 Atsushi Takeda*
Atsushi Takeda* Haruna Tamano
Haruna Tamano- Department of Bioorganic Chemistry, School of Pharmaceutical Sciences, University of Shizuoka, Shizuoka, Japan
Zinc is an essential component of physiological brain function. Vesicular zinc is released from glutamatergic (zincergic) neuron terminals and serves as a signal factor (Zn2+ signal) in both the intracellular (cytosol) compartment and the extracellular compartment. Synaptic Zn2+ signaling is dynamically linked to neurotransmission and is involved in processes of synaptic plasticity such as long-term potentiation and cognitive activity. On the other hand, the activity of the hypothalamic–pituitary–adrenal (HPA) axis, i.e., glucocorticoid secretion, which can potentiate glutamatergic neuron activity, is linked to cognitive function. HPA axis activity modifies synaptic Zn2+ dynamics at zincergic synapses. An increase in HPA axis activity, which occurs after exposure to stress, may induce excess intracellular Zn2+ signaling in the hippocampus, followed by hippocampus-dependent memory deficit. Excessive excitation of zincergic neurons in the hippocampus can contribute to cognitive decline under stressful and/or pathological conditions. This paper provides an overview of the “Hypothesis and Theory” of Zn2+-mediated modification of cognitive activity.
Introduction
Over 300 proteins require zinc to carry out their functions in microorganisms, plants, and animals. Zinc powerfully influences cell division and differentiation (Vallee and Falchuk, 1993; Maret and Sandstead, 2008; Prasad, 2008). Zinc is also essential for the growth and functioning of the brain. Zinc transport from the plasma to the brain’s extracellular fluid and cerebrospinal fluid is strictly regulated by the brain-barrier system, i.e., the blood–brain and blood-CSF barrier. The brain barrier system maintains zinc homeostasis in the brain (Takeda, 2000, 2001). Zinc homeostasis is critical for brain function (Capasso et al., 2005; Mocchegiani et al., 2005) and is spatiotemporally altered in the process of neurological diseases (Barnham and Bush, 2008).
Zinc is relatively concentrated in the hippocampus and amygdala (Takeda et al., 1995). Both regions are enriched with histochemically reactive zinc, as revealed by Timm’s sulfide-silver staining method (Frederickson, 1989; Frederickson and Danscher, 1990). Histochemically reactive zinc is found predominantly in the presynaptic vesicles and serves as a signal factor (Zn2+ signal) in both the cytosolic and extracellular compartments. Zn2+ is released with glutamate in a calcium-dependent and impulse-dependent manner from glutamatergic (zincergic) neuron terminals (Figure 1). Zn2+ released from these terminals modulates the activity of several important receptors, including the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptor, N-methyl-D-aspartate (NMDA) receptors, and γamino butyric acid (GABA) receptors in the extracellular compartment (Smart et al., 1994; Nakashima and Dyck, 2009), and is taken up into post-synaptic neurons to serve as an intracellular signal factor. Glutamatergic (zincergic) circuits play a key role in cognitive map building structures such as the hippocampus (Martinez-Guijarro et al., 1991; Nacher et al., 2000). It has been estimated that approximately 20% of total brain zinc is histochemically reactive, based on the finding that the removal of zinc transporter-3 (ZnT3) protein, which is responsible for the movement of zinc from the cytoplasm into synaptic vesicles (Palmiter et al., 1996), results in a 20% reduction of the total amount of zinc in the brain (Cole et al., 1999).
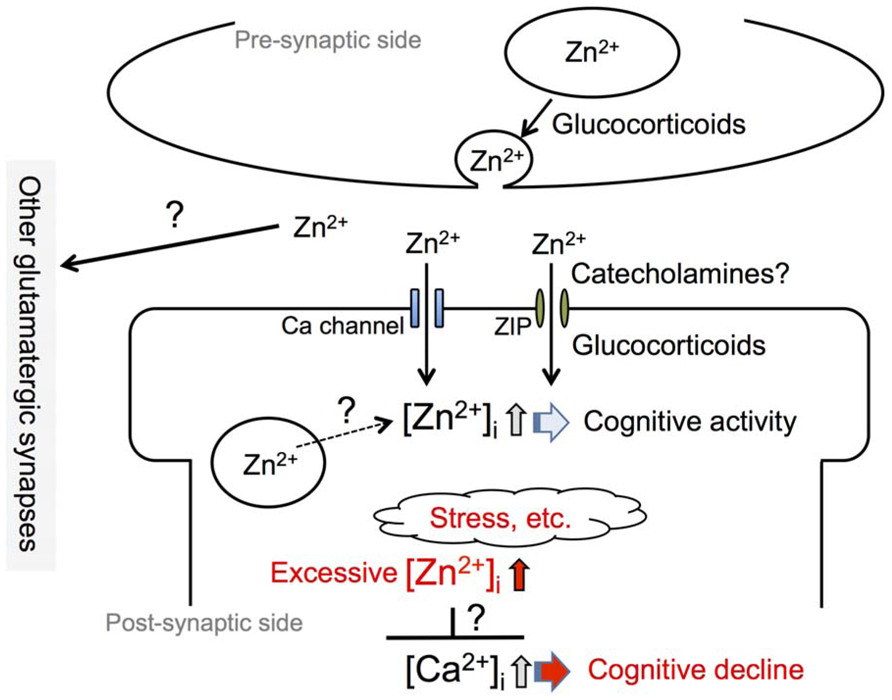
FIGURE 1. Involvement of synaptic Zn2+ dynamics in cognitive activity. An increase in intracellular Zn2+ concentration, [Zn2+]i, which is induced by an influx of extracellular Zn2+ at zincergic synapses in the hippocampus, is involved in cognitive activity. Presynaptic glucocorticoid signaling, a non-genomic action, and post-synaptic glucocorticoid signaling, a genomic action, modify the degree of increase in intracellular Zn2+. It is also possible that catecholamines modify the degree through the activity of the β-adrenergic system. The degree of increase in intracellular Zn2+ is linked to cognitive activity and excess intracellular Zn2+ signaling, which can be induced by stress, is involved in cognitive decline. The excess might affect intracellular Ca2+ signaling, which plays a key role for synaptic plasticity.
