 Robert A. Marr*
Robert A. Marr* Daniel M. Hafez
Daniel M. Hafez- Department of Neuroscience, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA
Accumulation of the amyloid-beta (Aβ) peptide is a central factor in Alzheimer’s disease (AD) pathogenesis as supported by continuing evidence. This review concisely summarizes this evidence supporting a critical role for Aβ in AD before discussing the clearance of this peptide. Mechanisms of clearance of Aβ are critical for preventing pathological elevations in Aβ concentration. Direct degradation of Aβ by endopeptidases has emerged as one important pathway for clearance. Of particular interest are endopeptidases that are sensitive to the neprilysin (NEP) inhibitors thiorphan and phosphoramidon (i.e., are “NEP-like”) as these inhibitors induce a dramatic increase in Aβ levels in rodents. This review will focus on neprilysin-2 (NEP2), a NEP-like endopeptidase which cooperates with NEP to control Aβ levels in the brain. The evidence for the involvement of NEP2 in AD is discussed as well as the therapeutic relevance with regards to gene therapy and the development of molecular markers for the disease.
The Amyloid Cascade Hypothesis
Amyloid-Beta Production
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder that leads to behavioral, cognitive, and memory deficits. Familial AD (FAD) is inherited in an autosomal dominant pattern with symptoms typically presenting in the 4th or 5th decade of life. Sporadic late onset AD (LOAD) has a much later age of onset, usually beginning in the 7th–8th decade. Confirmed post mortem, AD pathology shows accumulations of extracellular amyloid-beta (Aβ) containing plaques and intracellular neurofibrillary tau tangles in the brain. The involvement of Aβ in AD is a prerequisite to the significance of Aβ clearance to AD. Therefore, we will present a rationale for the clear link between Aβ and AD progression. The formation of Aβ has been well studied (Goedert and Spillantini, 2006; Roberson and Mucke, 2006). In the amyloidogenic pathway, APP is first cleaved by β-secretase (BACE1) at amino acid 1 (of Aβ). When the resulting C-terminal fragment, C99, undergoes γ-secretase cleavage, it releases the amyloidogenic Aβ peptide. Key proteolytic components of γ-secretase are presenilin-1 and 2 (PS1, PS2). While various Aβ peptide lengths are produced by γ-secretase, it is Aβ42 and Aβ40 that have received the most attention. The additional two hydrophobic residues in Aβ42 increase its ability to aggregate, providing the scaffold for oligomeric and fibrillar forms of Aβ (Jarrett and Lansbury, 1993; Iwatsubo, 1998). It should be noted that Aβ is a naturally occurring endogenous peptide that may have normal physiological functions. For example, it has been shown that picomolar concentrations of Aβ increased LTP resulting in improved synaptic plasticity and memory (Puzzo et al., 2008, 2012; Morley et al., 2010). Therefore, pathology associated with Aβ is related to its aberrant accumulation/aggregation.
Amyloid-Beta and Alzheimer’s Disease
Familial forms of early-onset AD are caused by mutations in APP, PS1, or PS2 or through increased copy number of APP (Wisniewski et al., 1985; Prasher et al., 1998; Rovelet-Lecrux et al., 2006). All of the roughly 180 mutations in PS1, 20 mutations in PS2, and 36 mutations in APP lead to elevations of total Aβ, the Aβ42: Aβ40 ratio, or its aggregation (Pimplikar, 2009). Within the Aβ sequence, point mutations, including the Arctic mutation, have been linked to increased aggregation of Aβ into protofibrils and fibrils (Clements et al., 1993, 1996; Nilsberth et al., 2001; Cheng et al., 2004; Walsh et al., 2007), as well as to reduced clearance of this peptide (Tsubuki et al., 2003; Kaden et al., 2012). However, the Arctic mutation does not affect cleavage and processing of APP; thus, this is an Aβ-only defect. Therefore, the only commonality between all the familial mutations is the effect on properties related to Aβ. To our knowledge, no other major neurodegenerative disease has familial forms that all genetically point to one common factor with the exception of monogenetic diseases like Huntington’s disease. This hypothesis is bolstered by the discovery of a protective mutation in APP in an Icelandic population that reduces BACE1 proteolysis and lowers Aβ levels (Jonsson et al., 2012). As would be consistent with the amyloid hypothesis, this mutation is associated with protection from developing AD as well as from normal cognitive decline with aging.
Unfortunately, therapies aimed at Aβ have been less than impressive in clinical trials. Most recently, Aβ-targeting monoclonal antibodies, such as bapineuzumab and solanezumab, have failed to reach their desired cognitive endpoints in trials of mild/moderate AD (Salloway et al., 2009, 2014; Farlow et al., 2012; Doody et al., 2014). Bapineuzumab did show trends for reduced Aβ42 levels and Aβ42: Aβ40 ratio, however, some subjects continued to demonstrate cognitive decline (Roher et al., 2013). Additionally, γ-secretase inhibitors (GSI) also failed to produce beneficial results, and were actually found to worsen cognitive function. It is believed that off-target effects, such as effects on the cleavage of notch by PS1 may have participated in producing this poor response and data suggest that the proper timing of GSIs is integral to their successful treatment of AD (Abramowski et al., 2008). In general it is unclear to what degree and for what period of time one would need to reduce Aβ levels to slow down, halt, or possibly reverse the pathology of AD. Likely, early intervention, before extensive pathological alterations occur, will be needed to effectively treat AD using anti-Aβ approaches.
