 Surjyadipta Bhattacharjee
Surjyadipta Bhattacharjee Yuhai Zhao
Yuhai Zhao Walter J. Lukiw
Walter J. Lukiw- Departments of Neurology, Neuroscience and Ophthalmology, Louisiana State University Neuroscience Center and Health Sciences Center, New Orleans, LA, USA
One characteristic feature of Alzheimer's disease (AD) neuropathology is the progressive generation, aggregation and deposition of the 42 amino acid amyloid beta (Aβ42) peptide, and other related amyloidogenic molecules, into dense clumps of insoluble, pro-inflammatory senile plaque cores in the extracellular space of the brain. It is not generally appreciated that the Aβ42 peptide, derived via tandem beta-gamma secretase cleavage of the larger ~770 amino acid transmembrane beta-amyloid precursor protein (βAPP) is one of the “stickiest” peptides known, due in part to its high content of lipophillic and hydrophobic peptide domains (21.4% valine-isoleucine; Mager, 1998; Watson et al., 2005). Aβ42 peptide monomers have tremendously high potential for relatively rapid self-aggregation into Aβ42 peptide dimers and higher order fibrillar structures via long-range, non-covalent hydrophobic forces that over time promote β-pleated sheet conformations (Mager, 1998; Watson et al., 2005; Boutajangout and Wisniewski, 2013; Chang et al., 2013). There is abundant evidence that under normal, homeostatic conditions, Aβ42 peptide monomers and perhaps other higher order Aβ42 peptides are effectively “cleared” from the brain's extracellular medium by highly active and efficient innate-immune surveillance and phagocytic systems that limit excessive Aβ42 peptide dimerization, accumulation and further self-aggregation into pathological senile plaque lesions. Recently described, one of the key phagocytosis sensor-receptors responsible for Aβ42 peptide clearance from the human central nervous system (CNS) is very likely the triggering receptor expressed in myeloid/microglial cells 2 (TREM2) enriched in myeloid cells and microglial cells of the CNS (Benitez et al., 2013; Forabosco et al., 2013; Gonzalez Murcia et al., 2013; Guerreiro et al., 2013; Neumann and Daly, 2013; Zhao et al., 2013; Jones et al., 2014; Figure 1). This short paper is an update on some very recent observations on TREM2 neurobiology, on newly discovered roles for miRNA-34a-mediated signaling in human degenerative disease, including miRNA-34a-mediated effects on TREM2 expression, and how dysfunctional TREM2 signaling may contribute to amyloidogenesis in AD and in related progressive, inflammatory neurodegenerative diseases of the human CNS.
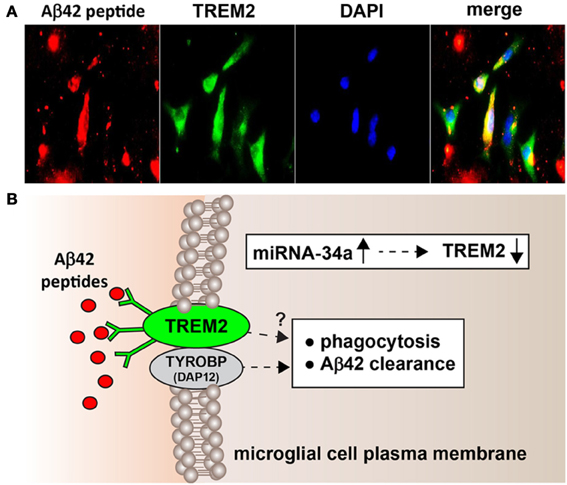
Figure 1. (A) Murine microglial cell mediated phagocytosis of Aβ42 peptides; 3 day old C8B4 murine microglia cells (ATCC CRL-2540; ATCC, Manassus VA) were treated with 5 μ M of Aβ42 for 24 h before staining; Aβ42 peptides (American Peptide Company, Sunnyvale, CA, cat # 62-0-80A) were prepared as described by Stine et al. (2003). Briefly, Aβ42 peptides were initially solubilized in hexafluoroisopropanol (HFIP; Fluka Chemical, cat# 52512; Sigma-Aldrich, St. Louis MO), aliquoted, and stored at −20°C as an HFIP film. After vacuum evaporation of HFIP, aliquoted peptide was re-suspended with DMSO to 5 mM and diluted to 5 μM into the cell culture media; cells were subsequently stained using a murine amyloid beta MABN10 (red fluorescence λmax ~650 nm; anti-Aβ antibody, clone W0-2; Millipore, Bellerica MA), a TREM-2 antibody (M-227): sc-48765 (green fluorescence; λmax ~510 nm; Santa Cruz, Santa