 J. Scott Miners
J. Scott Miners Jennifer C. Palmer
Jennifer C. Palmer Hannah Tayler
Hannah Tayler Laura E. Palmer
Laura E. Palmer Emma Ashby
Emma Ashby- Dementia Research Group, School of Clinical Sciences, Faculty of Medicine and Dentistry, University of Bristol, Bristol, UK
There is increasing evidence that deficient clearance of β-amyloid (Aβ) contributes to its accumulation in late-onset Alzheimer disease (AD). Several Aβ-degrading enzymes, including neprilysin (NEP), endothelin-converting enzyme (ECE), and angiotensin-converting enzyme (ACE) reduce Aβ levels and protect against cognitive impairment in mouse models of AD. In post-mortem human brain tissue we have found that the activity of these Aβ-degrading enzymes rise with age and increases still further in AD, perhaps as a physiological response that helps to minimize the build-up of Aβ. ECE-1/-2 and ACE are also rate-limiting enzymes in the production of endothelin-1 (ET-1) and angiotensin II (Ang II), two potent vasoconstrictors, increases in the levels of which are likely to contribute to reduced blood flow in AD. This review considers the possible interdependence between Aβ-degrading enzymes, ischemia and Aβ in AD: ischemia has been shown to increase Aβ production both in vitro and in vivo, whereas increased Aβ probably enhances ischemia by vasoconstriction, mediated at least in part by increased ECE and ACE activity. In contrast, NEP activity may help to maintain cerebral perfusion, by reducing the accumulation of Aβ in cerebral blood vessels and lessening its toxicity to vascular smooth muscle cells. In assessing the role of Aβ-degrading proteases in the pathogenesis of AD and, particularly, their potential as therapeutic agents, it is important to bear in mind the multifunctional nature of these enzymes and to consider their effects on other substrates and pathways.
Introduction
The accumulation of Aβ within the brain is central to the pathogenesis of AD. The level of Aβ depends not only on its rate of production but also on the rate of its removal via various clearance pathways including enzyme-mediated degradation. Mice with inactivation of the Mme (neprilysin, membrane metalloendopeptidase), Mmel1 (neprilysin-2, membrane metalloendopeptidase-like 1), Ide, Ece1, or Ece2 gene, all have a modest (1.5–2-fold) increase in Aβ levels. Overexpression of the human orthologs of these genes in transgenic mice expressing mutant forms of human amyloid-β precursor protein (hAPP) that cause familial AD, reduce Aβ accumulation and in most, but not all cases, improves motor and cognitive performance. Findings from large post-mortem human brain studies and in vitro experiments have, however, revealed that the level and activity of many Aβ-degrading proteases are increased in post-mortem brain tissue and are upregulated by Aβ, suggesting that the increases are secondary to Aβ accumulation, possibly representing physiological responses to the rise in concentration of substrate. Many of the Aβ-degrading enzymes are involved in other physiological systems. For example, ECE-1/-2 and ACE are rate-limiting enzymes in the production of ET-1 and Ang II, respectively. In this review we discuss the balance between beneficial and potential deleterious effects of upregulating these vasoactive enzyme systems, and the broader relationships between Aβ and cerebral perfusion in the context of AD.
Aβ Production and Clearance in Alzheimer's Disease
Amyloidogenic processing of APP to produce Aβ peptides results from the sequential actions of β- and γ-secretase (Selkoe, 2001; Evin and Weidemann, 2002; Mattson, 2004), yielding mostly Aβ1−40 and a lesser amount of Aβ1−42. Increased production of the less soluble Aβ1−42, or an increase in the ratio of Aβ1−42:Aβ1−40, is hypothesized to initiate a cascade of pathological processes leading to the development of AD (Hardy, 1997). According to the somewhat simplified view of the development of the hallmark pathological lesions of AD, extracellular Aβ1−42, which is more prone to aggregate (Jarrett et al., 1993) and more toxic than Aβ1−40, precipitates as “plaques” within the brain parenchyma and induces the development of neuritic and neurofibrillary tangle pathology, whereas much of the relatively soluble, less toxic Aβ1−40 reaches the cerebral blood vessels where some of it may precipitate, leading to cerebral amyloid angiopathy (CAA).
In all forms of AD, the level of Aβ in the brain is a reflection of the relative rates of Aβ production and clearance over time. In healthy individuals, the production and clearance of Aβ are rapid (estimated at ~7.6% and 8.3%, respectively, of the total volume of Aβ produced per hour (Bateman et al., 2006; Mawuenyega et al., 2010). These data suggest that even very small changes in the production or clearance of Aβ would soon cause abnormal accumulation in AD. Early-onset familial AD usually results from autosomal dominant mutations in the genes encoding APP (APP), presenilin-1 (PSEN1) or presenilin-2 (PSEN2) (reviewed in Tanzi and Bertram, 2005). These mutations alter the processing of APP, increasing the ratio of Aβ1−42:Aβ1−40 (Scheuner et al., 1996; Price et al., 1998). Early-onset AD can also be caused by increased production of APP, in trisomy-21 (Belza and Urich, 1986; Donahue et al., 1998), or duplication in the APP locus on chromosome 21 (Cabrejo et al., 2006; Rovelet-Lecrux et al., 2006). In sporadic AD (i.e., in over 90% of cases) there is less compelling evidence that increased amyloidogenic processing of APP is responsible for Aβ accumulation. Although studies on human post-mortem brain tissue reported increased β- and γ-secretase activities in sporadic AD (Tyler et al., 2002; Stockley et al., 2006; Miners et al., 2010a), studies in two strains of mouse transgenic for hAPP (one causing early and one causing late accumulation of Aβ) found that β-secretase activity increased only after the development of Aβ plaques (Zhao et al., 2007).
Over the last fifteen years, many studies have focused on possible abnormalities of clearance of Aβ in late-onset sporadic AD, and reported age- and disease-related deficiencies in numerous clearance pathways, including reductions in Aβ-degrading protease activities. Soluble Aβ is cleared from the brain by multiple, diverse pathways including drainage along perivascular basement membranes (Weller et al., 1998, 2000; Preston et al., 2003) and receptor-mediated transport of Aβ across the blood-brain barrier [including transport mediated by binding to low-density lipoprotein receptor-related protein (LRP-1) (Shibata et al., 2000) and through the activity of P-glycoprotein efflux pump (Lam et al., 2001; Vogelgesang et al., 2002; Kuhnke et al., 2007)]. For an in-depth review of pathways which mediate Aβ clearance, see Bell et al. (2007).
Multiple proteolytic enzymes have been identified that cleave Aβ at single or multiple sites (for comprehensive reviews, see Carson and Turner, 2002; Eckman and Eckman, 2005; Wang et al., 2006; Bell et al., 2007; Miners et al., 2011a; Nalivaeva et al., 2012). Amongst the enzymes that cleave Aβ in vitro are a number of zinc metalloendopeptidases including neprilysin (NEP), angiotensin-converting enzyme (ACE), and endothelin-converting enzyme-1 and -2 (ECE); thiol-metalloendopeptidases including insulin-degrading enzyme (IDE), matrix metalloproteinases [MMP-2, -9 and type-1 transmembrane MMP (MT1-MMP)]; serine proteases including myelin-basic protein (MBP), plasminogen and acyl peptide hydrolase (APEH), and cysteine proteases such as cathepsin B (for a more detailed description, refer to Miners et al., 2011a). Fragments of Aβ produced by proteolytic cleavage in vitro are generally considered to be less neurotoxic, and less likely to aggregate (and therefore predicted to be more easily cleared from the brain) (Mukherjee et al., 2000; Hu et al., 2001). Mice with inactivation of Mme (Iwata et al., 2001), Mmel1 (Hafez et al., 2011), Ece1, Ece2 (Eckman et al., 2003), or Ide (Farris et al., 2003; Miller et al., 2003) genes all have a moderate (1.5–2-fold) increase in endogenous Aβ. These KO mice did not display pathological deposition of endogenous Aβ compared to mice infused with thiorphan (Iwata et al., 2000), or phosphoramidon (Nisemblat et al., 2008), which probably reflects the overlapping substrate specificity of these inhibitors for multiple Aβ-degrading enzymes. Inactivation of NEP in hAPP mice was associated with impaired synaptic plasticity and cognitive performance (Huang et al., 2006) and was sufficient to cause plaque-like pathology (Farris et al., 2007). Conversely, overexpression of NEP (resulting in an 8- and 30-fold increase in protein level and enzyme activity, respectively) in hAPP transgenic (Swe/Ind) mice markedly reduced cerebral Aβ load, reversed pathological abnormalities, and improved cognitive performance and survival (Leissring et al., 2003; Poirier et al., 2006). Several other experimental strategies, aimed at increasing NEP within the CNS, by targeting either the CNS directly, or via the periphery, have suggested that upregulation of NEP might be used to treat AD (Iwata, 2003; Marr et al., 2003, 2004; El-Amouri et al., 2008; Spencer et al., 2008, 2011; Briyal et al., 2011). Induction of peripherally expressed NEP was reported to regulate Aβ level within the CNS perhaps by reducing plasma Aβ (Guan et al., 2009; Liu et al., 2009) but this, and the peripheral-sink hypothesis, has been refuted in recent studies (Walker et al., 2013; Henderson et al., 2014).
Earlier studies on human post-mortem brain tissue presented evidence to support the hypothesis that an age- and disease-related deficit in Aβ-degrading protease activity contributes to Aβ accumulation, particularly in late-onset AD. The studies reported reduced NEP and IDE mRNA levels and protein immunolabeling in AD compared to control brains (Akiyama et al., 2001; Yasojima et al., 2001; Russo et al., 2005; Miners et al., 2006; Hellstrom-Lindahl et al., 2008). However, they did not measure enzyme activity, the most relevant biological measure with respect to Aβ metabolism, were mostly based on small cohorts, in many cases did not adjust for neuronal content, and showed poor correlation between different methodologies (Russo et al., 2005; Miners et al., 2006; Hellstrom-Lindahl et al., 2008). We developed ELISA and immunocapture-based fluorogenic activity assays to measure the levels and/or specific enzyme activities of NEP, IDE, ECE-1, ECE-2, and ACE within biological tissue samples, including CSF, plasma, brain tissue homogenates and blood vessel preparations (Miners et al., 2008a,b), and used the concentration of neuron-specific enolase to adjust for neuronal content (Miners et al., 2009) in the case of NEP, IDE, ACE and ECE-2, and the concentration of factor VIII-related antigen to adjust for the endothelial cell content in the case of ECE-1 (Palmer et al., 2010). Contrary to our expectation, in a large series of well-characterized human post-mortem brains, we found that NEP, IDE, ACE, and ECE-1 activities and ECE-2 levels were all higher in AD than in age-matched controls (Miners et al., 2008c, 2009, 2010b; Palmer et al., 2009, 2010) and rose progressively with disease severity, as indicated by Braak tangle stage. The increase in the level and activity of NEP, IDE, and ACE was particularly pronounced after adjustment for neuronal loss in AD, and was directly related to the Braak tangle stage. Levels and activities of ACE and NEP were also elevated and correlated positively with Aβ1−42 level in Down's syndrome, which is associated with the development of AD-type neuropathology (Miners et al., 2011b). NEP, IDE, and ACE levels tended to rise with normal aging and were further increased in AD (Miners et al., 2010a). Other groups reported that other candidate Aβ-degrading proteases, including MMP-2 -3 and -9 (Yan et al., 2006; Bruno et al., 2009), cathepsin D (Gan et al., 2004; Mueller-Steiner et al., 2006; Sundelof et al., 2010) and APEH (Yamin et al., 2009), were also increased in human post-mortem brain tissue in AD, in most but not all studies (Baig et al., 2008; Barker et al., 2010). It should be noted we examined only the mid-frontal cortex, an area with a similar level of Aβ pathology to that in the temporal cortex (and more than that in the hippocampus) but affected by tau pathology only later in disease, and our approach did not take into account the relative contribution of various different cell types to overall NEP activity, an important topic for future research (see review by Saido, 2013).
