 George E. Barreto
George E. Barreto Francisco Capani2
Francisco Capani2 Ricardo Cabezas
Ricardo Cabezas- 1Departamento de Nutrición y Bioquímica, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá D.C., Colombia
- 2Laboratorio de Citoarquitectura y Plasticidad Neuronal, Facultad de Medicina, Instituto de Investigaciones Cardiológicas Prof. Dr. Alberto C. Taquini (ININCA), UBA-CONICET, Buenos Aires, Argentina
Chronic consumption of ethanol has a damaging effect in various organs and metabolic functions, including liver, kidney, heart, pancreas, and brain (Sun and Sun, 2001; Bezerra Rde et al., 2005). Additionally, it has been shown that both the acute and chronic ethanol intake induce alteration in voltage-gated channels causing behavioral and electrophysiological changes in the brain (Little, 1999). In the last few years, the effect of ethanol in the electrophysiological phenomenon, known as cortical spreading depression (CSD), has been widely assessed (Sonn and Mayevsky, 2001; Bezerra Rde et al., 2005; Abadie-Guedes et al., 2008) as an important model to predict the damaging effects of ethanol in both young and aged brains.
Cortical spreading depression is a neuronal and glial depolarization wave in gray matter, which is thought to be associated as an upstream event in migraine with aura, spontaneous intracerebral hemorrhage, ischemic injury and brain trauma (Zhou et al., 2010; Dreier, 2011). This phenomenon has a duration of approximately 1 min and very slow propagation (2–5 mm/min) when compared to action potentials, resulting in a suppression of electrical activity and disruption of ion homeostasis (Bennett et al., 2008; Seidel and Shuttleworth, 2011). This depolarization occurs when the extracellular K+ concentrations increase above a threshold of 40 mM, causing the CSD wave propagation into adjacent cells. CSD also requires the activation of the N-methyl-D-aspartate (NMDA) subtype of glutamate receptor, which facilitates the spread of the depolarization wave (Eikermann-Haerter and Ayata, 2010).
There are experimental evidences to suggest that various factors influence the brain CSD propagation, including pharmacological substances, age, ethanol abuse and the insufficient ingestion of antioxidants (Abadie-Guedes et al., 2008). To support this idea, previous report has shown that acute ethanol ingestion impairs the CSD propagation when compared to chronic administration of 7, or more days, in which this phenomenon is somehow stimulated (Bezerra Rde et al., 2005). Furthermore, it was previously reported that ethanol-treated rats had alterations in the NADH oxidation cycle and a decrease in energy production and CSD wave frequency (Sonn and Mayevsky, 2001). Acute ethanol administration has been also associated with the inhibition of the Na+-K+-ATPase pump function, the formation of free oxygen radicals and CSD regulation (Sonn and Mayevsky, 2001). In this aspect, ethanol increases lipid peroxidation in the brain and the serum NO (nitric oxide) levels, probably due to the activation of the inducible nitric oxide synthase (iNOS) by cytokines (Zima et al., 2001). Chronic ethanol administration affects the antioxidant defenses of the brain by reducing the levels of glutathione, peroxidase, α-tocopherol, glutathione reductase and catalase (Bezerra Rde et al., 2005), and the aged brain is particularly affected by this reduced antioxidant scavenging system (Reiter, 1995). Indeed, it is possible that cytoplasmic calcium levels may also be implicated in CSD deflagration (Torrente et al., 2014b). Altogether, these findings suggest that there are multiple mechanisms by which ethanol may damage the brain (Figure 1).