Cruz CA) or DAPI nuclear stain; magnification 60×; note self-aggregation of Aβ42 peptide after 24 h and Aβ42 peptide affinity for TREM2 containing cells (leftmost panels) and internalization (rightmost panel; yellow merge; λmax ~580 nm); additional relevant methods have been described (Griciuc et al., 2013; Zhao and Lukiw, 2013); (B) highly schematicized depiction of the possible actions of an NF-kB-regulated, miRNA-34a-mediated TREM2 sensor-phagocytosis protein down-regulated in AD brain; the triggering receptor for myeloid/microglial cells (TREM2) is a variably glycosylated transmembrane receptor known to be enriched in the microglial cell plasma membrane; signaling via the tyrosine kinase-binding protein (DNAX activation protein 12) [TYROBP (DAP12)] accessory receptor results in phagocytosis and ultimately, clearance of Aβ42 peptides (red ovals) from the extracellular space (Satoh et al., 2011; Benitez et al., 2013; Forabosco et al., 2013; Hickman and Khoury, 2014); interestingly, TREM2 knockout/knockdown mice have attenuated immunological and inflammatory responses and/or increases in age-related neuroinflammatory markers and cognitive deficiency (Jiang et al., 2013; Sieber et al., 2013); TYROBP knockout mice exhibit immune system deficits and an impairment in microglial cell differentiation (Nataf et al., 2005; Sieber et al., 2013); it is not clear what, if any, contribution TREM2 makes directly to phagocytosis and Aβ42 clearance (question mark) without TYROBP (DAP12); while no deficits in TYROBP (DAP12) have been observed in sporadic AD brain, insufficient TREM2 may be in part responsible for the inability to adequately phagocytose Aβ42 peptides, resulting in their buildup and self-aggregation in the extracellular space. Inset: miRNA-34a is found to be significantly increased in AD hippocampal CA1 and superior temporal lobe and in stressed microglial cells; miRNA-34a targeting of the TREM2 mRNA 3′-UTR appears to be in part responsible for this (see text); because miRNA-34a is encoded on an NF-kB-sensitive transcript, both anti-NF-kB and/or anti-miRNA strategies may be clinically useful in the restoration of homeostatic phagocytosis in the brain and CNS.
TREM2 (TREM-2, Trem2a), a variably glycosylated 230 amino acid type 1 transmembrane sensor-receptor protein of the immunoglobulin gene superfamily is translated from a ~1098 nucleotide (nt) linear mRNA transcribed at chr6p21.1, is highly expressed in microglial cells of the human CNS (NCBI GenBank NG_011561; BC032362; http://www.genecards.org/cgi-bin/carddisp.pl?gene=TREM2; Zhao et al., 2013; Jones et al., 2014). As a recently recognized myeloid/microglial cell surface phagocytosis sensor-receptor, TREM2 appears to play a critical function in immune surveillance, the sensing of extracellular debris and phagocytosis throughout the CNS, including the recognition and ingestion of neurotoxic Aβ42 peptides and other amyloidogenic extracellular debris (Benitez et al., 2013; Forabosco et al., 2013; Lattante et al., 2013; Neumann and Daly, 2013; Sieber et al., 2013; Hickman and Khoury, 2014; Jiang et al., 2014). TREM2 signaling is in part mediated through a tyrosine kinase-binding protein/DNAX activation adaptor protein of 12 kDa (TYROBP/DAP12; Sieber et al., 2013; Zhao et al., 2013; Hickman and Khoury, 2014; Figure 1). Deficiencies in TREM2 abundance and/or function are associated with a defective innate-immune system, bone fragility, deficits in phagocytosis and amyloidogenesis, neurological alterations leading to