Evidence for the Importance of Amyloid-Beta in Sporadic Alzheimer’s Disease
Regardless of the excuses for the failures of the many anti-Aβ clinical trials, these disappointments have understandably cast doubt over the amyloid hypothesis of AD. While the familial genetics clearly points to Aβ as a critical factor in the etiology of AD, it is conceivable that the much more common sporadic form of the disease (i.e., LOAD) has a distinct root cause(s) that is not dependent on Aβ. While this is a possibility, several lines of evidence suggest otherwise. First, these two forms of AD have very similar pathology (Lippa et al., 1996), supporting a common cause/progression. Second, if LOAD was not linked to Aβ, then polymorphisms associated with risk would be predicted to not affect Aβ. Indeed, there are many mutations affecting risk for LOAD and many of these polymorphisms are not obviously linked to Aβ; however, many others can be clearly linked to Aβ (Tanzi, 2012; Griciuc et al., 2013). What is even more important is that some of the most significant effectors of risk occur in genes strongly linked to Aβ. The most noteworthy of these are polymorphisms in the apolipoprotein E (apoE) gene with the ε4 mutation (cys112arg) multiplying risk by about 3-fold per inherited allele (Bu, 2009). ApoE can affect Aβ in multiple ways including its aggregation, clearance, and catabolism (Bales et al., 1999; Holtzman et al., 2000; Shibata et al., 2000; Fagan et al., 2002; Dolev and Michaelson, 2004; Koistinaho et al., 2004; Manelli et al., 2004; Dodart et al., 2005; Bu, 2009; Belinson et al., 2010; Hashimoto et al., 2012; Kline, 2012). Related to this, Jiang et al. (2008) showed that apoE promotes proteolytic degradation of Aβ by microglia. Another risk factor gene mutation has been discovered in ABCA7 which affects risk at a comparable level to the ε4 form of apoE (~3-fold) and has also been linked to the clearance of Aβ (Kim et al., 2013; Reitz et al., 2013). Third, if Aβ were not involved in sporadic AD then a mutation reducing Aβ production would not protect from LOAD. However, the Icelandic APP mutation (discussed above) reduces risk of the sporadic form of AD, again implicating Aβ in LOAD (Jonsson et al., 2012). These lines of evidence strongly support a critical role for Aβ in the pathogenesis of the more frequent sporadic form of the disease.
Perhaps the most common argument against the amyloid hypothesis is the fact that plaque burden correlates poorly with cognitive decline (Sorrentino et al., 2014). This discrepancy could be partially explained by current theories indicating soluble oligomeric forms of Aβ are the primary mediator of disease and not the insoluble fibrillar amyloid. However, there may be a more fundamental explanation. If Aβ is an initiator of the long and complex cascade of pathologic alterations that take place in AD, then a multitude of downstream effectors and modifying factors (including genetics, other medical conditions, and environment) would have a profound effect on the rate and severity of disease progression (Korf et al., 2004; Bennett et al., 2006; Barberger-Gateau et al., 2007; Ngandu et al., 2007; van Vliet et al., 2009; Chang et al., 2010; Rusanen et al., 2011; Reijmer et al., 2012; Tolppanen et al., 2013; Virta et al., 2013). In this scenario one would predict that Aβ would correlate more poorly (but still significantly) with cognitive decline while more downstream effectors (e.g., synaptic loss) would correlate better (Bennett et al., 2003, 2005).
Amyloid-Beta Clearance
In humans, Aβ is estimated to have a physiological production rate of 7.6% per hour and a clearance rate of 8.3% per hour (Bateman et al., 2006). The various mechanisms of removal provide greater Aβ clearance than production, thus limiting its accumulation. Interestingly, human data provide evidence that accumulation in LOAD results from impaired clearance rather than increased production of Aβ (Mawuenyega et al., 2010). Using a technique of in vivo labeling, Mawuenyega et al. found that the clearance rate of Aβ42 in AD individuals was reduced to 5.3% per hour from 7.6% per hour in controls. Likewise, the Aβ40 clearance rate was reduced to 5.2% per hour from 7.0% per hour in controls. This finding emphasizes the importance of Aβ clearance in AD.
The proteolytic degradation of Aβ is a major route of clearance. A variety of Aβ degrading enzymes have been found and this topic has been comprehensively reviewed (Miners et al., 2011a; Nalivaeva et al., 2012). Of these enzymes, neprilysin (NEP) is considered one of the most important for the control of cerebral Aβ levels. NEP is a member of the metalloprotease 13 (M13) family of zinc metalloproteases. This 97 kD cell surface-associated enzyme functions in the periphery and central nervous system where it has been shown to degrade small peptides (Turner et al., 2001). The 50 amino acid catalytic core cleaves on the N-terminal side of hydrophobic residues (Kerr and Kenny, 1974a,b; Howell et al., 1995). Using radiolableled Aβ, Iwata et al. (2000) showed that Aβ42 primarily underwent degradation by NEP in their in vivo assay. Furthermore, application of inhibitors to NEP in rat brain produced dramatic elevations of endogenous Aβ resulting in plaque deposition. This effect was independently replicated in mice (Dolev and Michaelson, 2004; Nisemblat et al., 2008). Further supporting NEP as a critical Aβ-degrading enzyme is the observation that NEP overexpression imparts significant reductions in Aβ plaque deposition in APP-transgenic mice (Marr et al., 2003), and in some experiments, improved cognitive performance (reviewed in Marr and Spencer, 2010). It has also been shown that NEP mRNA and protein expression levels are reduced in association with age or in AD subjects (Reilly, 2001; Yasojima et al., 2001a,b; Iwata et al., 2002; Apelt et al., 2003; Caccamo et al., 2005; Maruyama et al., 2005; Wang et al., 2005, 2010); however, this notion has been seriously challenged more recently. Miners and colleagues have used a highly specific enzyme-immunocapture/activity assay to show that NEP activity levels increase with age and during the progression of AD (Miners et al., 2009, 2010, 2011b). This is similar to the consensus on most endopeptidase expression levels in association with AD (Miners et al., 2011a), and may reflect a homeostatic response to the abundance of Aβ substrate and/or to the inflammatory environment occurring in AD. Regardless, these increased endogenous levels of Aβ-degrading enzymes are ultimately insufficient to prevent the accumulation and aggregation of Aβ in AD.