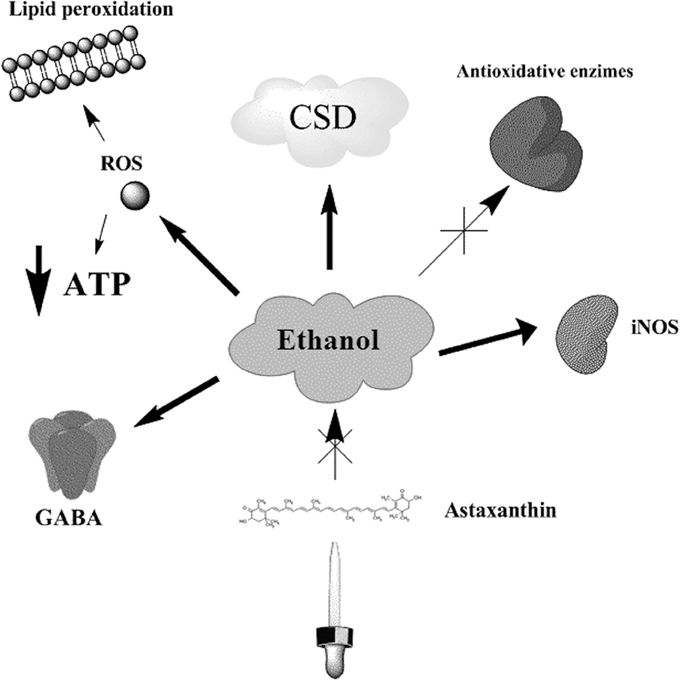
Figure 1. Ethanol effects on CSD related processes. Ethanol administration induces the inhibition of Na+-K+ -ATPase pump, depleting the ATP contents of the cell, and causing the generation of ROS and lipid peroxidation. Both chronic and acute administration of ethanol has impairing effects in CSD velocities, which in turn may reflect important alterations on the bioenergetic metabolism. Additionally, ethanol increases the expression of iNOS and the formation of NO, decreases the levels of antioxidant enzymes, like glutathione reductase and catalase, and promotes the activation of GABA receptor related responses. The administration of astaxanthin counteracts the effects induced by ethanol in CSD velocities.
Although one of the main mechanisms of ethanol-induced brain damage is due to an augmented production of reactive oxygen species (ROS), thus leading to increased CSD propagation, this statement has not been experimentally addressed. Most studies on CSD focused on the electrophysiological effects of ethanol abuse, and their reversion by some protective molecules, such as astaxanthin (Abadie-Guedes et al., 2008, 2012). To address this concern, it is important to point that (i) ethanol facilitates P450 2E1 activity, which metabolizes this compound and generates ROS in the process; and (ii) ethanol increases the levels of free iron in the cell, which also can promote ROS generation. It is possible that most antioxidants may reverse, scavenge, or even impair the production of free radicals and exert protection through this pathway. Ethanol affects the immune system in aged mice, suggesting that this might be one the main mechanisms by which ethanol induces neurodegeneration in aged brains (Drew and Kane, 2013; Kane et al., 2014).
Interestingly, astrocytes have been shown to be more resistant than neurons during ethanol-induced oxidative stress because of their greater content of glutathione (GSH) and other antioxidant enzymes (Watts et al., 2005; Barreto et al., 2011), and these cells may play an important role in CSD (Torrente et al., 2014a). On the other hand, the disruption of astrocyte oxidative metabolism, which results in ATP levels decline, has been shown to increase the rate of spreading depression in rat cortex (Largo et al., 1997; Seidel and Shuttleworth, 2011), suggesting that metabolic alterations of astrocytes are an important regulator of CSD events, especially during normal aging process (Farkas et al., 2011; Paradies et al., 2013; Yin et al., 2014). ATP depletion is likely to occur due to the oxidation of ethanol to acetaldehyde catalyzed by alcohol/dehydrogenase containing the coenzyme NAD+. The acetaldehyde is further oxidized to acetic acid and finally CO2 and water through the citric acid cycle. A number of metabolic effects from ethanol are directly linked to the production of an excess of both NADH and acetaldehyde. In this context, more NADH will dislocate the equilibrium of converting glucose to piruvate for energy production toward a overproduction of lactate and malate from piruvate. The increased accumulation of acetaldehyde by reduced conversion of this product into acetic acid—both processes impair mitochondrial functions and energetic metabolism—results in decreased ATP production. It is important to point that brain metabolism in the aged brain is somehow impaired, therefore the damaging effects of CSD are augmented (Farkas et al., 2011; Batista-De-Oliveira et al., 2012).
