 Clara De Palma
Clara De Palma Cristiana Perrotta
Cristiana Perrotta Paolo Pellegrino
Paolo Pellegrino Emilio Clementi
Emilio Clementi Davide Cervia
Davide Cervia- 1Unit of Clinical Pharmacology, Department of Biomedical and Clinical Sciences “L. Sacco”, National Research Council-Institute of Neuroscience, University Hospital “L. Sacco”, University of Milan, Milan, Italy
- 2Scientific Institute IRCCS Eugenio Medea, Bosisio Parini, Italy
- 3Department for Innovation in Biological, Agro-Food and Forest Systems, University of Tuscia, Viterbo, Italy
Muscular dystrophies are a group of genetic and heterogeneous neuromuscular disorders characterized by the primary wasting of skeletal muscle. In Duchenne muscular dystrophy (DMD), the most severe form of these diseases, the mutations in the dystrophin gene lead to muscle weakness and wasting, exhaustion of muscular regenerative capacity, and chronic local inflammation leading to substitution of myofibers by connective and adipose tissue. DMD patients suffer from continuous and progressive skeletal muscle damage followed by complete paralysis and death, usually by respiratory and/or cardiac failure. No cure is yet available, but several therapeutic approaches aiming at reversing the ongoing degeneration have been investigated in preclinical and clinical settings. Autophagy is an important proteolytic system of the cell and has a crucial role in the removal of proteins, aggregates, and organelles. Autophagy is constantly active in skeletal muscle and its role in tissue homeostasis is complex: at high levels, it can be detrimental and contribute to muscle wasting; at low levels, it can cause weakness and muscle degeneration, due to the unchecked accumulation of damaged proteins and organelles. The causal relationship between DMD pathogenesis and dysfunctional autophagy has been recently investigated. At molecular level, the Akt axis is one of the key dysregulated pathways, although the molecular events are not completely understood. The aim of this review is to describe and discuss the clinical relevance of the recent advances dissecting autophagy and its signaling pathway in DMD. The picture might pave the way for the development of interventions that are able to boost muscle growth and/or prevent muscle wasting.
Pathological Features of Dystrophin Deficiency
Duchenne muscular dystrophy (DMD) is the most frequent and lethal form of muscular dystrophy, affecting 1 out of 3,500 newborns (Govoni et al., 2013). DMD is an X-linked disorder caused by mutations of the largest gene of the human genome, the dystrophin gene. So far, more than 4,700 mutations have been identified and classified as deletions (65.8%), duplications (13.6%), and point mutations (micro-insertions, microdeletions, and nonsense, missense, and splicing mutations; 20.6%) (Magri et al., 2011). Mutations that change the reading frame of the gene generally result in premature stop codons with complete loss of dystrophin in DMD muscles. Other mutations can result in the generation of a smaller size-dystrophin or in a lesser amount of the protein being produced leading to the allelic dystrophinopathy disorder known as Becker muscular dystrophy, a disease milder than DMD, affecting 1 of 18,518 male births (Emery, 1991).
The diagnosis of DMD and Becker muscular dystrophy is based on careful analysis of the clinical features and confirmed by additional investigations including muscle biopsy and/or genetic testing. In particular, DMD is characterized by an early onset before 3 years of age, then ambulation loss occurs between 10 and 14 years of age, and finally death takes place between 20 and 30 years of age (Davies et al., 1988).
The most impaired muscle in DMD patients is the diaphragm, and its wasting is responsible for the respiratory failure (reduced wall and lung compliance, hypoventilation, hypercapnia, and hypoxemia) and thus death (Fayssoil et al., 2010; Mosqueira et al., 2013). DMD also affects cardiac muscle, and to a lesser extent, smooth muscle (Fayssoil et al., 2010; Mosqueira et al., 2013). More than 90% of DMD patients are affected by some degree of cardiomyopathy (Fayssoil et al., 2010; Mosqueira et al., 2013), which causes a progressive reduction in the ejection fraction, which may hesitate in heart failure with or without concomitant arrhythmias (Jefferies et al., 2005). The onset of cardiomyopathy is variable, starting from 18 years of age in DMD patients (Spurney, 2011; Politano and Nigro, 2012). Cardiac decline is observed also in Becker muscular dystrophy patients, which progresses faster than skeletal muscle decline (Bushby et al., 2010).
Functionally, dystrophic skeletal muscles are more susceptible to eccentric contraction than the healthy ones (Lynch et al., 2000). In response to this type of injury, altered muscle fibers undergo repeated cycles of necrosis and regeneration, during which satellite cells, the primary myogenic precursor cells of the muscle, activate muscle regeneration process (Le Grand and Rudnicki, 2007; Biressi and Rando, 2010). However, the regenerative process is rather inefficient and the repeated cycles of necrosis and regeneration lead to satellite cell depletion and gradual replacement of muscle by fat and connective tissue; this process is responsible for progressive muscle wasting and weakness. This may be attributed to a great extent to the loss of dystrophin.
Dystrophin is essential for maintenance of muscle membrane integrity; it is a scaffolding protein that recruits other structural and signaling proteins to the sarcolemma, forming a well-organized multimeric dystrophin-associated glycoprotein complex. The dystrophin-associated glycoprotein complex is composed of several transmembrane and peripheral proteins, depending on the tissue, in particular it encloses different sarcoplasmic proteins [α- and β-dystrobrevins, α1-, β1-, β2-, γ1-, and γ2-syntrophins, and neuronal nitric oxide synthase (nNOS)], transmembrane proteins (β-dystroglycan, α-, β-, γ-, δ-, ε-, and ζ-sarcoglycans, sarcospan, and caveolin-3), and extracellular proteins (α-dystroglycan and laminin). Dystrophin-associated glycoprotein complex plays a key role in the mechanical stabilization of the sarcolemma during muscle contractions (Straub and Campbell, 1997), and acts as a scaffold for different proteins implicated in intracellular signaling. The proteins inside dystrophin-associated glycoprotein complex are tightly connected to each other and abnormalities affecting one of these proteins lead to changes in the others impairing muscle function. Of importance, the disruption of the dystrophin-associated glycoprotein complex in DMD also causes the delocalization of the nNOS from sarcolemma, leading to a reduced generation of NO. NO is important in skeletal muscle physiology as it controls excitation–contraction coupling and energy balance (De Palma and Clementi, 2012). The loss of NO-dependent signaling pathways contributes significantly to impair muscle bulk and force generation with increased fatigability (De Palma and Clementi, 2012). Moreover, the loss of sarcolemmal nNOS contributes to increase the muscle fibers susceptibility to ischemia during exercise, and to mechanical damage resulting in a focal muscle injury, dependent on muscle use (Thomas, 2013).