It is well known that the hippocampus and amygdala are involved in cognitive and emotional behavior. Synaptic plasticity such as long-term potentiation (LTP) is believed to be a key cellular mechanism involved in learning and memory and has been widely studied in relation to glutamatergic synapses in the brain, especially in the hippocampus (Bliss and Collingridge, 2013). When information is processed in memory, glutamatergic neurons form a neural circuit in the hippocampus and the amygdala. Furthermore, it has been reported that plastic changes in hippocampal synapses occur activity-dependently during the performance of associative learning tasks (Gruart et al., 2006; Clarke et al., 2010).
On the other hand, the activity of the hypothalamic–pituitary–adrenal (HPA) axis, i.e., glucocorticoid secretion, is linked to cognitive and emotional functions and can potentiate glutamatergic neuron activity (Sandi, 2011). There is some evidence that the modification of synaptic Zn2+ signaling by HPA axis activity, which is enhanced by stress and aging, is linked to cognitive and emotional behavior, and that abnormal modification may induce cognitive decline (Takeda and Tamano, 2009, 2010, 2012). It is well known that abnormal Zn2+ influx into post-synaptic neurons, which is induced by abnormal glutamatergic (zincergic) neuron activity, induces neuronal death and is involved in neurological disorders such as stroke/ischemia and temporal lobe epilepsy (Frederickson et al., 2005; Sensi et al., 2011; Takeda, 2011a; Weiss, 2011). Therefore, the homeostasis of synaptic Zn2+ signaling is critical in both functional and pathological aspects (Takeda, 2011b; Takeda et al., 2013). On the basis of recent evidence that excessive excitation of zincergic neurons in the hippocampus can contribute to cognitive decline under stressful and/or pathological conditions (Takeda et al., 2009, 2011, 2012), this paper provides an overview of the “Hypothesis and Theory” of Zn2+-mediated modification of cognitive activity.
Synaptic Zn2+ Homeostasis
Total zinc concentration in the adult brain reaches around 200 μM (Markesbery et al., 1984). Extracellular zinc concentration in the adult brain is estimated to be less than 1 μM (Weiss et al., 2000). If zinc concentration in the brain’s extracellular fluid is equal to that in cerebrospinal fluid (Hershey et al., 1983), it is around 150 nM – approximately one thousandth of total brain zinc concentration. In zincergic synapses, Zn2+ concentration in the synaptic cleft is estimated to be higher than that in the brain’s (extrasynaptic) extracellular fluid, because under hippocampal-slice-experiment conditions the regions where zincergic synapses are found are intensely stained by ZnAF-2, a membrane-impermeable zinc indicator (Minami et al., 2006). The synaptic cleft is surrounded with the processes of astrocytes, which contribute to maintaining a steady concentration of zinc and neurotransmitters in the cleft. Interestingly, Zn2+ level in the brain’s extracellular fluid, which is estimated to be approximately 20 nM (Frederickson et al., 2006), is higher than that in the plasma (<1 nM; Magneson et al., 1987). In the brain’s extracellular fluid, the high ratio of Zn2+ concentration to total zinc concentration appears to be associated with the synaptic Zn2+ dynamics of the brain. There is some evidence that extracellular Zn2+ serves as a pool for the zinc in the synaptic vesicle and is involved in synaptic Zn2+ homeostasis (Takeda et al., 2006), although the chemical form of this vesicular zinc is unknown.
Basal Zn2+ concentration is extremely low in the intracellular (cytosol) compartment (<1 nM; Sensi et al., 1997; Colvin et al., 2008). ZnT proteins such as ZnT1, ZnT3, and ZnT10, and Zrt-Irt-like proteins (ZIP) such as ZIP4 and ZIP6 are involved in the control of Zn2+ levels in the cytosolic compartment, especially under static (basal) conditions (Emmetsberger et al., 2010). Some of these transporters transport cytosolic Zn2+ into a variety of subcellular organelles, including mitochondria, lysosomes, endosomes, and the Golgi apparatus, probably to maintain static Zn2+ levels in the cytosolic compartment (Sensi et al., 2003; Danscher and Stoltenberg, 2005; Colvin et al., 2006). On the other hand, it is possible that Zn2+ release from subcellular organelles, which might be induced by synaptic glutamate signaling, is involved in Zn2+ signaling (Stork and Li, 2010). Zn2+ levels other than vesicular zinc serving as Zn2+ are estimated to be less than 5% of the total amount of Zn2+ in the hippocampus and cerebral cortex (Lee et al., 2011). ZnT1 is a major Zn2+ transporter in the plasma membrane and may be involved in cytosolic Zn2+ homeostasis in neurons by transporting Zn2+ from the somata to the extracellular space (Sekler et al., 2002). It has been reported that ZnT1 prevents excessive accumulation of Zn2+ in the cytosolic compartment (Nolte et al., 2004), resulting in the protection of neurons from Zn2+ toxicity in neurological diseases such as transient forebrain ischemia (Aguilar-Alonso et al., 2008). Tissue plasminogen activator, a secreted serine protease, is excitotoxic and increases lysosomal sequestration of increased Zn2+ in the cytosolic compartment through interaction with ZIP4, which may also contribute to the protection of neurons from Zn2+ toxicity (Emmetsberger et al., 2010). The spatiotemporal control of Zn2+ signaling via ZIP and ZnT maintains a steady-state environment in both the extracellular and cytosolic compartments (Fukada and Kambe, 2011).
Functional and Neurotoxic Zn2+ Signaling
Zn2+ concentration is increased in the synaptic cleft during the excitation of zincergic synapses, followed by an increase in the cytosol (intracellular compartment; Figure 1). Released Zn2+ is quickly taken up into presynaptic and post-synaptic neurons and astrocytes. Calcium channels such as calcium-permeable AMPA/kainate receptors are involved in Zn2+ influx during neuronal excitation (Weiss et al., 2000; Jia et al., 2002; Takeda et al., 2007a). The increase in the extracellular concentration of Zn2+ is dependent on the frequency of depolarizing stimulation (Ueno et al., 2002). Therefore, the increase in intracellular concentration of Zn2+ serving as a signal factor is closely correlated to zincergic neuron excitation (Takeda et al., 2013).
