 Alan W. J. Morris
Alan W. J. Morris Roxana O. Carare
Roxana O. Carare Stefanie Schreiber
Stefanie Schreiber Cheryl A. Hawkes
Cheryl A. Hawkes- 1Faculty of Medicine, Clinical and Experimental Sciences, University of Southampton, Southampton, UK
- 2Department of Neurology, Otto-von-Guericke University, Magdeburg, Germany
- 3German Center for Neurodegenerative Diseases (DZNE), Helmholtz Association, Magdeburg, Germany
Cerebral amyloid angiopathy (CAA), the accumulation of β-amyloid (Aβ) peptides in the walls of cerebral blood vessels, is observed in the majority of Alzheimer’s disease (AD) brains and is thought to be due to a failure of the aging brain to clear Aβ. Perivascular drainage of Aβ along cerebrovascular basement membranes (CVBMs) is one of the mechanisms by which Aβ is removed from the brain. CVBMs are specialized sheets of extracellular matrix that provide structural and functional support for cerebral blood vessels. Changes in CVBM composition and structure are observed in the aged and AD brain and may contribute to the development and progression of CAA. This review summarizes the properties of the CVBM, its role in mediating clearance of interstitial fluids and solutes from the brain, and evidence supporting a role for CVBM in the etiology of CAA.
Alzheimer’s Disease and Cerebral Amyloid Angiopathy
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disease, currently estimated to affect 35.2 million people worldwide, making it the commonest form of dementia (Alzheimer’s Disease International, 2014). Life expectancy after diagnosis ranges from 4 to 6 years (Larson et al., 2004). The main risk factor is age, with the chance of developing AD doubling every 5 years after the age of 65 years (Gao et al., 1998). Other non-modifiable risk factors include mutations in genes encoding the amyloid precursor protein (APP) and presenilin-1 and -2 in familial AD cases (Tanzi, 2012) and possession of the apolipoprotein E (ApoE) ε4 allele in sporadic AD (Tanzi, 2012). Midlife hypertension, diabetes, and hypercholesterolemia are modifiable risk factors that increase the risk of developing AD in late life (Launer et al., 2000; Shah et al., 2012).
Alzheimer’s disease is characterized clinically by a slow and gradual deterioration in cognitive function as well as psychiatric symptoms and behavioral disturbances such as depression, delusions, and agitation (Burns, 1992; Bird and Miller, 2007). Neuropathologically, AD is associated with brain region-specific neuronal loss and synaptic degeneration (Bondareff et al., 1982; Whitehouse et al., 1982; Vogels et al., 1990; Aletrino et al., 1992), which begin in the entorhinal cortex and advance to the hippocampus and posterior temporal and parietal cortices (Begley et al., 1990; Braak and Braak, 1991). The end result of this degenerative neuronal loss is brain shrinkage and ventricular enlargement.
Two classical pathological features of AD are the accumulation of neurofibrillary tangles (NFTs) and senile plaques (Braak and Braak, 1991; Alafuzoff et al., 2008). NFTs are formed predominantly of ubiquitinated hyperphosphorylated tau (Esiri et al., 2004). Tau is a microtubule-associated protein that is present in mature neurons. In AD, tau becomes hyperphosphorylated and aggregates into NFTs of paired helical filaments (Iqbal et al., 2010). These aggregated bundles are seen pathologically as intraneuronal tangles in dystrophic neurites surrounding senile plaques and in the neuropil as neuropil threads (Braak et al., 1986; Grundke-Iqbal et al., 1986). Senile plaques are formed predominantly of amyloid β (Aβ). Amyloidogenic Aβ is produced from the sequential cleavage of APP by two proteases, β- and γ-secretase.
In addition to its deposition in the parenchyma, Aβ accumulates in the walls of cerebral blood vessels as cerebral amyloid angiopathy (CAA) (Miyakawa et al., 1982; Vinters, 1987; Weller et al., 2008). CAA is observed in 30–40% of non-demented elderly individuals and 60–95% of AD brains studied at autopsy (Vinters, 1987; Castano and Frangione, 1988; Haan et al., 1991; Jellinger, 2002). It affects predominantly leptomeningeal and cortical arteries but also occurs in cerebral capillaries (Vinters, 1987; Giannakopoulos et al., 1997; Roher et al., 2003). Unlike the parenchymal senile plaques that are composed principally of Aβ42 (Dickson et al., 1988), vascular Aβ deposits comprise predominantly Aβ40 (Suzuki et al., 1994). CAA is associated with capillary thinning and vessel tortuosity, inhibition of angiogenesis, and the death of pericytes, endothelial, and smooth muscle cells (Perlmutter et al., 1994; Miao et al., 2005; Haglund et al., 2006; Tian et al., 2006; Burger et al., 2009). Cerebral hypoperfusion, microhemorrhages, and cognitive impairment have also been shown to be associated with CAA severity (Natte et al., 2001; Pfeifer et al., 2002a,b; Shin et al., 2007; Chung et al., 2009).
The origin of the vascular Aβ deposits has been contested since the early twentieth century when it was first proposed that blood-borne Aβ was causing the phenomenon (Scholz, 1938). In the presiding half-century, other mechanisms were suggested, including the production of Aβ by vascular smooth muscle cells (VSMCs) (Wisniewski and Wegiel, 1994; Vinters et al., 1996; Wisniewski et al., 1996). However, the dichotomy between the specific localization of CAA to cerebral blood vessels and the ubiquitous expression of VSMCs throughout the body, suggested a brain-based origin of vascular Aβ. This hypothesis was supported by findings that transgenic mice producing mutant human Aβ only in the brain develop CAA (Herzig et al., 2004, 2006).