Mouse Models
In DMD, different mouse models with mutations in dystrophin gene have been generated. The mdx mouse on C57BL/10ScSn genetic background (C57BL/10ScSn-Dmdmdx, here referred as “mdx mouse”) (Coulton et al., 1988) shows a point mutation in exon 23, generating an early stop codon and leading to the absence of full-length dystrophin (Willmann et al., 2009). Other models bearing different mutations have been also generated, as for instance mdx2cv, mdx3cv, mdx4cv, or mdx5cv mouse (Im et al., 1996), mdx52 (with mutation on exon 52; Araki et al., 1997), and mdx/utr−/− (mdx mouse with additional knockout of utrophin) (Deconinck et al., 1997).
Among these models, the mdx mouse is the most commonly used (Nakamura and Takeda, 2011). The mutation on exon 23 is wide spread and affects one-third of DMD patients (Willmann et al., 2009). mdx mice have a shorter life compared to controls and display a decreased relative muscle force, whereas absolute muscle force is unaffected. Isolated muscles show a reduced force contraction, even if lacking standard condition of analysis is difficult to compare results from different groups.
Degeneration, regeneration, and necrosis are processes observed in young mdx mice (2–4 weeks) that result in increased number of regenerating-centronucleated fibers and in the heterogeneity of myofibers area (McGeachie et al., 1993). Necrotic fibers can be found at any age with very high frequency after 18 months (Nakamura and Takeda, 2011). Muscles undergo frequent cycles of necrosis/regeneration, associated with weakness and muscle loss. These cycles of regeneration lead to a muscle phenotype milder than the one observed in human patients. Fibrosis appears to be much less than in DMD patients, except for diaphragm muscle (Nakamura and Takeda, 2011). Although the mdx mouse diaphragm reproduces the degenerative changes of DMD, respiratory complications are visible only in 16-month-old mice (Stedman et al., 1991; Nakamura and Takeda, 2011). Muscle pathology can be worsened by forced exercise; this is a viable strategy to worsen the disease phenotype and better reveal the efficacy of new therapies.
Of importance, mdx mouse also displays cardiomyopathy, characterized by fibrosis and presence of necrosis and inflammation, thus sharing some aspects of DMD-patients cardiomyopathy and constitute a good model to investigate cardio-protective candidate molecules (Quinlan et al., 2004).
Autophagy and Its Role in Skeletal Muscle Homeostasis
Autophagy is a crucial mechanism involved in the turnover of cell components both in constitutive and catabolic (stress, nutrient deprivation, cytokines, amino acids deprivation) conditions. Autophagy classically functions as a physiological process to degrade cytoplasmic components, protein aggregates, and/or organelles, and as a regulator of cellular architecture. Autophagy in mammals generally protects the cells from death and defective autophagy can be associated with several diseases including cancer, neurodegenerative diseases, infectious diseases, and metabolic diseases (Cervia et al., 2013; Jiang and Mizushima, 2014; Schneider and Cuervo, 2014).
So far, three different mechanisms of autophagy have been described: macroautophagy, microautophagy, and chaperone-mediated autophagy. The most part of the information on autophagy in skeletal muscle is about macroautophagy, a process characterized by membranes that grow in size to generate double membrane-structures termed autophagosome, that enclose organelles, portion of cytoplasm, or protein aggregates. In this process, small ubiquitin-like proteins are required for the formation of autophagosomes and they are covalently bound to phosphatidylethanolamine.
Autophagy was considered initially as a non-specific degradation mechanism, but over the years selective forms of autophagy have been identified. For instance, selective removal of organelles, such as mitochondria or peroxisomes occurs via specific types of autophagy, termed mitophagy or peroxophagy, respectively.
Autophagic Signaling
The molecular signaling pathway leading to autophagy is very complex and regulated by autophagy-related genes (Atgs), which are connected with the formation of autophagosomes (Hurley and Schulman, 2014). The protein products of Atgs are organized in five functional groups, namely: (i) the Unc-51-like kinase (Ulk):Atg13:FIP200 initiation complex (Ganley et al., 2009; Hosokawa et al., 2009); (ii) the beclin1:hVps34[phosphatidylinositol 3 (PI3) kinase]:Atg14L nucleation complex (Itakura et al., 2008); (iii) the PI3–phosphate-binding WIPI-1/2 complex (Proikas-Cezanne et al., 2004; Vergne et al., 2009); (iv) the Atg5–Atg12 conjugation complex activated by Atg7 (Mizushima et al., 1998); and (v) the Atg8 (LC3) conjugation system (Kabeya et al., 2000). These protein complexes participate at specific stages in the autophagic process: initiation, formation, elongation, and fusion (Mehrpour et al., 2010; Awan and Deng, 2014); they are also controlled by several other signaling pathways that fine tune autophagy to regulate the pace of autophagosome formation.
