 Wei Qiao Qiu1,2,3*
Wei Qiao Qiu1,2,3* Haihao Zhu2
Haihao Zhu2- 1Department of Psychiatry, Boston University School of Medicine, Boston, MA, USA
- 2Department of Pharmacology and Experimental Therapeutics, Boston University School of Medicine, Boston, MA, USA
- 3Alzheimer's Disease Center, Boston University School of Medicine, Boston, MA, USA
Amylin, a gut-brain axis hormone, and amyloid-beta peptides (Aβ), a major component of the Alzheimer's disease (AD) brain, share several features, including similar β-sheet secondary structures, binding to the same receptor and being degraded by the same protease, insulin degrading enzyme (IDE). However, while amylin readily crosses the blood brain barrier (BBB) and mediates several activities including improving glucose metabolism, relaxing cerebrovascular structure, modulating inflammatory reaction and perhaps enhancing neural regeneration, Aβ has no known physiological functions. Thus, abundant Aβ in the AD brain could block or interfere with the binding of amylin to its receptor and hinder its functions. Recent studies using animal models for AD demonstrate that amylin and its analog reduce the AD pathology in the brain and improve cognitive impairment in AD. Given that, in addition to amyloid plaques and neurofibrillary tangles, perturbed cerebral glucose metabolism and cerebrovascular damage are the hallmarks of the AD brain, we propose that giving exogenous amylin type peptides have the potential to become a new avenue for the diagnosis and therapeutic of AD. Although amylin's property of self-aggregation may be a limitation to developing it as a therapeutic for AD, its clinical analog, pramlintide containing 3 amino acid differences from amylin, does not aggregate like human amylin, but more potently mediates amylin's activities in the brain. Pramlintide is an effective drug for diabetes with a favorable profile of safety. Thus a randomized, double-blind, placebo-controlled clinical trial should be conducted to examine the efficacy of pramlintide for AD. This review summarizes the knowledge and findings on amylin type peptides and discuss pros and cons for their potential for AD.
Introduction
As the number of Alzheimer's disease (AD) patients grows rapidly in the U.S and globally, the need to find effective treatments for the disease becomes more urgent. Currently there are only a few medications prescribed that delay cognitive decline in AD, but their effects are modest and do not modify the underlying disease process. From the perspective of drug discovery, while it is still important to target the core pathology of AD, e.g., amyloid plaques and tauopathy in the brain, it may also be beneficial and effective to treat the downstream of the pathological cascade, including perturbed glucose metabolism, damaged cerebrovasculature, and imbalanced inflammatory reaction, which causes neuronal death and inhibit neuronal regeneration. Our recent study found that treatment with amylin, a gut-brain axis peptide, reduces the AD pathology and improves cognitive impairment in animal models for AD. This review article will summarize the research data and knowledge including our own on amylin, and propose a hypothesis that amylin class peptides can be a potential treatment for AD. Meanwhile, we will discuss the tendency of amylin to form aggregation in type 2 diabetes. The goal of this review is to debate benefit vs. harm of amylin type peptides for the treatment of AD.
Pancreatic Peptide Amylin
Amylin (also known as islet amyloid polypeptide, or IAPP) is a 37–amino acid peptide hormone, and its gene is encoded on chromosome 12 and highly conserved in mammals during evolution (Nishi et al., 1989; Chang et al., 2004). While amylin is mainly produced and secreted by the β-cells in the pancreas (Westermark et al., 1987), its expression occurs in other locations such as the gut (Mulder et al., 1994) and in the sensory nervous system (Mulder et al., 1995).
Amylin peptide is stored together with insulin in dense core secretory granules in the pancreas (Lukinius et al., 1989). It is secreted in response to diet/nutrient intake (Vine et al., 1998) and exercise (Kraemer et al., 2002) stimuli, and displays a profile similar to that of insulin. The peptide circulates in a non-glycosylated (50%) and a glycosylated form (Nyholm et al., 1998), the former being the biological active compound. In healthy humans, fasting plasma amylin concentrations are in the range of 4–25 pmol/l, and amylin is distributed equally to insulin in plasma and interstitial fluids. Unlike insulin, amylin is not eliminated significantly in the liver (Kautzky-Willer et al., 1994) but mainly through renal metabolism (Hoppener et al., 2000).
Amylin belongs to the calcitonin gene peptide superfamily consisting of calcitonin (CT), calcitonin gene-related peptide (CGRP) and adrenomedullin in addition to amylin (Wimalawansa, 1997). These peptides bind to the calcitonin receptor (CTR) complexed with different receptor-activity-modifying protein (RAMPs) (Gebre-Medhin et al., 2000). As amylin readily crosses the blood brain barrier (BBB) (Banks et al., 1995; Banks and Kastin, 1998; Olsson et al., 2007), CTR and RAMPs are highly expressed in the brain. Individual RAMPs have been disrupted revealing a range of phenotypes. Mice with a disrupted RAMP1 gene were hypertensive and exhibited a dysregulated immune response, while removal of RAMP2 was lethal, and RAMP3 knockout mice appeared normal until old age when they were not as heavy as their wild-type littermates (Sexton et al., 2009). It is shown that herodimers between the CTR and RAMP1 or RAMP3 preferentially bind amylin (Christopoulos et al., 1999).
Amylin Reduces the AD Pathology and Improves Cognitive Impairment in the Animal Models for AD
Amyloid-β peptide (Aβ is a major component of AD pathology in the brain (Hardy and Selkoe, 2002). Amylin and Aβ share several features, including similar β-sheet secondary structures (Lim et al., 2008), binding to the same amylin receptor (Fu et al., 2012) and being degraded by Qiu et al. (1998); Bennett et al. (2003); Shen et al. (2006) or bound to insulin degrading enzyme (IDE) (de Tullio et al., 2013). Since amylin readily crosses the BBB (Banks et al., 1995; Banks and Kastin, 1998; Olsson et al., 2007), our team studied peripheral amylin's action on the amyloid pathology of AD in the brain.
Using amyloid precursor protein (APP) transgenic mice, we surprisingly found that chronic intraperitoneal (i.p.) injection of AD animals with both amylin and its analog, pramlintide, reduces the amyloid burden as well as lowers the concentrations of Aβ (Figure 1) (Zhu et al., 2014). These treatments significantly improve learning and memory in these mice as assessed by two behavioral tests, Y maze and Morris water maze. Similar to the finding by Adler et al. (2014), our unpublished data also found that elders who had mild cognitive impairment (MCI) or AD had lower concentrations of plasma amylin than controls in the absence diabetes (Table 1). Further, we found that increasing quartiles of plasma amylin were positively associated with the test scores of memory, visuospacial and executive function, but not with those of language and attention, after adjusting for demographic information, ApoE4 allele, diabetes, stroke, kidney function, and lipid profile (Table 2) (submitted and in revision). Given that impairments in these cognitive domains are signature symptoms of an early stage of AD (Weintraub et al., 2009), both mouse and human studies suggest that amylin, natural or synthetic, are likely to reduce the AD pathology in the brain and provides a new avenue of treatment for the disease.