presenile dementia in the autosomal recessive, inflammatory neurodegenerative disorder polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; MIM 221770; Jarvi-Hakola-Nasu disease), and more recently with Parkinson's disease (PD), AD and amyotrophic lateral sclerosis (ALS; Nataf et al., 2005; Satoh et al., 2011; Benitez et al., 2013; Forabosco et al., 2013; Guerreiro et al., 2013; Neumann and Daly, 2013; Sieber et al., 2013; Zhao et al., 2013; Abduljaleel et al., 2014; Cady et al., 2014). Conversely, acute brain injury-induced increases in TREM2 expression in microglia suggests that TREM2 may contribute to neurotrophic roles after brain ischemia and this may impart a long term neurological benefit in functional recovery (Kawabori et al., 2013; Abduljaleel et al., 2014). Genome-wide association studies and meta-analysis (GWAS/MA) for AD have recently identified an R47H (rs75932628) loss of function TREM2 variant as a significant risk factor for AD, conveying an increase for AD with an odds ratio of 1.3-8.8-fold (p = 0.008), an effect comparable to that of the e4 allele of the 299 amino acid APOE lipid transporter (Gonzalez Murcia et al., 2013; Guerreiro et al., 2013; Neumann and Daly, 2013; Zhao et al., 2013). However, TREM2 R47H mutations appear to be relatively rare occurrences and may predispose only highly selective human populations to increased risk for age-related, pro-inflammatory neurodegenerative disorders such as AD (Gonzalez Murcia et al., 2013; Guerreiro et al., 2013; Hampel and Lista, 2013; Lattante et al., 2013; Bagyinszky et al., 2014). Indeed, AD cases are each highly complex, heterogeneous and multigenic neurodegenerative disorders and can be divided into those with a genetic or familial component (~5% of all cases) or a sporadic or idiopathic form of AD with no known genetic cause (95% of all cases; Blennow et al., 2006; Lukiw, 2013b; Bagyinszky et al., 2014). AD can be further classified as either early onset (under 65 years of age), or late-onset AD (LOAD; over 65 years of age; Bagyinszky et al., 2014; Rosenthal and Kamboh, 2014). Evidence for the loss of function R47H mutation remains extremely rare in late onset AD in diverse human populations including relatively large, recent studies in Chinese Han (N = 625) or Japanese (N = 4688) groups (Ma et al., 2014; Miyashita et al., 2014; Rosenthal and Kamboh, 2014). Indeed, much recent, independently derived data support the contention that the incidence of genetic mutations and epigenetic factors in AD varies widely amongst different human populations with different evolutionary backgrounds (Raj et al., 2010; Olson, 2012; Lukiw, 2013a,b). Interestingly, genetic-based loss-of-function mutations in TREM2 in LOAD may have the same end effect as a deficiency of a functional TREM2 in sporadic AD, with both pathways leading to compromised TREM2-mediated signaling and defective Aβ42 sensing and removal by phagocytosis.
MicroRNAs (miRNAs) are small non-coding, single stranded RNAs (ssRNAs) that currently represent the smallest known carriers of highly selective genetic regulatory information in the human CNS (Lukiw, 2007; Hill et al., 2014a,b; Maffioletti et al., 2014). As highly mobile, autonomous genetic elements abundant in brain cell cytoplasm, the cerebrospinal fluid (CSF) and in the systemic circulation, miRNAs may be diagnostic for AD and other human CNS diseases (Alexandrov et al., 2012; Dorval et al., 2013). The primary mode of miRNA action is to recognize and bind to complementary ribonucleotide