Despite the data demonstrating the importance of NEP in enzymatic degradation of Aβ, other enzymes are clearly worthy of clinical study. For example, NEP knockout mice show only a moderate (1.5–2 fold) increase in Aβ levels that are far from the levels needed to induce plaque deposition, as observed with NEP inhibitors, until very advanced age (Iwata et al., 2001; Madani et al., 2006). This modest increase in Aβ raises the possibility of alternative Aβ degrading-enzymes that are likewise sensitive to NEP-inhibitors (i.e., are NEP-like).
Neprilysin-2
In the search for alternate Aβ degrading enzymes, NEP-like proteases are important because of their potential involvement in the spike in Aβ levels post treatment with NEP inhibitors. One such enzyme is neprilysin-2 (NEP2). NEP2 is also a zinc metalloendopeptidase belonging to the same M13 family as NEP. It has also been demonstrated in rodents that NEP2 is sensitive to the same NEP inhibitors, phosphoramidon and thiorphan (Ikeda et al., 1999; Ghaddar et al., 2000; Shirotani et al., 2001). NEP2 was first discovered while searching for an enzyme to degrade endothelin in endothelin converting enzyme-1 knockout mice and found to be a secreted peptide, termed soluble-secreted endopeptidase (SEP; Ikeda et al., 1999). Since then, it has also been referred to as neprilysin-like protein (NEPLP and Nl1) in mice and membrane-bound metalloendopeptidase-like enzyme 1 or 2 (MMEL1/2) in humans (Ghaddar et al., 2000; Bonvouloir et al., 2001; Shirotani et al., 2001). It is the closest homolog to NEP, with 55% sequence identity and similar catalytic sites. Structural modeling of NEP2 using sequence alignment and the crystal structure of NEP projects 97% identity in the active sites of these two enzymes (Voisin et al., 2004). Due to alternative splicing, murine NEP2 can exist in a membrane-bound form (mNEP2-α) or a secreted form (mNEP2-β; Figure 1). Alternative splicing also acts on the human form of NEP2 creating several isoforms. Human NEP2-β was found to be localized to both the extracellular surface and to be secreted, likely due to inefficient furan-like processing as a result of a proline residue near the processing site (P’2; Bonvouloir et al., 2001).
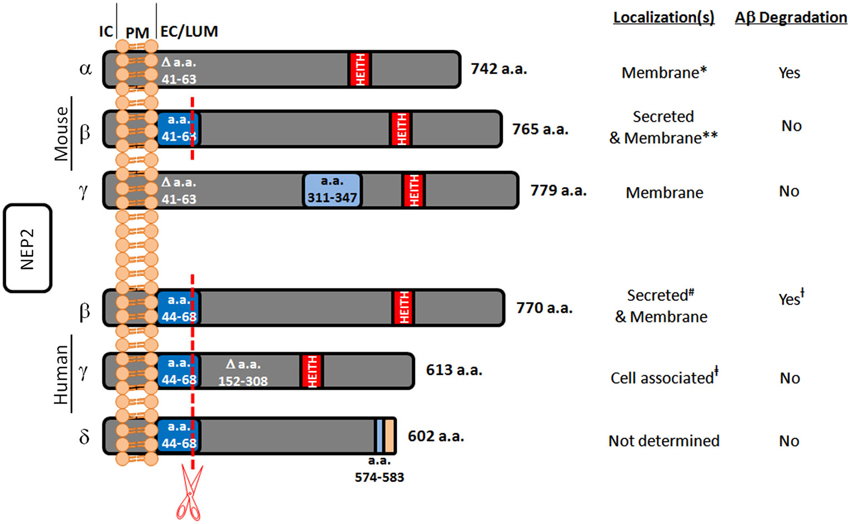
Figure 1. Summary of known murine and human NEP2 splice forms. This figure represents the protein sequences of NEP2 isoforms and summarizes their localizations and Aβ-degrading activity. The murine α-form is missing amino acids (a.a.) 41–63 which contains a furin-like processing site (red line) leaving this form membrane bound. The murine β-form contains this sequence and is secreted. The murine γ-form is missing a.a. 41–63 but contains an insertion at a.a. 311–347. All the known human forms of NEP2 contain the furin-like processing site. The β-form of human NEP2 is 77% identical to the murine β-form and is secreted. The human γ-form is missing a.a. 152–308 and is not secreted. Because of an alternate splice acceptor site, the δ-form of human NEP2 is knocked out of frame resulting in early termination and absence of the critical zinc-binding motif (HEITH). IC, intracellular; PM, plasma membrane; EC/LUM, extracellular or luminal; * associated with the membrane fraction and located inside the cell (Ikeda et al., 1999); however, in vitro expression did show the ability to degrade extracellular Aβ (Hafez et al., 2011); ** primarily secreted (Ghaddar et al., 2000) but also found in the membrane fraction (Ikeda et al., 1999; Raharjo et al., 2001; Shirotani et al., 2001). # associated with the membrane fraction and secreted (Whyteside and Turner, 2008); however, also shown to be expressed on the cell surface and to degrade extracellular Aβ (Huang et al., 2008); † Aβ-degrading activity found only at the extracellular surface (Huang et al., 2008); ‡ found in the total cell lysate (subcellular compartment localization not done) (Huang et al., 2008).
NEP2 Substrates and Localization
In a study by Shirotani et al. (2001) using membrane-bound fractions, mNEP2-α was shown to have slower and weaker Aβ40 degrading properties when compared to NEP (with little to no effect on Aβ42), and mNEP2-β/γ had nearly undetectable activity against Aβ. However, subsequent studies using live cell assays demonstrated that mNEP2-α and hNEP2-β are able to degrade Aβ40, and more importantly, Aβ42 with comparable efficiency with NEP (Huang et al., 2008; Hafez et al., 2011; Figure 1). Interestingly, this activity was also not secreted into the cell culture medium for hNEP2-β even though the protein can be found there. hNEP2-γ did not show secreted Aβ-degrading activity or activity at the cell surface. Despite the numerous similarities between NEP2 and NEP, differences do exist. While the substrate specificities are very similar (particularly in rodents) their localizations suggest divergent roles in the central nervous system. Studies in rodents have shown that, while NEP2 is expressed at variable levels in most brain regions, it is most highly expressed in the brain stem, hypothalamus and pituitary (Ouimet et al., 2000; Facchinetti et al., 2003). Considering this localization and the substrates cleaved (e.g., gonadotropin-releasing hormone) a role in the metabolism of neuropeptides of the hypothalamo-pituitary axis has been suggested (Rose et al., 2002). Unlike NEP, NEP2 is highly expressed in the testis and brain (Bonvouloir et al., 2001), and NEP2 knockout mice do show reduced sperm function (Carpentier et al., 2004). Furthermore, hNEP2 was found to have a more restricted substrate specificity compared to hNEP with less activity against several vasoactive peptides (Whyteside and Turner, 2008). More comprehensive reviews of NEP2 are available (Marr and Spencer, 2010; Marr, 2013).