Based in part on electron microscopy studies showing the deposition of Aβ within the cerebrovascular basement membrane (CVBM) of the arterial tunica media (Wisniewski and Wegiel, 1994), Weller et al. (1998) hypothesized that parenchymal Aβ is normally cleared from the brain along CVBM and that CAA results from the failure of this system in the aged brain.
This review will summarize the properties of the CVBM, its role in mediating clearance of interstitial fluid (ISF) and solutes from the brain, and evidence supporting a role for CVBM in the etiology of CAA.
The Basement Membrane of the Brain
Basement membranes (BMs) are specialized extracellular matrices 50–100 nm in size when visualized by transmission electron microscopy (TEM) that cover the basal aspect of all endothelial and epithelial cells and surround fat, muscle, and Schwann cells (Carlson et al., 1978; Yurchenco and Schittny, 1990; Weber, 1992; Timpl, 1996; Miner, 1999; Candiello et al., 2007; McKee et al., 2009; Rasi et al., 2010) (Figure 1). They are formed from a variety of intracellularly produced proteins that are secreted into the surrounding extracellular matrix (ECM). BMs provide structural support to tissues, separate cells from connective tissue, and are important in modulating cellular signaling pathways (Yurchenco and Schittny, 1990; Timpl and Brown, 1996; Kalluri, 2003). The majority of BMs contain the following proteins; type IV collagen, laminins, nidogen, and the major heparan sulfate proteoglycan (HSPG) perlecan (Timpl and Brown, 1996; Halfter et al., 2013). Each of these core BM proteins has a subset of isoforms, there are 6 isoforms of type IV collagen, 2 nidogen isoforms, 2 major HSPGs (perlecan and agrin), and 16 different isoforms of laminin (Sorokin, 2010). Variation in the structural composition of the core BM proteins results in protein heterogeneity. For example, collagen IV comprises six distinct polypeptide α-chains [α1(IV)–α6(IV)], which assemble with high specificity to form three distinct isoforms, α1α1α2, α3α4α5, and α5α5α6 (Khoshnoodi et al., 2008). Laminins are composed of α, β, and γ polypeptide chain (Miner and Yurchenco, 2004). Currently 5 distinct α chains, 3 distinct β chains, and 3 distinct γ chains have been characterized and they assemble in various combinations to form 16 different laminin isoforms, which are classified as α1β1γ1, which is abbreviated to 111, depending on which chains make up the final molecule (Aumailley et al., 2005). Auxiliary BM proteins including, type XV collagen, type XVIII collagen, agrin, osteonectin [also known as secreted protein acidic and rich in cysteine (SPARC) or BM protein 40 (BM40)], BM90, and fibulin contribute to the structural and functional diversity of the BM across different tissues (Timpl, 1996).
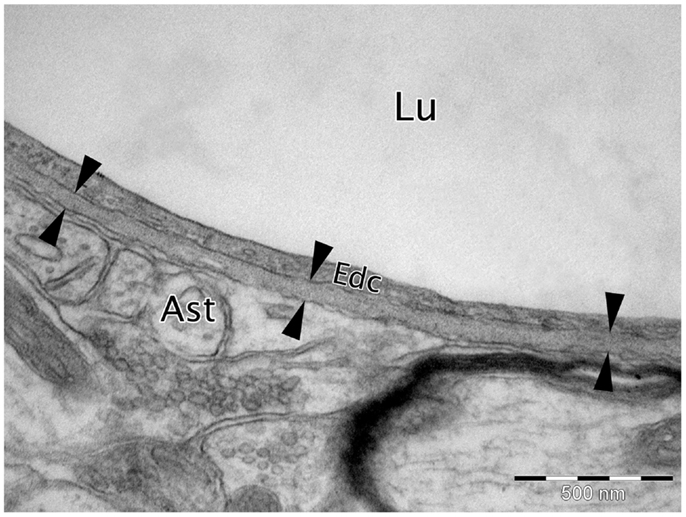
Figure 1. Micrograph of cortical Wistar Kyoto rat capillary. The cerebrovascular basement membrane is the electron dense area between the six arrow heads. Lu, capillary lumen; Edc, endothelial cell; Ast, astrocyte end foot. TEM, 50,000×, scale bar, 500 nm.
In the brain, BM proteins are expressed in the ECM of the parenchyma and within multiple layers of the cerebral blood vessels. Collagen IV (Khoshnoodi et al., 2008), laminin (Hagg et al., 1989; Hunter et al., 1992; Jucker et al., 1996a,b; Hallmann et al., 2005), nidogen -1 and -2 (Bader et al., 2005), HSPGs perlecan (Noonan and Hassell, 1993), fibronectin (Caffo et al., 2008), and agrin (Barber and Lieth, 1997), are the predominant BM constituents of the brain. However, there are variations in the composition of individual cerebral BMs, especially with regards to laminin. For example, the BM of the glia limitans contains both laminin α1 and α2 (Hallmann et al., 2005), whereas only laminin α1 is expressed in the meningeal epithelium and the astrocytic endothelium contains only laminin α2 (Hallmann et al., 2005). The different protein composition within each cerebral BM influences numerous cellular functions such as neural stem cell differentiation and migration, axon formation, apoptosis, myelination, and glial scar formation (Perris, 1997; Goldbrunner et al., 1999; Thyboll et al., 2002).