Nevertheless, there is evidence to suggest that proteolysis of Aβ is not deficient in AD. On the contrary, our findings in human post-mortem tissue indicate that Aβ-degrading activity correlates positively with Aβ concentration, as would be expected for a physiological response to increasing substrate. This is supported by a large number of cell culture studies showing that exposure of neuronal, glial and vascular cell lines to Aβ (particularly fibrillar Aβ) upregulates multiple Aβ-degrading proteases including NEP, ECE, ACE, ECE-2, IDE, MMP-2 -3 and -9 (Deb et al., 1999; Jung et al., 2003; Lee et al., 2003; Leal et al., 2006; Palmer et al., 2009; Wang et al., 2009a,b; Miners et al., 2010b). A number of studies have reported a rise in Aβ-degrading protease activity in aged hAPP transgenic mice, coinciding with the appearance of parenchymal deposits of Aβ (Tucker et al., 2000; Leal et al., 2006; Yin et al., 2006). A dose-dependent increase in NEP was also observed in hAPP mice following intracerebral injection of synthetic fibrillar Aβ (Mohajeri et al., 2002, 2004). Together, these data suggest that Aβ-mediated upregulation of protease activity is secondary to Aβ accumulation in late-onset AD and that Aβ accumulates in association with a failed compensatory response rather than an overall deficiency in Aβ-degrading enzyme activity. Even if this is the case it does not preclude the potential of enhancing or administering Aβ-degrading proteases as a therapeutic target for AD. However, whilst upregulation of Aβ-degrading enzymes probably slows the accumulation of Aβ, it may also increase the cleavage of other substrates of these multifunctional enzymes. Those substrates include several peptides that play important roles in regulating cerebral blood flow.
The Endothelin System in Alzheimer's Disease
Endothelin-1 (ET-1) is a potent, long-lasting vasoconstrictor in the brain (Haynes and Webb, 1994). It is produced predominantly by vascular endothelial cells, but also by neurons and macrophages in human brain tissue (Palmer et al., 2012). ET-1 is generated from the inactive precursor, big-ET-1, through cleavage by ECE-1 or -2 (Yanagisawa et al., 2000). ECE-1 is expressed predominantly within the cerebral vasculature within endothelial cells (Palmer et al., 2010) but is also expressed by smooth muscle cells (Maguire, 2002), and weak neuronal and astrocytic staining of ECE-1 was observed in human brain tissue (Palmer et al., 2010). ECE-2 is expressed by pyramidal neurons, and to a lesser extent glia, within the neocortex (Palmer et al., 2009), including cells in close association with blood vessels. ET-1 has potent and sustained vasoconstrictor effects both in vitro and in vivo (Brain et al., 1988; Yanagisawa et al., 1988; Borges et al., 1989; Brain, 1989). Chronic infusion of ET-1 in to Sprague–Dawley rats led to sustained elevation in mean arterial blood pressure (Mortensen et al., 1990) and reduced perfusion, with subsequent neuronal loss (Hughes et al., 2003).
There is dysregulation of the endothelin system in AD. Several studies have shown that the concentration of ET-1 is higher in AD than controls in both the cerebral cortex (Minami et al., 1995; Palmer et al., 2012) and in cerebral blood vessels (Luo and Grammas, 2010; Palmer et al., 2013). ECE-1 and ECE-2 degrade Aβ (Eckman et al., 2001), and ECE-1 heterozygous and ECE-2 homozygous knockout mice display significantly elevated endogenous Aβ1−40 and Aβ1−42 (Eckman et al., 2001, 2003). However, we found ECE-2 level and ECE-1 activity (Palmer et al., 2009, 2010, 2013) to be higher in AD than in age-matched controls, in human post-mortem cerebral cortex and leptomeninges. We did not observe similar elevations in post-mortem tissue from patients with vascular dementia (VaD).
Upregulation of ET-1 production in AD seems likely to be secondary to the accumulation of Aβ, since exposure of SH-SY5Y human neuroblastoma cells and human brain microvascular endothelial cells to Aβ caused upregulation of ECE-2 and ECE-1, respectively, resulting in increased production and release into the supernatant of ET-1 (Palmer et al., 2009, 2012, 2013). ET-1 production was also elevated in the cerebral vasculature of mice that had been infused with Aβ (Paris et al., 2003). The authors also showed that Aβ enhanced ET-1 induced vasoconstriction in isolated human middle cerebral and basilar arteries. Mice that overexpress hAPP showed no diminution in the contractile response of cerebral vessels to ET-1, despite an impaired response to vasodilators (Tong et al., 2005). Together, these studies suggest that upregulation of ECE-1 and -2 is a physiological feedback response to Aβ that probably increases degradation of Aβ and reduces its further accumulation but also results in increased production and release of ET-1 which mediates vasoconstriction (reviewed in Palmer and Love, 2011) (Figure 1).
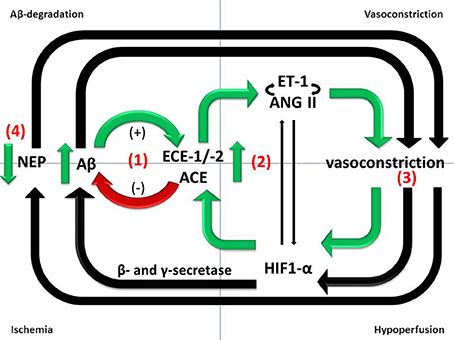
Figure 1. Divergent roles of Aβ-degrading enzymes in the pathogenesis of AD. (1) ECE-1/-2 and ACE cleave Aβ in vitro and ECE-1/-2 regulate endogenous Aβ level in mice. However, ECE-1/-2 and ACE activities are increased in the cerebral cortex in AD in human post-mortem brain tissue. The increases are likely to represent responses to accumulating Aβ and may mitigate the accumulation but are inadequate to prevent the level of Aβ from continuing to rise as the disease progresses. (2) Upregulation of ECE-1/2 and ACE by Aβ results in increased production of ET-1 and Ang II, both of which mediate vasoconstriction. (3) Reduced cerebral perfusion, as a result of vasoconstriction, probably increases Aβ production by promoting amyloidogenic processing of APP. (4) NEP protects vessels from Aβ-related pathology, including CAA, but NEP activity is reduced in conditions of hypoxia and oxidative stress.
It should be noted that hypoxia itself can upregulate components of the ET system. The EDN1 gene contains a HIF1-α binding site (Takanashi et al., 2004) and ET-1 expression is induced by hypoxia or ischemia (Li et al., 1994; Yamashita et al., 2001; Ao et al., 2002; Kang et al., 2011). In another study, ECE-1 protein level and ETA and ETB receptor expression were increased in response to hypoxia both in vitro and in vivo (Li et al., 1994; Kang et al., 2011). Furthermore, ET-1 was shown to induce HIF1-α expression dose-dependently in cultured pulmonary smooth muscle cells (Li et al., 2012). Upregulation of more than one pathway may therefore account for increased production of ET-1 in dementia, including in AD.
ETA receptor antagonists, but not dual ETA/ETB receptor antagonists significantly reduced the impairment in learning and memory induced by Aβ in rats (Briyal et al., 2011), as well as preventing the Aβ-induced increase in expression of ETA receptors and oxidative stress. Additionally, an ETB receptor agonist elevated CBF in normal rats and acted as an anti-apoptotic factor, counteracting the neurotoxicity and cognitive impairment induced by Aβ (Briyal et al., 2014). The findings suggest that ETA receptor antagonists, which are already used in the treatment of pulmonary hypertension (Palmer and Love, 2011), and potentially ETB receptor agonists, have therapeutic potential in AD.
The Renin-Angiotensin System in Alzheimer's Disease
The renin-angiotensin system (RAS) plays an important role in fluid homeostasis and regulation of blood pressure. ACE catalyzes the conversion of angiotensin I (non-vasoactive) to the vasoconstrictor, angiotensin II. ACE also cleaves and thereby inactivates the vasodilator, bradykinin. These vasopressor activities can be blocked either by ACE-inhibitors, a standard treatment for hypertension or by Ang II receptor antagonists that inhibit Ang II-mediated vasopressor effects. ACE is expressed by the endothelium and neurons within the brain (Turner and Hooper, 2002; Miners et al., 2008c).
The involvement of the RAS, particularly ACE, in the pathogenesis of AD has been studied extensively (reviewed in Kehoe et al., 2009). ACE cleaves Aβ in vitro (Hu et al., 2001; Hemming and Selkoe, 2005; Oba et al., 2005; Sun et al., 2008; Toropygin et al., 2008) and retards Aβ aggregation and neurotoxicity (Hu et al., 2001). A recent study indicated that ACE-mediated cleavage of Aβ1−42 resulted in the formation of the less neurotoxic Aβ1−40 species (Zou et al., 2007). The exact contribution of ACE to the regulation of Aβ in vivo remains unclear (reviewed in Kehoe et al., 2009). Eckman et al. (2006) reported that endogenous Aβ levels were unchanged in an ACE knockout mouse and in mice treated with ACE inhibitors. In support of the earlier studies, Hemming et al. (2007) found that captopril did not significantly affect Aβ levels in two human APP transgenic mouse lines. More recently, however, Wang et al. (Zou et al., 2007) showed that administration of captopril for 7 or 11 months to 6-month old tg2576 mice (transgenic for the Swedish double mutation in human APP) promoted Aβ deposition. The divergence of results could reflect a difference in the species and age of the animals used, and the respective dosage regimes, and highlights the need for further investigation.
ACE level and enzyme activity are increased within the cerebral cortex in AD. This has been demonstrated in multiple human post-mortem studies (Arregui et al., 1982; Barnes et al., 1991; He et al., 2006; Miners et al., 2008a). We showed a strong correlation between ACE activity and disease severity, as indicated by Braak tangle stage and parenchymal Aβ load (Miners et al., 2008a). Thus, ACE-mediated cleavage of Aβ is not impaired in AD. We also showed that ACE protein level and activity were upregulated in human neuronal SH-SY5Y cells in response to exposure to synthetic, aggregated Aβ1−42 (Miners et al., 2008a) suggesting that induction of ACE is a likely to be a physiological response to increasing Aβ level and may help to prevent further Aβ accumulation.
A common Alu (indel) insertion(I)/deletion(D) polymorphism (rs 1799752) in intron 16 of the ACE gene was reported to be associated with sporadic AD, the greatest risk being associated with homozygosity of the I allele (Kehoe et al., 1999). This was confirmed in several meta-analyses (Narain et al., 2000; Kehoe et al., 2003; Elkins et al., 2004; Lehmann et al., 2005) and was supported to some extent by some of the earlier whole-genome association studies (Li et al., 2008; Thornton-Wells et al., 2008) although a number of more recent and larger studies have failed to find any association. In the original studies, homozygosity of the D and I alleles was associated with the highest and lowest ACE protein level in the periphery, respectively, (Rigat et al., 1990) although this association only partly explains the total variance of ACE in the periphery (Terao et al., 2013). We found that the ACE II genotype was associated with reduced ACE protein level in the CSF (Miners et al., 2010b). Other studies showed significant association between ACE variants and Aβ1−42 concentration or the Aβ1−42:Aβ1−40 ratio in the CSF (Lehmann et al., 2005; Kauwe et al., 2009). These studies suggested that an association between the ACE polymorphism and AD might be mediated through reduced ACE-mediated proteolysis of Aβ. However, on analysis of post-mortem brain tissue, we subsequently found that the putative AD risk genotype, ACE II, was associated with increased rather than decreased ACE activity (Miners et al., 2010b). ACE expression within the brain did not mirror findings in the plasma and CSF; this finding is in keeping with other evidence that RAS within the brain functions in isolation from the periphery. In addition, ACE enzyme activity did not correlate with ACE protein level, possibly due to the effects of post-translational modification on ACE enzyme activity.