A key player in the control of autophagy is the mammalian target of rapamycin (mTOR), which together with raptor (regulatory associated protein of mTOR), G protein β-subunit-like protein (GβL) and proline-rich Akt substrate of 40 kDa (PRAS40) forms the multiprotein complex, mTORC1. The activity of mTOR, a negative regulator of autophagy, depends on several positive signals including normoxia, amino acid supply, high energy levels, or growth factors. Upon stimulation by growth factors or nutrients (glucose, amino acids), mTORC1 negatively regulates the macromolecular initiation complex Ulk:Atg13:FIP200 leading to autophagy suppression (Ganley et al., 2009; Hosokawa et al., 2009; Wong et al., 2013). Conversely, starvation and energy depletion, which stimulate autophagy, inhibit mTORC1, leading to the activation of Ulk (Jung et al., 2010; Kim et al., 2011; Mihaylova and Shaw, 2011).
The main signaling pathway controlling mTORC1 is the PI3 kinase/Akt pathway activated by the binding of growth factors or insulin to their cell surface receptors. Activated Akt in turn phosphorylates and inhibits the tuberous sclerosis complex 2 (TSC2), thus preventing the formation of the inhibitory TSC1/TSC2 heterodimer. This inhibition allows the small GTPase Rheb to activate directly mTORC1 (Long et al., 2005a,b; Huang and Manning, 2009) and to inhibit autophagy. Another signaling pathway controlling mTOR is the adenosine monophosphate-activated protein kinase (AMPK) pathway activated under energy-low conditions (Alers et al., 2012). Activated AMPK is known to regulate mTORC1 activity mainly through the phosphorylation and consequent activation of the negative regulator TSC2 (Inoki et al., 2003). However, the discovery that TSC2-deficient cells can respond to a decrease in energy levels has led to the investigation of additional mechanisms. Recently, it has been demonstrated that both mTORC1 and AMPK can act on the same substrate, Ulk1, with opposite effects: mTORC1 inhibits Ulk1 activation by phosphorylating Ser757; conversely, AMPK activates Ulk1 through its phosphorylation on Ser317 and Ser777 (Kim et al., 2011).
Additional proteins have been found to be associated with mTOR, i.e., rictor (rapamycin-insensitive companion of mTOR), GβL, SAPK-interacting protein 1 (SIN1), and protein observed with rictor (PROTOR), to form the multiprotein complex mTORC2 (Sarbassov et al., 2006). mTORC2 is involved in the phosphorylation and activation of Akt, thereby promoting its prosurvival action. Through this activation, mTOR determines the downregulation of the transcription factor Forkhead Box O3 (FoxO3), particularly important in muscle where it stimulates autophagy by enhancing the expression of genes including Atg12, Ulk1, Atg4b, and Gabarapl1 (Mammucari et al., 2007).
Autophagy Levels and Skeletal Muscle Impairment
The role of autophagy in skeletal muscle has been investigated extensively (Sandri, 2010; Neel et al., 2013). Muscle mass represents 40–50% of the human body and is one of the most important sites for the regulation of metabolism. Excessive protein degradation in the skeletal muscle is detrimental for the economy of the body and can lead to death. Studies in mice with muscle-specific inactivation of autophagic genes have been used to identify the role of autophagy in muscles (Sandri, 2010; Neel et al., 2013). Briefly, ablation of Atg7 gene led to an altered muscle structure, with sarcomere disorganization and myofibers degeneration due to activation of an unfolded protein response, accumulation of abnormal mitochondria, enhanced oxidative stress, and increased concentration of polyubiquitinated proteins (Masiero et al., 2009). Altogether, these effects accounted for muscle weakness, atrophy, and other signs of myopathy such as an irregular distribution of fibers’ shape as well as fibers with a vacuolated cytosol. In general, the blocking of basal autophagy in muscle enhanced the accumulation of damaged and dysfunctional mitochondria, suggesting that mitophagy was impaired and critical to maintain muscle homeostasis (Masiero et al., 2009). Further, muscle-specific Atg5−/− mice displayed an atrophic phenotype in the fibers of the fast type, associated to accumulation of autophagic substrates, for instance, ubiquitinated proteins (Raben et al., 2008). In particular, in fast muscles of Atg5−/− mice, the size and density of lysosomes were increased and their distribution on the microtubules altered. Microtubules displayed a more linear organization compared to that of wild-type mice, demonstrating that autophagy is crucial also for the correct arrangement of microtubules (Raben et al., 2008).
Finally, in studies in which the transcription of FoxO3 was upregulated an enhanced autophagy–lysosome system was observed, especially during muscle wasting and the factor itself was enough to enhance autophagy process and to trigger atrophy (Mammucari et al., 2007). Noteworthy, a recent study showed that mice lacking the nutrient-deprivation autophagy factor-1, displayed muscle weakness, associated with an increased autophagy, dysregulation of calcium and accumulation of enlarged mitochondria (Chang et al., 2012).
The lesson from these results is complex and indicates a dual role for autophagy in skeletal muscle homeostasis: at high levels, it can be detrimental and contribute to muscle atrophy; at low levels, it can cause weakness and muscle degeneration, due to the unchecked accumulation of damaged proteins and organelles (Sandri, 2010; Sandri et al., 2013). Thus, a proper autophagic process is vital for both functional skeletal muscle, which controls the support and movement of the skeleton, and muscle metabolism (Neel et al., 2013).
Deregulation of Autophagy in Duchenne Muscular Dystrophy
The fact that manipulation of animal models to dysregulate autophagy also leads to muscle pathology has prompted studies to investigate whether this process is altered in muscle diseases (Neel et al., 2013; Sandri et al., 2013). Of interest, our understanding on the role of detective autophagy in different forms of inherited muscular dystrophies, including Bethlem myopathy, Ullrich congenital muscular dystrophy, merosin-deficient congenital muscular dystrophy, and Emery–Dreifuss muscular dystrophy has emerged in the past 5 years (Sandri et al., 2013).
As initially postulated from indirect evidence, deficient autophagy was suggested to contribute to DMD pathogenesis. The presence of swollen and damaged mitochondria, protein aggregation, and distension of sarcoplasmic reticulum, which are cyto-pathological hallmarks of DMD, are often observed when autophagy is impaired (Culligan et al., 2002; Zhao et al., 2007). In addition, activation of Akt was significantly higher in muscles from mdx mice and dystrophin-deficient primary myotubes, thereby suggesting a defective autophagic process (Dogra et al., 2006; Peter and Crosbie, 2006). In line with these observations, DMD patients were found to exhibit a similar pattern of Akt activation (Peter and Crosbie, 2006).