NEP2 in Alzheimer’s Disease
NEP2 knockout experiments have demonstrated the importance of NEP2 in amyloid regulation (Hafez et al., 2011). Using mice deficient for the NEP2 gene (Carpentier et al., 2004), Hafez et al. (2011) reported significant elevations in total Aβ species in the hippocampus and brainstem/diencephalon (~1.5-fold). Increases in Aβ accumulation were more dramatic in NEP2 knockout mice crossbred with APP transgenic mice. In NEP/NEP2 double-knockout mice, Aβ levels were again increased (~1.5- to 2-fold), compared with NEP−/–/NEP2+/+ controls. Treatment of these double-knockout mice with phosphoramidon-infusing pumps resulted in significant elevations in Aβ. This significant elevation in Aβ levels was also observed with intranasal treatment of phosphoramidon in both wild-type and NEP/NEP2 double-knockout mice suggesting that yet other NEP-like Aβ-degrading endopeptidases are contributing to Aβ-catabolism (Hanson et al., 2010; Hafez et al., 2011).
The importance of NEP2 was further explored in human studies. Using brain tissue from various brain regions of non-impaired, mild-cognitive impaired (MCI), and AD subjects, NEP2 mRNA expression levels in the mid-temporal gyrus were found to be lowered in women with MCI compared to non-impaired women (Huang et al., 2012). Furthermore, this altered expression was found to have a sexually dimorphic change, with males with MCI having significantly elevated NEP2 mRNA levels compared to non-impaired males in the mid-temporal and mid-frontal gyri. However, levels of NEP2 mRNA in those with AD were not different from non-impaired controls. These expression profiles closely followed what was measured for NEP in the mid-temporal gyrus. These data suggest that there is a dysregulation of NEP-like enzyme mRNA levels early in the pathogenesis of AD. Huang et al. (2012) also used an immunodepletion/activity assay to detect NEP2 activity in brain specimens and found reduced activity in association with MCI and AD regardless of sex. Therefore, this initial study suggests that NEP2 activity is reduced in association with AD, which is contrary to most of the findings with other Aβ degrading proteases (discussed above). Finally, it was also shown that NEP2 mRNA can be detected in the peripheral blood of humans. These data suggest the potential of NEP2 assays to serve as preclinical mRNA expression or enzyme activity markers for AD if expression levels in the brain mimic what is present in the blood or cerebrospinal fluid. Many neural pathologies including AD can be reflected in changes in gene expression, splicing, and protein profiles in blood and CSF, providing precedent for examining gene expression in these body fluids (Tang et al., 2005; Blennow et al., 2010; Courtney et al., 2010; Wu et al., 2011; Leuner et al., 2012; Mapstone et al., 2014). The search for new preclinical biomarkers is important given that the identification of biomarkers in AD has been relatively unsuccessful. In a recent systematic review of 59 studies, McGhee et al. (2014) found insufficient evidence to recommend using any biomarker as an outcome measure in disease progression. This highlights the importance of finding tools like NEP2 expression to detect AD early in disease progression.
Conclusion
The impact that Alzheimer’s disease will have on the future of medicine over the next 40 years cannot be understated, and the accumulation of Aβ is currently the best theory to describe the main drive for the overall disease process. Therefore continued research into the mechanisms of Aβ clearance remains of upmost importance. The discovery of endopeptidases that degrade Aβ, such as NEP and NEP2 highlight an avenue of intervention via viral-mediated gene therapy. While NEP studies have produced encouraging results, studies utilizing NEP2 as a therapeutic agent are still warranted as NEP2 may be more selective for Aβ (Whyteside and Turner, 2008). Studies are also needed to evaluate the effectiveness of NEP2 assays as a potential molecular marker for the disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramowski, D., Wiederhold, K. H., Furrer, U., Jaton, A. L., Neuenschwander, A., Runser, M. J., et al. (2008). Dynamics of Abeta turnover and deposition in different beta-amyloid precursor protein transgenic mouse models following gamma-secretase inhibition. J. Pharmacol. Exp. Ther. 327, 411–424. doi: 10.1124/jpet.108.140327
Apelt, J., Ach, K., and Schliebs, R. (2003). Aging-related down-regulation of neprilysin, a putative beta-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of beta-amyloid plaques. Neurosci. Lett. 339, 183–186. doi: 10.1016/s0304-3940(03)00030-2
Bales, K. R., Verina, T., Cummins, D. J., Du, Y., Dodel, R. C., Saura, J., et al. (1999). Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 96, 15233–15238. doi: 10.1073/pnas.96.26.15233
Barberger-Gateau, P., Raffaitin, C., Letenneur, L., Berr, C., Tzourio, C., Dartigues, J. F., et al. (2007). Dietary patterns and risk of dementia: the three-city cohort study. Neurology 69, 1921–1930. doi: 10.1212/01.wnl.0000278116.37320.52
Bateman, R. J., Munsell, L. Y., Morris, J. C., Swarm, R., Yarasheski, K. E., and Holtzman, D. M. (2006). Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861. doi: 10.1038/nm1438