Cerebrovascular basement membranes play an important role in blood vessel development and health, formation and maintenance of the blood–brain barrier (BBB) and migration of peripheral cells including leukocytes into the brain (Sixt et al., 2001; Zlokovic, 2008; Wu et al., 2009). Endothelial cells, astrocytes and smooth muscle cells (Zlokovic, 2008) all contribute to the composition of the CVBM (Yousif et al., 2013) and this also receives contributions from pericytes (Dohgu et al., 2005; Bell et al., 2010) (Figure 2). The CVBM comprises collagen IV, laminin, perlecan, and nidogen as well as several minor glycoproteins (Hallmann et al., 2005). There is, however, variation in the expression of laminin isoforms within the CVBM of different vessel types (Sorokin, 2010). At the capillary level, the BM is fused between the endothelial cells and astrocyte end feet and contains laminin α2, α4, and α5 (Hallmann et al., 2005). Cerebral arterioles and arteries contain an endothelial BM (containing laminin α4 and/or α5), a BM that surrounds the VSMCs within tunica media (containing laminin α1 and α2) and an astrocytic BM (containing laminin α1 and α2) (Sixt et al., 2001). Veins comprise an endothelial BM (containing laminin α4 and a patchy distribution of laminin α5), a smooth muscle BM (containing laminin α1 and α2), and an astrocytic BM (containing laminin α2, α4, and α5) (Sixt et al., 2001; Yousif et al., 2013).
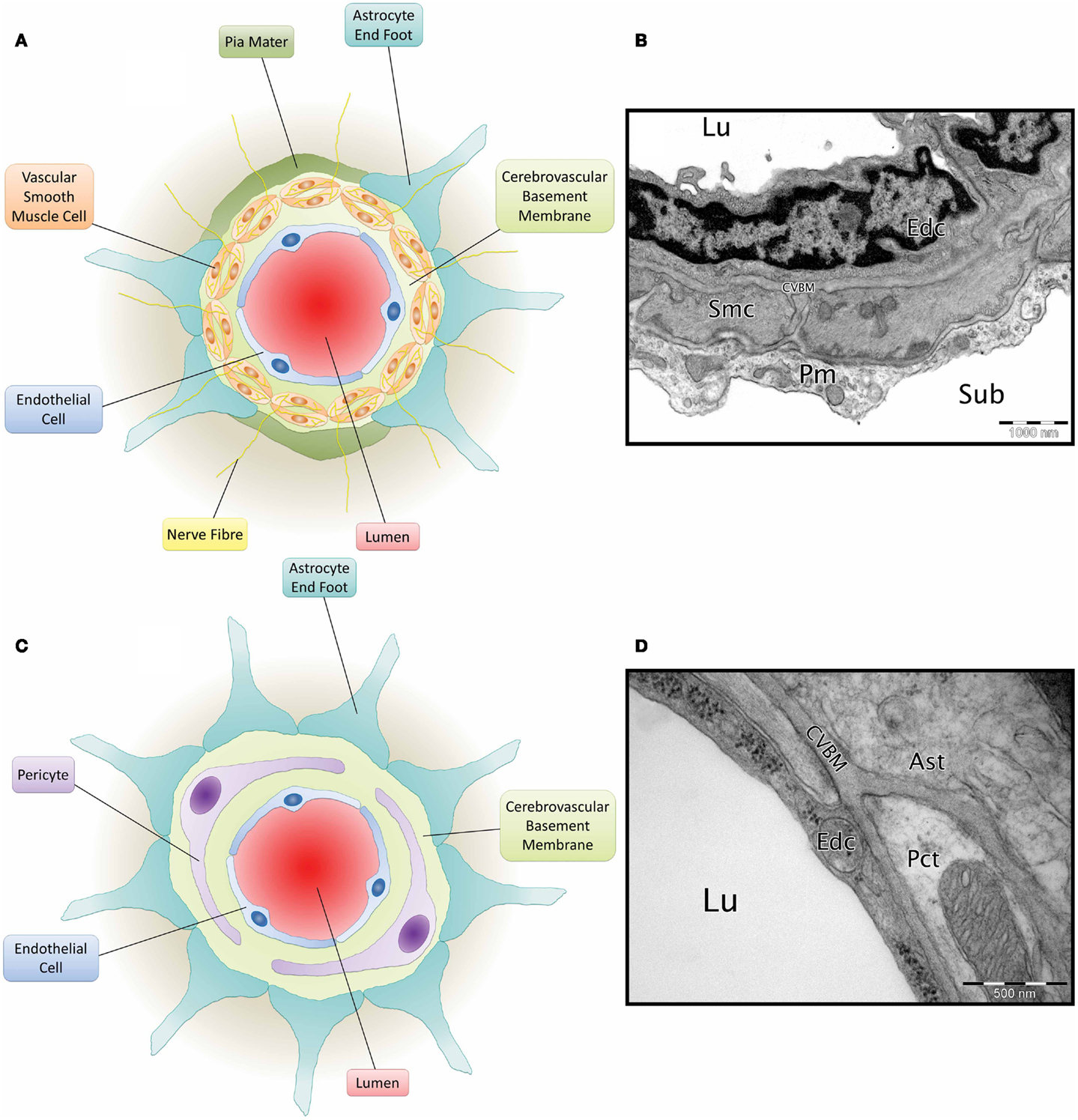
Figure 2. Composition of the neurovascular unit. (A) Diagram of leptomeningeal artery. Viewed from the center: arterial lumen (red), endothelial cell (blue), cerebrovascular basement membrane (light green), vascular smooth muscle cells (orange) have a cerebrovascular basement membrane running between them and are penetrated by nerve fibres (yellow), pia mater (dark green), and astrocyte end foot (teal). (B) Micrograph of C57BL/6 mouse cortical artery. Lu, lumen; Edc, endothelial cell; Smc, vascular smooth muscle cell; CVBM, cerebrovascular basement membrane; Pm, pia mater; sub, subarachnoid space. TEM, 25,000×, scale bar, 1000 nm, image credit: Matthew M. Sharp. (C) Diagram of a cerebral capillary. Viewed from the center; capillary lumen (red), endothelial cell (blue), cerebrovascular basement membrane (light green), pericyte (purple), and astrocyte end foot (teal). (D) Micrograph of Wistar Kyoto rat cerebral capillary. Lu, lumen; Edc, endothelial cell; Pct, pericyte; CVBM, cerebrovascular basement membrane; Ast, astrocyte end foot. TEM, 50,000×; scale bar, 500 nm.