Following a cortical spreading depression event, metabolic recovery is compromised, as glucose and glycogen levels are significantly decreased and lactate levels are found increased (anaerobic shuttle), thus leading to intense depolarization and cell death (Selman et al., 2004). In this context, we hypothesize that aging influences brain electrophysiological functioning due to reduced metabolic shuttle and antioxidant defense, and the impairment of both energy metabolism and redox homeostasis are a hallmark of brain aging (Yin et al., 2014). These observations are particularly seen in older people with alcoholism problems. A previous study reported that brain tissue loss was found increased in older subjects with alcoholism compared to younger subjects with alcoholism (Oscar-Berman et al., 1997), and this may accelerate normal aging process or cause premature aging of the brain. Importantly, ethanol was shown to impair vasodilatation enzymes such as nNOS (neuronal nitric oxide synthase) and eNOS (endothelial nitric oxide synthase) by the augmentation of ROS, and this was associated with an increase in ischemic brain damage in elder individuals (Sun et al., 2006).
To counteract this damaging effect of ethanol on aged brain, and following CSD, different antioxidant molecules and enzymes, such as α-tocopherol, ascorbic acid, peroxiredoxin, and astaxanthin are shown to be neuroprotective against ethanol intake in animal models (Bezerra Rde et al., 2005; Lee et al., 2010; Zhu et al., 2012). The carotenoid astaxanthin (3,3′-dihydroxy-β,β-carotene-4,4′-dione) has important metabolic functions, including its conversion to vitamin A, enhancement of immune response, inhibition of NO and lipopolysaccaride (LPS)-induced inflammation, scavenging action on reactive oxygen species, and protection of gastric mucosa against ethanol-induced injuries (Kim et al., 2005; Lee et al., 2010). Astaxanthin was also shown to protect against induced cerebral ischemia in rats by the inhibition on iNOS and induce the expression of Hsp32 and Hsp70, demonstrating a decrease in oxidative stress without affecting cell viability. This information supports the idea of an oxidative mechanism by which astaxanthin may be exerting a protective effect on ethanol induced damage. On this perspective, new findings on the antioxidant effects of astaxanthin and their relationship with CSD were previously reported (Abadie-Guedes et al., 2012). In this study, the antagonistic effects of acute ethanol consumption on CSD propagation in two groups of rats with different ages were assessed. Their main results showed that CSD outcome was counteracted by astaxanthin administration. Age of animals and time of ethanol/astaxantin administration are the key features, and age related effects of ethanol in the brain should be further addressed in additional experiments related with ROS production in both brain and blood of the treated animals, as well as the expression of antioxidant enzymes such as iNOs or SOD (superoxide dismutase). In this point of view, some hypotheses must be raised: (i) Perhaps, the antioxidant properties of astaxanthin on the scavenging of ROS could explain the reversion of ethanol effect on CSD velocities; and (ii) alternatively, it is possible that ethanol action on CSD involves an interaction with the GABA-chloride ion channels and the NMDA receptors, causing a suppression of nerve cell activity, through ionic modulation (Sonn and Mayevsky, 2001; Criswell et al., 2003). Whether or not this relationship between astaxanthin and the GABA is clear, this is a concern that deserves further attention. Moreover, it is important to address the possible neuroprotective effects of other antioxidants on the modulation of CSD in the setting of a pathophysiological condition, as is migraine with aura, stroke or brain injury, and also in the normal aging process.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abadie-Guedes, R., Guedes, R. C., and Bezerra, R. S. (2012). The impairing effect of acute ethanol on spreading depression is antagonized by astaxanthin in rats of 2 young-adult ages. Alcohol. Clin. Exp. Res. 36, 1563–1567. doi: 10.1111/j.1530-0277.2012.01766.x