Belinson, H., Kariv-Inbal, Z., Kayed, R., Masliah, E., and Michaelson, D. M. (2010). Following activation of the amyloid cascade, apolipoprotein E4 drives the in vivo oligomerization of amyloid-beta resulting in neurodegeneration. J. Alzheimers Dis. 22, 959–970. doi: 10.3233/JAD-2010-101008
Bennett, D. A., Schneider, J. A., Tang, Y., Arnold, S. E., and Wilson, R. S. (2006). The effect of social networks on the relation between Alzheimer’s disease pathology and level of cognitive function in old people: a longitudinal cohort study. Lancet Neurol. 5, 406–412. doi: 10.1016/S1474-4422(06)70417-3
Bennett, D. A., Schneider, J. A., Wilson, R. S., Bienias, J. L., Berry-Kravis, E., and Arnold, S. E. (2005). Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J. Neurol. Neurosurg. Psychiatry 76, 1194–1199. doi: 10.1136/jnnp.2004.054445
Bennett, D. A., Wilson, R. S., Schneider, J. A., Evans, D. A., Aggarwal, N. T., Arnold, S. E., et al. (2003). Apolipoprotein E epsilon4 allele, AD pathology and the clinical expression of Alzheimer’s disease. Neurology 60, 246–252. doi: 10.1212/01.wnl.0000042478.08543.f7
Blennow, K., Hampel, H., Weiner, M., and Zetterberg, H. (2010). Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 6, 131–144. doi: 10.1038/nrneurol.2010.4
Bonvouloir, N., Lemieux, N., Crine, P., Boileau, G., and DesGroseillers, L. (2001). Molecular cloning, tissue distribution and chromosomal localization of MMEL2, a gene coding for a novel human member of the neutral endopeptidase-24.11 family. DNA Cell Biol. 20, 493–498. doi: 10.1089/104454901316976127
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Caccamo, A., Oddo, S., Sugarman, M. C., Akbari, Y., and LaFerla, F. M. (2005). Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol. Aging 26, 645–654. doi: 10.1016/j.neurobiolaging.2004.06.013
Carpentier, M., Guillemette, C., Bailey, J. L., Boileau, G., Jeannotte, L., DesGroseillers, L., et al. (2004). Reduced fertility in male mice deficient in the zinc metallopeptidase NL1. Mol. Cell. Biol. 24, 4428–4437. doi: 10.1128/mcb.24.10.4428-4437.2004
Chang, M., Jonsson, P. V., Snaedal, J., Bjornsson, S., Saczynski, J. S., Aspelund, T., et al. (2010). The effect of midlife physical activity on cognitive function among older adults: AGES—Reykjavik study. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1369–1374. doi: 10.1093/gerona/glq152
Cheng, I. H., Palop, J. J., Esposito, L. A., Bien-Ly, N., Yan, F., and Mucke, L. (2004). Aggressive amyloidosis in mice expressing human amyloid peptides with the arctic mutation. Nat. Med. 10, 1190–1192. doi: 10.1038/nm1123
Clements, A., Allsop, D., Walsh, D. M., and Williams, C. H. (1996). Aggregation and metal-binding properties of mutant forms of the amyloid A beta peptide of Alzheimer’s disease. J. Neurochem. 66, 740–747. doi: 10.1046/j.1471-4159.1996.66020740.x
Clements, W. D., Halliday, M. I., McCaigue, M. D., Barclay, R. G., and Rowlands, B. J. (1993). Effects of extrahepatic obstructive jaundice on Kupffer cell clearance capacity. Arch. Surg. 128, 200–204; discussion 204–205. doi: 10.1001/archsurg.1993.01420140077012
Courtney, E., Kornfeld, S., Janitz, K., and Janitz, M. (2010). Transcriptome profiling in neurodegenerative disease. J. Neurosci. Methods 193, 189–202. doi: 10.1016/j.jneumeth.2010.08.018
Dodart, J. C., Marr, R. A., Koistinaho, M., Gregersen, B. M., Malkani, S., Verma, I. M., et al. (2005). Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 102, 1211–1216. doi: 10.1073/pnas.0409072102
Dolev, I., and Michaelson, D. M. (2004). A nontransgenic mouse model shows inducible amyloid-beta (Abeta) peptide deposition and elucidates the role of apolipoprotein E in the amyloid cascade. Proc. Natl. Acad. Sci. U S A 101, 13909–13914. doi: 10.1073/pnas.0404458101
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. doi: 10.1056/NEJMoa1312889
Facchinetti, P., Rose, C., Schwartz, J. C., and Ouimet, T. (2003). Ontogeny, regional and cellular distribution of the novel metalloprotease neprilysin 2 in the rat: a comparison with neprilysin and endothelin-converting enzyme-1. Neuroscience 118, 627–639. doi: 10.1016/s0306-4522(02)01002-3
Fagan, A. M., Watson, M., Parsadanian, M., Bales, K. R., Paul, S. M., and Holtzman, D. M. (2002). Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 9, 305–318. doi: 10.1006/nbdi.2002.0483
Farlow, M., Arnold, S. E., van Dyck, C. H., Aisen, P. S., Snider, B. J., Porsteinsson, A. P., et al. (2012). Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 8, 261–271. doi: 10.1016/j.jalz.2011.09.224
Ghaddar, G., Ruchon, A. F., Carpentier, M., Marcinkiewicz, M., Seidah, N. G., Crine, P., et al. (2000). Molecular cloning and biochemical characterization of a new mouse testis soluble-zinc-metallopeptidase of the neprilysin family. Biochem. J. 347, 419–429. doi: 10.1042/0264-6021:3470419
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer’s disease. Science 314, 777–781. doi: 10.1126/science.1132814
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Hafez, D., Huang, J. Y., Huynh, A. M., Valtierra, S., Rockenstein, E., Bruno, A. M., et al. (2011). Neprilysin-2 is an important beta-amyloid degrading enzyme. Am. J. Pathol. 178, 306–312. doi: 10.1016/j.ajpath.2010.11.012