Although it now seems unlikely that ACE contributes substantially to the degradation of Aβ in vivo, there is evidence that the RAS influences several other processes that contribute to the development of AD. Intracerebral infusion of Ang II in male Sprague–Dawley rats resulted in significant cognitive impairment, increased amyloid pathology [due to elevated β- and γ-secretase activity (Zhu et al., 2011)] and an increased level of phosphorylated tau (Tian et al., 2012). Ang II infusion also resulted in stimulation of type 1 Ang II receptors (AT1Rs) that mediate vasoconstriction. These effects were prevented by losartan, an AT1R receptor antagonist. Additionally, Ang II inhibits potassium-mediated release of ACh (Barnes et al., 1992), and influences tumor necrosis factor-α and transforming growth factor β signaling (Hamdi and Castellon, 2004) and blood-brain barrier maintenance (Wosik et al., 2007). Administration of the Ang II receptor antagonist valsartan in tg2576 mice (transgenic for the Swedish double mutation in human APP) resulted in lower Aβ deposition and improved cognitive performance (Wang et al., 2007). Danielyan and colleagues similarly showed that intra-nasal administration of losartan significantly reduced Aβ pathology (Danielyan et al., 2010). In view of these pleotropic actions of Ang II, that are associated with a number of AD pathological processes, and the recent observations in animal models of AD treated with losartan, AT1Rs have come under scrutiny as a potential therapeutic option for AD. Data from large retrospective clinical studies (Li et al., 2010; Davies et al., 2011) and secondary explorations of clinical trial studies (Gao et al., 2013; O'Caoimh et al., 2014) suggest that use of ARBs may be protective against the development and progression of AD (reviewed in Ashby and Kehoe, 2013). The finding in some studies that the reduction in risk is associated with ARBs but not (or not to the same extent) with ACE inhibitors (Kehoe et al., 2013) may reflect a beneficial effect of maintaining ACE-mediated Aβ degradation. Clinical trials are underway to determine whether losartan (ISRCTN93682878) or perindopril vs. telmisartan (NCT02085265) can reduce AD-associated pathology and cognitive decline in mild to moderate AD.
Chronic exposure to elevated ACE activity may increase the risk of dementia by promoting cerebrovascular disease. In a large prospective cohort of patients (the SMART-MR study) with symptomatic atherosclerotic disease, elevated serum ACE activity was associated with increased risk of ischemic stroke and high blood pressure (Jochemsen et al., 2012a) and increased progression of white matter ischemic damage (Jochemsen et al., 2012b). ACE expression is directly regulated by hypoxia via a HIF1-α-dependent mechanism. Krick et al. (2005) observed increased ACE expression in cultured human FBPA (advential fibroblasts) that was dependent on HIF1-α expression. Upregulation of more than one pathway may therefore account for increased production of Ang II in dementia, including AD.
Emerging studies highlight possible interactions between the ET-1 and RAS in AD. Ang II interacts with the ET system to increase ET-1 production from endothelial cells (Dohi et al., 1992) and vascular adventitial fibroblasts (An et al., 2006) via a mechanism involving NADPH oxidase (An et al., 2007). Ang II mediates the transcriptional regulation of the preproenothelin-1 (ppET-1) gene and induces the synthesis of ET-1 in vitro (Rossi et al., 1999). Ang II was also shown to increase tissue ET-1 in vivo (Moreau et al., 1997). In addition, ET-1 also contributes to the vascular actions of Ang II (Balakrishnan et al., 1996; Moreau et al., 1997) and ET-1 enhances the conversion of Ang I to Ang II in pulmonary artery endothelial cells (Kawaguchi et al., 1991).
Hypoperfusion and Vascular Dysfunction in Alzheimer's Disease
There is increasing evidence that vascular dysfunction, reduced cerebral blood flow and AD are interdependent pathological processes. In large epidemiological studies, cardiovascular risk factors (diabetes, hypertension, high cholesterol levels, atherosclerosis, obesity) have emerged as risk factors for AD (DeKosky et al., 1990; Johnson et al., 2005; Ruitenberg et al., 2005; Mak et al., 2012). Brain ischemia and stroke were reported to increase the risk of dementia and AD (de la Torre, 2005; Dickstein et al., 2010). Many AD patients have some degree of cerebrovascular pathology including white matter ischemic lesions (Brun and Englund, 1986a,b; Englund et al., 1988; Englund and Brun, 1990; Ellis et al., 1996; Kalaria, 2000) and most AD patients have CAA (Esiri and Wilcock, 1986; Love et al., 2003; Jellinger and Attems, 2006). Brain ischemia is the defining pathogenic process in most patients with VaD but in AD cerebral blood flow is also usually reduced (Sharp et al., 1986; Jagust et al., 1987; Schuff et al., 2009) and contributes to cognitive impairment. Reduced cerebral blood flow probably anticipates the development of dementia in AD (Ruitenberg et al., 2005), and occurs well before any pathological or neurological abnormalities in mouse models of AD (Niwa et al., 2002a,b; Iadecola, 2004).
There is evidence from some although not all studies (Chui et al., 2012) that ischemia is not only an additive cause of brain damage in AD but also contributes to the neurodegenerative disease processes. Transient brain ischemia resulting from cardiac arrest was shown to cause a marked increase in serum Aβ1−42 in humans (Zetterberg et al., 2011). Ischemia, or its in vitro simulation by combined deprivation of oxygen and glucose, is associated with increased production of Aβ in animal and cell culture models of AD (Zhang et al., 2007; Guglielmotto et al., 2009; Zhiyou et al., 2009). This probably reflects increased amyloidogenic processing of APP by γ- and β-secretase in response to hypoxia (Sun et al., 2006; Li et al., 2009), resulting in increased plaque formation (Garcia-Alloza et al., 2011). HIF-1α is the primary sensor of tissue hypoxia (Wenger, 2002). HIF-1α subunits are stabilized under conditions of hypoxia, dimerize with HIF-1β and translocate to the nucleus, where they associate with hypoxia-response elements and bind transcriptional co-activators to induce the expression of hypoxia-responsive genes. It is likely that Aβ also contributes indirectly to the reduction in CBF, by mediating vasoconstriction (Claassen and Zhang, 2011) (Figure 1).
We have reviewed (above) evidence that Aβ upregulates ET-1 and Ang II production. In addition, Aβ peptides were reported to enhance the vasoconstrictor action of a thromboxane analog (Niwa et al., 2001; Iadecola, 2003). In mouse models of AD, Aβ impaired cerebral autoregulation and functional hyperemia (Niwa et al., 2000, 2001, 2002a), although in studies to date, cerebral autoregulation has not been shown to be impaired in AD (Claassen and Zhang, 2011). The Iadecola group showed that oxidative stress plays a critical role in the attenuation of functional hyperemia by Aβ1−40 (Park et al., 2004). The in vitro finding by Palmer et al. (2013) that an Aβ1−40 induced increase in ET-1 production by endothelial cells could be blocked by co-administration of the antioxidant superoxide dismutase suggests that the attenuation of functional hyperemia by oxidative stress may be partly mediated by ET-1. Cerebral ischemia also induced tau phosphorylation, leading to the death of basal forebrain cholinergic neurons which are profoundly depleted in AD (Zheng et al., 2002; Li et al., 2011). Li et al. (2011) reported that ET-1 exacerbated Aβ deposition, tau phosphorylation and cognitive impairment after intracerebral injection of Aβ in rats.
Post-Mortem Assessment of Cerebral Hypoperfusion
Evaluation of the possible contribution of ischemia to the development or progression of AD and other neurological disorders has been impaired by the lack of objective, quantifiable markers of hypoperfusion in post-mortem brain tissue. Pathologists have relied on morphological assessment of paraffin sections, and although this approach has been informative it is limited by its subjectivity and relative insensitivity, relying on the identification of infarcts and regions of white matter rarefaction rather than more subtle manifestations of ischemia. One potential approach to quantifying ante-mortem hypoperfusion by examination of post-mortem brain tissue is the measurement of gene transcripts and proteins involved in the molecular responses to brain ischemia. As noted above, HIF-1α subunits are the principal sensors of tissue hypoxia (Acker and Acker, 2004). Under conditions of chronic or intermittent hypoxia there is an increase in HIF-1α transcription (Nanduri et al., 2008; Powell, 2009). Tissue hypoxia also causes upregulation of a large number of other genes. Some, like VEGF and heme-oxygenase (HO-1), are upregulated by HIFs (Liu et al., 1995; Shweiki et al., 1995; Pham et al., 2002); others, such as neuroglobin are upregulated independently of HIFs (Sun et al., 2001). Fernando et al. (2006) reported elevated immunolabeling of astrocytes or microglia for HIF-1α and HIF-2α within deep white matter lesions in the elderly. We measured VEGF by ELISA, in both cerebral cortex and white matter (Barker et al., 2013) from human post-mortem brains, and found it to be elevated in the cerebral cortex in AD independently of the severity of small vessel disease or CAA. VEGF was not elevated in the white matter in AD (Barker et al., 2014).
In a study of two independent post-mortem cohorts, we recently showed that the ratio of myelin-associated glycoprotein (MAG) to proteolipid protein 1 (PLP1) in the white matter correlated inversely with the severity of small vessel disease, and so presumably with the degree of ante-mortem ischemia (Barker et al., 2013). Our rationale for measuring this combination of myelin proteins was that the transport of myelin proteins from the oligodendrocyte cell body where they are synthesized, to their site of insertion in the myelin sheath, is an energy-dependent process and MAG, being expressed only adaxonally, far from the oligodendrocyte cell body, is more susceptible to ischemia than is PLP, expressed throughout the myelin sheath. We have found that MAG is also sensitive to ischemia in the gray matter and are currently using a combination of methods to assess the relationships between the activity of the endothelin and RAS, cerebral hypoperfusion, neurodegenerative pathology and white matter ischemic damage in AD and other dementias.
Our findings to date point toward different processes underlying ischemic changes in the cerebral cortex and white matter in AD, VaD and dementia with Lewy bodies (DLB) (Figure 2). Whereas ET-1 production is significantly increased in the cerebral cortex in AD, presumably reflecting locally elevated Aβ and oxidative stress, it is reduced in the white matter, as would be expected for a physiological adaptation to reduced blood flow. It should be noted that multiple, topographically remote processes are likely to contribute to white matter ischemia in AD. These include meningeal and cortical CAA, Ang II- and ET1-mediated constriction of perforating arterioles that traverse the cortex to supply the white matter, and in some patients, systemic processes such as postural hypotension (Ballard et al., 2000) and atrial fibrillation (Ott et al., 1997).
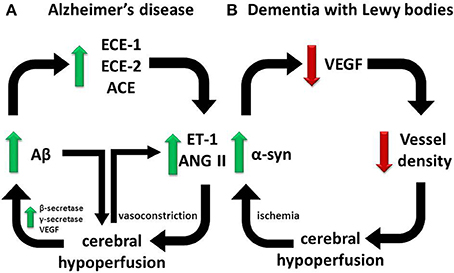
Figure 2. Divergent pathways responsible for cerebral hypoperfusion in dementia. Our observations on human post-mortem brain tissue suggest that (A) in AD, cerebral hypoperfusion is mediated in part by elevated ECE-1/-2 and ACE in the cerebral cortex in AD in response to accumulating Aβ, whereas (B) in DLB, cerebral hypoperfusion is mediated by reduced vessel density in association with a reduction in VEGF expression, possibly in response to α-syn accumulation mediated by ischemia. In each pathway a vicious cycle ensues that accelerates disease progression.
There is little published information on the possible contribution of cerebral hypoperfusion to other dementias. Patients who have DLB are particularly prone to neurocardiovascular instability (Ballard et al., 1998), which correlates with the severity of white matter ischemic lesions detectable by MRI (Kenny et al., 2004). Several other studies have demonstrated reduced perfusion of the parieto-occpital cortex in DLB (Ishii et al., 1999; Lobotesis et al., 2001; Colloby et al., 2002; Firbank et al., 2003; Fong et al., 2011). We recently reported that hypoperfusion in the occipital cortex of patients with DLB was associated with reduced microvessel density (as determined by measuring the level of factor VIII-related antigen) (Barker et al., 2014) and a reduction in VEGF (Miners et al., 2014). The VEGF level in DLB correlated positively with the ratio of MAG:PLP, in keeping with a direct relationship between reduced VEGF, reduced vascularity and hypoperfusion (leading to reduced MAG:PLP). The mechanism of VEGF reduction in the occipital cortex in DLB is unclear. However, it seems to be related to the underlying disease process, correlating negatively with the levels of total α-syn and of α-syn phosphorylated at serine-129 (Figure 2). VEGF level was reduced in SHSY-5Y cells overexpressing human α-syn. Conversely, oxygen-glucose deprivation increased α-syn level and aggregation. In contrast to AD, in DLB the level of ET-1 and activity of ACE were similar to levels in age-matched controls.