The causal relationship between DMD pathogenesis and dysfunctional autophagy has been investigated more recently in studies addressing this issue specifically. A severe impairment of autophagy was indeed demonstrated by biochemical and ultrastructural analyses in muscles from patients affected by DMD and mdx mice. In particular, these muscles display a significant reduction in the lipidated form of the protein LC3, which is a common marker of autophagy induction (De Palma et al., 2012; Bibee et al., 2014). The reduction in lipidated LC3 has been found to be accompanied by clear signs of impaired autophagy at the ultrastructural level, i.e., by the presence of damaged organelles, the increase in the signaling adaptor p62 protein (a marker inversely correlated with autophagic flux), and the decrease of Bnip3, a mitochondrial protein, which recruits LC3 to mitochondria (De Palma et al., 2012; Bibee et al., 2014). Recent studies have also shown a role for the TNF receptor-associated factor 6 (TRAF6) as an important regulator of autophagy. In skeletal muscle of mdx mice, the activity of TRAF6 is increased and its absence correlates with a reduced autophagy (Hindi et al., 2014). Strikingly, this study also reported that the inhibition of TRAF6 signaling deteriorates muscle pathology at later stages of disease progression. It has been thus hypothesized that the initial inhibition of autophagy in young mdx mice in the absence of TRAF6 gene may be a protective mechanism to preserve skeletal muscle mass. Autophagy emerges then as an essential process for the clearance of defunct cellular organelles and a continued inhibition of autophagy exaggerates dystrophic phenotype (Hindi et al., 2014). The beneficial effects of activating autophagy in mdx mice have been also confirmed by overexpressing peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1a), a transcriptional coactivator, which is a powerful mediator of muscle plasticity (Hollinger et al., 2013) and by pharmacological treatment with an agonist drug against the energy sensor AMPK, i.e., 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) (Pauly et al., 2012). In particular, AICAR treatment led to a significant activation of the autophagy, as indicated by the characteristic biochemical changes of increased lipidated LC3 content, an upregulation of other prototypical autophagy-associated proteins, and to significant improvements in both muscle structure and maximum force-generating capacity (Pauly et al., 2012).
From a functional point of view, it has been recently shown that voluntary exercise in mdx mice enhances markers of autophagy to or above the levels observed in healthy mice, thus suggesting that beneficial autophagy can be induced by exercise (Hulmi et al., 2013). In agreement with these data, fasting induced autophagy in wild-type and mdx mice diaphragm (Spitali et al., 2013). Interestingly in mdx mice, the ability of upregulating autophagy appears restricted to certain muscles: tibialis anterior muscle of mdx mice are unable to enhance autophagy in response to fasting, which induces autophagy in tibialis anterior muscle of wild-type mice.
Molecular Characteristics
The defect in autophagy of mdx mouse muscles is accompanied by persistent activation of the Akt–mTOR axis (Figure 1) and its related autophagy-inhibiting pathways, and the concomitant downregulation of several autophagy-inducing genes (Dogra et al., 2006; Peter and Crosbie, 2006; De Palma et al., 2012). In particular, Akt activation has been established to occur at very early, pre-necrotic stages of disease pathogenesis, with a progressive increase with disease worsening (Peter and Crosbie, 2006). This high activation of Akt stimulates in turn the mTOR-dependent pathways whereas the mTOR-independent axis is not significantly altered (Dogra et al., 2006; Peter and Crosbie, 2006). The involvement of the mTOR axis in the pathogenesis of mdx mice is demonstrated also by the effects of the mTOR-targeting, immunosuppressant drug rapamycin. Oral or injected rapamycin treatment has been shown to improve histopathological features of dystrophy (Eghtesad et al., 2011) and the treatment with rapamycin-loaded nanoparticles (RNPs) has been shown to increase skeletal muscle strength in both young and adult mice concomitantly to an increased mTOR-dependent autophagy (Bibee et al., 2014). Consistently, mdx mice treatment with a long-term low-protein diet reactivates autophagy in muscle fibers, with increased lipidation of LC3, reduced levels of p62, normalization of Akt and mTOR signaling, a reduced accumulation of damaged organelles, and a significant recovery of muscle inflammation, fibrosis, myofiber damage and muscle function (De Palma et al., 2012). Since the long-term low-protein diet was shown to preserve the regenerating ability of mdx mouse muscle, it is tempting to speculate that physiological levels of autophagy maintain the number and function of myogenic precursor cells.