Hanson, L. R., Hafez, D., Svitak, A. L., Burns, R. B., Li, X., Frey, W. H., et al. (2010). Intranasal phosphoramidon increases beta-amyloid levels in wild-type and NEP/NEP2-deficient mice. J. Mol. Neurosci. 43, 424–427. doi: 10.1007/s12031-010-9460-8
Hashimoto, T., Serrano-Pozo, A., Hori, Y., Adams, K. W., Takeda, S., Banerji, A. O., et al. (2012). Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 32, 15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012
Holtzman, D. M., Bales, K. R., Tenkova, T., Fagan, A. M., Parsadanian, M., Sartorius, L. J., et al. (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 97, 2892–2897. doi: 10.1073/pnas.050004797
Howell, S., Nalbantoglu, J., and Crine, P. (1995). Neutral endopeptidase can hydrolyze beta-amyloid (1–40) but shows no effect on beta-amyloid precursor protein metabolism. Peptides 16, 647–652. doi: 10.1016/0196-9781(95)00021-b
Huang, J. Y., Bruno, A. M., Patel, C. A., Huynh, A. M., Philibert, K. D., Glucksman, M. J., et al. (2008). Human membrane metallo-endopeptidase-like protein degrades both beta-amyloid 42 and beta-amyloid 40. Neuroscience 155, 258–262. doi: 10.1016/j.neuroscience.2008.05.006
Huang, J. Y., Hafez, D. M., James, B. D., Bennett, D. A., and Marr, R. A. (2012). Altered NEP2 expression and activity in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 28, 433–441. doi: 10.3233/JAD-2011-111307
Ikeda, K., Emoto, N., Raharjo, S. B., Nurhantari, Y., Saiki, K., Yokoyama, M., et al. (1999). Molecular identification and characterization of novel membrane-bound metalloprotease, the soluble secreted form of which hydrolyzes a variety of vasoactive peptides. J. Biol. Chem. 274, 32469–32477. doi: 10.1074/jbc.274.45.32469
Iwata, N., Takaki, Y., Fukami, S., Tsubuki, S., and Saido, T. C. (2002). Region-specific reduction of A beta-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J. Neurosci. Res. 70, 493–500. doi: 10.1002/jnr.10390
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., et al. (2000). Identification of the major Abeta1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150. doi: 10.1038/72237
Iwatsubo, T. (1998). Abeta42, presenilins and Alzheimer’s disease. Neurobiol. Aging 19, S11–S13. doi: 10.1016/S0197-4580(98)00027-X
Jarrett, J. T., and Lansbury, P. T. Jr. (1993). Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73, 1055–1058. doi: 10.1016/0092-8674(93)90635-4
Jiang, Q., Lee, C. Y., Mandrekar, S., Wilkinson, B., Cramer, P., Zelcer, N., et al. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693. doi: 10.1016/j.neuron.2008.04.010
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
Kaden, D., Harmeier, A., Weise, C., Munter, L. M., Althoff, V., Rost, B. R., et al. (2012). Novel APP/Aβ mutation K16N produces highly toxic heteromeric Abeta oligomers. EMBO Mol. Med. 4, 647–659. doi: 10.1002/emmm.201200239
Kerr, M. A., and Kenny, A. J. (1974a). The molecular weight and properties of a neutral metallo-endopeptidase from rabbit kidney brush border. Biochem. J. 137, 489–495.
Kerr, M. A., and Kenny, A. J. (1974b). The purification and specificity of a neutral endopeptidase from rabbit kidney brush border. Biochem. J. 137, 477–488.
Kim, W. S., Li, H., Ruberu, K., Chan, S., Elliott, D. A., Low, J. K., et al. (2013). Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer’s disease. J. Neurosci. 33, 4387–4394. doi: 10.1523/JNEUROSCI.4165-12.2013
Kline, A. (2012). Apolipoprotein E, amyloid-ss clearance and therapeutic opportunities in Alzheimer’s disease. Alzheimers Res. Ther. 4:32. doi: 10.1186/alzrt135
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., et al. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 10, 719–726. doi: 10.1038/nm1058
Korf, E. S., White, L. R., Scheltens, P., and Launer, L. J. (2004). Midlife blood pressure and the risk of hippocampal atrophy: the Honolulu Asia aging study. Hypertension 44, 29–34. doi: 10.1161/01.hyp.0000132475.32317.bb
Leuner, K., Schulz, K., Schutt, T., Pantel, J., Prvulovic, D., Rhein, V., et al. (2012). Peripheral mitochondrial dysfunction in Alzheimer’s disease: focus on lymphocytes. Mol. Neurobiol. 46, 194–204. doi: 10.1007/s12035-012-8300-y
Lippa, C. F., Saunders, A. M., Smith, T. W., Swearer, J. M., Drachman, D. A., Ghetti, B., et al. (1996). Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology 46, 406–412. doi: 10.1212/wnl.46.2.406
Madani, R., Poirier, R., Wolfer, D. P., Welzl, H., Groscurth, P., Lipp, H. P., et al. (2006). Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. J. Neurosci. Res. 84, 1871–1878. doi: 10.1002/jnr.21074
Manelli, A. M., Stine, W. B., Van Eldik, L. J., and Ladu, M. J. (2004). ApoE and Abeta1–42 interactions: effects of isoform and conformation on structure and function. J. Mol. Neurosci. 23, 235–246. doi: 10.1385/jmn:23:3:235
Mapstone, M., Cheema, A. K., Fiandaca, M. S., Zhong, X., Mhyre, T. R., MacArthur, L. H., et al. (2014). Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418. doi: 10.1038/nm.3466
Marr, R. A., Rockenstein, E., Mukherjee, A., Kindy, M. S., Hersh, L. B., Gage, F. H., et al. (2003). Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23, 1992–1996. doi: 10.1016/s0140-6736(03)12914-5