Neprilysin and Cerebral Amyloid Angiopathy
The most common form of CAA is caused by cerebrovascular accumulation of Aβ in the walls of leptomeningeal and cerebrocortical blood vessels, particularly arterioles (Revesz et al., 2002, 2003). CAA increases in prevalence with age, to about 45% in those over 80 years, and is present in over 90% of patients with AD (Love et al., 2003). Aβ1−40 is the most abundant isoform in vessel walls (Gravina et al., 1995). It is thought to be predominantly, if not exclusively, of neuronal origin (Calhoun et al., 1999; Burgermeister et al., 2000; van Dorpe et al., 2000). The Aβ accumulates initially in the basement membrane but eventually causes the death of the cerebrovascular smooth muscle cells (CVSMCs) and replaces the tunica media. Particularly in larger vessels, Aβ may also deposit in the adventitia (Weller et al., 2009a). Cerebrovascular accumulation of Aβ may be caused by increased production of this peptide, in familial forms of AD and CAA. However, in most sporadic cases, decreased removal, particularly as a result of the effects of age on vessel walls, is likely to be the major cause of CAA. Arteriosclerosis and age-related changes to the composition of the arterial basement membranes may impede the perivascular drainage of Aβ, as does preexisting CAA (Weller et al., 2009b; Hawkes et al., 2011).
Another determinant of the likelihood of vascular accumulation of Aβ is the activity of Aβ-degrading proteases within the tunica media. We and others showed that NEP is abundantly expressed by smooth muscle cells within larger arterioles in the cerebral cortex and leptomeninges (Carpentier et al., 2002; Miners et al., 2006) in human brain tissue, and that the level of NEP is reduced in AD patients with severe CAA. The reduction is not simply a consequence of the loss of smooth muscle cells. We demonstrated that the reduction of NEP protein in CAA affected both Aβ-laden and non-Aβ-laden vessels (Miners et al., 2006). We also showed that NEP enzyme activity is reduced in CAA, in both the leptomeninges (within which the only source of NEP is the smooth muscle cells in the blood vessels) and vessel-enriched preparations of cerebral cortex (Miners et al., 2011c). The reduction in NEP activity was still evident after adjustment for the smooth muscle actin content of the samples and was therefore not simply a consequence of death of CVSMCs, although loss of CVSMCs, whether from arteriosclerosis or as a consequence of CAA, would be expected to exacerbate any preexisting deficiency in vessel-associated NEP.
To model the role of NEP in CAA we manipulated NEP activity in cultured human brain-derived vascular smooth muscle cells, using siRNA knock-down of NEP, to reduce activity, or transfection of NEP cDNA to enhance activity, and measured cell death after adding recombinant Aβ1−42 (Miners et al., 2011c). We found that NEP protected against Aβ1−42-mediated cell death. We also analyzed the influence of APOE genotype on vessel-associated NEP activity. The relationship between APOE and vessel-associated NEP activity mirrored that between APOE genotype and risk of AD and CAA: after adjustment for smooth muscle actin, NEP activity was highest in blood vessels from patients with APOE ε2/3 genotype and decreased stepwise through APOE ε3/3, ε3/4, and ε4/4 genotypes. The possible contribution to CAA of changes in the activity of other vessel-associated Aβ-degrading enzymes, including ECE-1 and IDE (Morelli et al., 2004; Lynch et al., 2006) have yet to be assessed.
Concluding Remarks
Multiple Aβ-degrading proteases are upregulated in AD in response to the rising concentration of Aβ. This upregulation probably mitigates the accumulation and toxicity of Aβ, including in the context of CAA, but also enhances the degradation of other substrates of these proteases. Those substrates include the vasodilator bradykinin, and precursors of the potent vasoconstrictors ET-1 and Ang II. The resulting vasoconstriction is likely to contribute to reduced cerebral blood flow in AD and perhaps also to the neurodegenerative disease process itself, through increased production, accumulation and deposition of Aβ and increased phosphorylation of tau. Reduced cerebral blood flow also contributes to VaD and dementia with Lewy body diseases but the distribution and mechanisms of hypoperfusion are different from those in AD. Our findings underline the limitations of highly reductionist approaches for assessing the actions of multifunctional proteases, and the importance of validating experimental findings by analysis of human brain tissue.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Alzheimer's Research UK. The South West Dementia Brain Bank was also supported by BRACE (Bristol Research into Alzheimer's and Care of the Elderly) and ABBUK (Alzheimer's Brain Bank UK, supporting Brains for Dementia Research).
References
Acker, T., and Acker, H. (2004). Cellular oxygen sensing need in CNS function: physiological and pathological implications. J. Exp. Biol. 207, 3171–3188. doi: 10.1242/jeb.01075
Akiyama, H., Kondo, H., Ikeda, K., Kato, M., and McGeer, P. L. (2001). Immunohistochemical localization of neprilysin in the human cerebral cortex: inverse association with vulnerability to amyloid β-protein (Aβ) deposition. Brain Res. 902, 277–281. doi: 10.1016/S0006-8993(01)02390-3
An, S. J., Boyd, R., Wang, Y., Qiu, X., and Wang, H. D. (2006). Endothelin-1 expression in vascular adventitial fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 290, H700–H708. doi: 10.1152/ajpheart.00326.2005
An, S. J., Boyd, R., Zhu, M., Chapman, A., Pimentel, D. R., and Wang, H. D. (2007). NADPH oxidase mediates angiotensin II-induced endothelin-1 expression in vascular adventitial fibroblasts. Cardiovasc. Res. 75, 702–709. doi: 10.1016/j.cardiores.2007.02.015
Ao, Q., Hao, C., Xiong, M., and Wang, D. (2002). Expression of hypoxia-inducible factor-1 α and endothelin-1 gene in hypoxic pulmonary hypertension. Zhonghua Bing Li Xue Za Zhi 31, 140–142.
Arregui, A., Perry, E. K., Rossor, M., and Tomlinson, B. E. (1982). Angiotensin converting enzyme in Alzheimer's disease increased activity in caudate nucleus and cortical areas. J. Neurochem. 38, 1490–1492. doi: 10.1111/j.1471-4159.1982.tb07930.x
Ashby, E. L., and Kehoe, P. G. (2013). Current status of renin-aldosterone angiotensin system-targeting anti-hypertensive drugs as therapeutic options for Alzheimer's disease. Expert Opin. Investig. Drugs 22, 1229–1242. doi: 10.1517/13543784.2013.812631
Baig, S., Kehoe, P. G., and Love, S. (2008). MMP-2, -3 and -9 levels and activity are not related to Aβ load in the frontal cortex in Alzheimer's disease. Neuropathol. Appl. Neurobiol. 34, 205–215. doi: 10.1111/j.1365-2990.2007.00897.x
Balakrishnan, S. M., Wang, H. D., Gopalakrishnan, V., Wilson, T. W., and McNeill, J. R. (1996). Effect of an endothelin antagonist on hemodynamic responses to angiotensin II. Hypertension 28, 806–809. doi: 10.1161/01.HYP.28.5.806
Ballard, C., O'Brien, J., Barber, B., Scheltens, P., Shaw, F., McKeith, I., et al. (2000). Neurocardiovascular instability, hypotensive episodes, and MRI lesions in neurodegenerative dementia. Ann. N.Y. Acad. Sci. 903, 442–445. doi: 10.1111/j.1749-6632.2000.tb06396.x
Ballard, C. G., O'Brien, J., Lowery, K., Ayre, G. A., Harrison, R., Perry, R., et al. (1998). A prospective study of dementia with Lewy bodies. Age Ageing 27, 631–636. doi: 10.1093/ageing/27.5.631
Barker, R., Ashby, E. L., Wellington, D., Barrow, V. M., Palmer, J. C., Kehoe, P. G., et al. (2014). Pathophysiology of white matter perfusion in Alzheimer's disease and vascular dementia. Brain 137, 1524–1532. doi: 10.1093/brain/awu040
Barker, R., Love, S., and Kehoe, P. G. (2010). Plasminogen and plasmin in Alzheimer's disease. Brain Res. 1355, 7–15. doi: 10.1016/j.brainres.2010.08.025
Barker, R., Wellington, D., Esiri, M. M., and Love, S. (2013). Assessing white matter ischemic damage in dementia patients by measurement of myelin proteins. J. Cereb. Blood Flow Metab. 33, 1050–1057. doi: 10.1038/jcbfm.2013.46
Barnes, J. M., Barnes, N. M., Costall, B., Coughlan, J., Kelly, M. E., Naylor, R. J., et al. (1992). Angiotensin-converting enzyme inhibition, angiotensin, and cognition. J. Cardiovasc. Pharmacol. 19(Suppl. 6), S63–S71. doi: 10.1097/00005344-199219006-00011
Barnes, N. M., Cheng, C. H., Costall, B., Naylor, R. J., Williams, T. J., and Wischik, C. M. (1991). Angiotensin converting enzyme density is increased in temporal cortex from patients with Alzheimer's disease. Eur. J. Pharmacol. 200, 289–292. doi: 10.1016/0014-2999(91)90584-D
Bateman, R. J., Munsell, L. Y., Morris, J. C., Swarm, R., Yarasheski, K. E., and Holtzman, D. M. (2006). Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861. doi: 10.1038/nm1438
Bell, R. D., Sagare, A. P., Friedman, A. E., Bedi, G. S., Holtzman, D. M., Deane, R., et al. (2007). Transport pathways for clearance of human Alzheimer's amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 27, 909–918. doi: 10.1038/sj.jcbfm.9600419
Belza, M. G., and Urich, H. (1986). Cerebral amyloid angiopathy in Down's syndrome. Clin. Neuropathol. 5, 257–260.
Borges, R., Von Grafenstein, H., and Knight, D. E. (1989). Tissue selectivity of endothelin. Eur. J. Pharmacol. 165, 223–230. doi: 10.1016/0014-2999(89)90716-4
Brain, S. D. (1989). The direct observation of arteriolar constriction induced by endothelin in vivo. Eur. J. Pharmacol. 160, 401–403. doi: 10.1016/0014-2999(89)90096-4
Brain, S. D., Tippins, J. R., and Williams, T. J. (1988). Endothelin induces potent microvascular constriction. Br. J. Pharmacol. 95, 1005–1007. doi: 10.1111/j.1476-5381.1988.tb11731.x
Briyal, S., Philip, T., and Gulati, A. (2011). Endothelin-A receptor antagonists prevent amyloid-β-induced increase in ETA receptor expression, oxidative stress, and cognitive impairment. J. Alzheimers Dis. 23, 491–503. doi: 10.3233/JAD-2010-101245
Briyal, S., Shepard, C., and Gulati, A. (2014). Endothelin receptor type B agonist, IRL-1620, prevents β amyloid (Aβ) induced oxidative stress and cognitive impairment in normal and diabetic rats. Pharmacol. Biochem. Behav. 120, 65–72. doi: 10.1016/j.pbb.2014.02.008
Brun, A., and Englund, E. (1986a). Brain changes in dementia of Alzheimer's type relevant to new imaging diagnostic methods. Prog. Neuropsychopharmacol. Biol. Psychiatry 10, 297–308. doi: 10.1016/0278-5846(86)90009-6
Brun, A., and Englund, E. (1986b). A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann. Neurol. 19, 253–262. doi: 10.1002/ana.410190306
Bruno, M. A., Mufson, E. J., Wuu, J., and Cuello, A. C. (2009). Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. 68, 1309–1318. doi: 10.1097/NEN.0b013e3181c22569
Burgermeister, P., Calhoun, M. E., Winkler, D. T., and Jucker, M. (2000). Mechanisms of cerebrovascular amyloid deposition. Lessons from mouse models. Ann. N.Y. Acad. Sci. 903, 307–316. doi: 10.1111/j.1749-6632.2000.tb06381.x
Cabrejo, L., Guyant-Marechal, L., Laquerriere, A., Vercelletto, M., De la Fourniere, F., Thomas-Anterion, C., et al. (2006). Phenotype associated with APP duplication in five families. Brain 129, 2966–2976. doi: 10.1093/brain/awl237
Calhoun, M. E., Burgermeister, P., Phinney, A. L., Stalder, M., Tolnay, M., Wiederhold, K. H., et al. (1999). Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. U.S.A. 96, 14088–14093. doi: 10.1073/pnas.96.24.14088
Carpentier, M., Robitaille, Y., DesGroseillers, L., Boileau, G., and Marcinkiewicz, M. (2002). Declining expression of neprilysin in Alzheimer disease vasculature: possible involvement in cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 849–856.