Perivascular Elimination Pathway of ISF
The brain only accounts for around 2% of total body mass, but it is responsible for approximately a quarter of the body’s total oxygen and glucose consumption (Zlokovic, 2008). This metabolic output produces large quantities of waste neurotoxins that must be transported, along with other solutes, out of the brain for eventual excretion from the body. Unlike peripheral organs that communicate with the lymphatic system to remove ISF, the brain does not have conventional lymphatics. Solutes that cannot be eliminated across the BBB or into the CSF must therefore be removed via alternative routes.
Transmission electron microscopy analyses indicate continuity between the extracellular spaces of the brain and the endothelial capillary BM (Figure 3), suggesting a putative pathway by which parenchymal ISF may be cleared. Results from early drainage studies found that large molecular weight compounds injected into the brain parenchyma spread along perivascular spaces and could be observed in the walls of leptomeningeal arteries and in cervical lymph nodes (Szentistvanyi et al., 1984). Further, the clearance of small and large molecules occurred at similar rates (Cserr et al., 1981), suggesting that movement of ISF was dependent on bulk flow rather than diffusion, with an estimated clearance rate of 0.15–0.29 μL/min/g in the rat brain and 0.10–0.15 μL/min/g in the rabbit (Szentistvanyi et al., 1984; Abbott, 2004; Carare et al., 2008).
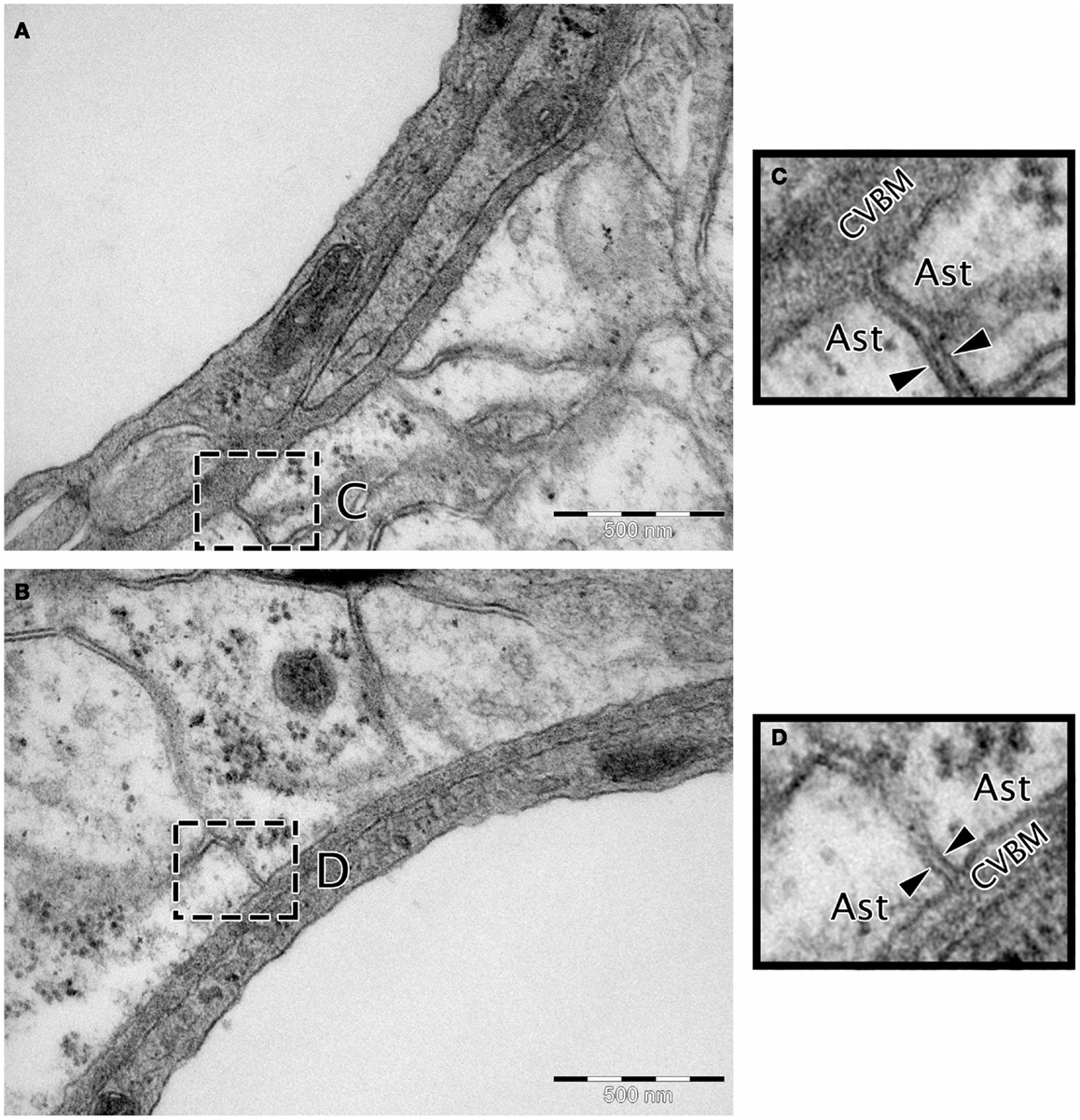
Figure 3. Micrographs showing the continuity between the extracellular matrix and the cerebrovascular basement membrane. (A,B) Wistar Kyoto rat cerebral capillary, TEM, 50,000× scale bar 500 nm. (C,D) Magnified insert highlighting continuity between the BM and extracellular matrix. The extracellular matrix is the electron dense area between the two arrow heads in (C,D). Ast, astrocyte end foot; CVBM, cerebrovascular basement membrane.