Abadie-Guedes, R., Santos, S. D., Cahu, T. B., Guedes, R. C., and De Souza Bezerra, R. (2008). Dose-dependent effects of astaxanthin on cortical spreading depression in chronically ethanol-treated adult rats. Alcohol. Clin. Exp. Res. 32, 1417–1421. doi: 10.1111/j.1530-0277.2008.00710.x
Barreto, G. E., Gonzalez, J., Torres, Y., and Morales, L. (2011). Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci. Res. 71, 107–113. doi: 10.1016/j.neures.2011.06.004
Batista-De-Oliveira, M., Lopes, A. A., Mendes-Da-Silva, R. F., and Guedes, R. C. (2012). Aging-dependent brain electrophysiological effects in rats after distinct lactation conditions, and treadmill exercise: a spreading depression analysis. Exp. Gerontol. 47, 452–457. doi: 10.1016/j.exger.2012.03.016
Bennett, M. R., Farnell, L., and Gibson, W. G. (2008). A quantitative model of cortical spreading depression due to purinergic and gap-junction transmission in astrocyte networks. Biophys. J. 95, 5648–5660. doi: 10.1529/biophysj.108.137190
Bezerra Rde, S., Abadie-Guedes, R., Melo, F. R., Paiva, A. M., Amancio-Dos-Santos, A., and Guedes, R. C. (2005). Shrimp carotenoids protect the developing rat cerebral cortex against the effects of ethanol on cortical spreading depression. Neurosci. Lett. 391, 51–55. doi: 10.1016/j.neulet.2005.08.040
Criswell, H. E., Ming, Z., Griffith, B. L., and Breese, G. R. (2003). Comparison of effect of of ethanol on N-methyl-D-aspartate- and GABA-gated currents from acutely dissociated neurons: absence of regional differences in sensitivity to ethanol. J. Pharmacol. Exp. Ther. 304, 192–199. doi: 10.1124/jpet.102.041590
Dreier, J. P. (2011). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 17, 439–447. doi: 10.1038/nm.2333nm.2333
Drew, P. D., and Kane, C. J. (2013). “Neuroimmune mechanisms of glia and their interplay with alcohol exposure across the lifespan,” in Neural-Immune Interactions in Brain Function and Alcohol Related Disorders, ed C. Cui (New York, NY: Springer Science + Business Media), 359–386.
Eikermann-Haerter, K., and Ayata, C. (2010). Cortical spreading depression and migraine. Curr. Neurol. Neurosci. Rep. 10, 167–173. doi: 10.1007/s11910-010-0099-1
Farkas, E., Obrenovitch, T. P., Institoris, A., and Bari, F. (2011). Effects of early aging and cerebral hypoperfusion on spreading depression in rats. Neurobiol. Aging 32, 1707–1715. doi: 10.1016/j.neurobiolaging.2009.10.002
Kane, C. J., Phelan, K. D., Douglas, J. C., Wagoner, G., Johnson, J. W., Xu, J., et al. (2014). Effects of ethanol on immune response in the brain: region-specific changes in adolescent versus adult mice. Alcohol. Clin. Exp. Res. 38, 384–391. doi: 10.1111/acer.12244
Kim, J. H., Choi, S. K., Choi, S. Y., Kim, H. K., and Chang, H. I. (2005). Suppressive effect of astaxanthin isolated from the Xanthophyllomyces dendrorhous mutant on ethanol-induced gastric mucosal injury in rats. Biosci. Biotechnol. Biochem. 69, 1300–1305. doi: 10.1271/bbb.69.1300
Largo, C., Ibarz, J. M., and Herreras, O. (1997). Effects of the gliotoxin fluorocitrate on spreading depression and glial membrane potential in rat brain in situ. J. Neurophysiol. 78, 295–307.
Lee, D. H., Lee, Y. J., and Kwon, K. H. (2010). Neuroprotective effects of astaxanthin in oxygen-glucose deprivation in sh-sy5y cells and global cerebral ischemia in rat. J. Clin. Biochem. Nutr. 47, 121–129. doi: 10.3164/jcbn.10-29
Little, H. J. (1999). The contribution of electrophysiology to knowledge of the acute and chronic effects of ethanol. Pharmacol. Ther. 84, 333–353. doi: 10.1016/S0163-7258(99)00040-6
Oscar-Berman, M., Shagrin, B., Evert, D. L., and Epstein, C. (1997). Impairments of brain and behavior: the neurological effects of alcohol. Alcohol Health Res. World 21, 65–75.
