. (2011). Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr. Opin. Cell Biol. 23, 239–252.
Meacham, G. C., Patterson, C., Zhang, W., Younger, J. M., and Cyr, D. M. (2001). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105.
Meusser, B., Hirsch, C., Jarosch, E., and Sommer, T. (2005). ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772.
Michallet, A. S., Mondiere, P., Taillardet, M., Leverrier, Y., Genestier, L., and Defrance, T. (2011). Compromising the unfolded protein response induces autophagy-mediated cell death in multiple myeloma cells. PLoS One 6:e25820. doi: 10.1371/journal.pone.0025820
Milenkovic, D., Muller, J., Stojanovski, D., Pfanner, N., and Chacinska, A. (2007). Diverse mechanisms and machineries for import of mitochondrial proteins. Biol. Chem. 388, 891–897.
Moisoi, N., Klupsch, K., Fedele, V., East, P., Sharma, S., Renton, A., Plun-Favreau, H., Edwards, R. E., Teismann, P., Esposti, M. D., Morrison, A. D., Wood, N. W., Downward, J., and Martins, L. M. (2009). Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 16, 449–464.
Mokranjac, D., and Neupert, W. (2009). Thirty years of protein translocation into mitochondria: unexpectedly complex and still puzzling. Biochim. Biophys. Acta 1793, 33–41.
Morais, V. A., and de Strooper, B. (2010). Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J. Alzheimers Dis. 20(Suppl. 20), S255–S263.
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A., and Yuan, J. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403, 98–103.
Olanow, C. W., and Tatton, W. G. (1999). Etiology and pathogenesis of Parkinson's disease. Annu. Rev. Neurosci. 22, 123–144.
Papa, L., and Germain, D. (2011). Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 124, 1396–1402.
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 65, 1272–1284.
Park, H. J., Seong, Y. M., Choi, J. Y., Kang, S., and Rhim, H. (2004). Alzheimer's disease-associated amyloid beta interacts with the human serine protease HtrA2/Omi. Neurosci. Lett. 357, 63–67.
Pimenta de Castro, I., Costa, A. C., Lam, D., Tufi, R., Fedele, V., Moisoi, N., Dinsdale, D., Deas, E., Loh, S. H., and Martins, L. M. (2012). Genetic analysis of mitochondrial protein misfolding in Drosophila melanogaster. Cell Death Differ. [Epub ahead of print].
Plun-Favreau, H., Klupsch, K., Moisoi, N., Gandhi, S., Kjaer, S., Frith, D., Harvey, K., Deas, E., Harvey, R. J., McDonald, N., Wood, N. W., Martins, L. M., and Downward, J. (2007). The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat. Cell Biol. 9, 1243–1252.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C., Di Iorio, G., Golbe, L. I., and Nussbaum, R. L. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047.
Pridgeon, J. W., Olzmann, J. A., Chin, L. S., and Li, L. (2007). PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5:e172. doi: 10.1371/journal.pbio.0050172
Priller, C., Bauer, T., Mitteregger, G., Krebs, B., Kretzschmar, H. A., and Herms, J. (2006). Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 26, 7212–7221.
Radke, S., Chander, H., Schafer, P., Meiss, G., Kruger, R., Schulz, J. B., and Germain, D. (2008). Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 283, 12681–12685.
Rakovic, A., Grunewald, A., Voges, L., Hofmann, S., Orolicki, S., Lohmann, K., and Klein, C. (2011). PINK1-Interacting proteins: proteomic analysis of overexpressed PINK1. Parkinsons Dis. 2011, 153979.
Ramsay, R. R., Krueger, M. J., Youngster, S. K., Gluck, M. R., Casida, J. E., and Singer, T. P. (1991). Interaction of 1-methyl-4-phenylpyridinium ion (MPPC) and its analogs with the rotenone/piericidin binding site of NADH dehydrogenase. J. Neurochem. 56, 1184–1190.
Rapoport, T. A. (2007). Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669.
Rath, E., Berger, E., Messlik, A., Nunes, T., Liu, B., Kim, S. C., Hoogenraad, N., Sans, M., Sartor, R. B., and Haller, D. (2011). Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. [Epub ahead of print].