Glutamate accumulates in the extracellular compartment due to excessive excitation of glutamatergic (zincergic) neurons. Excessive activation of glutamate receptors caused by excess extracellular glutamate leads to a number of deleterious consequences, including impairment of calcium buffering, generation of free radicals, activation of mitochondrial permeability transition, and secondary excitotoxicity (Danbolt, 2001; Dong et al., 2009). Glutamate excitotoxicity, a final common pathway for neuronal death, is observed in numerous pathological processes such as stroke/ischemia, temporal lobe epilepsy, Alzheimer’s disease, and amyotrophic lateral sclerosis. An excess of extracellular Zn2+, which is induced under glutamate excototoxicity, is harmful; excessive Zn2+ influx into post-synaptic neurons is involved in neurodegeneration under pathological conditions. Calcium-permeable AMPA receptors may play a key role in this Zn2+ influx (Liu et al., 2004; Noh et al., 2005; Weiss, 2011).
Zn2+ also plays a neuroprotective role in glutamate-induced excitotoxicity by activating pre-synaptic ATP-sensitive potassium channels and by inhibiting GABA transporter 4 (Bancila et al., 2004; Cohen-Kfir et al., 2005). It is estimated that the neuroprotective action of Zn2+ occurs under conditions in which zincergic neurons are not excessively excited. Zn2+ released from zincergic neuron terminals may also serve as a negative feedback factor against glutamate release (Minami et al., 2006; Takeda et al., 2007b). Therefore, the degree of increase in extracellular Zn2+ is critical in both functional and neurotoxic aspects.
Zn2+ Signaling and Cognition
Synaptic Zn2+ signaling is involved in processes of synaptic plasticity such as LTP in the hippocampus and amygdala. Enhanced plasticity in zincergic synapses is associated with cortical modification after exposure to an enriched environment (Nakashima and Dyck, 2008). The enhanced plasticity of zincergic synapses in the hippocampus underlies the acquisition of new motor and cognitive abilities (Delgado-García and Gruart, 2006; Jurado-Parras et al., 2013). These findings suggest that synaptic Zn2+ signaling is involved in cognitive and emotional behavior through the modulation of synaptic plasticity such as LTP (Figure 1).
Targeted deletion of the ZnT3 prevents vesicular Zn2+ uptake (Cole et al., 1999) and ablates Zn2+ release into the extracellular space by action potentials. There is a correlation between vesicular Zn2+ levels and ZnT3 protein expression (Palmiter et al., 1996). Zn2+ transport into the synaptic vesicle is ZnT3-dependent and is important for amassing the large pool of Zn2+ used in signaling (Lee et al., 2011). It has been reported that Zn2+ signaling is involved in cognitive and emotional behavior even in ZnT3KO (Adlard et al., 2010; Martel et al., 2010, 2011; Sindreu et al., 2011). The pool of Zn2+ may be located in other subcellular organelles (Figure 1) and/or zinc-binding proteins such as metallothionein in ZnT3KO mice. On the other hand, memory deficit and the changes in emotional (freezing) behavior have been observed in wild-type animals when acute loss or chelation of synaptic Zn2+ is induced by treatment with zinc chelators (Takeda et al., 2010a, b). The amount of Zn2+ functioning as a signal factor seems to be lower in ZnT3KO mice than in wild-type mice.
Saito et al. (2000) report that age-dependent reduction of Zn2+ levels in the synaptic vesicles of the mossy fibers induced by low ZnT3 expression causes both glutamatergic excitotoxicity in hippocampal neurons and the deterioration of learning and memory in senescence-accelerated mouse prone 10 (SAMP10). There are also reports of age-dependent reductions in ZnT3 expression and synaptic Zn2+ levels in the hippocampal mossy fibers of human amyloid precursor protein-transgenic (Tg2576) mice, suggesting that extensive modifications of the brain’s Zn2+ pool, particularly synaptic (vesicular) Zn2+, underlie the neuronal dysfunction characteristic of Alzheimer’s disease (Lee et al., 2012). Furthermore, there is a significant age-related decline in cortical ZnT3 levels from age 48 to 91 in healthy people (Adlard et al., 2010) and ZnT3 levels are more markedly decreased in the cortex in Alzheimer’s disease. It is likely that the increase in extracellular Zn2+ induced by the physiological excitation of zincergic neurons requires cognitive activity (Figure 1) and that an insufficient increase is involved in the pathophysiology of Alzheimer’s disease.
On the other hand, Zn2+ released from zincergic neurons is known to mediate parenchymal and cerebrovascular amyloid formation in Tg2576 mice (Lee et al., 2002; Friedlich et al., 2004; Stoltenberg et al., 2007). The transsynaptic movement of Zn2+ may be severely compromised in Alzheimer’s disease, both by lack of ZnT3 expression and by sequestration in amyloid. Adlard et al. (2010) report that the genetic ablation of ZnT3 may represent a phenocopy for memory deficits in Alzheimer’s disease. Deshpande et al. (2009) postulate that the sequestration of Zn2+ in oligomeric amyloid-β (Aβ)–Zn complexes may lead to a reduction in Zn2+ availability at the synapses, resulting in a loss of the modulatory activity of Zn2+, and leading to the cognitive decline of Alzheimer’s disease. Such changes in synaptic Zn2+ availability may participate in modifying cognitive activity and also in cognitive decline (Figure 1; Linkous et al., 2009; Bush, 2013; Bosomworth et al., 2013).