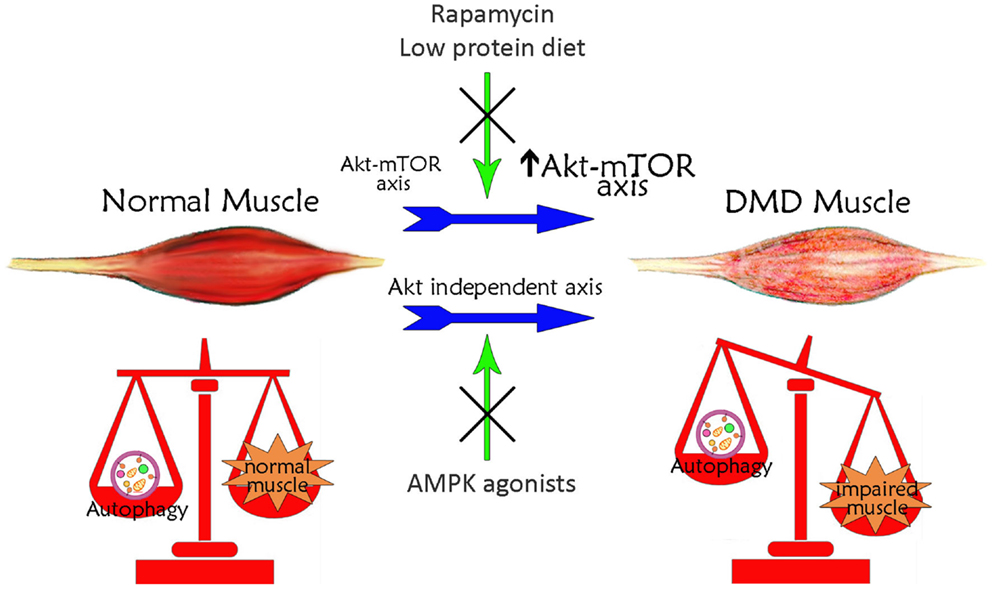
Figure 1. Schematic illustration of the correlation between autophagy and Duchenne muscular dystrophy. Basal autophagy levels are required for muscle homeostasis and for the maintenance of healthy myofibers. In DMD, muscle autophagy is impaired contributing to muscle degeneration. This autophagy inhibition is dependent on the iper-activation of Akt–mTOR axis. Treatments with rapamycin or low-protein diet, acting on Akt pathway, restore autophagy ameliorating DMD muscle phenotype and function. The impaired autophagy may also occur independently of Akt. Treatments with AMPK agonists, which increase AMPK activation, counteract the Akt-independent axes enhancing autophagy and inducing a positive effect in DMD muscle.
The picture is not completely clear and some controversies still exist and issues remain to be solved. Spitali et al. (2013) reported that autophagy levels and Akt axis activation in mdx mice is similar to wild-type control mice. Likewise, a study on Akt–mTOR axis activation in very old mdx mice correlated the advancing of age with a reduction in mTOR signaling in dystrophic muscles (Mouisel et al., 2010). In another study, however, it was also reported that mTOR activation is increased by age in diaphragm muscle of wild-type mice, but not in mdx mice, while similar levels of mTOR activation were found in tibialis anterior muscles (Eghtesad et al., 2011). Although these data are apparently difficult to reconcile, different confounding factors may explain them, as for instance the fact that mdx mice undergo extensive muscle regeneration between 6 and 12 weeks. Furthermore, the fact that the regulation of autophagy in mdx mice was indicated to differ depending on the muscle type (Spitali et al., 2013) should be taken into consideration, with the glycolytic muscles showing a process of vesicles formation significantly greater than oxidative muscles (Mizushima et al., 2004).
Autophagy can be modulated in mdx mice also through mTOR-independent mechanisms (Figure 1). Chronic AICAR administration induced autophagy in mdx muscles, with a significant upregulation of Ulk1 and an increased expression of Bnip3 in the absence of mTOR inhibition (Pauly et al., 2012). In addition, it has been suggested that TRAF6 regulation of Akt signaling is independent of TRAF6 regulation of autophagy (Hindi et al., 2014). The evidence for potentially augmented autophagic signaling may thus be suggestive of an adaptive response to attempt to rid dystrophic myofibers of defective and/or detrimental constituents, such as dysfunctional mitochondria (Ljubicic and Jasmin, 2013). The complex and apparently contrasting evidence on the role of authophagy in mdx mice suggests that the autophagic machinery in these mice is more complex than in wild-type mice and that a systematic analysis taking into account the variable of time has to be undertaken to clarify each single aspect in distinct muscles and during disease progression.
Therapeutic Implications of Corrective Autophagy in Duchenne Muscular Dystrophy
At present, there is no satisfactory therapy for DMD. The treatment with corticosteroids is the actual gold standard. Treatment with glucocorticoids significantly delays the impairment of muscle force and function, extends ambulation period, and retards the onset of pulmonary failure, cardiomyopathy, and scoliosis (Bushby et al., 2010). These effects, however, are often only temporary and associated with severe side effects. More than 25% of patients are not treated with glucocorticoids due to adverse effects, such as obesity, immune suppression, bone demineralization, associated with negative behavioral changes, or lack of response (Bushby et al., 2010). This strongly indicates an urgent requirement of new clinical intervention for DMD patients.
The genetic approaches [exon skipping, viral vector-mediated gene delivery, and cell therapy (Pichavant et al., 2011; Mendell et al., 2012)], currently being investigated, show some degree of success and some of them have been recently granted orphan status by either the European Medicines Agency or the Food and Drug Administration (http://orphandruganaut.wordpress.com/2013/12/14/duchenne-muscular-dystrophy-2013-fda-orphan-drug-designations-2/). They, however, are directed to specific subsets of population and cannot restore fully the damage already caused by the disease to the muscle. Alternative strategies are therefore needed and to this end identification of suitable therapeutic targets is necessary.
In this context, pharmacological modulation of autophagy can be considered a possible strategy aimed at delaying muscle degeneration (Figure 1). As mentioned above, the AICAR activating AMPK, ameliorated phenotypic and functional features of dystrophic muscle (Pauly et al., 2012). AMPK pharmacological agonists exist; they are clinically approved and AICAR has been tested for ischemic damage in the heart and used in clinical trial for metabolic disorders (Ljubicic and Jasmin, 2013). The beneficial effects of AICAR in dystrophic muscle could be due to a well-known effect in stimulating the slow, oxidative phenotype (Ljubicic et al., 2011). It could also be due to induction of autophagic pathways, since AICAR stimulates the removal of damaged mitochondria via mitophagy (Pauly et al., 2012).
A second pharmacological approach that targets autophagy has been proposed by Bibee et al. (2014) in which RNPs were used to successfully enhance grip strength and the left-ventricular ejection fraction in mdx mice, ameliorating both physical and cardiac performances. These effects with RNPs have been achieved after administration of only eight doses of RNPs, with the final dose within the range of recommended oral doses for immune-suppressive therapy in patients. Interestingly, oral administration of rapamycin at pharmacological doses was of no efficacy on muscle strength. It appears that the way RNPs worked was via autophagy induction, dependent on nanoparticles delivery of the drug: rapamycin was locally delivered at high concentration thus becoming able to trigger autophagy (Bibee et al., 2014). Interestingly, at least in the mdx mice also corticosteroids induce autophagy and this may contribute to explain their beneficial effects.