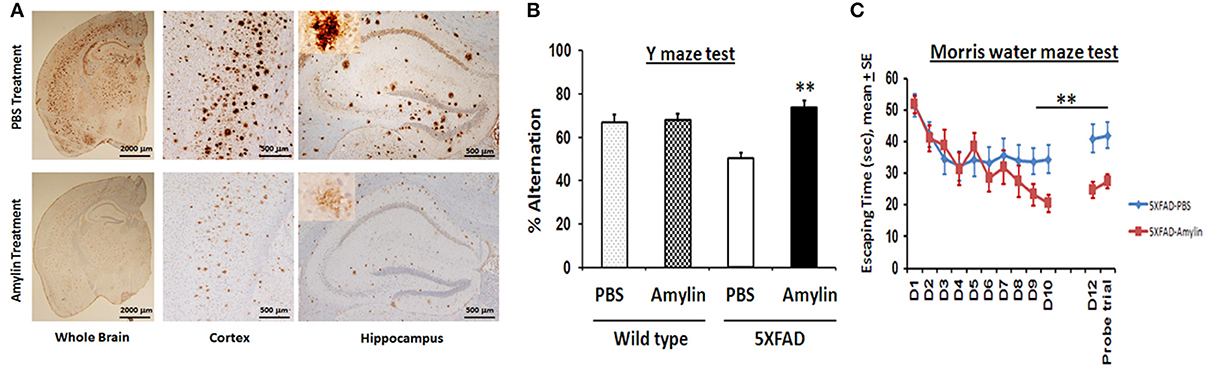
Figure 1. Amylin treatment of 5XFAD mice reduces the amyloid burden and improves their learning and memory. At 3.5 months of age, 5XFAD mice were treated by i.p. injection of PBS or amylin (200 pg/kg) daily for 10 weeks (n = 10 per group). (A) Dense-cored Aβ plaque burden is reduced in the whole brain including the cerebral cortex and the hippocampus. The amylin treated 5XFAD mice illustrated improved cognition by showing increased percent alternation in the Y maze test (B) (p = 0.001) and by showing shortened times in Morris water maze test (C) in finding the hidden platform at day 10 (D10) (p = 0.005), in memory at day 12 (D12) after the completion of training and skipping day 11 (p = 0.002), and in the probe trial (p = 0.03). Mean ± SE was used with **p < 0.01.
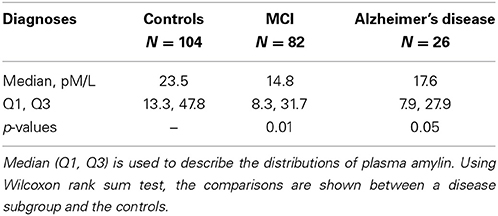
Table 1. Comparisons of plasma amylin in humans in the absence of diabetes.
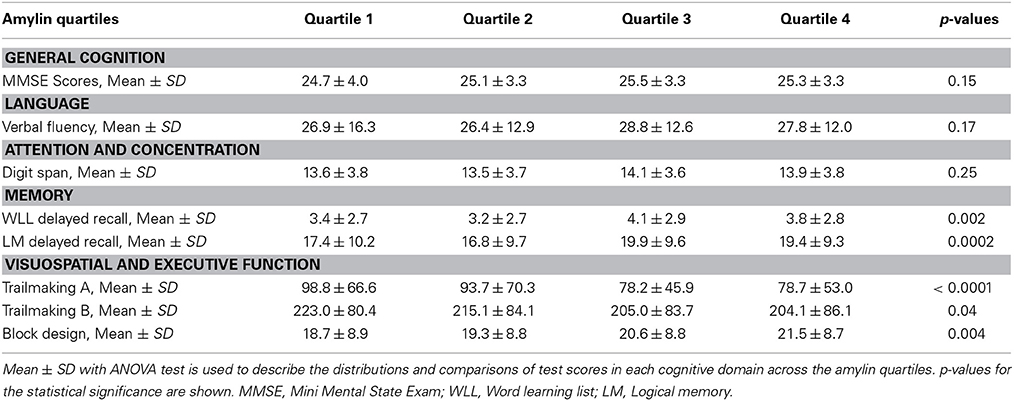
Table 2. Comparisons of functions in cognitive domains across amylin quartiles.
To understand how amylin type peptides improve cognition in the AD mice, our mechanistic study revealed that amylin type peptides enhance the removal of neurotoxic Aβ out of the brain. Both amylin and pramlintide treatments increase the concentrations of Aβ1-42 in cerebral spinal fluid (CSF) while reducing the amount of Aβ in the brain. A single i.p. injection of either amylin or pramlintide induces a surge of Aβ in serum, the magnitude of which is proportionate to the amount of Aβ in the brain tissue. One intracerebroventricular (i.c.v.) injection of amylin induces a more significant surge in serum Aβ than one i.p. injection of the peptide. Thus amylin treatment results in the translocation of Aβ from the brain into the CSF and blood probably through its effects on cerebral vasculature (Westfall and Curfman-Falvey, 1995; Edvinsson et al., 2001). If our mouse findings that amylin removes Aβ from the brain were relevant to humans, we would expect amylin to be positively associated with Aβ in human plasma samples. Indeed, using human fasting plasma samples, we found that concentrations of Aβ1-42 (P < 0.0001) and Aβ1-40 (P < 0.0001) increased with each quartile increase of amylin (Qiu et al., 2014) after adjusting for age, gender, ethnicity, ApoE4, BMI, diabetes, stroke, kidney function and lipid profile. Given that there is abundant Aβ in the AD brain, a stronger positive association between amylin and Aβ1-42 as well as Aβ1-40 was found in patients with AD or amnestic MCI than the one found in elderly with normal cognition (Table 3). Figure 2 shows our hypothesis that abundant Aβ, in either monomeric, oligomeric or fibriller form, in the AD brain may block the ability of amylin to bind to its receptor and interefere with normal amylin functions in the brain; giving exogenous amylin class peptides could rescue the amylin activities in the brain as well as removing Aβ out of the brain.
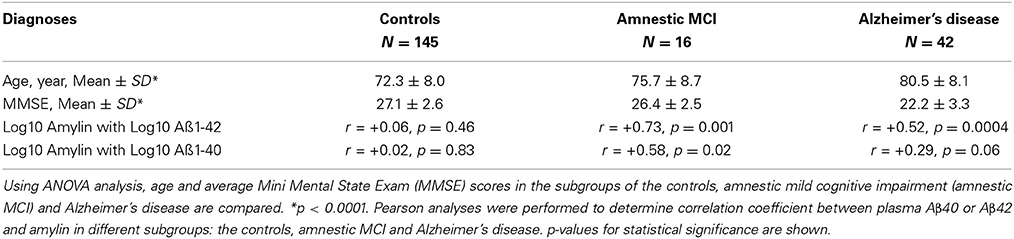
Table 3. Correlations between Aβ and Amylin in Plasma in Humans.