Raza, H., Robin, M. A., Fang, J. K., and Avadhani, N. G. (2002). Multiple isoforms of mitochondrial glutathione S-transferases and their differential induction under oxidative stress. Biochem. J. 366, 45–55.
Reddy, P. H. (2009). Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer's disease. CNS Spectr. 14, 8–13; discussion 16–18.
Reddy, P. H., Manczak, M., Mao, P., Calkins, M. J., Reddy, A. P., and Shirendeb, U. (2010). Amyloid-beta and mitochondria in aging and Alzheimer's disease: implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 20(Suppl. 2), S499–S512.
Rohe, C. F., Montagna, P., Breedveld, G., Cortelli, P., Oostra, B. A., and Bonifati, V. (2004). Homozygous PINK1 C-terminus mutation causing early-onset parkinsonism. Ann. Neurol. 56, 427–431.
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529.
Ryu, E. J., Angelastro, J. M., and Greene, L. A. (2005). Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol. Dis. 18, 54–74.
Schroder, M., and Kaufman, R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789.
Selkoe, D. J. (2011). Alzheimer's disease. Cold Spring Harb. Perspect. Biol. 3. doi: 10.1101/cshperspect.a004457
Seubert, P., Vigo-Pelfrey, C., Esch, F., Lee, M., Dovey, H., Davis, D., Sinha, S., Schlossmacher, M., Whaley, J., Swindlehurst, C., McCormack, R., Wolfert, R., Selkoe, D. J., Lieberburg, I., and Schenk, D. (1992). Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature 359, 325–327.
Sha, D., Chin, L. S., and Li, L. (2010). Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum. Mol. Genet. 19, 352–363.
Shavali, S., Brown-Borg, H. M., Ebadi, M., and Porter, J. (2008). Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci. Lett. 439, 125–128.
Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K. S., and Selkoe, D. J. (2001). Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293, 263–269.
Shoji, M., Golde, T. E., Ghiso, J., Cheung, T. T., Estus, S., Shaffer, L. M., Cai, X. D., McKay, D. M., Tintner, R., Frangione, B., and Younkin, S. G. (1992). Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258, 126–129.
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039.
Siegelin, M. D., Dohi, T., Raskett, C. M., Orlowski, G. M., Powers, C. M., Gilbert, C. A., Ross, A. H., Plescia, J., and Altieri, D. C. (2011). Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J. Clin. Invest. 121, 1349–1360.
Silva, R. M., Ries, V., Oo, T. F., Yarygina, O., Jackson-Lewis, V., Ryu, E. J., Lu, P. D., Marciniak, S. J., Ron, D., Przedborski, S., Kholodilov, N., Greene, L. A., and Burke, R. E. (2005). CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 95, 974–986.
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., Paisan-Ruiz, C., Lichtner, P., Scholz, S. W., Hernandez, D. G., Kruger, R., Federoff, M., Klein, C., Goate, A., Perlmutter, J., Bonin, M., Nalls, M. A., Illig, T., Gieger, C., Houlden, H., Steffens, M., Okun, M. S., Racette, B. A., Cookson, M. R., Foote, K. D., Fernandez, H. H., Traynor, B. J., Schreiber, S., Arepalli, S., Zonozi, R., Gwinn, K., van der Brug, M., Lopez, G., Chanock, S. J., Schatzkin, A., Park, Y., Hollenbeck, A., Gao, J., Huang, X., Wood, N. W., Lorenz, D., Deuschl, G., Chen, H., Riess, O., Hardy, J. A., Singleton, A. B., and Gasser, T. (2009). Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312.
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., Lincoln, S., Crawley, A., Hanson, M., Maraganore, D., Adler, C., Cookson, M. R., Muenter, M., Baptista, M., Miller, D., Blancato, J., Hardy, J., and Gwinn-Hardy, K. (2003). alpha-Synuclein locus triplication causes Parkinson's disease. Science 302, 841.
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., Dawson, V. L., Dawson, T. M., and Ross, C. A. (2005). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 14, 3801–3811.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Strauss, K. M., Martins, L. M., Plun-Favreau, H., Marx, F. P., Kautzmann, S., Berg, D., Gasser, T., Wszolek, Z., Muller, T., Bornemann, A., Wolburg, H., Downward, J., Riess, O., Schulz, J. B., and Kruger, R. (2005). Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet. 14, 2099–2111.