Marr, R. A., and Spencer, B. J. (2010). NEP-like endopeptidases and Alzheimer’s disease. Curr. Alzheimer Res. 7, 223–229. doi: 10.2174/156720510791050849
Maruyama, M., Higuchi, M., Takaki, Y., Matsuba, Y., Tanji, H., Nemoto, M., et al. (2005). Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer’s disease. Ann. Neurol. 57, 832–842. doi: 10.1002/ana.20494
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774. doi: 10.1126/science.1197623
McGhee, D. J., Ritchie, C. W., Thompson, P. A., Wright, D. E., Zajicek, J. P., and Counsell, C. E. (2014). A systematic review of biomarkers for disease progression in Alzheimer’s disease. PLoS One 9:e88854. doi: 10.1371/journal.pone.0088854
Miners, J. S., Baig, S., Tayler, H., Kehoe, P. G., and Love, S. (2009). Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J. Neuropathol. Exp. Neurol. 68, 902–914. doi: 10.1097/NEN.0b013e3181afe475
Miners, J. S., Barua, N., Kehoe, P. G., Gill, S., and Love, S. (2011a). Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 70, 944–959. doi: 10.1097/NEN.0b013e3182345e46
Miners, J. S., Morris, S., Love, S., and Kehoe, P. G. (2011b). Accumulation of insoluble amyloid-beta in down’s syndrome is associated with increased BACE-1 and neprilysin activities. J. Alzheimers Dis. 23, 101–108. doi: 10.3233/JAD-2010-101395
Miners, J. S., van Helmond, Z., Kehoe, P. G., and Love, S. (2010). Changes with age in the activities of beta-secretase and the Abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 20, 794–802. doi: 10.1111/j.1750-3639.2010.00375.x
Morley, J. E., Farr, S. A., Banks, W. A., Johnson, S. N., Yamada, K. A., and Xu, L. (2010). A physiological role for amyloid-beta protein:enhancement of learning and memory. J. Alzheimers Dis. 19, 441–449. doi: 10.3233/JAD-2009-1230
Nalivaeva, N. N., Beckett, C., Belyaev, N. D., and Turner, A. J. (2012). Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 120(Suppl. 1), 167–185. doi: 10.1111/j.1471-4159.2011.07510.x
Ngandu, T., von Strauss, E., Helkala, E. L., Winblad, B., Nissinen, A., Tuomilehto, J., et al. (2007). Education and dementia: what lies behind the association? Neurology 69, 1442–1450. doi: 10.1212/01.wnl.0000277456.29440.16
Nilsberth, C., Westlind-Danielsson, A., Eckman, C. B., Condron, M. M., Axelman, K., Forsell, C., et al. (2001). The ’Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 4, 887–893. doi: 10.1038/nn0901-887
Nisemblat, Y., Belinson, H., Dolev, I., and Michaelson, D. M. (2008). Activation of the amyloid cascade by intracerebroventricular injection of the protease inhibitor phosphoramidon. Neurodegener. Dis. 5, 166–169. doi: 10.1159/000113692
Ouimet, T., Facchinetti, P., Rose, C., Bonhomme, M. C., Gros, C., and Schwartz, J. C. (2000). Neprilysin II: a putative novel metalloprotease and its isoforms in CNS and testis. Biochem. Biophys. Res. Commun. 271, 565–570. doi: 10.1006/bbrc.2000.2664
Pimplikar, S. W. (2009). Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int. J. Biochem. Cell Biol. 41, 1261–1268. doi: 10.1016/j.biocel.2008.12.015
Prasher, V. P., Farrer, M. J., Kessling, A. M., Fisher, E. M., West, R. J., Barber, P. C., et al. (1998). Molecular mapping of Alzheimer-type dementia in down’s syndrome. Ann. Neurol. 43, 380–383. doi: 10.1002/ana.410430316
Puzzo, D., Privitera, L., Leznik, E., Fa, M., Staniszewski, A., Palmeri, A., et al. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008
Puzzo, D., Privitera, L., and Palmeri, A. (2012). Hormetic effect of amyloid-beta peptide in synaptic plasticity and memory. Neurobiol. Aging 33, 1484.e15–1484.e24. doi: 10.1016/j.neurobiolaging.2011.12.020
Raharjo, S. B., Emoto, N., Ikeda, K., Sato, R., Yokoyama, M., and Matsuo, M. (2001). Alternative splicing regulates the endoplasmic reticulum localization or secretion of soluble secreted endopeptidase. J. Biol. Chem. 276, 25612–25620. doi: 10.1074/jbc.m101703200
Reijmer, Y. D., van den Berg, E., Dekker, J. M., Nijpels, G., Stehouwer, C. D., Kappelle, L. J., et al. (2012). Development of vascular risk factors over 15 years in relation to cognition: the Hoorn study. J. Am. Geriatr. Soc. 60, 1426–1433. doi: 10.1111/j.1532-5415.2012.04081.x
Reilly, C. E. (2001). Neprilysin content is reduced in Alzheimer brain areas. J. Neurol. 248, 159–160. doi: 10.1007/s004150170259
Reitz, C., Jun, G., Naj, A., Rajbhandary, R., Vardarajan, B. N., Wang, L. S., et al. (2013). Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492. doi: 10.1001/jama.2013.2973
Roberson, E. D., and Mucke, L. (2006). 100 years and counting: prospects for defeating Alzheimer’s disease. Science 314, 781–784. doi: 10.1126/science.1132813
Roher, A. E., Cribbs, D. H., Kim, R. C., Maarouf, C. L., Whiteside, C. M., Kokjohn, T. A., et al. (2013). Bapineuzumab alters abeta composition: implications for the amyloid cascade hypothesis and anti-amyloid immunotherapy. PLoS One 8:e59735. doi: 10.1371/journal.pone.0059735
Rose, C., Voisin, S., Gros, C., Schwartz, J. C., and Ouimet, T. (2002). Cell-specific activity of neprilysin 2 isoforms and enzymic specificity compared with neprilysin. Biochem. J. 363, 697–705. doi: 10.1042/0264-6021:3630697
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerriere, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi: 10.1038/ng1718