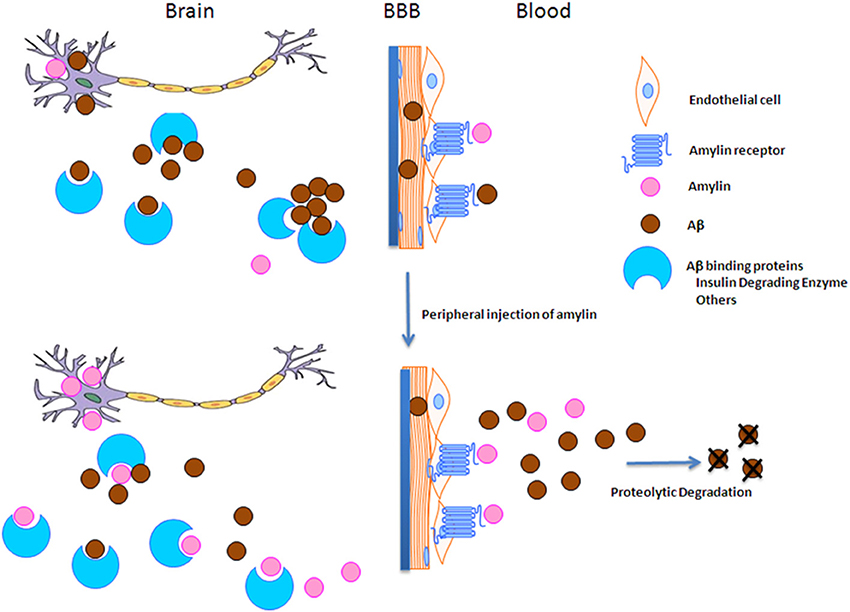
Figure 2. The hypothesis of amylin type peptides as a novel therapeutic and a diagnostic tool for AD. Since amylin and Aβ share a secondary β-sheet structure, we hypothesize that abundant Aβ in the AD brain may block amylin from binding to its receptor and hinder normal amylin functions in the brain. Thus giving extra amylin type peptides not only removes Aβ out of the brain to reduce the AD pathology in the brain, but also may restore/repair the impaired functions in the downstream of Aβ pathway in AD.
Independently, Adler et al., used pramlintide to treat another AD mouse model, SAMP8, which have increased amount of Aβ and other AD pathology (Adler et al., 2014). They found that pramlintide treatment improved the performance of these mice in the novel object recognition task. Their study demonstrated that the pramlintide-treated mice had increased expression of the synaptic marker synapsin I and the kinase cyclin-dependent kinase-5 in the hippocampus, as well as decreased oxidative stress and inflammatory markers in the hippocampus. Thus the effects of amylin type peptides for AD extend beyond just the removal of Aβ from the brain, distinct from the targets of current AD drug discovery (Dunkel et al., 2012).
Other Amylin Activities that are Potentially Beneficial for AD
Since the first description of amylin 25 years ago (Cooper et al., 1987; Westermark et al., 1987), a large body of data has documented the physiological actions of this peptide in the brain (Roth, 2013). In contrast to amylin, there is no known physiological function of Aβ in brain. Aβ in the AD brain might interfere with the following functions mediated by amylin, and thus giving extra amylin type peptides may restore/repair them.
Amylin improves glucose metabolism
Multiple imaging studies have demonstrated perturbed cerebral glucose metabolism in the AD brain (Johnson et al., 2012). Amylin is an important hormone regulating glucose metabolism in the body (Min et al., 2011; Roth, 2013). Since it readily crosses the BBB, and the brain accounts for approximately 50% of the body's glucose use (Bryant et al., 2002), amylin may play a similar role in the brain. Abundant Aβ in the AD brain may block or interfere with this activity of amylin.
Amylin regulates glucose metabolism in the body through three primary mechanisms in the brain, (1) slowing the rate of gastric emptying, (2) suppressing post-meal glucagon secretion and (3) reducing food intake (Roth et al., 2012; Roth, 2013). Amylin acts on the area postrema (AP), nucleus of the solitary tract (NTS), lateral parabrachial nucleus and the central nucleus of the amygdala in the brain to mediate these activities (Boyle and Lutz, 2011; Roth, 2013). Aspiration of the AP abolishes amylin's effects on gastric emptying (Young, 2005; Wickbom et al., 2008). Antagonism of amylin receptors with AC187 increases food intake and abrogates the anorexigenic effects of exogenous peripheral amylin (Mollet et al., 2004). Amylin's activity in these brain areas is mediated through inducing c-Fos expression (Rowland et al., 1997).
Amylin knock-out mice do not show any differences in basal insulin and glucose concentrations compared to wild types throughout development (Gebre-Medhin et al., 1998a), and there are also no differences in body weight, body composition or plasma leptin concentrations observed when mice are maintained on a low-fat diet (6 weeks) (Turek et al., 2010). Consistently, global deletion of the CTR has no effects on body weight or metabolic disease-related endpoints (Davey et al., 2008). Some studies reported that amylin stimulates insulin secretion (Fehmann et al., 1990), while another one demonstrated that amylin inhibits insulin secretion when the concentration of insulin is high (Gebre-Medhin et al., 2000). It should be noted that in response to changes in ambient glucose concentrations in vivo, amylin and insulin mRNA expressions appear to be regulated in parallel (Alam et al., 1992). Using amylin null mice (Gebre-Medhin et al., 1998a) and amylin transgenic mice (Ahren et al., 1998), it is shown that amylin plays a major role in inhibiting insulin secretion by pancreas and decreasing glucose tolerance after a glucose loading. All these researches suggest that amylin is probably not a homeostasis peptide to maintain metabolism in the body, but it is more likely a regulatory peptide responding to metabolic or other environmental stimuli.
Amylin promotes cell growth
It has been proposed that amylin may play a role as a trophic factor (Potes and Lutz, 2010). Amylin is shown to stimulate osteoblast growth (Cornish et al., 1998). After injection of alloxan, more severe diabetes and β cell damage/dysfunction were developed in amylin null mice than in the wild type mice (Mulder et al., 2000), probably due to the lack of amylin to promote growth and regeneration of cells after the insult (Gebre-Medhin et al., 2000). Amylin deficient mice showed reduced pain reaction when challenged with formalin, suggesting an essential role of amylin in the function of sensory neurons (Gebre-Medhin et al., 1998b; Mulder et al., 1999). Additionally, amylin infusion enhanced neurogenesis in the hippocampus and the area postrema in the brain of rodents with ovariectomy and improved their immobility in the forced swim test (Trevaskis et al., 2010). Amylin may be involved in the formation of synapsis by inducing the expression of synapsis I and cdk5 (Adler et al., 2014). The neuronal protective activity of amylin under pathological insults can be beneficial to reversing the effects of neuronal degeneration.