The role of the CVBM in the perivascular clearance of parenchymal solutes was shown experimentally by Carare et al., who found that fluorescent tracers were localized to the BM of both capillaries and arteries within 5 min of injection into the deep gray matter (Carare et al., 2008). In arterial walls, the tracers were specifically located in the BM of the tunica media, but not in the endothelial BM or the outer most BM between the arterial wall and brain parenchyma (Carare et al., 2008). At 3 h after injection, the tracer had cleared the CVBMs and tracers were only visible within perivascular macrophages (Carare et al., 2008). More recent studies using multiphoton imaging have confirmed the drainage of intracerebrally injected tracers along perivascular BMs (Arbel-Ornath et al., 2013). This pathway appears specific to small solutes, as large molecules, including immune complexes, Indian ink, and 20 nm fluorospheres are unable to enter the CVBM (Zhang et al., 1992; Barua et al., 2012; Teeling et al., 2012).
The motive force for the perivascular elimination pathway is still unclear. It has been hypothesized that cerebral blood flow plays a major role because clearance ceases immediately after cardiac arrest (Carare et al., 2008). Mathematical models have also provided insight into potential mechanisms by highlighting the potential for reflection wave driven flow (Schley et al., 2006; Wang and Olbricht, 2011). Each arterial pulsation is followed by a contrary (reflection) wave that passes along the vessel wall in the opposite direction to blood flow, which matches the observed pattern of distribution of solutes injected into the gray matter (Schley et al., 2006; Wang and Olbricht, 2011). This reflection wave could be aided by conformational changes in the CVBMs occurring during constriction and relaxation of the vessel wall in arteries (Carare et al., 2013) and of pericytes in capillaries. BM proteins may also adopt a valve-like configuration to prevent the ISF back flow (Schley et al., 2006).
Role of CVBM and Perivascular Drainage in CAA
Since the initial studies into perivascular elimination of solutes, it has been shown that soluble Aβ follows the same route after injection (Figure 4) (Hawkes et al., 2011, 2012, 2013). The pattern of distribution closely matches with that of human CAA, supporting the hypothesis that CAA outlines the natural route of elimination of cerebral Aβ. Multiple additional lines of evidence suggest an important role for CVBMs in the clearance of Aβ and its failure in CAA:
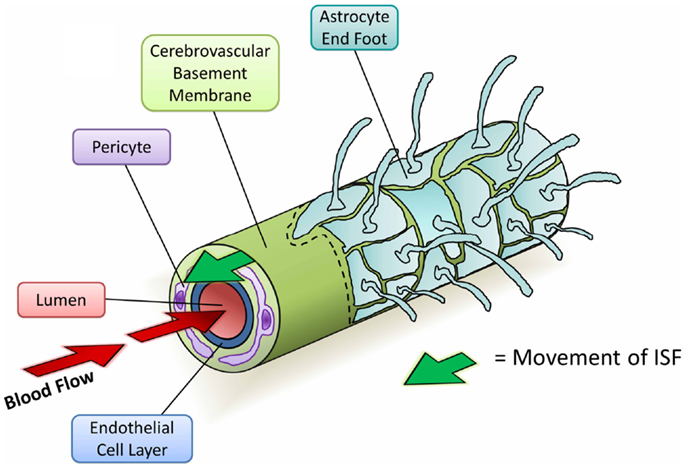
Figure 4. Diagram depicting the perivascular elimination of solutes along the cerebrovascular basement membrane of a cerebral capillary. The arterial pulsatile wave driving blood flow into the brain (red arrows) is followed by a refractory wave, which may drive the movement of interstitial fluid (ISF) out of the brain along the cerebrovascular basement membrane (green arrow). Conformational changes in cerebrovascular basement membranes during the refractory wave may provide a valve-like mechanism that promotes unidirectional flow of ISF. Viewed from the center: capillary lumen (red), endothelial cell layer (blue), cerebrovascular basement membrane (light green), pericyte (purple), and astrocyte end foot (teal) with a section removed beyond dotted line.
Morphologic Changes in the Cerebrovasculature and CVBM
Thickening, splitting, duplication, and the presence of abnormal inclusions in the CVBM have been reported in the brains of aged animals and humans and to a greater degree in the AD brain (Perlmutter, 1994; Kalaria, 1996; Farkas and Luiten, 2001; Shimizu et al., 2009). Thickening of the CVBM in both rodents and humans appears to be most predominant in brain areas that are susceptible to AD and CAA pathology (Zarow et al., 1997; Hawkes et al., 2013) and precedes CAA onset in TGF-β transgenic mice (Wyss-Coray et al., 2000). Further, increasing age is associated with arterial rigidity, elongation, and tortuosity, as well as a reduction in the vascular content of smooth muscle (Dahl, 1976). Such changes may diminish the force of arterial contraction and thereby diminish the driving force for perivascular clearance of Aβ.
Changes in the Biochemical Composition of the CVBM
Laminin, nidogens, and collagen IV inhibit the aggregation of Aβ and destabilize pre-formed fibrils of Aβ in vitro, while HSPG agrin and perlecan accelerate its aggregation (Aumailley and Krieg, 1996; Bronfman et al., 1996, 1998; Castillo et al., 1997; Cotman et al., 2000; Kiuchi et al., 2002a,b). Decreased amounts of collagen IV have been reported in small diameter vessels (<50 μm) from AD patients compared to aged-matched controls (Christov et al., 2008). Conversely, the levels of HSPGs were increased in AD brains (Berzin et al., 2000; Shimizu et al., 2009). Similar alterations have also been observed in the levels of collagen IV, laminin, nidogen 2, fibronectin, and perlecan in CAA-vulnerable brain regions of aged mice (Hawkes et al., 2013). These data suggest a change in the BM composition of the aged brain toward a pro-amyloidogenic environment. In addition, mice expressing the human ApoE ε4 allele show greater changes in CVBM expression over the lifecourse than mice expressing human Apo ε3 allele, in association with disrupted perivascular drainage of Aβ40 (Hawkes et al., 2012).