Autophagy cannot certainly be envisaged as a stand-alone therapeutic option. Low-protein diet regimen has been recently hypothesized to be safely usable in the treatment of DMD patients in combination with pharmacological treatment and cell and gene therapies (De Palma et al., 2012). RPNs and low-protein diet might also be used in combination with corticosteroids to act as steroids sparing drugs, reducing their toxic effects. Low-protein diet has been demonstrated safe and useful also in Col6a1-deficient mouse, considered an animal model of Bethlem myopathy (Grumati et al., 2010). Of interest, a very recent clinical study showed a clear induction of autophagy with a low-protein diet regimen in muscles from Becker and Ulrich patients (Merlini and Nishino, 2014), further suggesting that also in humans a strategy of autophagy reactivation is applicable.
Conclusion
Autophagy has emerged as a key process whose dysregulation contributes to the pathogenesis of several muscular dystrophies. The relevance of this process is that its normalization by pharmacological approaches leads to an amelioration of the dystrophic phenotype. While drugs targeting autophagy have good perspective in terms of therapy, we still need to refine them, by identifying appropriate targets in the autophagic pathway against which to design selected modulating drugs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by: “Ministero della Salute” “Giovani Ricercatori 2011–2012” grant to Clara De Palma and “Ricerca corrente 2014” grant to Emilio Clementi; “Ministero dell’Istruzione, Università e Ricerca,” PRIN 2010–2011 grants to Emilio Clementi and Davide Cervia; European Community’s framework program FP7/2007–2013 under the agreement no. 223098 (OPTISTEM) and no. 241440 (ENDOSTEM) to Emilio Clementi.
References
Alers, S., Loffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11. doi: 10.1128/MCB.06159-11
Araki, E., Nakamura, K., Nakao, K., Kameya, S., Kobayashi, O., Nonaka, I., et al. (1997). Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 238, 492–497. doi:10.1006/bbrc.1997.7328
Awan, M. U., and Deng, Y. (2014). Role of autophagy and its significance in cellular homeostasis. Appl. Microbiol. Biotechnol. 98, 5319–5328. doi:10.1007/s00253-014-5721-8
Bibee, K. P., Cheng, Y. J., Ching, J. K., Marsh, J. N., Li, A. J., Keeling, R. M., et al. (2014). Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 28, 2047–2061. doi:10.1096/fj.13-237388
Biressi, S., and Rando, T. A. (2010). Heterogeneity in the muscle satellite cell population. Semin. Cell Dev. Biol. 21, 845–854. doi:10.1016/j.semcdb.2010.09.003
Bushby, K., Finkel, R., Birnkrant, D. J., Case, L. E., Clemens, P. R., Cripe, L., et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93. doi:10.1016/S1474-4422(09)70271-6
Cervia, D., Perrotta, C., Moscheni, C., De Palma, C., and Clementi, E. (2013). Nitric oxide and sphingolipids control apoptosis and autophagy with a significant impact on Alzheimer’s disease. J. Biol. Regul. Homeost. Agents 27, 11–22.
Chang, N. C., Nguyen, M., Bourdon, J., Risse, P. A., Martin, J., Danialou, G., et al. (2012). Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 21, 2277–2287. doi:10.1093/hmg/dds048
Coulton, G. R., Curtin, N. A., Morgan, J. E., and Partridge, T. A. (1988). The mdx mouse skeletal muscle myopathy: II. Contractile properties. Neuropathol. Appl. Neurobiol. 14, 299–314. doi:10.1111/j.1365-2990.1988.tb00890.x
Culligan, K., Banville, N., Dowling, P., and Ohlendieck, K. (2002). Drastic reduction of calsequestrin-like proteins and impaired calcium binding in dystrophic mdx muscle. J. Appl. Physiol. 92, 435–445. doi:10.1152/japplphysiol.00903.2001
Davies, K. E., Smith, T. J., Bundey, S., Read, A. P., Flint, T., Bell, M., et al. (1988). Mild and severe muscular dystrophy associated with deletions in Xp21 of the human X chromosome. J. Med. Genet. 25, 9–13. doi:10.1136/jmg.25.1.9
De Palma, C., and Clementi, E. (2012). Nitric oxide in myogenesis and therapeutic muscle repair. Mol. Neurobiol. 46, 682–692. doi:10.1007/s12035-012-8311-8
De Palma, C., Morisi, F., Cheli, S., Pambianco, S., Cappello, V., Vezzoli, M., et al. (2012). Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 3, e418. doi:10.1038/cddis.2012.159
Deconinck, A. E., Rafael, J. A., Skinner, J. A., Brown, S. C., Potter, A. C., Metzinger, L., et al. (1997). Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90, 717–727. doi:10.1016/S0092-8674(00)80532-2
Dogra, C., Changotra, H., Wergedal, J. E., and Kumar, A. (2006). Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J. Cell. Physiol. 208, 575–585. doi:10.1002/jcp.20696
Eghtesad, S., Jhunjhunwala, S., Little, S. R., and Clemens, P. R. (2011). Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol. Med. 17, 917–924. doi:10.2119/molmed.2010.00256
Emery, A. E. (1991). Population frequencies of inherited neuromuscular diseases – a world survey. Neuromuscul. Disord. 1, 19–29. doi:10.1016/0960-8966(91)90039-U
Fayssoil, A., Nardi, O., Orlikowski, D., and Annane, D. (2010). Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail. Rev. 15, 103–107. doi:10.1007/s10741-009-9156-8
Ganley, I. G., Lam du, H., Wang, J., Ding, X., Chen, S., and Jiang, X. (2009). ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305. doi:10.1074/jbc.M900573200
Govoni, A., Magri, F., Brajkovic, S., Zanetta, C., Faravelli, I., Corti, S., et al. (2013). Ongoing therapeutic trials and outcome measures for Duchenne muscular dystrophy. Cell. Mol. Life Sci. 70, 4585–4602. doi:10.1007/s00018-013-1396-z
Grumati, P., Coletto, L., Sabatelli, P., Cescon, M., Angelin, A., Bertaggia, E., et al. (2010). Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 16, 1313–1320. doi:10.1038/nm.2247
Hindi, S. M., Sato, S., Choi, Y., and Kumar, A. (2014). Distinct roles of TRAF6 at early and late stages of muscle pathology in the mdx model of Duchenne muscular dystrophy. Hum. Mol. Genet. 23, 1492–1505. doi:10.1093/hmg/ddt536
Hollinger, K., Gardan-Salmon, D., Santana, C., Rice, D., Snella, E., and Selsby, J. T. (2013). Rescue of dystrophic skeletal muscle by PGC-1alpha involves restored expression of dystrophin-associated protein complex components and satellite cell signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R13–R23. doi:10.1152/ajpregu.00221.2012