Amylin modulates inflammatory process
Human amylin has been shown to be a modulator of inflammation, especially activation of the NLRP3 inflammasome, peripherally (Masters et al., 2010). Mice with a disrupted RAMP1 gene exhibited a dysregulated immune response (Sexton et al., 2009). Amylin induces production of interleukin 1β (IL-1β) through CD36 receptor (Sheedy et al., 2013). Human amylin, but not rat amylin or Aβ, stimulates the release of the gradulocyte-macrophage colony-stimulating factor (G-MCSF) in eosinophils, and inhibits the in vitro interleukin-5 (IL-5)-mediated survival of eosinophils (Hom et al., 1995). After unilateral adjuvant-induced inflammation, expression of amylin is upregulated in innervating sensory neurons and is involved in the the inflammatory response (Mulder et al., 1997). In inflammatory models characterized with a vascular component including mouse ear oedema induced by croton oil and acetic acid-induced peritonitis, amylin exerts anti-inflammatory activity (Clementi et al., 1995). All these data demonstrate that amylin is a modulator in peripheral inflammation. Whether or not and how amylin modulates neuroinflammation in the brain are not yet known.
Amylin and Aβ mediate different intracellular signal transduction
Despite the fact that amylin and Aβ bind to the same amylin receptor (Fu et al., 2012), a recent study shows that while amylin and pramlintide increase intracellular cAMP, an important secondary messenger for learning, memory and mood, Aβ1-42 does not influence intracellular cAMP (Gingell et al., 2014). The data suggest that amylin and Aβ do not mediate the same physiological function within cells. Additionally, amylin activates the extracellular-signal regulated kinase ½ (ERK1/2) pathway by inducing the phosphorylation of ERK1/2 (pERK) (Potes et al., 2012); in contrast, Aβ decreases pERK in neurons, leading to the generation of toxic tau phosphorylation and fragments (Reifert et al., 2011). A recent study shows that amylin treatment can increase glutamate release enough to cause cell firing (Fukuda et al., 2013), which may be necessary for restoring learning and memory in AD (Danysz and Parsons, 2012).
Amylin's Self Aggregation Feature Under Pathological Conditions
The amylin amyloid deposits were first found in the pancreas in diabetic patients in Opie (1901). These amylin aggregates disrupt islet structure and contribute to the β cell dysfunction in most type 2 diabetes patients (Hoppener et al., 1994; Hull et al., 2004). On the other hand, the Aβ aggregation and amyloid plaques are identified as a hallmark pathology in the AD brain and have been thought to be a key element in the AD pathogenesis (Hardy and Selkoe, 2002). Although amylin and Aβ have little or no amino acid sequence homology, the core structure of the fibrils from their amyloids is essentially the same (Sunde et al., 1997). A recent study found an accumulation of amylin amyloid in the cerebrovascular system in the AD brain (Jackson et al., 2013). It is possible that the pathological environment of the AD brain causes any amyloidgenic peptides, including amylin, to aggregate and become cell toxic.
The mechanism(s) responsible for amylin amyloid formation in type 2 diabetes is still unclear, but it appears that an increase in the secretion of amylin, per se, is not sufficient to form aggregates and amyloids. The heterozygous transgenic mice that produce a large amount of human amylin do not develop islet amyloids, and only some, but not all, homozygous mice develop this pancreatic pathology (Hoppener et al., 1999), which requires extrapancreatic and environmental factors such as high-glucose or high-fat feeding to occur (Hull et al., 2003; Andrikopoulos et al., 2004). These factors, which can promote the formation of amylin amyloids, include the following: (1) insulin resistance like hyperglycemia (de Koning et al., 1994), (2) the ob gene introduction (Hoppener et al., 1999), or (3) glycosylation of amylin (Kapurniotu et al., 1998). Notably, the changed ratio of amylin to insulin in plasma induced by diet or other experimental stimulations is related to the formation amylin amyloid in the pancreas (Gebre-Medhin et al., 2000).
Future Perspectives in Developing Amylin Type Peptides as a Treatment for AD
Although amylin's self-aggregation property under the pathlogical conditions (Pillay and Govender, 2013) may affect its development as a drug for AD, several studies demonstrate that amylin can inhibit Aβ aggregate, as they can form the cross-interactions (Andreetto et al., 2010; Seeliger et al., 2012). Monomeric amylin and its analogs inhibit the formation of Aβ aggregation in vitro (Yan et al., 2007, 2013, 2014; Sellin et al., 2010; Andreetto et al., 2011). It is shown that amylin with the methylation at N-terminal region is highly soluble and inhibits the aggregation of Aβ40 (Yan et al., 2007, 2013; Sellin et al., 2010). N-terminal region of amylin is critical for inhibiting Aβ fribrillogenesis and cell toxicity through Aβ-amylin interaction (Andreetto et al., 2011). As the Aβ oligomer is a key element in the AD pathogenesis (Selkoe, 2008), that Aβ-amylin hetero-oligomers are not cytotoxic (Yan et al., 2014) may be another mechanism to reduce amyloid pathology in the brain. Rodent amylin also shows an ability to inhibit formation of fibrils from human amylin (Westermark et al., 2000), and can not induce cell apoptosis like human amylin after incubating for 48 h (Ritzel et al., 2007).
The debate of over 10 years ago on whether amylin is beneficial or harmful to the treatment of type 2 diabetes (Gebre-Medhin et al., 2000) may provide a lesson for drug development for AD. Since the clinical utility of human amylin is limited by a propensity for self-aggregation despite its activity of inhibiting appetite and regulating glucose metabolism, that limitation was overcome by the substitution of prolines at positions 25, 28 and 29 of human amylin based on rat amylin sequences (Colburn et al., 1996; Moriarty and Raleigh, 1999). This resulted in a synthetic amylinomimetic peptide, pramlintide, with improved stability and decreased potential for aggregation and pramlintide has become a potent anti-diabetic drug (Pencek et al., 2010). Pharmacokinetic studies show that the terminal half-life of amylin in rats is ~13 min and that the half-life for pramlintide in humans is ~20–45 min (Colburn et al., 1996; Young, 2005). Pramlintide has a favorable safety profile in clinical use, and only nausea is the most common tolerability-related adverse event (Aronne et al., 2007).
Since pramlintide is a relatively new drug for diabetes, there are no available data on the association between pramlintide use and AD yet. However, some medications, which are shown to influence the concentration of amylin in blood, are associated with cognitive function in humans. For example, metformin is shown to lower serum amylin concentrations in patients with type 2 diabetes (Zapecka-Dubno et al., 1999), and use of metformin may be associated with cognitive impairment (Moore et al., 2013) and increased risk of AD development (Imfeld et al., 2012). In contrast, another drug sulfonylurea does not affect serum amylin concentrations (Rachman et al., 1998), and is found not associated with the risk of AD. One animal study shows that sulfonylurea treatment reduces the AD pathology in the brain (Baraka and ElGhotny, 2010).