Relationship between Parenchymal and Vascular Aβ
Increased CAA severity has been noted in the brains of both mice and humans following anti-Aβ immunotherapy in association with decreased cerebral plaque load (Pfeifer et al., 2002b; Wilcock et al., 2004; Patton et al., 2006; Boche et al., 2008). These vascular Aβ deposits contained high levels of Aβ42, suggesting that following immunization, parenchymal Aβ became solubilized, drained along CVBMs, and became entrapped in the perivascular drainage pathways.
Collectively, these findings indicate that alterations in the composition, level of expression, or morphology of CVBMs may disturb perivascular drainage of Aβ from the parenchyma and initiate the deposition of Aβ within the walls of the cerebral vasculature, leading to a feed-forward mechanism of Aβ accumulation as CAA.
Conclusion
Increasing evidence supports a role for the cerebrovasculature in the development of AD. Preventative strategies targeting cardiovascular risk factors have been shown to be effective in reducing the incidence of AD (Guo et al., 1999; in’t Veld et al., 2001; Ohrui et al., 2004; Khachaturian et al., 2006; Haag et al., 2009a,b; Sink et al., 2009; Chuang et al., 2014), supporting the importance of maintaining cerebral vascular health across the lifespan in the prevention of disease. The CVBM plays an important role in cerebrovascular health, including mediating the clearance of Aβ. Therapeutic strategies targeted at improving and/or facilitating perivascular drainage of Aβ along the CVBM of the elderly brain may therefore represent a novel approach to the treatment of AD and CAA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Age UK and the BBSRC for funding.
References
Abbott, N. J. (2004). Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem. Int. 45, 545–552. doi:10.1016/j.neuint.2003.11.006
Alafuzoff, I., Arzberger, T., Al-Sarraj, S., Bodi, I., Bogdanovic, N., Braak, H., et al. (2008). Staging of neurofibrillary pathology in Alzheimer’s disease: a study of the BrainNet Europe Consortium. Brain Pathol. 18, 484–496. doi:10.1111/j.1750-3639.2008.00147.x
Aletrino, M. A., Vogels, O. J., Van Domburg, P. H., and Ten Donkelaar, H. J. (1992). Cell loss in the nucleus raphes dorsalis in Alzheimer’s disease. Neurobiol. Aging 13, 461–468. doi:10.1016/0197-4580(92)90073-7
Alzheimer’s Disease International. (2014). Dementia Statistics. Available from: http://www.alz.co.uk/research/statistics
Arbel-Ornath, M., Hudry, E., Eikermann-Haerter, K., Hou, S., Gregory, J. L., Zhao, L., et al. (2013). Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 126, 353–364. doi:10.1007/s00401-013-1145-2
Aumailley, M., Bruckner-Tuderman, L., Carter, W. G., Deutzmann, R., Edgar, D., Ekblom, P., et al. (2005). A simplified laminin nomenclature. Matrix Biol. 24, 326–332. doi:10.1016/j.matbio.2005.05.006
Aumailley, M., and Krieg, T. (1996). Laminins: a family of diverse multifunctional molecules of basement membranes. J. Invest. Dermatol. 106, 209–214. doi:10.1111/1523-1747.ep12340471
Bader, B. L., Smyth, N., Nedbal, S., Miosge, N., Baranowsky, A., Mokkapati, S., et al. (2005). Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol. Cell. Biol. 25, 6846–6856. doi:10.1128/MCB.25.15.6846-6856.2005
Barber, A. J., and Lieth, E. (1997). Agrin accumulates in the brain microvascular basal lamina during development of the blood-brain barrier 23. Dev. Dyn. 208, 62–74.