Carson, J. A., and Turner, A. J. (2002). B-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 81, 1–8. doi: 10.1046/j.1471-4159.2002.00855.x
Chui, H. C., Zheng, L., Reed, B. R., Vinters, H. V., and Mack, W. J. (2012). Vascular risk factors and Alzheimer's disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res. Ther. 4, 1. doi: 10.1186/alzrt98
Claassen, J. A., and Zhang, R. (2011). Cerebral autoregulation in Alzheimer's disease. J. Cereb. Blood Flow Metab. 31, 1572–1577. doi: 10.1038/jcbfm.2011.69
Colloby, S. J., Fenwick, J. D., Williams, E. D., Paling, S. M., Lobotesis, K., Ballard, C., et al. (2002). A comparison of (99m)Tc-HMPAO SPET changes in dementia with Lewy bodies and Alzheimer's disease using statistical parametric mapping. Eur. J. Nucl. Med. Mol. Imaging 29, 615–622. doi: 10.1007/s00259-002-0778-5
Danielyan, L., Klein, R., Hanson, L. R., Buadze, M., Schwab, M., Gleiter, C. H., et al. (2010). Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res. 13, 195–201. doi: 10.1089/rej.2009.0944
Davies, N. M., Kehoe, P. G., Ben-Shlomo, Y., and Martin, R. M. (2011). Associations of anti-hypertensive treatments with Alzheimer's disease, vascular dementia, and other dementias. J. Alzheimers Dis. 26, 699–708. doi: 10.3233/JAD-2011-110347
de la Torre, J. C. (2005). Is Alzheimer's disease preceded by neurodegeneration or cerebral hypoperfusion? Ann. Neurol. 57, 783–784. doi: 10.1002/ana.20516
Deb, S., Zhang, J. W., and Gottschall, P. E. (1999). Activated isoforms of MMP-2 are induced in U87 human glioma cells in response to β-amyloid peptide. J. Neurosci. Res. 55, 44–53.
DeKosky, S. T., Shih, W. J., Schmitt, F. A., Coupal, J., and Kirkpatrick, C. (1990). Assessing utility of single photon emission computed tomography (SPECT) scan in Alzheimer disease: correlation with cognitive severity. Alzheimer Dis. Assoc. Disord. 4, 14–23. doi: 10.1097/00002093-199040100-00002
Dickstein, D. L., Walsh, J., Brautigam, H., Stockton, S. D. Jr., Gandy, S., and Hof, P. R. (2010). Role of vascular risk factors and vascular dysfunction in Alzheimer's disease. Mt. Sinai J. Med. 77, 82–102. doi: 10.1002/msj.20155
Dohi, Y., Hahn, A. W., Boulanger, C. M., Buhler, F. R., and Luscher, T. F. (1992). Endothelin stimulated by angiotensin II augments contractility of spontaneously hypertensive rat resistance arteries. Hypertension 19, 131–137. doi: 10.1161/01.HYP.19.2.131
Donahue, J. E., Khurana, J. S., and Adelman, L. S. (1998). Intracerebral hemorrhage in two patients with Down's syndrome and cerebral amyloid angiopathy. Acta Neuropathol. 95, 213–216. doi: 10.1007/s004010050789
Eckman, E. A., Adams, S. K., Troendle, F. J., Stodola, B. A., Kahn, M. A., Fauq, A. H., et al. (2006). Regulation of steady-state β-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J. Biol. Chem. 281, 30471–30478. doi: 10.1074/jbc.M605827200
Eckman, E. A., and Eckman, C. B. (2005). Aβ-degrading enzymes: modulators of Alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochem. Soc. Trans. 33, 1101–1105. doi: 10.1042/BST20051101
Eckman, E. A., Reed, D. K., and Eckman, C. B. (2001). Degradation of the Alzheimer's amyloid β peptide by endothelin-converting enzyme. J. Biol. Chem. 276, 24540–24548. doi: 10.1074/jbc.M007579200
Eckman, E. A., Watson, M., Marlow, L., Sambamurti, K., and Eckman, C. B. (2003). Alzheimer's disease β-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 278, 2081–2084. doi: 10.1074/jbc.C200642200
El-Amouri, S. S., Zhu, H., Yu, J., Marr, R., Verma, I. M., and Kindy, M. S. (2008). Neprilysin: an enzyme candidate to slow the progression of Alzheimer's disease. Am. J. Pathol. 172, 1342–1354. doi: 10.2353/ajpath.2008.070620
Elkins, J. S., Douglas, V. C., and Johnston, S. C. (2004). Alzheimer disease risk and genetic variation in ACE: a meta-analysis. Neurology 62, 363–368. doi: 10.1212/01.WNL.0000106823.72493.FF
Ellis, R. J., Olichney, J. M., Thal, L. J., Mirra, S. S., Morris, J. C., Beekly, D., et al. (1996). Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology 46, 1592–1596. doi: 10.1212/WNL.46.6.1592
Englund, E., and Brun, A. (1990). White matter changes in dementia of Alzheimer's type: the difference in vulnerability between cell compartments. Histopathology 16, 433–439. doi: 10.1111/j.1365-2559.1990.tb01542.x
Englund, E., Brun, A., and Alling, C. (1988). White matter changes in dementia of Alzheimer's type. Biochemical and neuropathological correlates. Brain 111(pt 6), 1425–39. doi: 10.1093/brain/111.6.1425
Esiri, M. M., and Wilcock, G. K. (1986). Cerebral amyloid angiopathy in dementia and old age. J. Neurol. Neurosurg. Psychiatr. 49, 1221–1226. doi: 10.1136/jnnp.49.11.1221
Evin, G., and Weidemann, A. (2002). Biogenesis and metabolism of Alzheimer's disease Aβ amyloid peptides. Peptides 23, 1285–1297. doi: 10.1016/S0196-9781(02)00063-3
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Farris, W., Schutz, S. G., Cirrito, J. R., Shankar, G. M., Sun, X., George, A., et al. (2007). Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. 171, 241–251. doi: 10.2353/ajpath.2007.070105
Fernando, M. S., Simpson, J. E., Matthews, F., Brayne, C., Lewis, C. E., Barber, R., et al. (2006). White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37, 1391–1398. doi: 10.1161/01.STR.0000221308.94473.14
Firbank, M. J., Colloby, S. J., Burn, D. J., McKeith, I. G., and O'Brien, J. T. (2003). Regional cerebral blood flow in Parkinson's disease with and without dementia. Neuroimage 20, 1309–1319. doi: 10.1016/S1053-8119(03)00364-1
Fong, T. G., Inouye, S. K., Dai, W., Press, D. Z., and Alsop, D. C. (2011). Association cortex hypoperfusion in mild dementia with Lewy bodies: a potential indicator of cholinergic dysfunction? Brain Imaging Behav. 5, 25–35. doi: 10.1007/s11682-010-9108-x
Gan, L., Ye, S., Chu, A., Anton, K., Yi, S., Vincent, V. A., et al. (2004). Identification of cathepsin B as a mediator of neuronal death induced by Aβ-activated microglial cells using a functional genomics approach. J. Biol. Chem. 279, 5565–5572. doi: 10.1074/jbc.M306183200
Gao, Y., O'Caoimh, R., Healy, L., Kerins, D. M., Eustace, J., Guyatt, G., et al. (2013). Effects of centrally acting ACE inhibitors on the rate of cognitive decline in dementia. BMJ Open 3:e002881. doi: 10.1136/bmjopen-2013-002881
Garcia-Alloza, M., Gregory, J., Kuchibhotla, K. V., Fine, S., Wei, Y., Ayata, C., et al. (2011). Cerebrovascular lesions induce transient β-amyloid deposition. Brain 134, 3697–3707. doi: 10.1093/brain/awr300
Gravina, S. A., Ho, L., Eckman, C. B., Long, K. E., Otvos, L. Jr., Younkin, L. H., et al. (1995). Amyloid β protein (Aβ) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A β 40 or A β 42(43). J. Biol. Chem. 270, 7013–7016.
Guan, H., Liu, Y., Daily, A., Police, S., Kim, M. H., Oddo, S., et al. (2009). Peripherally expressed neprilysin reduces brain amyloid burden: a novel approach for treating Alzheimer's disease. J. Neurosci. Res. 87, 1462–1473. doi: 10.1002/jnr.21944
Guglielmotto, M., Aragno, M., Autelli, R., Giliberto, L., Novo, E., Colombatto, S., et al. (2009). The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1α. J. Neurochem. 108, 1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x
Hafez, D., Huang, J. Y., Huynh, A. M., Valtierra, S., Rockenstein, E., Bruno, A. M., et al. (2011). Neprilysin-2 is an important β-amyloid degrading enzyme. Am. J. Pathol. 178, 306–312. doi: 10.1016/j.ajpath.2010.11.012
Hamdi, H. K., and Castellon, R. (2004). A genetic variant of ACE increases cell survival: a new paradigm for biology and disease. Biochem. Biophys. Res. Commun. 318, 187–191. doi: 10.1016/j.bbrc.2004.04.004
Hardy, J. (1997). Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 20, 154–159. doi: 10.1016/S0166-2236(96)01030-2
Hawkes, C. A., Hartig, W., Kacza, J., Schliebs, R., Weller, R. O., Nicoll, J. A., et al. (2011). Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 121, 431–443. doi: 10.1007/s00401-011-0801-7
Haynes, W. G., and Webb, D. J. (1994). Contribution of endogenous generation of endothelin-1 to basal vascular tone. Lancet 344, 852–854. doi: 10.1016/S0140-6736(94)92827-4
He, M., Ohrui, T., Maruyama, M., Tomita, N., Nakayama, K., Higuchi, M., et al. (2006). ACE activity in CSF of patients with mild cognitive impairment and Alzheimer disease. Neurology 67, 1309–1310. doi: 10.1212/01.wnl.0000238102.04582.ec
Hellstrom-Lindahl, E., Ravid, R., and Nordberg, A. (2008). Age-dependent decline of neprilysin in Alzheimer's disease and normal brain: inverse correlation with A β levels. Neurobiol. Aging 29, 210–221. doi: 10.1016/j.neurobiolaging.2006.10.010
Hemming, M. L., and Selkoe, D. J. (2005). Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J. Biol. Chem. 280, 37644–37650. doi: 10.1074/jbc.M508460200
Hemming, M. L., Selkoe, D. J., and Farris, W. (2007). Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid β-protein metabolism in mouse models of Alzheimer disease. Neurobiol. Dis. 26, 273–281. doi: 10.1016/j.nbd.2007.01.004
Henderson, S. J., Andersson, C., Narwal, R., Janson, J., Goldschmidt, T. J., Appelkvist, P., et al. (2014). Sustained peripheral depletion of amyloid-β with a novel form of neprilysin does not affect central levels of amyloid-β. Brain 137, 553–564. doi: 10.1093/brain/awt308
Hu, J., Igarashi, A., Kamata, M., and Nakagawa, H. (2001). Angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide (Aβ); retards A β aggregation, deposition, fibril formation; and inhibits cytotoxicity. J. Biol. Chem. 276, 47863–47868. doi: 10.1074/jbc.M104068200
Huang, S. M., Mouri, A., Kokubo, H., Nakajima, R., Suemoto, T., Higuchi, M., et al. (2006). Neprilysin-sensitive synapse-associated amyloid-β peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 281, 17941–17951. doi: 10.1074/jbc.M601372200
Hughes, P. M., Anthony, D. C., Ruddin, M., Botham, M. S., Rankine, E. L., Sablone, M., et al. (2003). Focal lesions in the rat central nervous system induced by endothelin-1. J. Neuropathol. Exp. Neurol. 62, 1276–1286.