Glucocorticoid Signaling, Zn2+ Signaling, and Cognition
The hippocampus is enriched with corticosteroid receptors and is the major target region of corticosteroids (Joëls, 2008). Mineralocorticoid receptors and glucocorticoid receptors are colocalized in CA1 and CA2 pyramidal cells and in dentate gyrus granule cells. In CA3 pyramidal cells, on the other hand, mineralocorticoid receptors are abundantly expressed, while glucocorticoid receptors are expressed at much lower levels (Ozawa, 2005). Mineralocorticoid receptors are extensively occupied with low levels of corticosterone, and glucocorticoid receptors are particularly activated after exposure to stress (Joëls, 2008; Sandi, 2011).
An increase in serum corticosterone level induces a rapid increase in hippocampal corticosterone level, in parallel with an increase in extracellular glutamate level (Venero and Borrell, 1999). Corticosterone-induced increase in extracellular glutamate levels in the hippocampus appears to be exerted through the action of membrane-associated mineralocorticoid receptors and/or glucocorticoid receptors, which increase the probability of glutamate release in synaptic activation (Karst et al., 2005; Musazzi et al., 2010). The rapid effects of corticosterone on glutamatergic transmission appear to be linked to diverse effects on synaptic plasticity and memory processes in the hippocampus (Figure 1). An increase in the probability of glutamate release through the action of corticosterone leads to increases both in the amount of glutamate released during learning and in the degree of activation of post-synaptic glutamate receptors. Corticosterone can contribute to an increase in the efficacy of glutamatergic transmission by AMPA receptor insertion at synaptic sites, through both the rapid and the delayed (genomic) effects. These effects are of advantage to processes of synaptic plasticity such as LTP and memory acquisition (Sandi, 2011). Therefore, it is estimated that corticosterone increases the probability of Zn2+ release from zincergic neuron terminals through the rapid non-genomic effect in the hippocampus (Figure 1; Takeda et al., 2012). Futhermore, corticosterone requires intracellular Zn2+ signaling for the genomic effect, possibly followed by the delayed influx of extracellular Zn2+ through zinc transport systems such as ZIP (Figure 1). Although the evidence is limited, it is likely that synaptic Zn2+ signaling cooperates with corticosteroid signaling in learning and memory.
In contrast, glutamate accumulates in the extracellular compartment at high levels through a corticosterone-mediated blockade of glutamate transporter activity when corticosterone is abnormally secreted under conditions of severe stress. Abnormal corticosterone secretion also contributes to abnormal glutamate release from neuron terminals (Wong et al., 2007; Howland and Wang, 2008). The extracellular spillover of glutamate impairs spatial memory retrieval. Furthermore, Wong et al. (2007) demonstrate that hippocampal long-term depression (LTD) is both necessary and sufficient to cause acute stress-induced impairment of spatial memory retrieval. Excess intracellular Zn2+ signaling induced by corticosterone and/or stress is also involved in the impairment of hippocampal LTP (Takeda et al., 2009, 2012), possibly followed by the impairment of learning and memory (Takeda et al., 2011; Figure 1). In hippocampal CA3, on the other hand, an increase in intracellular Zn2+ via a zinc ionophore not only decreases basal Ca2+ level but also suppresses increases in Ca2+ level via metabotropic glutamate receptors (Takeda et al., 2007a). Such excess intracellular Zn2+ signaling may lead to negative crosstalk in intracellular Ca2+ signaling, which plays a key role in LTP and LTD (Figure 1).
A selective increase in the nocturnal levels of corticol has been observed in aged humans (Landfield and Eldridge, 1994). Furthermore, high levels of cortisol are found in Alzheimer’s disease as well as in depression. In Alzheimer’s disease patients, cognitive deficits (such as in memory) and psychological symptoms (such as anxiety) are associated with an early deregulation of the HPA axis (Swanwick et al., 1998; Brureau et al., 2013). Therefore, it is possible that excess intracellular Zn2+ signaling through abnormal cortisol secretion is involved in cognitive deficits in both normal aging and neurological diseases such as dementia.
On the other hand, corticotrophin releasing hormone (CRH) drives the HPA axis and is considered to be the central coordinator of behavioral, autonomic, and neuroendocrine stress responses. The stress mediators activated by CRH are organized in the sympathetic nervous system, as well as in the HPA axis (de Kloet, 2008). Adrenaline, along with norepinephrine, is largely responsible for the immediate reactions that are felt under conditions of stress. Responses of adrenaline and norepinephrine, such as an increase in heart rate, occur more quickly than those of glucocorticoids. Catecholamines are released from the sympathetic nerve system and the adrenal glad. It has been reported that the enhanced memory associated with emotional experiences involves activation of the β-adrenergic system (Cahill et al., 1994; McEwen and Sapolsky, 1995). β-Adrenergic receptor activation facilitates the induction of a protein synthesis-dependent late phase in LTP in the hippocampus (Gelinas and Nguyen, 2005). Learning-facilitated LTD and LTP at mossy fiber-CA3 synapses requires activation of β-adrenergic receptors (Hagena and Manahan-Vaughan, 2012). The above evidence suggests that synaptic Zn2+ signaling is modified by the β-adrenergic system and is involved in cognitive activity associated with emotional experiences. The relationship between synaptic Zn2+ signaling and the β-adrenergic system is an issue which requires further clarification. Stress is a known precipitant for metabolic and neurological diseases (Koenig et al., 2011) and synaptic Zn2+ signaling is likely to be involved in the diverse effects of stress through the stress mediators activated by CRH.
Perspective
Synaptic Zn2+ homeostasis is critical for synaptic function, and seems to be controlled by two Zn2+ pools, one in the synaptic vesicle and the other in the extracellular compartment. Synaptic Zn2+ signaling is involved in cognitive activity, and both its lack and its excess are involved in cognitive decline (Figure 1). HPA axis activity increases with aging, and this increase is superimposed on neurological diseases such as depression and Alzheimer’s disease. It is likely that synaptic Zn2+ signaling through the HPA axis activity is involved in cognitive decline in both normal aging and dementia, and it is possible that sympathetic nervous system activity is also involved. However, evidence related to synaptic Zn2+ dynamics is very limited, not only under physiological conditions, but also under stressful and pathological conditions. The molecular mechanisms of abnormal Zn2+ signaling in cognitive decline also remain to be clarified.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to thank Philip Hawke of the University of Shizuoka Scientific English Program for his comments on the English in the manuscript.