Paradies, G., Paradies, V., Ruggiero, F. M., and Petrosillo, G. (2013). Changes in the mitochondrial permeability transition pore in aging and age-associated diseases. Mech. Ageing Dev. 134, 1–9. doi: 10.1016/j.mad.2012.12.006
Reiter, R. J. (1995). Oxidative processes and antioxidative defense mechanisms in the aging brain. FASEB J. 9, 526–533.
Seidel, J. L., and Shuttleworth, C. W. (2011). Contribution of astrocyte glycogen stores to progression of spreading depression and related events in hippocampal slices. Neuroscience 192, 295–303. doi: 10.1016/j.neuroscience.2011.05.006
Selman, W. R., Lust, W. D., Pundik, S., Zhou, Y., and Ratcheson, R. A. (2004). Compromised metabolic recovery following spontaneous spreading depression in the penumbra. Brain Res. 999, 167–174. doi: 10.1016/j.brainres.2003.11.016
Sonn, J., and Mayevsky, A. (2001). The effect of ethanol on metabolic, hemodynamic and electrical responses to cortical spreading depression. Brain Res. 908, 174–186. doi: 10.1016/S0006-8993(01)02643-9
Sun, A. Y., and Sun, G. Y. (2001). Ethanol and oxidative mechanisms in the brain. J. Biomed. Sci. 8, 37–43. doi: 10.1159/000054011
Sun, H., Molacek, E., Zheng, H., Fang, Q., Patel, K. P., and Mayhan, W. G. (2006). Alcohol-induced impairment of neuronal nitric oxide synthase (nNOS)-dependent dilation of cerebral arterioles: role of NAD(P)H oxidase. J. Mol. Cell. Cardiol. 40, 321–328. doi: 10.1016/j.yjmcc.2005.11.004
Torrente, D., Cabezas, R., Avila, M. F., Garcia-Segura, L. M., Barreto, G. E., and Guedes, R. C. (2014a). Cortical spreading depression in traumatic brain injuries: is there a role for astrocytes? Neurosci. Lett. 565C, 2–6. doi: 10.1016/j.neulet.2013.12.058
Torrente, D., Mendes-Da-Silva, R. F., Lopes, A. A., Gonzalez, J., Barreto, G. E., and Guedes, R. C. (2014b). Increased calcium influx triggers and accelerates cortical spreading depression in vivo in male adult rats. Neurosci. Lett. 558, 87–90. doi: 10.1016/j.neulet.2013.11.004
Watts, L. T., Rathinam, M. L., Schenker, S., and Henderson, G. I. (2005). Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death. J. Neurosci. Res. 80, 655–666. doi: 10.1002/jnr.20502
Yin, F., Boveris, A., and Cadenas, E. (2014). Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 20, 353–371. doi: 10.1089/ars.2012.4774
Zhou, N., Gordon, G. R., Feighan, D., and Macvicar, B. A. (2010). Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb. Cortex 20, 2614–2624. doi: 10.1093/cercor/bhq018bhq018
Zhu, H., Santo, A., and Li, Y. (2012). The antioxidant enzyme peroxiredoxin and its protective role in neurological disorders. Exp. Biol. Med. (Maywood) 237, 143–149. doi: 10.1258/ebm.2011.011152
Keywords: cortical spreading depression, ethanol, mitochondria dysfunction, antioxidants, aging
Citation: Barreto GE, Capani F and Cabezas R (2014) Cortical spreading depression and mitochondrial dysfunction with aging: lessons from ethanol abuse. Front. Aging Neurosci. 6:117. doi: 10.3389/fnagi.2014.00117
Received: 15 April 2014; Accepted: 25 May 2014;
Published online: 10 June 2014.
Edited by:
Merce Pallas, University of Barcelona, SpainReviewed by:
Enrico Sanna, University of Cagliari, ItalyRubem C. A. Guedes, Universidade Federal de Pernambuco, Brazil
Copyright © 2014 Barreto, Capani and Cabezas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:Z3NhbXBhaW9AamF2ZXJpYW5hLmVkdS5jbw==