Iadecola, C. (2003). Cerebrovascular effects of amyloid-β peptides: mechanisms and implications for Alzheimer's dementia. Cell. Mol. Neurobiol. 23, 681–689. doi: 10.1023/A:1025092617651
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Ishii, K., Yamaji, S., Kitagaki, H., Imamura, T., Hirono, N., and Mori, E. (1999). Regional cerebral blood flow difference between dementia with Lewy bodies and AD. Neurology 53, 413–416. doi: 10.1212/WNL.53.2.413
Iwata, N. (2003). [Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma]. Seikagaku 75, 97–107.
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Aβ by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., et al. (2000). Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150. doi: 10.1038/77399
Jagust, W. J., Budinger, T. F., and Reed, B. R. (1987). The diagnosis of dementia with single photon emission computed tomography. Arch. Neurol. 44, 258–262. doi: 10.1001/archneur.1987.00520150014011
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jellinger, K. A., and Attems, J. (2006). Prevalence and impact of cerebrovascular pathology in Alzheimer's disease and parkinsonism. Acta Neurol. Scand. 114, 38–46. doi: 10.1111/j.1600-0404.2006.00665.x
Jochemsen, H. M., Geerlings, M. I., Grool, A. M., Vincken, K. L., Mali, W. P., van der Graaf, Y., et al. (2012b). Angiotensin-converting enzyme and progression of white matter lesions and brain atrophy–the SMART-MR study. J. Alzheimers Dis. 29, 39–49. doi: 10.3233/JAD-2012-111772
Jochemsen, H. M., Geerlings, M. I., Visseren, F. L., Algra, A., van der Graaf, Y., Muller, M., et al. (2012a). Serum angiotensin-converting enzyme and recurrent vascular events. The SMART-MR study. Atherosclerosis 224, 486–491. doi: 10.1016/j.atherosclerosis.2012.07.010
Johnson, N. A., Jahng, G. H., Weiner, M. W., Miller, B. L., Chui, H. C., Jagust, W. J., et al. (2005). Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 234, 851–859. doi: 10.1148/radiol.2343040197
Jung, S. S., Zhang, W., and Van Nostrand, W. E. (2003). Pathogenic A β induces the expression and activation of matrix metalloproteinase-2 in human cerebrovascular smooth muscle cells. J. Neurochem. 85, 1208–1215. doi: 10.1046/j.1471-4159.2003.01745.x
Kalaria, R. N. (2000). The role of cerebral ischemia in Alzheimer's disease. Neurobiol. Aging 21, 321–330. doi: 10.1016/S0197-4580(00)00125-1
Kang, B. Y., Kleinhenz, J. M., Murphy, T. C., and Hart, C. M. (2011). The PPARγ ligand rosiglitazone attenuates hypoxia-induced endothelin signaling in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 301, L881–L891. doi: 10.1152/ajplung.00195.2011
Kauwe, J. S., Wang, J., Mayo, K., Morris, J. C., Fagan, A. M., Holtzman, D. M., et al. (2009). Alzheimer's disease risk variants show association with cerebrospinal fluid amyloid β. Neurogenetics 10, 13–17. doi: 10.1007/s10048-008-0150-4
Kawaguchi, H., Sawa, H., and Yasuda, H. (1991). Effect of endothelin on angiotensin converting enzyme activity in cultured pulmonary artery endothelial cells. J. Hypertens. 9, 171–174. doi: 10.1097/00004872-199102000-00012
Kehoe, P. G., Davies, N. M., Martin, R. M., and Ben-Shlomo, Y. (2013). Associations of angiotensin targeting antihypertensive drugs with mortality and hospitalization in primary care patients with dementia. J. Alzheimers Dis. 33, 999–1008. doi: 10.3233/JAD-2012-121090
Kehoe, P. G., Katzov, H., Feuk, L., Bennet, A. M., Johansson, B., Wiman, B., et al. (2003). Haplotypes extending across ACE are associated with Alzheimer's disease. Hum. Mol. Genet. 12, 859–867. doi: 10.1093/hmg/ddg094
Kehoe, P. G., Miners, S., and Love, S. (2009). Angiotensins in Alzheimer's disease - friend or foe? Trends Neurosci. 32, 619–628. doi: 10.1016/j.tins.2009.07.006
Kehoe, P. G., Russ, C., McIlory, S., Williams, H., Holmans, P., Holmes, C., et al. (1999). Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat. Genet. 21, 71–72. doi: 10.1038/5009
Kenny, R. A., Shaw, F. E., O'Brien, J. T., Scheltens, P. H., Kalaria, R., and Ballard, C. (2004). Carotid sinus syndrome is common in dementia with Lewy bodies and correlates with deep white matter lesions. J. Neurol. Neurosurg. Psychiatr. 75, 966–971. doi: 10.1136/jnnp.2003.023812
Krick, S., Hanze, J., Eul, B., Savai, R., Seay, U., Grimminger, F., et al. (2005). Hypoxia-driven proliferation of human pulmonary artery fibroblasts: cross-talk between HIF-1α and an autocrine angiotensin system. FASEB J. 19, 857–859. doi: 10.1096/fj.04-2890fje
Kuhnke, D., Jedlitschky, G., Grube, M., Krohn, M., Jucker, M., Mosyagin, I., et al. (2007). MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer's amyloid-β peptides–implications for the mechanisms of Aβ clearance at the blood-brain barrier. Brain Pathol. 17, 347–353. doi: 10.1111/j.1750-3639.2007.00075.x
Lam, F. C., Liu, R., Lu, P., Shapiro, A. B., Renoir, J. M., Sharom, F. J., et al. (2001). β-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 76, 1121–1128. doi: 10.1046/j.1471-4159.2001.00113.x
Leal, M. C., Dorfman, V. B., Gamba, A. F., Frangione, B., Wisniewski, T., Castano, E. M., et al. (2006). Plaque-associated overexpression of insulin-degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with Alzheimer pathology. J. Neuropathol. Exp. Neurol. 65, 976–987. doi: 10.1097/01.jnen.0000235853.70092.ba
Lee, J. M., Yin, K. J., Hsin, I., Chen, S., Fryer, J. D., Holtzman, D. M., et al. (2003). Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann. Neurol. 54, 379–382. doi: 10.1002/ana.10671
Lehmann, D. J., Cortina-Borja, M., Warden, D. R., Smith, A. D., Sleegers, K., Prince, J. A., et al. (2005). Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer's disease. Am. J. Epidemiol. 162, 305–317. doi: 10.1093/aje/kwi202
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. doi: 10.1016/S0896-6273(03)00787-6
Li, H., Chen, S. J., Chen, Y. F., Meng, Q. C., Durand, J., Oparil, S., et al. (1994). Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J. Appl. Physiol. (1985) 77, 1451–1459.
Li, H., Wetten, S., Li, L., St Jean, P. L., Upmanyu, R., Surh, L., et al. (2008). Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 65, 45–53. doi: 10.1001/archneurol.2007.3
Li, J., Wang, Y. J., Zhang, M., Fang, C. Q., and Zhou, H. D. (2011). Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer's disease. Life Sci. 89, 86–92. doi: 10.1016/j.lfs.2011.04.024
Li, L., Zhang, X., Yang, D., Luo, G., Chen, S., and Le, W. (2009). Hypoxia increases Aβ generation by altering β- and γ-cleavage of APP. Neurobiol. Aging 30, 1091–1098. doi: 10.1016/j.neurobiolaging.2007.10.011
Li, M., Liu, Y., Jin, F., Sun, X., Li, Z., Liu, Y., et al. (2012). Endothelin-1 induces hypoxia inducible factor 1α expression in pulmonary artery smooth muscle cells. FEBS Lett. 586, 3888–3893. doi: 10.1016/j.febslet.2012.08.036
Li, N. C., Lee, A., Whitmer, R. A., Kivipelto, M., Lawler, E., Kazis, L. E., et al. (2010). Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ 340:b5465. doi: 10.1136/bmj.b5465
Liu, Y., Cox, S. R., Morita, T., and Kourembanas, S. (1995). Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5' enhancer. Circ. Res. 77, 638–643. doi: 10.1161/01.RES.77.3.638
Liu, Y., Studzinski, C., Beckett, T., Guan, H., Hersh, M. A., Murphy, M. P., et al. (2009). Expression of neprilysin in skeletal muscle reduces amyloid burden in a transgenic mouse model of Alzheimer disease. Mol. Ther. 17, 1381–1386. doi: 10.1038/mt.2009.115
Lobotesis, K., Fenwick, J. D., Phipps, A., Ryman, A., Swann, A., Ballard, C., et al. (2001). Occipital hypoperfusion on SPECT in dementia with Lewy bodies but not AD. Neurology 56, 643–649. doi: 10.1212/WNL.56.5.643
Love, S., Nicoll, J. A., Hughes, A., and Wilcock, G. K. (2003). APOE and cerebral amyloid angiopathy in the elderly. Neuroreport 14, 1535–1536. doi: 10.1097/00001756-200308060-00027
Luo, J., and Grammas, P. (2010). Endothelin-1 is elevated in Alzheimer's disease brain microvessels and is neuroprotective. J. Alzheimers Dis. 21, 887–896. doi: 10.3233/JAD-2010-091486
Lynch, J. A., George, A. M., Eisenhauer, P. B., Conn, K., Gao, W., Carreras, I., et al. (2006). Insulin degrading enzyme is localized predominantly at the cell surface of polarized and unpolarized human cerebrovascular endothelial cell cultures. J. Neurosci. Res. 83, 1262–1270. doi: 10.1002/jnr.20809
Maguire, J. J. (2002). Endothelin-converting enzyme activity in vascular smooth muscle preparations in vitro. Methods Mol. Biol. 206, 165–177. doi: 10.1385/1-59259-289-9:165
Mak, H. K., Chan, Q., Zhang, Z., Petersen, E. T., Qiu, D., Zhang, L., et al. (2012). Quantitative assessment of cerebral hemodynamic parameters by QUASAR arterial spin labeling in Alzheimer's disease and cognitively normal Elderly adults at 3-tesla. J. Alzheimers Dis. 31, 33–44. doi: 10.3233/JAD-2012-111877
Marr, R. A., Guan, H., Rockenstein, E., Kindy, M., Gage, F. H., Verma, I., et al. (2004). Neprilysin regulates amyloid B peptide levels. J. Mol. Neurosci. 22, 5–11. doi: 10.1385/JMN:22:1-2:5
Marr, R. A., Rockenstein, E., Mukherjee, A., Kindy, M. S., Hersh, L. B., Gage, F. H., et al. (2003). Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23, 1992–1996.
Mattson, M. P. (2004). Pathways towards and away from Alzheimer's disease. Nature 430, 631–639. doi: 10.1038/nature02621
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science 330, 1774. doi: 10.1126/science.1197623
Miller, B. C., Eckman, E. A., Sambamurti, K., Dobbs, N., Chow, K. M., Eckman, C. B., et al. (2003). Amyloid-β peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 6221–6226. doi: 10.1073/pnas.1031520100
Minami, M., Kimura, M., Iwamoto, N., and Arai, H. (1995). Endothelin-1-like immunoreactivity in cerebral cortex of Alzheimer-type dementia. Prog. Neuropsychopharmacol. Biol. Psychiatry 19, 509–513. doi: 10.1016/0278-5846(95)00031-P
Miners, J. S., Ashby, E., Van Helmond, Z., Chalmers, K. A., Palmer, L. E., Love, S., et al. (2008c). Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer's disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 34, 181–193. doi: 10.1111/j.1365-2990.2007.00885.x
Miners, J. S., Baig, S., Tayler, H., Kehoe, P. G., and Love, S. (2009). Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J. Neuropathol. Exp. Neurol. 68, 902–914. doi: 10.1097/NEN.0b013e3181afe475
Miners, J. S., Barua, N., Kehoe, P. G., Gill, S., and Love, S. (2011a). Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 70, 944–959. doi: 10.1097/NEN.0b013e3182345e46
Miners, J. S., Kehoe, P., and Love, S. (2011c). Neprilysin protects against cerebral amyloid angiopathy and Aβ-induced degeneration of cerebrovascular smooth muscle cells. Brain Pathol. 21, 594–605. doi: 10.1111/j.1750-3639.2011.00486
Miners, J. S., Kehoe, P. G., and Love, S. (2008a). Immunocapture-based fluorometric assay for the measurement of insulin-degrading enzyme activity in brain tissue homogenates. J. Neurosci. Methods 169, 177–181. doi: 10.1016/j.jneumeth.2007.12.003
Miners, J. S., Morris, S., Love, S., and Kehoe, P. G. (2011b). Accumulation of insoluble amyloid-β in down's syndrome is associated with increased BACE-1 and neprilysin activities. J. Alzheimers Dis. 23, 101–108. doi: 10.3233/JAD-2010-101395
Miners, J. S., van Helmond, Z., Chalmers, K., Wilcock, G., Love, S., and Kehoe, P. G. (2006). Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 65, 1012–1021. doi: 10.1097/01.jnen.0000240463.87886.9a
Miners, J. S., van Helmond, Z., Kehoe, P. G., and Love, S. (2010a). Changes with age in the activities of β-secretase and the Aβ-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 20, 794–802. doi: 10.1111/j.1750-3639.2010.00375.x
Miners, J. S., van Helmond, Z., Raiker, M., Love, S., and Kehoe, P. G. (2010b). ACE variants and association with brain Aβ levels in Alzheimer's disease. Am. J. Transl. Res. 3, 73–80.