References
Adlard, P. A., Parncutt, J. M., Finkelstein, D. I., and Bush, A. I. (2010). Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 30, 1631–1636. doi: 10.1523/JNEUROSCI.5255-09.2010
Aguilar-Alonso, P., Martinez-Fong, D., Pazos-Salazar, N. G., Brambila, E., Gonzalez-Barrios, J. A., Mejorada, A., et al. (2008). The increase in zinc levels and upregulation of zinc transporters are mediated by nitric oxide in the cerebral cortex after transient ischemia in the rat. Brain Res. 1200, 89–98. doi: 10.1016/j.brainres.2007.11.077
Bancila, V., Nikonenko, I., Dunant, Y., and Bloc, A. (2004). Zinc inhibits glutamate release via activation of pre-synaptic KATP channels and reduces ischaemic damage in rat hippocampus. J. Neurochem. 90, 1243–1250. doi: 10.1111/j.1471-4159.2004.02587.x
Barnham, K. J., and Bush, A. I. (2008). Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 12, 222–228. doi: 10.1016/j.cbpa.2008.02.019
Bliss, T. V., and Collingridge, G. L. (2013). Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol. Brain 6, 5. doi: 10.1186/1756-6606-6-5
Bosomworth, H. J., Adlard, P. A., Ford, D., and Valentine, R. A. (2013). Altered expression of ZnT10 in Alzheimer’s disease brain. PLoS ONE 8:e65475. doi: 10.1371/journal.pone.0065475
Brureau, A., Zussy, C., Delair, B., Ogier, C., Ixart, G., Maurice, T., et al. (2013). Deregulation of hypothalamic-pituitary-adrenal axis functions in an Alzheimer’s disease rat model. Neurobiol. Aging 34, 1426–1439. doi: 10.1016/j.neurobiolaging.2012.11.015
Bush, A. I. (2013). The metal theory of Alzheimer’s disease. J. Alzheimers Dis. 33, S277–S281. doi: 10.3233/JAD-2012-129011
Cahill, L., Prins, B., Weber, M., and McGaugh, J. L. (1994). Beta-adrenergic activation and memory for emotional events. Nature 371, 702–704. doi: 10.1038/371702a0
Capasso, M., Jeng, J. M., Malavolta, M., Mocchegiani, E., and Sensi, S. L. (2005). Zinc dyshomeostasis: a key modulator of neuronal injury. J. Alzheimers Dis. 8, 93–108.
Clarke, J. R., Cammarota, M., Gruart, A., Izquierdo, I., and Delgado-García, J. M. (2010). Plastic modifications induced by object recognition memory processing. Proc. Natl. Acad. Sci. U.S.A. 107, 2652–2657. doi: 10.1073/pnas.0915059107
Cohen-Kfir, E., Lee, W., Eskandari, S., and Nelson, N. (2005). Zinc inhibition of gamma-aminobutyric acid transporter 4 (GAT4) reveals a link between excitatory and inhibitory neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 102, 6154–6159. doi: 10.1073/pnas.0501431102
Cole, T. B., Wenzel, H. J., Kafer, K. E., Schwartzkroin, P. A., and Palmiter, R. D. (1999). Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. U.S.A. 96, 1716–1721. doi: 10.1073/pnas.96.4.1716
Colvin, R. A., Bush, A. I., Volitakis, I., Fontaine, C. P., Thomas, D., Kikuchi, K., et al. (2008). Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am. J. Physiol. Cell Physiol. 294, C726–C742. doi: 10.1152/ajpcell.00541.2007
Colvin, R. A., Laskowski, M., and Fontaine, C. P. (2006). Zinquin identifies subcellular compartmentalization of zinc in cortical neurons. Relation to the trafficking of zinc and the mitochondrial compartment. Brain Res. 1085, 1–10. doi: 10.1016/j.brainres.2006.02.043
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
Danscher, G., and Stoltenberg, M. (2005). Zinc-specific autometallographic in vivo selenium methods: tracing of zinc-enriched (ZEN) terminals, ZEN pathways, and pools of zinc ions in a multitude of other ZEN cells. J. Histochem. Cytochem. 53, 141–153. doi: 10.1369/jhc.4R6460.2005
de Kloet, E. R. (2008). Commentary: neuroendocrine basis. Prog. Brain Res. 167, 53–62. doi: 10.1016/S0079-6123(07)67004-6
Delgado-García, J. M., and Gruart, A. (2006). Building new motor responses: eyelid conditioning revisited. Trends Neurosci. 29, 330–338. doi: 10.1016/j.tins.2006.05.003
Deshpande, A., Kawai, H., Metherate, R., Glabe, C. G., and Busciglio, J. (2009). A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 29, 4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009
Dong, X. X., Wang, Y., and Qin, Z. H. (2009). Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 30, 379–387. doi: 10.1038/aps.2009.24
Emmetsberger, J., Mirrione, M. M., Zhou, C., Fernandez-Monreal, M., Siddiq, M. M., Ji, K., et al. (2010). Tissue plasminogen activator alters intracellular sequestration of zinc through interaction with the transporter ZIP4. J. Neurosci. 30, 6538–6547. doi: 10.1523/JNEUROSCI.6250-09.2010
Frederickson, C. J. (1989). Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 31, 145–238.