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991. doi:10.1091/mbc.E08-12-1248
Huang, J., and Manning, B. D. (2009). A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 37, 217–222. doi:10.1042/BST0370217
Hulmi, J. J., Oliveira, B. M., Silvennoinen, M., Hoogaars, W. M., Pasternack, A., Kainulainen, H., et al. (2013). Exercise restores decreased physical activity levels and increases markers of autophagy and oxidative capacity in myostatin/activin-blocked mdx mice. Am. J. Physiol. Endocrinol. Metab. 305, E171–E182. doi:10.1152/ajpendo.00065.2013
Hurley, J. H., and Schulman, B. A. (2014). Atomistic autophagy: the structures of cellular self-digestion. Cell 157, 300–311. doi:10.1016/j.cell.2014.01.070
Im, W. B., Phelps, S. F., Copen, E. H., Adams, E. G., Slightom, J. L., and Chamberlain, J. S. (1996). Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 5, 1149–1153. doi:10.1093/hmg/5.8.1149
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi:10.1016/S0092-8674(03)00929-2
Itakura, E., Kishi, C., Inoue, K., and Mizushima, N. (2008). Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19, 5360–5372. doi:10.1091/mbc.E08-01-0080
Jefferies, J. L., Eidem, B. W., Belmont, J. W., Craigen, W. J., Ware, S. M., Fernbach, S. D., et al. (2005). Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 112, 2799–2804. doi:10.1161/CIRCULATIONAHA.104.528281
Jiang, P., and Mizushima, N. (2014). Autophagy and human diseases. Cell Res. 24, 69–79. doi:10.1038/cr.2013.161
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., and Kim, D. H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. doi:10.1016/j.febslet.2010.01.017
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi:10.1093/emboj/19.21.5720
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi:10.1038/ncb2152
Le Grand, F., and Rudnicki, M. A. (2007). Skeletal muscle satellite cells and adult myogenesis. Curr. Opin. Cell Biol. 19, 628–633. doi:10.1016/j.ceb.2007.09.012
Ljubicic, V., and Jasmin, B. J. (2013). AMP-activated protein kinase at the nexus of therapeutic skeletal muscle plasticity in Duchenne muscular dystrophy. Trends Mol. Med. 19, 614–624. doi:10.1016/j.molmed.2013.07.002
Ljubicic, V., Miura, P., Burt, M., Boudreault, L., Khogali, S., Lunde, J. A., et al. (2011). Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum. Mol. Genet. 20, 3478–3493. doi:10.1093/hmg/ddr265
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005a). Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713. doi:10.1016/j.cub.2005.02.053
Long, X., Ortiz-Vega, S., Lin, Y., and Avruch, J. (2005b). Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 280, 23433–23436. doi:10.1074/jbc.C500169200
Lynch, G. S., Rafael, J. A., Chamberlain, J. S., and Faulkner, J. A. (2000). Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am. J. Physiol. Cell Physiol. 279, C1290–C1294.
Magri, F., Govoni, A., D’Angelo, M. G., Del Bo, R., Ghezzi, S., Sandra, G., et al. (2011). Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 258, 1610–1623. doi:10.1007/s00415-011-5979-z
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi:10.1016/j.cmet.2007.11.001
Masiero, E., Agatea, L., Mammucari, C., Blaauw, B., Loro, E., Komatsu, M., et al. (2009). Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515. doi:10.1016/j.cmet.2009.10.008
McGeachie, J. K., Grounds, M. D., Partridge, T. A., and Morgan, J. E. (1993). Age-related changes in replication of myogenic cells in mdx mice: quantitative autoradiographic studies. J. Neurol. Sci. 119, 169–179. doi:10.1016/0022-510X(93)90130-Q
Mehrpour, M., Esclatine, A., Beau, I., and Codogno, P. (2010). Overview of macroautophagy regulation in mammalian cells. Cell Res. 20, 748–762. doi:10.1038/cr.2010.82
Mendell, J. R., Rodino-Klapac, L., Sahenk, Z., Malik, V., Kaspar, B. K., Walker, C. M., et al. (2012). Gene therapy for muscular dystrophy: lessons learned and path forward. Neurosci. Lett. 527, 90–99. doi:10.1016/j.neulet.2012.04.078
Merlini, L., and Nishino, I. (2014). 201St ENMC international workshop: autophagy in muscular dystrophies – translational approach, 1-3 November 2013, Bussum, The Netherlands. Neuromuscul. Disord. 24, 546–561. doi:10.1016/j.nmd.2014.03.009
Mihaylova, M. M., and Shaw, R. J. (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023. doi:10.1038/ncb2329
Mizushima, N., Sugita, H., Yoshimori, T., and Ohsumi, Y. (1998). A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 273, 33889–33892. doi:10.1074/jbc.273.51.33889
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T., and Ohsumi, Y. (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111. doi:10.1091/mbc.E03-09-0704
Mosqueira, M., Zeiger, U., Forderer, M., Brinkmeier, H., and Fink, R. H. (2013). Cardiac and respiratory dysfunction in Duchenne muscular dystrophy and the role of second messengers. Med. Res. Rev. 33, 1174–1213. doi:10.1002/med.21279
Mouisel, E., Vignaud, A., Hourde, C., Butler-Browne, G., and Ferry, A. (2010). Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in mTOR signaling in skeletal muscles of venerable (18-24-month-old) dystrophic mdx mice. Muscle Nerve 41, 809–818. doi:10.1002/mus.21624
Nakamura, A., and Takeda, S. (2011). Mammalian models of Duchenne Muscular Dystrophy: pathological characteristics and therapeutic applications. J. Biomed. Biotechnol. 2011, 184393. doi:10.1155/2011/184393
Neel, B. A., Lin, Y., and Pessin, J. E. (2013). Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol. Metab. 24, 635–643. doi:10.1016/j.tem.2013.09.004
Pauly, M., Daussin, F., Burelle, Y., Li, T., Godin, R., Fauconnier, J., et al. (2012). AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 181, 583–592. doi:10.1016/j.ajpath.2012.04.004
Peter, A. K., and Crosbie, R. H. (2006). Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp. Cell Res. 312, 2580–2591. doi:10.1016/j.yexcr.2006.04.024
Pichavant, C., Aartsma-Rus, A., Clemens, P. R., Davies, K. E., Dickson, G., Takeda, S., et al. (2011). Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol. Ther. 19, 830–840. doi:10.1038/mt.2011.59
Politano, L., and Nigro, G. (2012). Treatment of dystrophinopathic cardiomyopathy: review of the literature and personal results. Acta Myol. 31, 24–30.