sequences in the 3-prime un-translated region (3′-UTR) of target messenger RNAs (mRNAs) and in doing so, down-regulate their expression (Lagos-Quintana et al., 2001; Lukiw et al., 2008; Guo et al., 2010). Several independent laboratories have recently reported the highly selective up-regulation of specific pathology-related miRNAs in AD brain including: (1) a significant up-regulation in the pro-inflammatory miRNA-34a (encoded at chr1p36.22) in AD and in Aβ42 peptide- and cytokine-stressed human primary brain cells (Cogswell et al., 2008; Lukiw, 2012; Zhao et al., 2013); (2) a significant miRNA-34a up-regulation in amyloid overexpressing transgenic mouse models of AD (Wang et al., 2009; Zhao et al., 2013); (3) a productive and CNS-relevant miRNA-34a-TREM2-3′-UTR interaction resulting in TREM2 deficits in cellular models of AD (Zhao and Lukiw, 2013; Zhao et al., 2013; Jones et al., 2014); and (4) an NF-kB-mediated up-regulation of miRNA-34a coupled to a down-regulation of TREM2 in human neuronal-glial co-cultures (Alexandrov et al., 2013). The miRNA-34a-mediated down-regulation of TREM2 appears to be due to an unusually strong miRNA-34a recognition feature within the 299 nt TREM2 mRNA 3′-UTR region (energy of association, EA <-16 kcal/mol; http://www.genecards.org/cgi-bin/carddisp.pl?gene=TREM2; Zhao et al., 2013; Abduljaleel et al., 2014; Jiang et al., 2014; Jones et al., 2014). Interestingly, the stress- and inflammation-induced transcription factor NF-kB, a driver for miRNA-34a expression, is also selectively up-regulated in AD-affected brain regions, and both NF-kB inhibitors and stabilized anti-miRNA-34a appear to be effective in restoring TREM2 back to homeostatic levels, at least in in vitro primary cell models of inflammatory neurodegeneration (Crampton and O'Keeffe, 2013; Lukiw, 2013a; Zhao and Lukiw, 2013; Zhao et al., 2013). Pathologically up-regulated miRNA-34a-signaling has also been recently associated (1) with the repression of expression of several selected genes involved in cell survival and oxidative defense pathways such as Bcl2 and SIRT1 (Bhatnagar et al., 2014); (2) with spinal cord tissues in ALS (Cady et al., 2014); (3) with altered immunity associated with multiple sclerosis (Junker et al., 2009); (4) with progressive neurotrophic deficits including dysfunctional Bcl-2 signaling in transgenic murine models of AD (Wang et al., 2009); (5) with altered synaptogenesis (Agostini et al., 2011); (6) with deficient immune and phagocytotic responses in progressive inflammatory degeneration in cardiovascular disease (Boon et al., 2013); (7) with aging of the murine brain (Li et al., 2011); (8) with vasculature aging and cellular senescence (Boon et al., 2013; Agostini and Knight, 2014); (9) with blood mononuclear cells in sporadic AD patients (Schipper et al., 2007; Bhatnagar et al., 2014); (10) with lower mini-mental state examination (MMSE) scores when detected in the blood plasma of AD patients (Bhatnagar et al., 2014) and (11) with the progressive inflammatory neurodegeneration and epileptiform activities associated with epilepsy and the early stages of AD (Zhao et al., 2013; Henshall, 2014). The role of the CNS-enriched miRNA-34a and other pro-inflammatory miRNAs in epilepsy and AD is particularly interesting due to the overlapping neuropathology of these two neurological disorders with respect to the incidence of seizures and cognitive decline first apparent in the earliest stages of each disease (Vossel et al., 2013; Hill et al., 2014a,b).