Miners, J. S., Verbeek, M. M., Rikkert, M. O., Kehoe, P. G., and Love, S. (2008b). Immunocapture-based fluorometric assay for the measurement of neprilysin-specific enzyme activity in brain tissue homogenates and cerebrospinal fluid. J. Neurosci. Methods 167, 229–236. doi: 10.1016/j.jneumeth.2007.08.012
Miners, S., Moulding, H., de Silva, R., and Love, S. (2014). Reduced vascular endothelial growth factor and capillary density in the occipital cortex in dementia with lewy bodies. Brain Pathol. 24, 334–343. doi: 10.1111/bpa.12130
Mohajeri, M. H., Kuehnle, K., Li, H., Poirier, R., Tracy, J., and Nitsch, R. M. (2004). Anti-amyloid activity of neprilysin in plaque-bearing mouse models of Alzheimer's disease. FEBS Lett. 562, 16–21. doi: 10.1016/S0014-5793(04)00169-3
Mohajeri, M. H., Wollmer, M. A., and Nitsch, R. M. (2002). Aβ 42-induced increase in neprilysin is associated with prevention of amyloid plaque formation in vivo. J. Biol. Chem. 277, 35460–35465. doi: 10.1074/jbc.M202899200
Moreau, P., d'Uscio, L. V., Shaw, S., Takase, H., Barton, M., and Luscher, T. F. (1997). Angiotensin II increases tissue endothelin and induces vascular hypertrophy: reversal by ET(A)-receptor antagonist. Circulation 96, 1593–1597. doi: 10.1161/01.CIR.96.5.1593
Morelli, L., Llovera, R. E., Mathov, I., Lue, L. F., Frangione, B., Ghiso, J., et al. (2004). Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid β vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J. Biol. Chem. 279, 56004–56013. doi: 10.1074/jbc.M407283200
Mortensen, L. H., Pawloski, C. M., Kanagy, N. L., and Fink, G. D. (1990). Chronic hypertension produced by infusion of endothelin in rats. Hypertension 15, 729–733. doi: 10.1161/01.HYP.15.6.729
Mueller-Steiner, S., Zhou, Y., Arai, H., Roberson, E. D., Sun, B., Chen, J., et al. (2006). Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron 51, 703–714. doi: 10.1016/j.neuron.2006.07.027
Mukherjee, A., Song, E., Kihiko-Ehmann, M., Goodman, J. P. Jr., Pyrek, J. S., Estus, S., et al. (2000). Insulysin hydrolyzes amyloid β peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J. Neurosci. 20, 8745–8749.
Nalivaeva, N. N., Belyaev, N. D., Zhuravin, I. A., and Turner, A. J. (2012). The Alzheimer's amyloid-degrading peptidase, neprilysin: can we control it? Int. J. Alzheimers Dis. 2012, 383796. doi: 10.1155/2012/383796
Nanduri, J., Yuan, G., Kumar, G. K., Semenza, G. L., and Prabhakar, N. R. (2008). Transcriptional responses to intermittent hypoxia. Respir. Physiol. Neurobiol. 164, 277–281. doi: 10.1016/j.resp.2008.07.006
Narain, Y., Yip, A., Murphy, T., Brayne, C., Easton, D., Evans, J. G., et al. (2000). The ACE gene and Alzheimer's disease susceptibility. J. Med. Genet. 37, 695–697. doi: 10.1136/jmg.37.9.695
Nisemblat, Y., Belinson, H., Dolev, I., and Michaelson, D. M. (2008). Activation of the amyloid cascade by intracerebroventricular injection of the protease inhibitor phosphoramidon. Neurodegener. Dis. 5, 166–169. doi: 10.1159/000113692
Niwa, K., Carlson, G. A., and Iadecola, C. (2000). Exogenous Aβ1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J. Cereb. Blood Flow Metab. 20, 1659–1668. doi: 10.1097/00004647-200012000-00005
Niwa, K., Kazama, K., Younkin, L., Younkin, S. G., Carlson, G. A., and Iadecola, C. (2002a). Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am. J. Physiol. Heart Circ. Physiol. 283, H315–H323. doi: 10.1152/ajpheart.00022.2002
Niwa, K., Kazama, K., Younkin, S. G., Carlson, G. A., and Iadecola, C. (2002b). Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol. Dis. 9, 61–68. doi: 10.1006/nbdi.2001.0460
Niwa, K., Porter, V. A., Kazama, K., Cornfield, D., Carlson, G. A., and Iadecola, C. (2001). A β-peptides enhance vasoconstriction in cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 281, H2417–H2424.
Oba, R., Igarashi, A., Kamata, M., Nagata, K., Takano, S., and Nakagawa, H. (2005). The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide. Eur. J. Neurosci. 21, 733–740. doi: 10.1111/j.1460-9568.2005.03912.x
O'Caoimh, R., Healy, L., Gao, Y., Svendrovski, A., Kerins, D. M., Eustace, J., et al. (2014). Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with Alzheimer's disease. J. Alzheimers Dis. 40, 595–603. doi: 10.3233/JAD-131694
Ott, A., Breteler, M. M., de Bruyne, M. C., van Harskamp, F., Grobbee, D. E., and Hofman, A. (1997). Atrial fibrillation and dementia in a population-based study. The Rotterdam Study. Stroke 28, 316–321. doi: 10.1161/01.STR.28.2.316
Palmer, J., and Love, S. (2011). Endothelin receptor antagonists: potential in Alzheimer's disease. Pharmacol. Res. 63, 525–531. doi: 10.1016/j.phrs.2010.12.008
Palmer, J. C., Baig, S., Kehoe, P. G., and Love, S. (2009). Endothelin-converting enzyme-2 is increased in Alzheimer's disease and up-regulated by Aβ. Am. J. Pathol. 175, 262–270. doi: 10.2353/ajpath.2009.081054
Palmer, J. C., Barker, R., Kehoe, P. G., and Love, S. (2012). Endothelin-1 is elevated in Alzheimer's disease and upregulated by amyloid-β. J. Alzheimers Dis. 29, 853–861. doi: 10.3233/JAD-2012-111760
Palmer, J. C., Kehoe, P. G., and Love, S. (2010). Endothelin-converting enzyme-1 in Alzheimer's disease and vascular dementia. Neuropathol. Appl. Neurobiol. 36, 487–497. doi: 10.1111/j.1365-2990.2010.01084.x
Palmer, J. C., Tayler, H. M., and Love, S. (2013). Endothelin-converting enzyme-1 activity, endothelin-1 production, and free radical-dependent vasoconstriction in Alzheimer's disease. J. Alzheimers Dis. 36, 577–587. doi: 10.3233/JAD-130383
Paris, D., Humphrey, J., Quadros, A., Patel, N., Crescentini, R., Crawford, F., et al. (2003). Vasoactive effects of A β in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer's disease: role of inflammation. Neurol. Res. 25, 642–651. doi: 10.1179/016164103101201940
Park, L., Anrather, J., Forster, C., Kazama, K., Carlson, G. A., and Iadecola, C. (2004). Aβ-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J. Cereb. Blood Flow Metab. 24, 334–342. doi: 10.1097/01.WCB.0000105800.49957.1E
Pham, I., Uchida, T., Planes, C., Ware, L. B., Kaner, R., Matthay, M. A., et al. (2002). Hypoxia upregulates VEGF expression in alveolar epithelial cells in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L1133–L1142. doi: 10.1152/ajplung.00464.2001
Poirier, R., Wolfer, D. P., Welzl, H., Tracy, J., Galsworthy, M. J., Nitsch, R. M., et al. (2006). Neuronal neprilysin overexpression is associated with attenuation of Aβ-related spatial memory deficit. Neurobiol. Dis. 24, 475–483. doi: 10.1016/j.nbd.2006.08.003
Powell, F. L. (2009). Adaptation to chronic hypoxia involves immune cell invasion and increased expression of inflammatory cytokines in rat carotid body. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L156–L157. doi: 10.1152/ajplung.90597.2008
Preston, S. D., Steart, P. V., Wilkinson, A., Nicoll, J. A., and Weller, R. O. (2003). Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid β from the human brain. Neuropathol. Appl. Neurobiol. 29, 106–117. doi: 10.1046/j.1365-2990.2003.00424.x
Price, D. L., Tanzi, R. E., Borchelt, D. R., and Sisodia, S. S. (1998). Alzheimer's disease: genetic studies and transgenic models. Annu. Rev. Genet. 32, 461–493. doi: 10.1146/annurev.genet.32.1.461
Revesz, T., Ghiso, J., Lashley, T., Plant, G., Rostagno, A., Frangione, B., et al. (2003). Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J. Neuropathol. Exp. Neurol. 62, 885–898.
Revesz, T., Holton, J. L., Lashley, T., Plant, G., Rostagno, A., Ghiso, J., et al. (2002). Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 12, 343–357. doi: 10.1111/j.1750-3639.2002.tb00449.x
Rigat, B., Hubert, C., Alhenc-Gelas, F., Cambien, F., Corvol, P., and Soubrier, F. (1990). An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Invest. 86, 1343–1346. doi: 10.1172/JCI114844
Rossi, G. P., Sacchetto, A., Cesari, M., and Pessina, A. C. (1999). Interactions between endothelin-1 and the renin-angiotensin-aldosterone system. Cardiovasc. Res. 43, 300–307. doi: 10.1016/S0008-6363(99)00110-8
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerriere, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi: 10.1038/ng1718
Ruitenberg, A., den Heijer, T., Bakker, S. L., van Swieten, J. C., Koudstaal, P. J., Hofman, A., et al. (2005). Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann. Neurol. 57, 789–794. doi: 10.1002/ana.20493
Russo, R., Borghi, R., Markesbery, W., Tabaton, M., and Piccini, A. (2005). Neprylisin decreases uniformly in Alzheimer's disease and in normal aging. FEBS Lett. 579, 6027–6030. doi: 10.1016/j.febslet.2005.09.054
Saido, T. C. (2013). Metabolism of amyloid β peptide and pathogenesis of Alzheimer's disease. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 89, 321–339. doi: 10.2183/pjab.89.321
Scheuner, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., et al. (1996). Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 2, 864–870. doi: 10.1038/nm0896-864
Schuff, N., Matsumoto, S., Kmiecik, J., Studholme, C., Du, A., Ezekiel, F., et al. (2009). Cerebral blood flow in ischemic vascular dementia and Alzheimer's disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimers Dement. 5, 454–462. doi: 10.1016/j.jalz.2009.04.1233
Sharp, P. F., Smith, F. W., Gemmell, H. G., Lyall, D., Evans, N. T., Gvozdanovic, D., et al. (1986). Technetium-99m HM-PAO stereoisomers as potential agents for imaging regional cerebral blood flow: human volunteer studies. J. Nucl. Med. 27, 171–177.