Rusanen, M., Kivipelto, M., Quesenberry, C. P. Jr., Zhou, J., and Whitmer, R. A. (2011). Heavy smoking in midlife and long-term risk of Alzheimer disease and vascular dementia. Arch. Intern. Med. 171, 333–339. doi: 10.1001/archinternmed.2010.393
Salloway, S., Sperling, R., Fox, N. C., Blennow, K., Klunk, W., Raskind, M., et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. doi: 10.1056/NEJMoa1304839
Salloway, S., Sperling, R., Gilman, S., Fox, N. C., Blennow, K., Raskind, M., et al. (2009). A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73, 2061–2070. doi: 10.1212/WNL.0b013e3181c67808
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/jci10498
Shirotani, K., Tsubuki, S., Iwata, N., Takaki, Y., Harigaya, W., Maruyama, K., et al. (2001). Neprilysin degrades both amyloid beta peptides 1–40 and 1-42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 276, 21895–21901. doi: 10.1074/jbc.m008511200
Sorrentino, P., Iuliano, A., Polverino, A., Jacini, F., and Sorrentino, G. (2014). The dark sides of amyloid in Alzheimer’s disease pathogenesis. FEBS Lett. 588, 641–652. doi: 10.1016/j.febslet.2013.12.038
Tang, Y., Gilbert, D. L., Glauser, T. A., Hershey, A. D., and Sharp, F. R. (2005). Blood gene expression profiling of neurologic diseases: a pilot microarray study. Arch. Neurol. 62, 210–215. doi: 10.1001/archneur.62.2.210
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006296. doi: 10.1101/cshperspect.a006296
Tolppanen, A. M., Lavikainen, P., Solomon, A., Kivipelto, M., Uusitupa, M., Soininen, H., et al. (2013). History of medically treated diabetes and risk of Alzheimer disease in a nationwide case-control study. Diabetes Care 36, 2015–2019. doi: 10.2337/dc12-1287
Tsubuki, S., Takaki, Y., and Saido, T. C. (2003). Dutch, Flemish, Italian and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet 361, 1957–1958. doi: 10.1016/s0140-6736(03)13555-6
Turner, A. J., Isaac, R. E., and Coates, D. (2001). The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays 23, 261–269. doi: 10.1002/1521-1878(200103)23:3<261::aid-bies1036>3.0.co;2-k
van Vliet, P., van de Water, W., de Craen, A. J., and Westendorp, R. G. (2009). The influence of age on the association between cholesterol and cognitive function. Exp. Gerontol. 44, 112–122. doi: 10.1016/j.exger.2008.05.004
Virta, J. J., Heikkila, K., Perola, M., Koskenvuo, M., Raiha, I., Rinne, J. O., et al. (2013). Midlife cardiovascular risk factors and late cognitive impairment. Eur. J. Epidemiol. 28, 405–416. doi: 10.1007/s10654-013-9794-y
Voisin, S., Rognan, D., Gros, C., and Ouimet, T. (2004). A three-dimensional model of the neprilysin 2 active site based on the X-ray structure of neprilysin. Identification of residues involved in substrate hydrolysis and inhibitor binding of neprilysin 2. J. Biol. Chem. 279, 46172–46181. doi: 10.1074/jbc.m407333200
Walsh, D. M., Minogue, A. M., Sala Frigerio, C., Fadeeva, J. V., Wasco, W., and Selkoe, D. J. (2007). The APP family of proteins: similarities and differences. Biochem. Soc. Trans. 35, 416–420. doi: 10.1042/bst0350416
Wang, D. S., Lipton, R. B., Katz, M. J., Davies, P., Buschke, H., Kuslansky, G., et al. (2005). Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J. Neuropathol. Exp. Neurol. 64, 378–385.
Wang, S., Wang, R., Chen, L., Bennett, D. A., Dickson, D. W., and Wang, D. S. (2010). Expression and functional profiling of neprilysin, insulin-degrading enzyme and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J. Neurochem. 115, 47–57. doi: 10.1111/j.1471-4159.2010.06899.x
Whyteside, A. R., and Turner, A. J. (2008). Human neprilysin-2 (NEP2) and NEP display distinct subcellular localisations and substrate preferences. FEBS Lett. 582, 2382–2386. doi: 10.1016/j.febslet.2008.05.046
Wisniewski, K. E., Dalton, A. J., McLachlan, C., Wen, G. Y., and Wisniewski, H. M. (1985). Alzheimer’s disease in down’s syndrome: clinicopathologic studies. Neurology 35, 957–961. doi: 10.1212/wnl.35.7.957
Wu, Y., Le, W., and Jankovic, J. (2011). Preclinical biomarkers of Parkinson disease. Arch. Neurol. 68, 22–30. doi: 10.1001/archneurol.2010.321
Yasojima, K., Akiyama, H., McGeer, E. G., and McGeer, P. L. (2001a). Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci. Lett. 297, 97–100. doi: 10.1016/s0304-3940(00)01675-x
Keywords: neprilysin, NEP, neprilysin-2, NEP2, amyloid hypothesis, clearance, amyloid-beta degradation, Alzheimer’s disease
Citation: Marr RA and Hafez DM (2014) Amyloid-beta and Alzheimer’s disease: the role of neprilysin-2 in amyloid-beta clearance. Front. Aging Neurosci. 6:187. doi: 10.3389/fnagi.2014.00187
Received: 02 April 2014; Paper pending published: 26 June 2014;
Accepted: 09 July 2014; Published online: 13 August 2014.
Edited by:
Eliezer Masliah, University of California, San Diego, USAReviewed by:
Junming Wang, University of Mississippi Medical Center, USAGianluigi Zanusso, University of Verona, Italy
Copyright © 2014 Marr and Hafez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert A. Marr, Department of Neuroscience, Rosalind Franklin University of Medicine and Science, 3333 Green Bay Road, North Chicago, IL 60064, USA e-mail:Um9iZXJ0Lk1hcnJAcm9zYWxpbmRmcmFua2xpbi5lZHU=