Strengthening evidence continues to support the hypothesis that multiple genes, through multiple genetic processes, drive the initiation, propagation and course of sporadic AD. Epigenetic mechanisms involving NF-κB-mediated, miRNA-34a up-regulation and consequent down-regulation of TREM2 expression may drive the progressive extinction of the phagocytic response that in turn contributes to dysfunctional innate-immunity, amyloidogenesis and inflammatory neurodegeneration. Current data also suggest that the orchestrated interaction of at least two independent gene products on two different chromosomes—miRNA-34a at chr1p36.22 and TREM2 at chr6p21.1—modulate TREM2 activities, the sensing of potentially hazardous waste molecules in the extracellular space, and the phagocytosis and clearance of this neurotoxic debris to maintain functional homeostasis in the CNS. Importantly, defective regulation of miRNA-34a and TREM2 signaling and other epigenetic effects on gene expression in sporadic AD would not be detectable via classical GWAS/MA or single nucleotide polymorphism (SNP) analysis of the genome (Hampel and Lista, 2013; Lukiw, 2013b). While dysfunction along the miRNA-34a-TREM2-TYOBP(DAP12) axis may be a particularly strong contributor to phagocytosis deficits and amyloidogenesis in AD it is important to note that other miRNA-mRNA pairings may also be involved in Aβ42 clearance and altered innate-immune responses in this complex genetic regulatory network. AD-relevant stress-mediated increases in miRNA-34a in cultured brain cells, subsequent down-regulation in the expression of TREM2-3'-UTR reporter vectors, and rescue by anti-NF-kB or anti-miRNA-34a pharmacological strategies indicates that TREM2 and accessory genetic signaling components that drive defective Aβ42 peptide sensing and phagocytosis can be effectively quenched, at least in in vitro studies (Lukiw, 2013b; Zhao et al., 2013; Jones et al., 2014). There is currently a great deal of pharmacological interest in the use of miRNA-34a mimics and their potential role in treating degenerative disease, including CNS disease and cancer, and miRNA-34a mimics have become the first half-life stabilized miRNAs to reach phase 1 clinical trials (Boon et al., 2013; Zhao et al., 2013; Agostini and Knight, 2014). Indeed anti-NF-kB, anti-miRNA-34a and/or analogous pharmacological molecular strategies may be useful in the future clinical management of AD and other multi-pathway neurological diseases with an amyloidogenic component, including novel combinatorial therapeutic approaches that have not yet been considered.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Part of this study was presented at the “Non-Coding RNAs as Biomarkers and Diagnostics Symposium, Cambridge Healthtech Institute's 10th Annual Biomarkers & Diagnostics World Congress,” April 30-May 2, 2014 in Philadelphia PA, USA. Sincere thanks are extended to Drs. L. Carver, F. Culicchia, C. Eicken, E. Head and C. Hebel for short post-mortem interval (PMI) human brain biopsies, tissues or extracts, miRNA array analysis and preliminary data interpretation, and to D Guillot and AI Pogue for expert technical assistance. Thanks are also extended to the many physicians and neuropathologists of Canada and the USA who have provided high quality, short post-mortem interval human brain tissues for study. Additional human temporal lobe and other control and AD brain tissues were provided by the Memory Impairments and Neurological Disorders (MIND) Institute and the University of California, Irvine Alzheimer's Disease Research Center (UCI-ADRC; NIA P50 AG16573). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Research on miRNA in the Lukiw laboratory involving the innate-immune response in AD, amyloidogenesis and neuroinflammation was supported through a COBRE III Pilot Project, a Translational Research Initiative Grant from LSUHSC, the Louisiana Biotechnology Research Network (LBRN), an unrestricted departmental grant from Research to Prevent Blindness (RPB) and NIH Grants NEI EY006311, NIA AG18031 and NIA AG038834.
References
Abduljaleel, Z., Al-Allaf, F. A., Khan, W., Athar, M., Shahzad, N., Taher, M. M., et al. (2014). Evidence of TREM2 variant associated with triple risk of Alzheimer's disease. PLoS ONE 9:e92648. doi: 10.1371/journal.pone.0092648
Agostini, M., Tucci, P., Killick, R., Candi, E., Sayan, B. S., Rivetti di Val Cervo, P., et al. (2011). Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc. Natl. Acad. Sci. U.S.A. 108, 21093–21098. doi: 10.1073/pnas.1112061109
Alexandrov, P. N., Dua, P., Hill, J. M., Bhattacharjee, S., Zhao, Y., and Lukiw, W. J. (2012). microRNA (miRNA) speciation in Alzheimer's disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 3, 365–373.