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/JCI10498
Shweiki, D., Neeman, M., Itin, A., and Keshet, E. (1995). Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: implications for tumor angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 92, 768–772. doi: 10.1073/pnas.92.3.768
Spencer, B., Marr, R. A., Gindi, R., Potkar, R., Michael, S., Adame, A., et al. (2011). Peripheral delivery of a CNS targeted, metalo-protease reduces aβ toxicity in a mouse model of Alzheimer's disease. PLoS ONE 6:e16575. doi: 10.1371/journal.pone.0016575
Spencer, B., Marr, R. A., Rockenstein, E., Crews, L., Adame, A., Potkar, R., et al. (2008). Long-term neprilysin gene transfer is associated with reduced levels of intracellular Aβ and behavioral improvement in APP transgenic mice. BMC Neurosci. 9:109. doi: 10.1186/1471-2202-9-109
Stockley, J. H., Ravid, R., and O'Neill, C. (2006). Altered β-secretase enzyme kinetics and levels of both BACE1 and BACE2 in the Alzheimer's disease brain. FEBS Lett. 580, 6550–6560. doi: 10.1016/j.febslet.2006.10.076
Sun, X., He, G., Qing, H., Zhou, W., Dobie, F., Cai, F., et al. (2006). Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 18727–18732. doi: 10.1073/pnas.0606298103
Sun, X., Wiesner, B., Lorenz, D., Papsdorf, G., Pankow, K., Wang, P., et al. (2008). Interaction of angiotensin-converting enzyme (ACE) with membrane-bound carboxypeptidase M (CPM) - a new function of ACE. Biol. Chem. 389, 1477–1485. doi: 10.1515/BC.2008.168
Sun, Y., Jin, K., Mao, X. O., Zhu, Y., and Greenberg, D. A. (2001). Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc. Natl. Acad. Sci. U.S.A. 98, 15306–15311. doi: 10.1073/pnas.251466698
Sundelof, J., Sundstrom, J., Hansson, O., Eriksdotter-Jonhagen, M., Giedraitis, V., Larsson, A., et al. (2010). Higher cathepsin B levels in plasma in Alzheimer's disease compared to healthy controls. J. Alzheimers Dis. 22, 1223–1230. doi: 10.3233/JAD-2010-101023
Takanashi, M., Miyauchi, T., Kakinuma, Y., Goto, K., and Yamaguchi, I. (2004). Establishment of hypoxia inducible factor-1α overexpressing cells that produce endothelin-1. J. Cardiovasc. Pharmacol. 44(Suppl. 1), S268–S273. doi: 10.1097/01.fjc.0000166282.01034.8f
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Terao, C., Bayoumi, N., McKenzie, C. A., Zelenika, D., Muro, S., Mishima, M., et al. (2013). Quantitative variation in plasma angiotensin-I converting enzyme activity shows allelic heterogeneity in the ABO blood group locus. Ann. Hum. Genet. 77, 465–471. doi: 10.1111/ahg.12034
Thornton-Wells, T. A., Moore, J. H., Martin, E. R., Pericak-Vance, M. A., and Haines, J. L. (2008). Confronting complexity in late-onset Alzheimer disease: application of two-stage analysis approach addressing heterogeneity and epistasis. Genet. Epidemiol. 32, 187–203. doi: 10.1002/gepi.20294
Tian, M., Zhu, D., Xie, W., and Shi, J. (2012). Central angiotensin II-induced Alzheimer-like tau phosphorylation in normal rat brains. FEBS Lett. 586, 3737–3745. doi: 10.1016/j.febslet.2012.09.004
Tong, X. K., Nicolakakis, N., Kocharyan, A., and Hamel, E. (2005). Vascular remodeling versus amyloid β-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J. Neurosci. 25, 11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005
Toropygin, I. Y., Kugaevskaya, E. V., Mirgorodskaya, O. A., Elisseeva, Y. E., Kozmin, Y. P., Popov, I. A., et al. (2008). The N-domain of angiotensin-converting enzyme specifically hydrolyzes the Arg-5-His-6 bond of Alzheimer's Aβ-(1-16) peptide and its isoAsp-7 analogue with different efficiency as evidenced by quantitative matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 22, 231–239. doi: 10.1002/rcm.3357
Tucker, H. M., Kihiko, M., Caldwell, J. N., Wright, S., Kawarabayashi, T., Price, D., et al. (2000). The plasmin system is induced by and degrades amyloid-β aggregates. J. Neurosci. 20, 3937–3946.
Turner, A. J., and Hooper, N. M. (2002). The angiotensin-converting enzyme gene family: genomics and pharmacology. Trends Pharmacol. Sci. 23, 177–183. doi: 10.1016/S0165-6147(00)01994-5
Tyler, S. J., Dawbarn, D., Wilcock, G. K., and Allen, S. J. (2002). α- and β-secretase: profound changes in Alzheimer's disease. Biochem. Biophys. Res. Commun. 299, 373–376. doi: 10.1016/S0006-291X(02)02635-9
van Dorpe, J., Smeijers, L., Dewachter, I., Nuyens, D., Spittaels, K., Van Den Haute, C., et al. (2000). Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am. J. Pathol. 157, 1283–1298. doi: 10.1016/S0002-9440(10)64644-5
Vogelgesang, S., Cascorbi, I., Schroeder, E., Pahnke, J., Kroemer, H. K., Siegmund, W., et al. (2002). Deposition of Alzheimer's β-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 12, 535–541. doi: 10.1097/00008571-200210000-00005
Walker, J. R., Pacoma, R., Watson, J., Ou, W., Alves, J., Mason, D. E., et al. (2013). Enhanced proteolytic clearance of plasma Aβ by peripherally administered neprilysin does not result in reduced levels of brain Aβ in mice. J. Neurosci. 33, 2457–2464. doi: 10.1523/JNEUROSCI.3407-12.2013
Wang, D. S., Dickson, D. W., and Malter, J. S. (2006). β-Amyloid degradation and Alzheimer's disease. J. Biomed. Biotechnol. 2006:58406. doi: 10.1155/JBB/2006/58406
Wang, J., Ho, L., Chen, L., Zhao, Z., Zhao, W., Qian, X., et al. (2007). Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J. Clin. Invest. 117, 3393–3402. doi: 10.1172/JCI31547
Wang, R., Wang, S., Malter, J. S., and Wang, D. S. (2009a). Effects of HNE-modification induced by Aβ on neprilysin expression and activity in SH-SY5Y cells. J. Neurochem. 108, 1072–1082. doi: 10.1111/j.1471-4159.2008.05855.x
Wang, R., Wang, S., Malter, J. S., and Wang, D. S. (2009b). Effects of 4-hydroxy-nonenal and Amyloid-β on expression and activity of endothelin converting enzyme and insulin degrading enzyme in SH-SY5Y cells. J. Alzheimers Dis. 17, 489–501. doi: 10.3233/JAD-2009-1066
Weller, R. O., Boche, D., and Nicoll, J. A. (2009b). Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol. 118, 87–102. doi: 10.1007/s00401-009-0498-z
Weller, R. O., Massey, A., Kuo, Y. M., and Roher, A. E. (2000). Cerebral amyloid angiopathy: accumulation of A β in interstitial fluid drainage pathways in Alzheimer's disease. Ann. N.Y. Acad. Sci. 903, 110–117. doi: 10.1111/j.1749-6632.2000.tb06356.x
Weller, R. O., Massey, A., Newman, T. A., Hutchings, M., Kuo, Y. M., and Roher, A. E. (1998). Cerebral amyloid angiopathy: amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am. J. Pathol. 153, 725–733. doi: 10.1016/S0002-9440(10)65616-7
Weller, R. O., Preston, S. D., Subash, M., and Carare, R. O. (2009a). Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res. Ther. 1, 6. doi: 10.1186/alzrt6
Wenger, R. H. (2002). Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 16, 1151–1162. doi: 10.1096/fj.01-0944rev
Wosik, K., Cayrol, R., Dodelet-Devillers, A., Berthelet, F., Bernard, M., Moumdjian, R., et al. (2007). Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J. Neurosci. 27, 9032–9042. doi: 10.1523/JNEUROSCI.2088-07.2007
Yamashita, K., Discher, D. J., Hu, J., Bishopric, N. H., and Webster, K. A. (2001). Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J. Biol. Chem. 276, 12645–12653. doi: 10.1074/jbc.M011344200
Yamin, R., Zhao, C., O'Connor, P. B., McKee, A. C., and Abraham, C. R. (2009). Acyl peptide hydrolase degrades monomeric and oligomeric amyloid-β peptide. Mol. Neurodegener. 4, 33. doi: 10.1186/1750-1326-4-33
Yan, P., Hu, X., Song, H., Yin, K., Bateman, R. J., Cirrito, J. R., et al. (2006). Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J. Biol. Chem. 281, 24566–24574. doi: 10.1074/jbc.M602440200
Yanagisawa, H., Hammer, R. E., Richardson, J. A., Emoto, N., Williams, S. C., Takeda, S., et al. (2000). Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J. Clin. Invest. 105, 1373–1382. doi: 10.1172/JCI7447
Yanagisawa, M., Kurihara, H., Kimura, S., Goto, K., and Masaki, T. (1988). A novel peptide vasoconstrictor, endothelin, is produced by vascular endothelium and modulates smooth muscle Ca2+ channels. J. Hypertens. Suppl. 6, S188–S191. doi: 10.1097/00004872-198812040-00056
Yasojima, K., Akiyama, H., McGeer, E. G., and McGeer, P. L. (2001). Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci. Lett. 297, 97–100. doi: 10.1016/S0304-3940(00)01675-X
Yin, K. J., Cirrito, J. R., Yan, P., Hu, X., Xiao, Q., Pan, X., et al. (2006). Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-β peptide catabolism. J. Neurosci. 26, 10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006
Zetterberg, H., Mortberg, E., Song, L., Chang, L., Provuncher, G. K., Patel, P. P., et al. (2011). Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 6:e28263. doi: 10.1371/journal.pone.0028263
Zhang, X., Zhou, K., Wang, R., Cui, J., Lipton, S. A., Liao, F. F., et al. (2007). Hypoxia-inducible factor 1α (HIF-1α)-mediated hypoxia increases BACE1 expression and β-amyloid generation. J. Biol. Chem. 282, 10873–10880. doi: 10.1074/jbc.M608856200
Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O'Connor, T., et al. (2007). B-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J. Neurosci. 27, 3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007
Zheng, W. H., Bastianetto, S., Mennicken, F., Ma, W., and Kar, S. (2002). Amyloid β peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115, 201–211. doi: 10.1016/S0306-4522(02)00404-9
Zhiyou, C., Yong, Y., Shanquan, S., Jun, Z., Liangguo, H., Ling, Y., et al. (2009). Upregulation of BACE1 and β-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer's disease. Neurochem. Res. 34, 1226–1235. doi: 10.1007/s11064-008-9899-y
Zhu, D., Shi, J., Zhang, Y., Wang, B., Liu, W., Chen, Z., et al. (2011). Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. PLoS ONE 6:e16037. doi: 10.1371/journal.pone.0016037
Keywords: endothelin-1, angiotensin-converting enzyme, neprilysin, cerebral hypoperfusion, cerebral amyloid angiopathy, Alzheimer's disease
Citation: Miners JS, Palmer JC, Tayler H, Palmer LE, Ashby E, Kehoe PG and Love S (2014) Aβ degradation or cerebral perfusion? Divergent effects of multifunctional enzymes. Front. Aging Neurosci. 6:238. doi: 10.3389/fnagi.2014.00238
Received: 09 July 2014; Paper pending published: 28 July 2014;
Accepted: 20 August 2014; Published online: 11 September 2014.
Edited by:
Robert Marr, Rosalind Franklin University of Medicine and Science, USAReviewed by:
Anthony J. Turner, University of Leeds, UKElizabeth A. Eckman, Biomedical Research Institute of New Jersey, USA
Copyright © 2014 Miners, Palmer, Tayler, Palmer, Ashby, Kehoe and Love. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Scott Miners, Dementia Research Group, School of Clinical Sciences, Faculty of Medicine and Dentistry, University of Bristol, Level 1, Learning and Research, Southmead Hospital, Bristol BS10 5NB, UK e-mail:c2NvdHQubWluZXJzQGJyaXN0b2wuYWMudWs=