While many papers in the AD field have been focused on amylin's self-aggregation like Aβ (Gotz et al., 2009; DeToma et al., 2012), probably equal attention needs to be given to the potential benefits of soluble amylin or its non-amyloidgenic analogs for the AD brain. The research findings that amylin readily crosses the BBB, mediates important brain functions and mimics the Aβ structure so that they can antagonize each other are hard to ignore, taken together. Based on ours and Adler et al.,'s studies, it is hypothesized that this old foe, amylin, or its analogs may become a new friend for AD. More basic researches are needed to understand the mechanism of amylin's effects in the AD brain and probably to search for better analogs of it. Ultimately, whether amylin type peptides can be a new and novel avenue of therapeutic for AD should only be concluded through a double blind, placebo controlled clinical trial in humans.
Conflict of Interest Statement
Wei Qiao Qiu and Boston University have filed a patent application for their amylin findings. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from NIA, AG-022476 and Ignition Award (Wei Qiao Qiu) and BU ADC pilot grant (Haihao Zhu). Support was also provided through P30 AG13864 for the Alzheimer's Disease Center at Boston University.
References
Adler, B. L., Yarchoan, M., Hwang, H. M., Louneva, N., Blair, J. A., Palm, R., et al. (2014). Neuroprotective effects of the amylin analogue pramlintide on Alzheimer's disease pathogenesis and cognition. Neurobiol Aging 35, 793–801. doi: 10.1016/j.neurobiolaging.2013.10.076
Ahren, B., Oosterwijk, C., Lips, C. J., and Hoppener, J. W. (1998). Transgenic overexpression of human islet amyloid polypeptide inhibits insulin secretion and glucose elimination after gastric glucose gavage in mice. Diabetologia 41, 1374–1380. doi: 10.1007/s001250051079
Alam, T., Chen, L., Ogawa, A., Leffert, J. D., Unger, R. H., and Luskey, K. L. (1992). Coordinate regulation of amylin and insulin expression in response to hypoglycemia and fasting. Diabetes 41, 508–514. doi: 10.2337/diab.41.4.508
Andreetto, E., Yan, L. M., Caporale, A., and Kapurniotu, A. (2011). Dissecting the role of single regions of an IAPP mimic and IAPP in inhibition of Abeta40 amyloid formation and cytotoxicity. Chembiochem 12, 1313–1322. doi: 10.1002/cbic.201100192
Andreetto, E., Yan, L. M., Tatarek-Nossol, M., Velkova, A., Frank, R., and Kapurniotu, A. (2010). Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew. Chem. Int. Ed Engl. 49, 3081–3085. doi: 10.1002/anie.200904902
Andrikopoulos, S., Hull, R. L., Verchere, C. B., Wang, F., Wilbur, S. M., Wight, T. N., et al. (2004). Extended life span is associated with insulin resistance in a transgenic mouse model of insulinoma secreting human islet amyloid polypeptide. Am. J. Physiol. Endocrinol. Metab. 286, E418–E424. doi: 10.1152/ajpendo.00137.2003
Aronne, L., Fujioka, K., Aroda, V., Chen, K., Halseth, A., Kesty, N. C., et al. (2007). Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: a phase 2, randomized, placebo-controlled, dose-escalation study. J. Clin. Endocrinol. Metab. 92, 2977–2983. doi: 10.1210/jc.2006-2003
Banks, W. A., and Kastin, A. J. (1998). Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19, 883–889. doi: 10.1016/S0196-9781(98)00018-7
Banks, W. A., Kastin, A. J., Maness, L. M., Huang, W., and Jaspan, J. B. (1995). Permeability of the blood-brain barrier to amylin. Life Sci. 57, 1993–2001. doi: 10.1016/0024-3205(95)02197-Q
Baraka, A., and ElGhotny, S. (2010). Study of the effect of inhibiting galanin in Alzheimer's disease induced in rats. Eur. J. Pharmacol. 641, 123–127. doi: 10.1016/j.ejphar.2010.05.030
Bennett, R. G., Hamel, F. G., and Duckworth, W. C. (2003). An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amylin-induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes 52, 2315–2320. doi: 10.2337/diabetes.52.9.2315
Boyle, C. N., and Lutz, T. A. (2011). Amylinergic control of food intake in lean and obese rodents. Physiol. Behav. 105, 129–137. doi: 10.1016/j.physbeh.2011.02.015
Bryant, N. J., Govers, R., and James, D. E. (2002). Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 3, 267–277. doi: 10.1038/nrm782
Chang, C. L., Roh, J., and Hsu, S. Y. (2004). Intermedin, a novel calcitonin family peptide that exists in teleosts as well as in mammals: a comparison with other calcitonin/intermedin family peptides in vertebrates. Peptides 25, 1633–1642. doi: 10.1016/j.peptides.2004.05.021
Christopoulos, G., Perry, K. J., Morfis, M., Tilakaratne, N., Gao, Y., Fraser, N. J., et al. (1999). Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol. Pharmacol. 56, 235–242.
Clementi, G., Caruso, A., Cutuli, V. M., Prato, A., de Bernardis, E., Fiore, C. E., et al. (1995). Anti-inflammatory activity of amylin and CGRP in different experimental models of inflammation. Life Sci. 57, PL193–PL197. doi: 10.1016/0024-3205(95)02100-W
Colburn, W. A., Gottlieb, A. B., Koda, J., and Kolterman, O. G. (1996). Pharmacokinetics and pharmacodynamics of AC137 (25,28,29 tripro-amylin, human) after intravenous bolus and infusion doses in patients with insulin-dependent diabetes. J. Clin. Pharmacol. 36, 13–24. doi: 10.1002/j.1552-4604.1996.tb04147.x
Cooper, G. J., Willis, A. C., Clark, A., Turner, R. C., Sim, R. B., and Reid, K. B. (1987). Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. U.S.A. 84, 8628–8632. doi: 10.1073/pnas.84.23.8628
Cornish, J., Callon, K. E., King, A. R., Cooper, G. J., and Reid, I. R. (1998). Systemic administration of amylin increases bone mass, linear growth, and adiposity in adult male mice. Am. J. Physiol. 275(4 Pt 1), E694–E699.