Alexandrov, P. N., Zhao, Y., Jones, B. M., Bhattacharjee, S., and Lukiw, W. J. (2013). Expression of the phagocytosis-essential protein TREM2 is down-regulated by an aluminum-induced miRNA-34a in a murine microglial cell line. J. Inorg. Biochem. 128, 267–269. doi: 10.1016/j.jinorgbio.2013.05.010
Bagyinszky, E., Youn, Y. C., An, S. S., and Kim, S. (2014). The genetics of Alzheimer's disease. Clin. Interv. Aging 9, 535–551. doi: 10.2147/CIA.S51571
Benitez, B. A., Cruchaga, C., and United States–Spain Parkinson's Disease Research Group. (2013). TREM2 and neurodegenerative disease. N. Engl. J. Med. 369, 1567–1568. doi: 10.1056/NEJMc1306509#SA4
Bhatnagar, S., Chertkow, H., Schipper, H. M., Yuan, Z., Shetty, V., Jenkins, S., et al. (2014). Increased microRNA-34c abundance in Alzheimer's disease circulating blood plasma. Front. Mol. Neurosci. 7:2. doi: 10.3389/fnmol.2014.00002
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer's disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Boon, R. A., Lekushi, K., Lechner, S., Seeger, T., Fischer, A., Heydt, S., et al. (2013). MicroRNA-34a regulates cardiac ageing and function. Nature 495, 107–110. doi: 10.1038/nature11919
Boutajangout, A., and Wisniewski, T. (2013). The innate immune system in Alzheimer's disease. Int. J. Cell Biol. 2013:576383. doi: 10.1155/2013/576383
Cady, J., Koval, E. D., Benitez, B. A., Zaidman, C., Jockel-Balsarotti, J., Allred, P., et al. (2014). TREM2 Variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 71, 449–453. doi: 10.1001/jamaneurol.2013.6237
Chang, C. C., Althaus, J. C., Carruthers, C. J., Sutton, M. A., Steel, D. G., and Gafni, A. (2013). Synergistic interactions between Alzheimer's Aβ40 and Aβ42 on the surface of primary neurons revealed by single molecule microscopy. PLoS ONE 8:e82139. doi: 10.1371/journal.pone.0082139
Cogswell, J. P., Ward, J., Taylor, I. A., Waters, M., Shi, Y., Cannon, B., et al. (2008). Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 14, 27–41. doi: 10.1016/j.jalz.2008.05.420
Crampton, S. J., and O'Keeffe, G. W. (2013). NF-κB: emerging roles in hippocampal development and function. Int. J. Biochem. Cell Biol. 45, 1821–1824. doi: 10.1016/j.biocel.2013.05.037
Dorval, V., Nelson, P. T., and Hébert, S. S. (2013). Circulating microRNAs in Alzheimer's disease: the search for novel biomarkers. Front. Mol. Neurosci. 6:24. doi: 10.3389/fnmol.2013.00024
Forabosco, P., Ramasamy, A., Trabzuni, D., Walker, R., Smith, C., Bras, J., et al. (2013). Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol. Aging 34, 2699–2714. doi: 10.1016/j.neurobiolaging.2013.05.001
Gonzalez Murcia, J. D., Schmutz, C., Munger, C., Perkes, A., Gustin, A., Peterson, M., et al. (2013). Assessment of TREM2 rs75932628 association with Alzheimer's disease in a population-based sample: the Cache County Study. Neurobiol. Aging 34, 2889.e11–2889.e13. doi: 10.1016/j.neurobiolaging.2013.06.004
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guo, H., Ingolia, N. T., Weissman, J. S., and Bartel, D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. doi: 10.1038/nature09267
Hampel, H., and Lista, S. (2013). Have we learnt all we need to know from genetic studies - is genetics over in Alzheimer's disease? Alzheimers Res. Ther. 5:11. doi: 10.1186/alzrt165
Henshall, D. C. (2014). MicroRNA and epilepsy: profiling, functions and potential clinical applications. Curr. Opin. Neurol. 27, 199–205. doi: 10.1097/WCO.0000000000000079
Hickman, S. E., and El Khoury, J. (2014). TREM2 and the neuroimmunology of Alzheimer's disease. Biochem. Pharmacol. 88, 495–498. doi: 10.1016/j.bcp.2013.11.021