Frederickson, C. J., and Danscher, G. (1990). Zinc-containing neurons in hippocampus and related CNS structures. Prog. Brain Res. 83, 71–84. doi: 10.1016/S0079-6123(08)61242-X
Frederickson, C. J., Giblin, L. J., Krezel, A., McAdoo, D. J., Muelle, R. N., Zeng, Y., et al. (2006). Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 198, 285–293. doi: 10.1016/j.expneurol.2005.08.030
Frederickson, C. J., Koh, J. Y., and Bush, A. I. (2005). The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 6, 449–462. doi: 10.1038/nrn1671
Friedlich, A. L., Lee, J. Y., van Groen, T., Cherny, R. A., Volitakis, I., Cole, T. B., et al. (2004). Neuronal zinc exchange with the blood vessel wall promotes cerebral amyloid angiopathy in an animal model of Alzheimer’s disease. J. Neurosci. 24, 3453–3459. doi: 10.1523/JNEUROSCI.0297-04.2004
Fukada, T., and Kambe, T. (2011). Molecular and genetic features of zinc transporters in physiology and pathogenesis. Metallomics 3, 662–674. doi: 10.1039/c1mt00011j
Gelinas, J. N., and Nguyen, P. V. (2005). Beta-adrenergic receptor activation facilitates induction of a protein synthesis-dependent late phase of long-term potentiation. J. Neurosci. 25, 3294–3303. doi: 10.1523/JNEUROSCI.4175-04.2005
Gruart, A., Muñoz, M. D., and Delgado-García, J. M. (2006). Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J. Neurosci. 26, 1077–1087. doi: 10.1523/JNEUROSCI.2834-05.2006
Hagena, H., and Manahan-Vaughan, D. (2012). Learning-facilitated long-term depression and long-term potentiation at mossy fiber-CA3 synapses requires activation of β-adrenergic receptors. Front. Integr. Neurosci. 6:23. doi: 10.3389/fnint.2012.00023
Hershey, C. O., Hershey, L. A., Varnes, A., Vibhakar, S. D., Lavin, P., and Strain, W. H. (1983). Cerebrospinal fluid trace element content in dementia: clinical, radiologic, and pathologic correlations. Neurology 33, 1350–1353. doi: 10.1212/WNL.33.10.1350
Howland, J. G., and Wang, Y. T. (2008). Synaptic plasticity in learning and memory: stress effects in the hippocampus. Prog. Brain Res. 169, 145–158. doi: 10.1016/S0079-6123(07)00008-8
Jia, Y., Jeng, J. M., Sensi, S., and Weiss, J. H. (2002). Zn2+ currents are mediated by calcium-permeable AMPA/kainite channels in cultured murine hippocampal neurons. J. Physiol. (Lond.) 543, 35–48. doi: 10.1113/jphysiol.2002.020172
Joëls, M. (2008). Functional actions of corticosteroids in the hippocampus. Eur. J. Pharmacol. 583, 312-321 doi: 10.1016/j.ejphar.2007.11.064
Joëls, M., Karst, H., DeRijk, R., and de Kloet, E. R. (2008). The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 31, 1–7. doi: 10.1016/j.tins.2007.10.005
Jurado-Parras, M. T., Sánchez-Campusano, R., Castellanos, N. P., del-Pozo, F., Gruart, A., and Delgado-García, J. M. (2013). Differential contribution of hippocampal circuits to appetitive and consummatory behaviors during operant conditioning of behaving mice. J. Neurosci. 33, 2293–2304. doi: 10.1523/JNEUROSCI.1013-12.2013
Karst, H., Berger, S., Turiault, M., Tronche, F., Schütz, G., and Joëls, M. (2005). Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. U.S.A. 102, 19204–19207. doi: 10.1073/pnas.0507572102
Koenig, J. I., Walker, C. D., Romeo, R. D., and Lupien, S. J. (2011). Effects of stress across the lifespan. Stress 14, 475–480. doi: 10.3109/10253890.2011.604879
Landfield, P. W., and Eldridge, J. C. (1994). Evolving aspects of the glucocorticoid hypothesis of brain aging: hormonal modulation of neuronal calcium homeostasis. Neurobiol. Aging 15, 579–588. doi: 10.1016/0197-4580(94)90101-5
Lee, J. Y., Cho, E., Seo, J. W., Hwang, J. J., and Koh, J. Y. (2012). Alteration of the cerebral zinc pool in a mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 71, 211–222. doi: 10.1097/NEN.0b013e3182417387
Lee, J. Y., Cole, T. B., Palmiter, R. D., Suh, S. W., and Koh, J. Y. (2002). Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 7705–7710. doi: 10.1073/pnas.092034699
Lee, J. Y., Kim, J. S., Byun, H. R., Palmiter, R. D., and Koh, J. Y. (2011). Dependence of the histofluorescently reactive zinc pool on zinc transporter-3 in the normal brain. Brain Res. 1418, 12–22. doi: 10.1016/j.brainres.2011.08.055
Linkous, D. H., Adlard, P. A., Wanschura, P. B., Conko, K. M., and Flinn, J. M. (2009). The effects of enhanced zinc on spatial memory and plaque formation in transgenic mice. J. Alzheimers Dis. 18, 565–579. doi: 10.3233/JAD-2009-1162
Liu, S., Lau, L., Wei, J., Zhu, D., Zou, S., Sun, H. S., et al. (2004). Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron 43, 43–55. doi: 10.1016/j.neuron.2004.06.017
Magneson, G. R., Puvathingal, J. M., and Ray, W. J. Jr. (1987). The concentrations of free Mg2+ and free Zn2+ in equine blood plasma. J. Biol. Chem. 262, 11140–11148.