Danysz, W., and Parsons, C. G. (2012). Alzheimer's disease, beta-amyloid, glutamate, NMDA receptors and memantine–searching for the connections. Br. J. Pharmacol. 167, 324–352. doi: 10.1111/j.1476-5381.2012.02057.x
Davey, R. A., Turner, A. G., McManus, J. F., Chiu, W. S., Tjahyono, F., Moore, A. J., et al. (2008). Calcitonin receptor plays a physiological role to protect against hypercalcemia in mice. J. Bone Miner. Res. 23, 1182–1193. doi: 10.1359/jbmr.080310
de Koning, E. J., Morris, E. R., Hofhuis, F. M., Posthuma, G., Hoppener, J. W., Morris, J. F., et al. (1994). Intra- and extracellular amyloid fibrils are formed in cultured pancreatic islets of transgenic mice expressing human islet amyloid polypeptide. Proc. Natl. Acad. Sci. U.S.A. 91, 8467–8471. doi: 10.1073/pnas.91.18.8467
DeToma, A. S., Salamekh, S., Ramamoorthy, A., and Lim, M. H. (2012). Misfolded proteins in Alzheimer's disease and type II diabetes. Chem. Soc. Rev. 41, 608–621. doi: 10.1039/c1cs15112f
de Tullio, M. B., Castelletto, V., Hamley, I. W., Martino Adami, P. V., Morelli, L., and Castano, E. M. (2013). Proteolytically inactive insulin-degrading enzyme inhibits amyloid formation yielding non-neurotoxic abeta peptide aggregates. PLoS ONE 8:e59113. doi: 10.1371/journal.pone.0059113
Dunkel, P., Chai, C. L., Sperlagh, B., Huleatt, P. B., and Matyus, P. (2012). Clinical utility of neuroprotective agents in neurodegenerative diseases: current status of drug development for Alzheimer's, Parkinson's and Huntington's diseases, and amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 21, 1267–1308. doi: 10.1517/13543784.2012.703178
Edvinsson, L., Goadsby, P. J., and Uddman, R. (2001). Amylin: localization, effects on cerebral arteries and on local cerebral blood flow in the cat. ScientificWorldJournal 1, 168–180. doi: 10.1100/tsw.2001.23
Fehmann, H. C., Weber, V., Goke, R., Goke, B., and Arnold, R. (1990). Cosecretion of amylin and insulin from isolated rat pancreas. FEBS Lett. 262, 279–281. doi: 10.1016/0014-5793(90)80210-A
Fu, W., Ruangkittisakul, A., MacTavish, D., Shi, J. Y., Ballanyi, K., and Jhamandas, J. H. (2012). Amyloid beta (Abeta) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J. Biol. Chem. 287, 18820–18830. doi: 10.1074/jbc.M111.331181
Fukuda, T., Hirai, Y., Maezawa, H., Kitagawa, Y., and Funahashi, M. (2013). Electrophysiologically identified presynaptic mechanisms underlying amylinergic modulation of area postrema neuronal excitability in rat brain slices. Brain Res. 1494, 9–16. doi: 10.1016/j.brainres.2012.11.051
Gebre-Medhin, S., Mulder, H., Pekny, M., Westermark, G., Tornell, J., Westermark, P., et al. (1998a). Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem. Biophys. Res. Commun. 250, 271–277. doi: 10.1006/bbrc.1998.9308
Gebre-Medhin, S., Mulder, H., Zhang, Y., Sundler, F., and Betsholtz, C. (1998b). Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Brain Res. Mol. Brain Res. 63, 180–183. doi: 10.1016/S0169-328X(98)00269-1
Gebre-Medhin, S., Olofsson, C., and Mulder, H. (2000). Islet amyloid polypeptide in the islets of Langerhans: friend or foe? Diabetologia 43, 687–695. doi: 10.1007/s001250051364
Gingell, J. J., Burns, E. R., and Hay, D. L. (2014). Activity of pramlintide, rat and human amylin but not Aβ1-42 at human amylin receptors. Endocrinology 155, 21–26. doi: 10.1210/en.2013-1658
Gotz, J., Ittner, L. M., and Lim, Y. A. (2009). Common features between diabetes mellitus and Alzheimer's disease. Cell. Mol. Life Sci. 66, 1321–1325. doi: 10.1007/s00018-009-9070-1
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hom, J. T., Estridge, T., Pechous, P., and Hyslop, P. A. (1995). The amyloidogenic peptide human amylin augments the inflammatory activities of eosinophils. J. Leukoc. Biol. 58, 526–532.
Hoppener, J. W., Ahren, B., and Lips, C. J. (2000). Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 343, 411–419. doi: 10.1056/NEJM200008103430607
Hoppener, J. W., Oosterwijk, C., Nieuwenhuis, M. G., Posthuma, G., Thijssen, J. H., Vroom, T. M., et al. (1999). Extensive islet amyloid formation is induced by development of Type II diabetes mellitus and contributes to its progression: pathogenesis of diabetes in a mouse model. Diabetologia 42, 427–434. doi: 10.1007/s001250051175
Hoppener, J. W., Oosterwijk, C., van Hulst, K. L., Verbeek, J. S., Capel, P. J., de Koning, E. J., et al. (1994). Molecular physiology of the islet amyloid polypeptide (IAPP)/amylin gene in man, rat, and transgenic mice. J. Cell. Biochem. 55(Suppl), 39–53. doi: 10.1002/jcb.240550006
Hull, R. L., Andrikopoulos, S., Verchere, C. B., Vidal, J., Wang, F., Cnop, M., et al. (2003). Increased dietary fat promotes islet amyloid formation and beta-cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes 52, 372–379. doi: 10.2337/diabetes.52.2.372
Hull, R. L., Westermark, G. T., Westermark, P., and Kahn, S. E. (2004). Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J. Clin. Endocrinol. Metab. 89, 3629–3643. doi: 10.1210/jc.2004-0405
Imfeld, P., Bodmer, M., Jick, S. S., and Meier, C. R. (2012). Metformin, other antidiabetic drugs, and risk of Alzheimer's disease: a population-based case-control study. J. Am. Geriatr. Soc. 60, 916–921. doi: 10.1111/j.1532-5415.2012.03916.x
Jackson, K., Barisone, G. A., Diaz, E., Jin, L. W., Decarli, C., and Despa, F. (2013). Amylin deposition in the brain: a second amyloid in Alzheimer disease? Ann. Neurol. 74, 517–526. doi: 10.1002/ana.23956
Johnson, K. A., Fox, N. C., Sperling, R. A., and Klunk, W. E. (2012). Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006213. doi: 10.1101/cshperspect.a006213
Kapurniotu, A., Bernhagen, J., Greenfield, N., Al-Abed, Y., Teichberg, S., Frank, R. W., et al. (1998). Contribution of advanced glycosylation to the amyloidogenicity of islet amyloid polypeptide. Eur. J. Biochem. 251, 208–216. doi: 10.1046/j.1432-1327.1998.2510208.x
Kautzky-Willer, A., Thomaseth, K., Pacini, G., Clodi, M., Ludvik, B., Streli, C., et al. (1994). Role of islet amyloid polypeptide secretion in insulin-resistant humans. Diabetologia 37, 188–194. doi: 10.1007/s001250050092
Kraemer, R. R., Acevedo, E. O., Synovitz, L. B., Durand, R. J., Johnson, L. G., Petrella, E., et al. (2002). Glucoregulatory endocrine responses to intermittent exercise of different intensities: plasma changes in a pancreatic beta-cell peptide, amylin. Metab. Clin. Exp. 51, 657–663. doi: 10.1053/meta.2002.32023
Lim, Y. A., Ittner, L. M., Lim, Y. L., and Gotz, J. (2008). Human but not rat amylin shares neurotoxic properties with Abeta42 in long-term hippocampal and cortical cultures. FEBS Lett. 582, 2188–2194. doi: 10.1016/j.febslet.2008.05.006
Lukinius, A., Wilander, E., Westermark, G. T., Engstrom, U., and Westermark, P. (1989). Co-localization of islet amyloid polypeptide and insulin in the B cell secretory granules of the human pancreatic islets. Diabetologia 32, 240–244. doi: 10.1007/BF00285291
Masters, S. L., Dunne, A., Subramanian, S. L., Hull, R. L., Tannahill, G. M., Sharp, F. A., et al. (2010). Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 11, 897–904. doi: 10.1038/ni.1935
Min, D. K., Tuor, U. I., Koopmans, H. S., and Chelikani, P. K. (2011). Changes in differential functional magnetic resonance signals in the rodent brain elicited by mixed-nutrient or protein-enriched meals. Gastroenterology 141, 1832–1841. doi: 10.1053/j.gastro.2011.07.034
Mollet, A., Gilg, S., Riediger, T., and Lutz, T. A. (2004). Infusion of the amylin antagonist AC 187 into the area postrema increases food intake in rats. Physiol. Behav. 81, 149–155. doi: 10.1016/j.physbeh.2004.01.006
Moore, E. M., Mander, A. G., Ames, D., Kotowicz, M. A., Carne, R. P., Brodaty, H., et al. (2013). Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 36, 2981–2987. doi: 10.2337/dc13-0229
Moriarty, D. F., and Raleigh, D. P. (1999). Effects of sequential proline substitutions on amyloid formation by human amylin20-29. Biochemistry 38, 1811–1818. doi: 10.1021/bi981658g
Mulder, H., Gebre-Medhin, S., Betsholtz, C., Sundler, F., and Ahren, B. (2000). Islet amyloid polypeptide (amylin)-deficient mice develop a more severe form of alloxan-induced diabetes. Am. J. Physiol. Endocrinol. Metab. 278, E684–E691.