Szegezdi, E., Fitzgerald, U., and Samali, A. (2003). Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann. N.Y. Acad. Sci. 1010, 186–194.
Tabas, I., and Ron, D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190.
Terro, F., Czech, C., Esclaire, F., Elyaman, W., Yardin, C., Baclet, M. C., Touchet, N., Tremp, G., Pradier, L., and Hugon, J. (2002). Neurons overexpressing mutant presenilin-1 are more sensitive to apoptosis induced by endoplasmic reticulum-Golgi stress. J. Neurosci. Res. 69, 530–539.
Trombetta, E. S., and Parodi, A. J. (2003). Quality control and protein folding in the secretory pathway. Annu. Rev. Cell Dev. Biol. 19, 649–676.
Tsang, W. Y., and Lemire, B. D. (2002). Mitochondrial genome content is regulated during nematode development. Biochem. Biophys. Res. Commun. 291, 8–16.
Umeda, T., Tomiyama, T., Sakama, N., Tanaka, S., Lambert, M. P., Klein, W. L., and Mori, H. (2011). Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 89, 1031–1042.
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A. R., Healy, D. G., Albanese, A., Nussbaum, R., Gonzalez-Maldonado, R., Deller, T., Salvi, S., Cortelli, P., Gilks, W. P., Latchman, D. S., Harvey, R. J., Dallapiccola, B., Auburger, G., and Wood, N. W. (2004a). Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160.
Valente, E. M., Salvi, S., Ialongo, T., Marongiu, R., Elia, A. E., Caputo, V., Romito, L., Albanese, A., Dallapiccola, B., and Bentivoglio, A. R. (2004b). PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 56, 336–341.
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., May, J., Tocilescu, M. A., Liu, W., Ko, H. S., Magrane, J., Moore, D. J., Dawson, V. L., Grailhe, R., Dawson, T. M., Li, C., Tieu, K., and Przedborski, S. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383.
Westerlund, M., Behbahani, H., Gellhaar, S., Forsell, C., Belin, A. C., Anvret, A., Zettergren, A., Nissbrandt, H., Lind, C., Sydow, O., Graff, C., Olson, L., Ankarcrona, M., and Galter, D. (2011). Altered enzymatic activity and allele frequency of OMI/HTRA2 in Alzheimer's disease. FASEB J. 25, 1345–1352.
Yang, Y., Turner, R. S., and Gaut, J. R. (1998). The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J. Biol. Chem. 273, 25552–25555.
Yao, J., Du, H., Yan, S., Fang, F., Wang, C., Lue, L. F., Guo, L., Chen, D., Stern, D. M., Gunn Moore, F. J., Xi Chen, J., Arancio, O., and Yan, S. S. (2011). Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J. Neurosci. 31, 2313–2320.
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 14670–14675.
Yoneda, T., Benedetti, C., Urano, F., Clark, S. G., Harding, H. P., and Ron, D. (2004). Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 117, 4055–4066.
Yoneda, T., Imaizumi, K., Oono, K., Yui, D., Gomi, F., Katayama, T., and Tohyama, M. (2001). Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940.
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891.
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., Vidal, L., Hoenicka, J., Rodriguez, O., Atares, B., Llorens, V., Gomez Tortosa, E., Del Ser, T., Munoz, D. G., and De Yebenes, J. G. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173.
Keywords: unfolded protein response, endoplasmic reticulum, mitochondria, Alzheimer's disease, Parkinson's disease, neurodegeneration
Citation: Bernales S, Soto MM, and McCullagh E (2012) Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Front. Ag. Neurosci. 4:5. doi: 10.3389/fnagi.2012.00005
Received: 08 March 2012; Accepted: 10 April 2012;
Published online: 25 April 2012.
Edited by:
P. Hemachandra Reddy, Oregen Health and Science University, USAReviewed by:
Ana I. Duarte, University of Coimbra, PortugalPeizhong Mao, Oregon Health and Science University, USA
Copyright: © 2012 Bernales, Soto and McCullagh. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Emma McCullagh, Fundación Ciencia y Vida, Protein Folding and Disease Lab, Avenida Zañartu 1482, Ñuñoa, Santiago, Chile. e-mail:ZW1tYW1jY3VsbGFnaEBnbWFpbC5jb20=