Barua, N. U., Bienemann, A. S., Hesketh, S., Wyatt, M. J., Castrique, E., Love, S., et al. (2012). Intrastriatal convection-enhanced delivery results in widespread perivascular distribution in a pre-clinical model. Fluids Barriers CNS 9, 2. doi:10.1186/2045-8118-9-2
Begley, D. J., Squires, L. K., Zlokovic, B. V., Mitrovic, D. M., Hughes, C. C., Revest, P. A., et al. (1990). Permeability of the blood-brain barrier to the immunosuppressive cyclic peptide cyclosporin A. J. Neurochem. 55, 1222–1230. doi:10.1111/j.1471-4159.1990.tb03128.x
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., LaRue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi:10.1016/j.neuron.2010.09.043
Berzin, T. M., Zipser, B. D., Rafii, M. S., Kuo-Leblanc, V., Yancopoulos, G. D., Glass, D. J., et al. (2000). Agrin and microvascular damage in Alzheimer’s disease. Neurobiol. Aging 21, 349–355. doi:10.1016/S0197-4580(00)00121-4
Boche, D., Zotova, E., Weller, R. O., Love, S., Neal, J. W., Pickering, R. M., et al. (2008). Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 131(Pt 12), 3299–3310. doi:10.1093/brain/awn261
Bondareff, W., Mountjoy, C. Q., and Roth, M. (1982). Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 32, 164–168. doi:10.1212/WNL.32.2.164
Braak, H., and Braak, E. (1991). Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi:10.1007/BF00308809
Braak, H., Braak, E., Grundke-Iqbal, I., and Iqbal, K. (1986). Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci. Lett. 65, 351–355. doi:10.1016/0304-3940(86)90288-0
Bronfman, F. C., Alvarez, A., Morgan, C., and Inestrosa, N. C. (1998). Laminin blocks the assembly of wild-type A beta and the Dutch variant peptide into Alzheimer’s fibrils. Amyloid 5, 16–23. doi:10.3109/13506129809007285
Bronfman, F. C., Garrido, J., Alvarez, A., Morgan, C., and Inestrosa, N. C. (1996). Laminin inhibits amyloid-beta-peptide fibrillation. Neurosci. Lett. 218, 201–203. doi:10.1016/S0304-3940(96)13147-5
Burger, S., Noack, M., Kirazov, L. P., Kirazov, E. P., Naydenov, C. L., Kouznetsova, E., et al. (2009). Vascular endothelial growth factor (VEGF) affects processing of amyloid precursor protein and beta-amyloidogenesis in brain slice cultures derived from transgenic Tg2576 mouse brain. Int. J. Dev. Neurosci. 27, 517–523. doi:10.1016/j.ijdevneu.2009.06.011
Burns, A. (1992). Psychiatric phenomena in dementia of the Alzheimer type. Int. Psychogeriatr. 4(Suppl. 1), 43–54. doi:10.1017/S1041610292001145
Caffo, M., Caruso, G., Galatioto, S., Meli, F., Cacciola, F., Germano, A., et al. (2008). Immunohistochemical study of the extracellular matrix proteins laminin, fibronectin and type IV collagen in secretory meningiomas. J. Clin. Neurosci. 15, 806–811. doi:10.1016/j.jocn.2007.05.029
Candiello, J., Balasubramani, M., Schreiber, E. M., Cole, G. J., Mayer, U., Halfter, W., et al. (2007). Biomechanical properties of native basement membranes. FEBS J. 274, 2897–2908. doi:10.1111/j.1742-4658.2007.05823.x
Carare, R. O., Bernardes-Silva, M., Newman, T. A., Page, A. M., Nicoll, J. A., Perry, V. H., et al. (2008). Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 34, 131–144. doi:10.1111/j.1365-2990.2007.00926.x
Carare, R. O., Hawkes, C. A., Jeffrey, M., Kalaria, R. N., and Weller, R. O. (2013). Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol. Appl. Neurobiol. 39, 593–611. doi:10.1111/nan.12042
Carlson, E. C., Brendel, K., Hjelle, J. T., and Meezan, E. (1978). Ultrastructural and biochemical analyses of isolated basement membranes from kidney glomeruli and tubules and brain and retinal microvessels. J. Ultrastruct. Res. 62, 26–53. doi:10.1016/S0022-5320(78)80028-8
Castano, E. M., and Frangione, B. (1988). Human amyloidosis, Alzheimer disease and related disorders. Lab. Invest. 58, 122–132.
Castillo, G. M., Ngo, C., Cummings, J., Wight, T. N., and Snow, A. D. (1997). Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem. 69, 2452–2465. doi:10.1046/j.1471-4159.1997.69062452.x
Christov, A., Ottman, J., Hamdheydari, L., and Grammas, P. (2008). Structural changes in Alzheimer’s disease brain microvessels. Curr. Alzheimer Res. 5, 392–395. doi:10.2174/156720508785132334
Chuang, Y. F., Breitner, J. C., Chiu, Y. L., Khachaturian, A., Hayden, K., Corcoran, C., et al. (2014). Use of diuretics is associated with reduced risk of Alzheimer’s disease: the Cache County Study. Neurobiol. Aging. doi:10.1016/j.neurobiolaging.2014.05.002
Chung, Y. A., JH, O., Kim, J. Y., Kim, K. J., and Ahn, K. J. (2009). Hypoperfusion and ischemia in cerebral amyloid angiopathy documented by 99mTc-ECD brain perfusion SPECT. J. Nucl. Med. 50, 1969–1974. doi:10.2967/jnumed.109.062315
Cotman, S. L., Halfter, W., and Cole, G. J. (2000). Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer’s disease brain. Mol. Cell. Neurosci. 15, 183–198. doi:10.1006/mcne.1999.0816
Cserr, H. F., Cooper, D. N., Suri, P. K., and Patlak, C. S. (1981). Efflux of radiolabeled polyethylene glycols and albumin from rat brain. Am. J. Physiol. 240, F319–F328.
Dahl, E. (1976). “Microscopic observations on cerebral arteries,” in The Cerebral Vessels Wall, eds J. Cervos-Navarro, E. Betz, F. Matakas, and R. Wüllenweber (New York: Raven Press), p. 15–21.
Dickson, D. W., Farlo, J., Davies, P., Crystal, H., Fuld, P., and Yen, S. H. (1988). Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am. J. Pathol. 132, 86–101.
Dohgu, S., Takata, F., Yamauchi, A., Nakagawa, S., Egawa, T., Naito, M., et al. (2005). Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production 1. Brain Res. 1038, 208–215. doi:10.1016/j.brainres.2005.01.027
Esiri, M., Lee, V. M.-Y., and Trojanowski, J. Q. (2004). The Neuropathology of Dementia, 2nd Edn. Cambridge: Cambridge University Press.