Mulder, H., Jongsma, H., Zhang, Y., Gebre-Medhin, S., Sundler, F., and Danielsen, N. (1999). Pituitary adenylate cyclase-activating polypeptide and islet amyloid polypeptide in primary sensory neurons: functional implications from plasticity in expression on nerve injury and inflammation. Mol. Neurobiol. 19, 229–253. doi: 10.1007/BF02821715
Mulder, H., Leckstrom, A., Uddman, R., Ekblad, E., Westermark, P., and Sundler, F. (1995). Islet amyloid polypeptide (amylin) is expressed in sensory neurons. J. Neurosci. 15, 7625–7632.
Mulder, H., Lindh, A. C., Ekblad, E., Westermark, P., and Sundler, F. (1994). Islet amyloid polypeptide is expressed in endocrine cells of the gastric mucosa in the rat and mouse. Gastroenterology 107, 712–719. doi: 10.1016/0016-5085(94)90118-X
Mulder, H., Zhang, Y., Danielsen, N., and Sundler, F. (1997). Islet amyloid polypeptide and calcitonin gene-related peptide expression are upregulated in lumbar dorsal root ganglia after unilateral adjuvant-induced inflammation in the rat paw. Brain Res. Mol. Brain Res. 50, 127–135. doi: 10.1016/S0169-328X(97)00178-2
Nishi, M., Sanke, T., Seino, S., Eddy, R. L., Fan, Y. S., Byers, M. G., et al. (1989). Human islet amyloid polypeptide gene: complete nucleotide sequence, chromosomal localization, and evolutionary history. Mol. Endocrinol. 3, 1775–1781. doi: 10.1210/mend-3-11-1775
Nyholm, B., Fineman, M. S., Koda, J. E., and Schmitz, O. (1998). Plasma amylin immunoreactivity and insulin resistance in insulin resistant relatives of patients with non-insulin-dependent diabetes mellitus. Horm. Metab. Res. 30, 206–212. doi: 10.1055/s-2007-978867
Olsson, M., Herrington, M. K., Reidelberger, R. D., Permert, J., and Arnelo, U. (2007). Comparison of the effects of chronic central administration and chronic peripheral administration of islet amyloid polypeptide on food intake and meal pattern in the rat. Peptides 28, 1416–1423. doi: 10.1016/j.peptides.2007.06.011
Opie, E. L. (1901). On the relation of chronic interstitial pancreatitis to the islands of langerhans and to diabetes melutus. J. Exp. Med. 5, 397–428. doi: 10.1084/jem.5.4.397
Pencek, R., Roddy, T., Peters, Y., De Young, M. B., Herrmann, K., Meller, L., et al. (2010). Safety of pramlintide added to mealtime insulin in patients with type 1 or type 2 diabetes: a large observational study. Diabetes Obes. Metab. 12, 548–551. doi: 10.1111/j.1463-1326.2010.01201.x
Pillay, K., and Govender, P. (2013). Amylin uncovered: a review on the polypeptide responsible for type II diabetes. Biomed Res. Int. 2013, 826706. doi: 10.1155/2013/826706
Potes, C. S., Boyle, C. N., Wookey, P. J., Riediger, T., and Lutz, T. A. (2012). Involvement of the extracellular signal-regulated kinase 1/2 signaling pathway in amylin's eating inhibitory effect. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R340–351. doi: 10.1152/ajpregu.00380.2011
Potes, C. S., and Lutz, T. A. (2010). Brainstem mechanisms of amylin-induced anorexia. Physiol. Behav. 100, 511–518. doi: 10.1016/j.physbeh.2010.03.001
Qiu, W. Q., Wallack, M., Dean, M., Liebson, E., Mwamburi, M., and Zhu, H. (2014). Association between amylin and amyloid-beta peptides in plasma in the context of apolipoprotein E4 allele. PLoS ONE 9:e88063. doi: 10.1371/journal.pone.0088063
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., et al. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 273, 32730–32738. doi: 10.1074/jbc.273.49.32730
Rachman, J., Payne, M. J., Levy, J. C., Barrow, B. A., Holman, R. R., and Turner, R. C. (1998). Changes in amylin and amylin-like peptide concentrations and beta-cell function in response to sulfonylurea or insulin therapy in NIDDM. Diabetes Care 21, 810–816. doi: 10.2337/diacare.21.5.810
Reifert, J., Hartung-Cranston, D., and Feinstein, S. C. (2011). Amyloid beta-mediated cell death of cultured hippocampal neurons reveals extensive Tau fragmentation without increased full-length tau phosphorylation. J. Biol. Chem. 286, 20797–20811. doi: 10.1074/jbc.M111.234674
Ritzel, R. A., Meier, J. J., Lin, C. Y., Veldhuis, J. D., and Butler, P. C. (2007). Human islet amyloid polypeptide oligomers disrupt cell coupling, induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes 56, 65–71. doi: 10.2337/db06-0734
Roth, J. D. (2013). Amylin and the regulation of appetite and adiposity: recent advances in receptor signaling, neurobiology and pharmacology. Curr. Opin. Endocrinol. Diabetes Obes. 20, 8–13. doi: 10.1097/MED.0b013e32835b896f
Roth, J. D., Erickson, M. R., Chen, S., and Parkes, D. G. (2012). GLP-1R and amylin agonism in metabolic disease: complementary mechanisms and future opportunities. Br. J. Pharmacol. 166, 121–136. doi: 10.1111/j.1476-5381.2011.01537.x
Rowland, N. E., Crews, E. C., and Gentry, R. M. (1997). Comparison of Fos induced in rat brain by GLP-1 and amylin. Regul. Pept. 71, 171–174. doi: 10.1016/S0167-0115(97)01034-3
Seeliger, J., Evers, F., Jeworrek, C., Kapoor, S., Weise, K., Andreetto, E., et al. (2012). Cross-amyloid interaction of Abeta and IAPP at lipid membranes. Angew. Chem. Int. Ed Engl. 51, 679–683. doi: 10.1002/anie.201105877
Selkoe, D. J. (2008). Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer's disease. Handb. Clin. Neurol. 89, 245–260. doi: 10.1016/S0072-9752(07)01223-7