Maret, W., and Sandstead, H. H. (2008). Possible roles of zinc nutriture in the fetal origins of disease. Exp. Gerontol. 43, 378–381. doi: 10.1016/j.exger.2007.10.005
Markesbery, W. R., Ehmann, W. D., Alauddin, M., and Hossain, T. I. M. (1984). Brain trace element concentrations in aging. Neurobiol. Aging 5, 19–28. doi: 10.1016/0197-4580(84)90081-2
Martel, G., Hevi, C., Friebely, O., Baybutt, T., and Shumyatsky, G. P. (2010). Zinc transporter 3 is involved in learned fear and extinction, but not in innate fear. Learn. Mem. 17, 582–590. doi: 10.1101/lm.1962010
Martel, G., Hevi, C., Kane-Goldsmith, N., and Shumyatsky, G. P. (2011). Zinc transporter 3 is involved in learned fear and extinction, but not in innate fear. Behav. Brain Res. 223, 233–238. doi: 10.1016/j.bbr.2011.04.020
Martinez-Guijarro, F. J., Soriano, E., Del Rio, J. A., and Lopez-Garcia, C. (1991). Zinc-positive boutons in the cerebral cortex of lizards show glutamate immunoreactivity. J. Neurocytol. 20, 834–843. doi: 10.1007/BF01191734
Minami, A., Sakurada, N., Fuke, S., Kikuchi, K., Nagano, T., Oku, N., et al. (2006). Inhibition of presynaptic activity by zinc released from mossy fiber terminals during tetanic stimulation. J. Neurosci. Res. 83, 167–176. doi: 10.1002/jnr.20714
McEwen, B. S., and Sapolsky, R. M. (1995). Stress and cognitive function. Curr. Opin. Neurobiol. 5, 205–216. doi: 10.1016/0959-4388(95)80028-X
Mocchegiani, E., Bertoni-Freddari, C., Marcellini, F., and Malavolta, M. (2005). Brain, aging and neurodegeneration: role of zinc ion availability. Prog. Neurobiol. 75, 367–390. doi: 10.1016/j.pneurobio.2005.04.005
Musazzi, L., Milanese, M., Farisello, P., Zappettini, S., Tardito, D., Barbiero, V. S., et al. (2010). Acute stress increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: the dampening action of antidepressants. PLoS ONE 5:e8566. doi: 10.1371/journal.pone.0008566
Nacher, J., Palop, J. J., Ramirez, C., Molowny, A., and Lopez-Garcia, C. (2000). Early histological maturation in the hippocampus of the guinea pig. Brain Behav. Evol. 56, 38–44. doi: 10.1159/000006676
Nakashima, A. S., and Dyck, R. H. (2008). Enhanced plasticity in zincergic, cortical circuits after exposure to enriched environments. J. Neurosci. 28, 13995–13999. doi: 10.1523/JNEUROSCI.4645-08.2008
Nakashima, A. S., and Dyck, R. H. (2009). Zinc and cortical plasticity. Brain Res. Rev. 59, 347–373. doi: 10.1016/j.brainresrev.2008.10.003
Noh, K. M., Yokota, H., Mashiko, T., Castillo, P. E., Zukin, R. S., and Bennett, M. V. (2005). Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc. Natl. Acad. Sci. U.S.A. 102, 12230–12235. doi: 10.1073/pnas.0505408102
Nolte, C., Gore, A., Sekler, I., Kresse, W., Hershfinkel, M., Hoffmann, A., et al. (2004). ZnT-1 expression in astroglial cells protects against zinc toxicity and slows the accumulation of intracellular zinc. Glia 48, 145–155. doi: 10.1002/glia.20065
Ozawa, H. (2005). Steroid hormones, their receptors and neuroendocrine system. J. Nippon Med. Sch. 72, 316–325. doi: 10.1272/jnms.72.316
Palmiter, R. D., Cole, T. B., Quaife, C. J., and Findley, S. D. (1996). ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 93, 14934–14939. doi: 10.1073/pnas.93.25.14934
Prasad, A. S. (2008). Zinc in human health: effect of zinc on immune cells. Mol. Med. 14, 353–357. doi: 10.2119/2008-00033.Prasad
Saito, T., Takahashi, K., Nakagawa, N., Hosokawa, T., Kurasaki, M., Yamanoshita, O., et al. (2000). Deficiencies of hippocampal Zn and ZnT3 accelerate brain aging of Rat. Biochem. Biophys. Res. Commun. 279, 505–511. doi: 10.1006/bbrc.2000.3946
Sandi, C. (2011). Glucocorticoids act on glutamatergic pathways to affect memory processes. Trends Neurosci. 34, 165–176. doi: 10.1016/j.tins.2011.01.006
Sekler, I., Moran, A., Hershfinkel, M., Dori, A., Margulis, A., Birenzweig, N., et al. (2002). Distribution of the zinc transporter ZnT-1 in comparison with chelatable zinc in the mouse brain. J. Comp. Neurol. 447, 201–209. doi: 10.1002/cne.10224
Sensi, S. L., Canzoniero, L. M. T., Yu, S. P., Ying, H. S., Koh, J. Y., Kerchner, G. A., et al. (1997). Measurement of intracellular free zinc in living cortical neurons: routes of entry. J. Neurosci. 15, 9554–9564.
Sensi, S. L., Paoletti, P., Koh, J. Y., Aizenman, E., Bush, A. I., and Hershfinkel, M. (2011). The neurophysiology and pathology of brain zinc. J. Neurosci. 31, 16076–16085. doi: 10.1523/JNEUROSCI.3454-11.2011
Sensi, S. L., Ton-That, D., Sullivan, P. G., Jonas, E. A., Gee, K. R., Kaczmarek, L. K., et al. (2003). Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl. Acad. Sci. U.S.A. 100, 6157–6162. doi: 10.1073/pnas.1031598100
Sindreu, C., Palmiter, R. D., and Storm, D. R. (2011). Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc. Natl. Acad. Sci. U.S.A. 108, 3366–3370. doi: 10.1073/pnas.1019166108
Smart, T. G., Xie, X., and Krishek, B. J. (1994). Modulation of inhibitory and excitatory amino acid receptor ion channels by zinc. Prog. Neurobiol. 42, 393–441. doi: 10.1016/0301-0082(94)90082-5
Stoltenberg, M., Bush, A. I., Bach, G., Smidt, K., Larsen, A., Rungby, J., et al. (2007). Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 150, 357–369. doi: 10.1016/j.neuroscience.2007.09.025
Stork, C. J., and Li, Y. V. (2010). Zinc release from thapsigargin/IP3-sensitive stores in cultured cortical neurons. J. Mol. Signal. 5, 5. doi: 10.1186/1750-2187-5-5
Swanwick, G. R. J., Kirby, M., Bruce, I., Buggy, F., Coen, R. F., Coakley, D., et al. (1998). Hypothalamic-pituitary-adrenal axis dysfunction in Alzheimer’s disease:lack of association between longitudinal and cross-sectional findings. Am. J. Psychiatry 155, 286e289.