Hill, J. M., Bhattacharjee, S., Pogue, A. I., and Lukiw, W. J. (2014b). The gastrointestinal (GI) tract microbiome and potential link to Alzheimer's disease (AD). Front. Neurol. 5:43. doi: 10.3389/fneur.2014.00043
Hill, J. M., Zhao, Y., Bhattacharjee, S., and Lukiw, W. J. (2014a). miRNAs and viroids utilize common strategies in genetic signal transfer. Front. Mol. Neurosci. 7:10. doi: 10.3389/fnmol.2014.00010
Jiang, T., Yu, J. T., Zhu, X. C., and Tan, L. (2013). TREM2 in Alzheimer's disease. Mol. Neurobiol. 48, 180–185. doi: 10.1007/s12035-013-8424-8
Jiang, T., Yu, J. T., Zhu, X. C., Tan, M. S., Gu, L. Z., Zhang, Y. D., et al. (2014). Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol. Aging 35, 1243–1251. doi: 10.1016/j.neurobiolaging.2013.11.026
Jones, B. M., Bhattacharjee, S., Dua, P., Hill, J. M., Zhao, Y., and Lukiw, W. J. (2014). Regulating amyloidogenesis through the natural triggering receptor expressed in myeloid/microglial cells 2 (TREM2). Front. Cell. Neurosci. 8:94. doi: 10.3389/fncel.2014.00094
Junker, A., Krumbholz, M., Eisele, S., Mohan, H., Augstein, F., Bittner, R., et al. (2009). MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 132, 3342–3352. doi: 10.1093/brain/awp300
Kawabori, M., Hokari, M., Zheng, Z., Kim, J. Y., Calosing, C., Hsieh, C. L., et al. (2013). Triggering receptor expressed on myeloid cells-2 correlates to hypothermic neuroprotection in ischemic stroke. Ther. Hypothermia Temp. Manag. 3, 189–198. doi: 10.1089/ther.2013.0020
Lagos-Quintana, M., Rauhut, R., Lendeckel, W., and Tuschl, T. (2001). Identification of novel genes coding for small expressed RNAs. Science 294, 853–858. doi: 10.1126/science.1064921
Lattante, S., Le Ber, I., Camuzat, A., Dayan, S., Godard, C., Van Bortel, I., et al. (2013). TREM2 mutations are rare in a French cohort of patients with frontotemporal dementia. Neurobiol. Aging 34, 2443.e1–2443.e2. doi: 10.1016/j.neurobiolaging.2013.04.030
Li, X., Khanna, A., Li, N., and Wang, E. (2011). Circulatory miR34a as an RNA-based, noninvasive biomarker for brain aging. Aging (Albany NY) 3, 985–1002.
Lukiw, W. J. (2007). Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport 18, 297–300. doi: 10.1097/WNR.0b013e3280148e8b
Lukiw, W. J. (2012). NF-κB-regulated, proinflammatory miRNAs in Alzheimer's disease. Alzheimers Res. Ther. 4, 47. doi: 10.1186/alzrt150
Lukiw, W. J. (2013a). Antagonism of NF-κB-up-regulated micro RNAs (miRNAs) in sporadic Alzheimer's disease (AD): anti-NF-κB vs. anti-miRNA strategies. Front. Genet. 4:77. doi: 10.3389/fgene.2013.00077
Lukiw, W. J. (2013b) Variability in micro RNA (miRNA) abundance, speciation and complexity amongst different human populations and potential relevance to Alzheimer's disease (AD). Front. Cell. Neurosci. 7:133. doi: 10.3389/fncel.2013.00133
Lukiw, W. J., Zhao, Y., and Cui, J. G. (2008). An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 283, 31315–31322. doi: 10.1074/jbc.M805371200
Ma, J., Zhou, Y., Xu, J., Liu, X., Wang, Y., Deng, Y., et al. (2014). Association study of TREM2 polymorphism rs75932628 with late-onset Alzheimer's disease in Chinese Han population. Neurol. Res. 13:1743132814Y0000000376. doi: 10.1179/1743132814Y.0000000376
Maffioletti, E., Tardito, D., Gennarelli, M., and Bocchio-Chiavetto, L. (2014). Micro spies from the brain to the periphery: new clues from studies on microRNAs in neuropsychiatric disorders. Front. Cell. Neurosci. 8:75. doi: 10.3389/fncel.2014.00075
Mager, P. P. (1998). Molecular simulation of the primary and secondary structures of the Abeta (1-42)-peptide of Alzheimer's disease. Med. Res. Rev. 18, 403–430.