Sellin, D., Yan, L. M., Kapurniotu, A., and Winter, R. (2010). Suppression of IAPP fibrillation at anionic lipid membranes via IAPP-derived amyloid inhibitors and insulin. Biophys. Chem. 150, 73–79. doi: 10.1016/j.bpc.2010.01.006
Sexton, P. M., Poyner, D. R., Simms, J., Christopoulos, A., and Hay, D. L. (2009). Modulating receptor function through RAMPs: can they represent drug targets in themselves? Drug Discov. Today 14, 413–419. doi: 10.1016/j.drudis.2008.12.009
Sheedy, F. J., Grebe, A., Rayner, K. J., Kalantari, P., Ramkhelawon, B., Carpenter, S. B., et al. (2013). CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820. doi: 10.1038/ni.2639
Shen, Y., Joachimiak, A., Rosner, M. R., and Tang, W. J. (2006). Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 443, 870–874. doi: 10.1038/nature05143
Sunde, M., Serpell, L. C., Bartlam, M., Fraser, P. E., Pepys, M. B., and Blake, C. C. (1997). Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 273, 729–739. doi: 10.1006/jmbi.1997.1348
Trevaskis, J. L., Turek, V. F., Wittmer, C., Griffin, P. S., Wilson, J. K., Reynolds, J. M., et al. (2010). Enhanced amylin-mediated body weight loss in estradiol-deficient diet-induced obese rats. Endocrinology 151, 5657–5668. doi: 10.1210/en.2010-0590
Turek, V. F., Trevaskis, J. L., Levin, B. E., Dunn-Meynell, A. A., Irani, B., Gu, G., et al. (2010). Mechanisms of amylin/leptin synergy in rodent models. Endocrinology 151, 143–152. doi: 10.1210/en.2009-0546
Vine, W., Blase, E., Koda, J., and Young, A. (1998). Plasma amylin concentrations in fasted and fed rats quantified by a monoclonal immunoenzymometric assay. Horm. Metab. Res. 30, 581–585. doi: 10.1055/s-2007-978937
Weintraub, S., Salmon, D., Mercaldo, N., Ferris, S., Graff-Radford, N. R., Chui, H., et al. (2009). The alzheimer's disease centers' uniform data set (UDS): the neuropsychologic test battery. Alzheimer Dis. Assoc. Disord. 23, 91–101. doi: 10.1097/WAD.0b013e318191c7dd
Westermark, G. T., Gebre-Medhin, S., Steiner, D. F., and Westermark, P. (2000). Islet amyloid development in a mouse strain lacking endogenous islet amyloid polypeptide (IAPP) but expressing human IAPP. Mol. Med. 6, 998–1007.
Westermark, P., Wernstedt, C., Wilander, E., Hayden, D. W., O'Brien, T. D., and Johnson, K. H. (1987). Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. U.S.A. 84, 3881–3885. doi: 10.1073/pnas.84.11.3881
Westfall, T. C., and Curfman-Falvey, M. (1995). Amylin-induced relaxation of the perfused mesenteric arterial bed: meditation by calcitonin gene-related peptide receptors. J. Cardiovasc. Pharmacol. 26, 932–936. doi: 10.1097/00005344-199512000-00012
Wickbom, J., Herrington, M. K., Permert, J., Jansson, A., and Arnelo, U. (2008). Gastric emptying in response to IAPP and CCK in rats with subdiaphragmatic afferent vagotomy. Regul. Pept. 148, 21–25. doi: 10.1016/j.regpep.2008.03.010
Wimalawansa, S. J. (1997). Amylin, calcitonin gene-related peptide, calcitonin, and adrenomedullin: a peptide superfamily. Crit. Rev. Neurobiol. 11, 167–239. doi: 10.1615/CritRevNeurobiol.v11.i2-3.40
Yan, L. M., Velkova, A., and Kapurniotu, A. (2014). Molecular characterization of the hetero-assembly of β-amyloid peptide with islet amyloid polypeptide. Curr. Pharm. Des. 20, 1182–1191.
Yan, L. M., Velkova, A., Tatarek-Nossol, M., Andreetto, E., and Kapurniotu, A. (2007). IAPP mimic blocks Abeta cytotoxic self-assembly: cross-suppression of amyloid toxicity of Abeta and IAPP suggests a molecular link between Alzheimer's disease and type II diabetes. Angew. Chem. Int. Ed Engl. 46, 1246–1252. doi: 10.1002/anie.200604056
Yan, L. M., Velkova, A., Tatarek-Nossol, M., Rammes, G., Sibaev, A., Andreetto, E., et al. (2013). Selectively N-Methylated Soluble IAPP Mimics as Potent IAPP receptor agonists and nanomolar inhibitors of cytotoxic self-assembly of both IAPP and Abeta40. Angew. Chem. Int. Ed Engl. 52, 10378–10383. doi: 10.1002/anie.201302840
Young, A. (2005). Central nervous system and other effects. Adv. Pharmacol. 52, 281–288. doi: 10.1016/S1054-3589(05)52017-9
Zapecka-Dubno, B., Czyzyk, A., Dworak, A., and Bak, M. I. (1999). Effect of oral antidiabetic agents on plasma amylin level in patients with non-insulin-dependent diabetes mellitus (type 2). Arzneimittelforschung 49, 330–334.
Zhu, H., Wang, X., Wallack, M., Li, H., Carreras, I., Dedeoglu, A., et al. (2014). Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer's disease. Mol. Psychiatry. doi: 10.1038/mp.2014.17. [Epub ahead of print].
Keywords: amylin, amylin analogs, Alzheimer's disease, treatment, diagnosis, animal models, humans
Citation: Qiu WQ and Zhu H (2014) Amylin and its analogs: a friend or foe for the treatment of Alzheimer's disease? Front. Aging Neurosci. 6:186. doi: 10.3389/fnagi.2014.00186
Received: 27 March 2014; Accepted: 09 July 2014;
Published online: 29 July 2014.
Edited by:
Eliezer Masliah, UC San Diego, USAReviewed by:
Junming Wang, University of Mississippi Medical Center, USAFernanda Laezza, University of Texas Medical Branch, USA
Copyright © 2014 Qiu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Qiao Qiu, Boston University School of Medicine, Boston University Medical Campus, 72 East Concord Street, R-623D, Boston, MA 02118, USA e-mail:d3FpdTY3QGJ1LmVkdQ==