Takeda, A. (2000). Movement of zinc and its functional significance in the brain. Brain Res. Rev. 34, 137–148. doi: 10.1016/S0165-0173(00)00044-8
Takeda, A. (2001). Zinc homeostasis and functions of zinc in the brain. Biometals 14, 343–352. doi: 10.1023/A:1012982123386
Takeda, A. (2011a). Insight into glutamate excitotoxicity from synaptic zinc homeostasis. Int. J. Alzheimers Dis. 2011, 491597. doi: 10.4061/2011/491597
Takeda, A. (2011b). Zinc signaling in the hippocampus and its relation to pathogenesis of depression. Mol. Neurobiol. 44, 167–174. doi: 10.1007/s12035-010-8158-9
Takeda, A., Ando, M., Kanno, S., and Oku, N. (2009). Unique response of zinc in the hippocampus to behavioral stress and attenuation of subsequent mossy fiber long-term potentiation. Neurotoxicology 30, 712–717. doi: 10.1016/j.neuro.2009.05.009
Takeda, A., Fuke, S., Minami, A., and Oku, N. (2007a). Role of zinc influx via AMPA/kainate receptor activation in metabotropic glutamate receptor-mediated calcium release. J. Neurosci. Res. 85, 1310–1317. doi: 10.1002/jnr.21233
Takeda, A., Fuke, S., Tsutsumi, W., and Oku, N. (2007b). Negative modulation of presynaptic activity by zinc released from Schaffer collaterals. J. Neurosci. Res. 85, 3666–3672. doi: 10.1002/jnr.21449
Takeda, A., Itoh, H., Tamano, H., and Oku, N. (2006). Responsiveness to kainate in young rats after 2-week zinc deprivation. Biometals 19, 565–572. doi: 10.1007/s10534-005-6145-9
Takeda, A., Nakamura, M., Fujii, H., and Tamano, H. (2013). Synaptic Zn2+ homeostasis and its significance. Metallomics 5, 417–423. doi: 10.1039/c3mt20269k
Takeda, A., Sawashita, J., and Okada, S. (1995). Biological half-lives of zinc and manganese in rat brain. Brain Res. 695, 53–58. doi: 10.1016/0006-8993(95)00916-E
Takeda, A., Suzuki, M., Tamano, H., Takada, S., Ide, K., and Oku, N. (2012). Involvement of glucocorticoid-mediated Zn2+ signaling in attenuation of hippocampal CA1 LTP by acute stress. Neurochem. Int. 60, 394–399. doi: 10.1016/j.neuint.2012.01.021
Takeda, A., Takada, S., Ando, M., Itagaki, K., Tamano, H., Suzuki, M., et al. (2010a). Impairment of recognition memory and hippocampal long-term potentiation after acute exposure to clioquinol. Neuroscience 171, 443–450. doi: 10.1016/j.neuroscience.2010.09.017
Takeda, A., Tamano, H., Imano, S., and Oku, N. (2010b). Increases in extracellular zinc in the amygdala in acquisition and recall of fear experience and their roles in response to fear. Neuroscience 168, 715–722. doi: 10.1016/j.neuroscience.2010.04.017
Takeda, A., Takada, S., Nakamura, M., Suzuki, M., Tamano, H., Ando, M., et al. (2011). Transient increase in Zn2+ in hippocampal CA1 pyramidal neurons causes reversible memory deficit. PLoS ONE 6:e28615. doi: 10.1371/journal.pone.0028615
Takeda, A., and Tamano, H. (2009). Insight into zinc signaling from dietary zinc deficiency. Brain Res. Rev. 62, 33–34. doi: 10.1016/j.brainresrev.2009.09.003
Takeda, A., and Tamano, H. (2010). Zinc signaling through glucocorticoid and glutamate signaling in stressful circumstances. J. Neurosci. Res. 88, 3002–3010. doi: 10.1002/jnr.22456
Takeda, A., and Tamano, H. (2012). Proposed glucocorticoid-mediated zinc signaling in the hippocampus. Metallomics 4, 614–618. doi: 10.1039/c2mt20018j
Ueno, S., Tsukamoto, M., Hirano, T., Kikuchi, K., Yamada, M. K., Nishiyama, N., et al. (2002). Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. J. Cell Biol. 158, 215–220. doi: 10.1083/jcb.200204066
Vallee, B. L., and Falchuk, K. H. (1993). The biological basis of zinc physiology. Physiol. Rev. 73, 79–118.
Venero, C., and Borrell, J. (1999). Rapid glucocorticoid effects on excitatory amino acid levels in the hippocampus: a microdialysis study in freely moving rats. Eur. J. Neurosci. 11, 2465–2473. doi: 10.1046/j.1460-9568.1999.00668.x
Weiss, J. H. (2011). Ca permeable AMPA channels in diseases of the nervous system. Front. Mol. Neurosci. 4:42. doi: 10.3389/fnmol.2011.00042
Weiss, J. H., Sensi, S. L., and Koh, J. Y. (2000). Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci. 21, 395–401. doi: 10.1016/S0165-6147(00)01541-8
Keywords: Zn2+ signal, hippocampus, cognition, glucocorticoid, glutamate
Citation: Takeda A and Tamano H (2014) Cognitive decline due to excess synaptic Zn2+ signaling in the hippocampus. Front. Aging Neurosci. 6:26. doi: 10.3389/fnagi.2014.00026
Received: 02 December 2013; Accepted: 13 February 2014;
Published online: 27 February 2014.
Edited by:
Paul Adlard, The Mental Health Research Institute, AustraliaReviewed by:
José M. Delgado-García, University Pablo de Olavide, SpainEric Blalock, University of Kentucky, USA
Copyright © 2014 Takeda and Tamano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Takeda, Department of Bioorganic Chemistry, School of Pharmaceutical Sciences, University of Shizuoka, 52-1 Yada, Suruga-ku, Shizuoka 422-8526, Japan e-mail:dGFrZWRhYUB1LXNoaXp1b2thLWtlbi5hYy5qcA==