Miyashita, A., Wen, Y., Kitamura, N., Matsubara, E., Kawarabayashi, T., Shoji, M., et al. (2014). Lack of genetic association between TREM2 and late-onset Alzheimer's disease in a Japanese population. J. Alzheimers Dis. doi: 10.3233/JAD-140225. [Epub ahead of print].
Nataf, S., Anginot, A., Vuaillat, C., Malaval, L., Fodi, L. N., Chereul, E., et al. (2005). Brain and bone damage in KARAP/DAP12 loss-of-function mice correlate with alterations in microglia and osteoclast lineages. Am. J. Pathol. 166, 275–286. doi: 10.1016/S0002-9440(10)62251-1
Neumann, H., and Daly, M. J. (2013). Variant TREM2 as risk factor for Alzheimer's disease. N. Engl. J. Med. 368, 182–184. doi: 10.1056/NEJMe1213157
Olson, M. V. (2012). Human genetic individuality. Annu. Rev. Genomics Hum. Genet. 13, 1–27. doi: 10.1146/annurev-genom-090711-163825
Raj, A., Rifkin, S. A., Andersen, E., and van Oudenaarden, A. (2010). Variability in gene expression underlies incomplete penetrance. Nature 463, 913–918. doi: 10.1038/nature08781
Rosenthal, S. L., and Kamboh, M. I. (2014). Late-onset Alzheimer's disease genes and the potentially implicated pathways. Curr. Genet. Med. Rep. 2, 85–101. doi: 10.1007/s40142-014-0034-x
Satoh, J., Shimamura, Y., and Tabunoki, H. (2011). Gene expression profile of THP-1 monocytes following knockdown of DAP12, a causative gene for Nasu-Hakola disease. Cell. Mol. Neurobiol. 32, 337–343. doi: 10.1007/s10571-011-9769-z
Schipper, H. M., Maes, O. C., Chertkow, H. M., and Wang, E. (2007). MicroRNA expression in Alzheimer blood mononuclear cells. Gene Regul. Syst. Bio. 1, 263–274.
Sieber, M. W., Jaenisch, N., Brehm, M., Guenther, M., Linnartz-Gerlach, B., and Neumann, H. (2013). Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS ONE 8:e52982. doi: 10.1371/journal.pone.0052982
Stine, W. B. Jr. Dahlgren, K. N., Krafft, G. A., and LaDu, M. J. (2003). In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622. doi: 10.1074/jbc.M210207200
Vossel, K. A., Beagle, A. J., Rabinovici, G. D., Shu, H., Lee, S. E., Naasan, G., et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. doi: 10.1001/jamaneurol.2013.136
Wang, X., Liu, P., Zhu, H., Xu, Y., Ma, C., Dai, X., et al. (2009). miRNA-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer's disease, inhibits bcl2 translation. Brain Res. Bull. 80, 268–273. doi: 10.1016/j.brainresbull.2009.08.006
Watson, D., Castaño, E., Kokjohn, T. A., Kuo, Y. M., Lyubchenko, Y., Pinsky, D., et al. (2005). Physicochemical characteristics of soluble oligomeric Aβ and their pathologic role in Alzheimer's disease. Neurol. Res. 27, 869–881. doi: 10.1179/016164105X49436
Zhao, Y., Bhattacharjee, S., Jones, B. M., Dua, P., Alexandrov, P. N., Hill, J. M., et al. (2013). Regulation of TREM2 expression by an NF-êB-sensitive miRNA-34a. Neuroreport 24, 318–323. doi: 10.1097/WNR.0b013e32835fb6b0
Keywords: Alzheimer's disease, miRNA-34a, TREM2, phagocytosis, innate-immune response, amyloidosis
Citation: Bhattacharjee S, Zhao Y and Lukiw WJ (2014) Deficits in the miRNA-34a-regulated endogenous TREM2 phagocytosis sensor-receptor in Alzheimer's disease (AD); an update. Front. Aging Neurosci. 6:116. doi: 10.3389/fnagi.2014.00116
Received: 06 April 2014; Accepted: 24 May 2014;
Published online: 17 June 2014.
Edited and reviewed by: Robert Marr, Rosalind Franklin University of Medicine and Science, USA
Copyright © 2014 Bhattacharjee, Zhao and Lukiw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:d2x1a2l3QGxzdWhzYy5lZHU=