Proikas-Cezanne, T., Waddell, S., Gaugel, A., Frickey, T., Lupas, A., and Nordheim, A. (2004). WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 23, 9314–9325. doi:10.1038/sj.onc.1208331
Quinlan, J. G., Hahn, H. S., Wong, B. L., Lorenz, J. N., Wenisch, A. S., and Levin, L. S. (2004). Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul. Disord. 14, 491–496. doi:10.1016/j.nmd.2004.04.007
Raben, N., Hill, V., Shea, L., Takikita, S., Baum, R., Mizushima, N., et al. (2008). Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 17, 3897–3908. doi:10.1093/hmg/ddn292
Sandri, M. (2010). Autophagy in skeletal muscle. FEBS Lett. 584, 1411–1416. doi:10.1016/j.febslet.2010.01.056
Sandri, M., Coletto, L., Grumati, P., and Bonaldo, P. (2013). Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell. Sci. 126, 5325–5333. doi:10.1242/jcs.114041
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. doi:10.1016/j.molcel.2006.03.029
Schneider, J. L., and Cuervo, A. M. (2014). Autophagy and human disease: emerging themes. Curr. Opin. Genet. Dev. 26C, 16–23. doi:10.1016/j.gde.2014.04.003
Spitali, P., Grumati, P., Hiller, M., Chrisam, M., Aartsma-Rus, A., and Bonaldo, P. (2013). Autophagy is impaired in the tibialis anterior of dystrophin null mice. PLoS Curr. 5. doi:10.1371/currents.md.e1226cefa851a2f079bbc406c0a21e80
Spurney, C. F. (2011). Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve 44, 8–19. doi:10.1002/mus.22097
Stedman, H. H., Sweeney, H. L., Shrager, J. B., Maguire, H. C., Panettieri, R. A., Petrof, B., et al. (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539. doi:10.1038/352536a0
Straub, V., and Campbell, K. P. (1997). Muscular dystrophies and the dystrophin-glycoprotein complex. Curr. Opin. Neurol. 10, 168–175. doi:10.1097/00019052-199704000-00016
Thomas, G. D. (2013). Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front. Physiol. 4:381. doi:10.3389/fphys.2013.00381
Vergne, I., Roberts, E., Elmaoued, R. A., Tosch, V., Delgado, M. A., Proikas-Cezanne, T., et al. (2009). Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 28, 2244–2258. doi:10.1038/emboj.2009.159
Willmann, R., Possekel, S., Dubach-Powell, J., Meier, T., and Ruegg, M. A. (2009). Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul. Disord. 19, 241–249. doi:10.1016/j.nmd.2008.11.015
Wong, P. M., Puente, C., Ganley, I. G., and Jiang, X. (2013). The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9, 124–137. doi:10.4161/auto.23323
Keywords: skeletal muscle, Duchenne muscular dystrophy, muscle wasting, autophagy, Akt–mTOR axis, mdx mice, therapeutic target
Citation: De Palma C, Perrotta C, Pellegrino P, Clementi E and Cervia D (2014) Skeletal muscle homeostasis in Duchenne muscular dystrophy: modulating autophagy as a promising therapeutic strategy. Front. Aging Neurosci. 6:188. doi: 10.3389/fnagi.2014.00188
Received: 16 June 2014; Paper pending published: 27 June 2014;
Accepted: 10 July 2014; Published online: 24 July 2014.
Edited by:
Luciano Merlini, Istituto Ortopedico Rizzoli IRCCS, ItalyReviewed by:
Gabriella Minchiotti, Institute of Genetics and Biophysics-CNR, ItalyFrancesca Tagliavini, Institute of Molecular Genetics-CNR, Italy
Copyright: © 2014 De Palma, Perrotta, Pellegrino, Clementi and Cervia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emilio Clementi, Department of Biomedical and Clinical Sciences “L. Sacco”, Università di Milano, Via G.B. Grassi 74, Milano 20157, Italy e-mail:ZW1pbGlvLmNsZW1lbnRpQHVuaW1pLml0;
Davide Cervia, Department for Innovation in Biological, Agro-Food and Forest Systems, Università della Tuscia, Largo dell’Università snc, Blocco D, Viterbo 01100, Italy e-mail:ZC5jZXJ2aWFAdW5pdHVzLml0