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 64, 575–611. doi:10.1016/S0301-0082(00)00068-X
Gao, S., Hendrie, H. C., Hall, K. S., and Hui, S. (1998). The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch. Gen. Psychiatry 55, 809–815. doi:10.1001/archpsyc.55.9.809
Giannakopoulos, P., Hof, P. R., Michel, J. P., Guimon, J., and Bouras, C. (1997). Cerebral cortex pathology in aging and Alzheimer’s disease: a quantitative survey of large hospital-based geriatric and psychiatric cohorts. Brain Res. Brain Res. Rev. 25, 217–245. doi:10.1016/S0165-0173(97)00023-4
Goldbrunner, R. H., Bernstein, J. J., and Tonn, J. C. (1999). Cell-extracellular matrix interaction in glioma invasion. Acta Neurochir. 141, 295–305, discussion 4-5. doi:10.1007/s007010050301
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi:10.1073/pnas.83.13.4913
Guo, Z., Fratiglioni, L., Zhu, L., Fastbom, J., Winblad, B., and Viitanen, M. (1999). Occurrence and progression of dementia in a community population aged 75 years and older: relationship of antihypertensive medication use. Arch. Neurol. 56, 991–996. doi:10.1001/archneur.56.8.991
Haag, M. D., Hofman, A., Koudstaal, P. J., Breteler, M. M., and Stricker, B. H. (2009a). Duration of antihypertensive drug use and risk of dementia: a prospective cohort study. Neurology 72, 1727–1734. doi:10.1212/01.wnl.0000345062.86148.3f
Haag, M. D., Hofman, A., Koudstaal, P. J., Stricker, B. H., and Breteler, M. M. (2009b). Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J. Neurol. Neurosurg. Psychiatr. 80, 13–17. doi:10.1136/jnnp.2008.150433
Haan, J., Hardy, J. A., and Roos, R. A. (1991). Hereditary cerebral hemorrhage with amyloidosis – Dutch type: its importance for Alzheimer research. Trends Neurosci. 14, 231–234. doi:10.1016/0166-2236(91)90120-J
Hagg, T., Muir, D., Engvall, E., Varon, S., and Manthorpe, M. (1989). Laminin-like antigen in rat CNS neurons: distribution and changes upon brain injury and nerve growth factor treatment. Neuron 3, 721–732. doi:10.1016/0896-6273(89)90241-9
Haglund, M., Passant, U., Sjobeck, M., Ghebremedhin, E., and Englund, E. (2006). Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int. J. Geriatr. Psychiatry 21, 681–687. doi:10.1002/gps.1550
Halfter, W., Candiello, J., Hu, H., Zhang, P., Schreiber, E., and Balasubramani, M. (2013). Protein composition and biomechanical properties of in vivo-derived basement membranes. Cell Adh. Migr. 7, 64–71. doi:10.4161/cam.22479
Hallmann, R., Horn, N., Selg, M., Wendler, O., Pausch, F., and Sorokin, L. M. (2005). Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 85, 979–1000. doi:10.1152/physrev.00014.2004
Hawkes, C. A., Gatherer, M., Sharp, M. M., Dorr, A., Yuen, H. M., Kalaria, R., et al. (2013). Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-beta from the mouse brain. Aging Cell 12, 224–236. doi:10.1111/acel.12045
Hawkes, C. A., Hartig, W., Kacza, J., Schliebs, R., Weller, R. O., Nicoll, J. A., et al. (2011). Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 121, 431–443. doi:10.1007/s00401-011-0801-7
Hawkes, C. A., Sullivan, P. M., Hands, S., Weller, R. O., Nicoll, J. A., and Carare, R. O. (2012). Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PLoS ONE 7:e41636. doi:10.1371/journal.pone.0041636
Herzig, M. C., Van Nostrand, W. E., and Jucker, M. (2006). Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol. 16, 40–54. doi:10.1111/j.1750-3639.2006.tb00560.x
Herzig, M. C., Winkler, D. T., Burgermeister, P., Pfeifer, M., Kohler, E., Schmidt, S. D., et al. (2004). Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 7, 954–960. doi:10.1038/nn1302
Hunter, D. D., Llinas, R., Ard, M., Merlie, J. P., and Sanes, J. R. (1992). Expression of s-laminin and laminin in the developing rat central nervous system. J. Comp. Neurol. 323, 238–251. doi:10.1002/cne.903230208
in’t Veld, B. A., Ruitenberg, A., Hofman, A., Stricker, B. H., and Breteler, M. M. (2001). Antihypertensive drugs and incidence of dementia: the Rotterdam Study. Neurobiol. Aging 22, 407–412. doi:10.1016/S0197-4580(00)00241-4
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi:10.2174/156720510793611592
Jellinger, K. A. (2002). Alzheimer disease and cerebrovascular pathology: an update. J. Neural Trans. 109, 813–836. doi:10.1007/s007020200068
Jucker, M., Tian, M., and Ingram, D. K. (1996a). Laminins in the adult and aged brain. Mol. Chem. Neuropathol. 28, 209–218. doi:10.1007/BF02815224
Jucker, M., Tian, M., Norton, D. D., Sherman, C., and Kusiak, J. W. (1996b). Laminin alpha 2 is a component of brain capillary basement membrane: reduced expression in dystrophic dy mice. Neuroscience 71, 1153–1161. doi:10.1016/0306-4522(95)00496-3
Kalaria, R. N. (1996). Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol. Ther. 72, 193–214. doi:10.1016/S0163-7258(96)00116-7
Kalluri, R. (2003). Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 3, 422–433. doi:10.1038/nrc1094
Khachaturian, A. S., Zandi, P. P., Lyketsos, C. G., Hayden, K. M., Skoog, I., Norton, M. C., et al. (2006). Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch. Neurol. 63, 686–692. doi:10.1001/archneur.63.5.noc60013
Khoshnoodi, J., Pedchenko, V., and Hudson, B. G. (2008). Mammalian collagen IV. Microsc. Res. Tech. 71, 357–370. doi:10.1002/jemt.20564
Kiuchi, Y., Isobe, Y., Fukushima, K., and Kimura, M. (2002a). Disassembly of amyloid beta-protein fibril by basement membrane components. Life Sci. 70, 2421–2431. doi:10.1016/S0024-3205(02)01501-1
Kiuchi, Y., Isobe, Y., and Fukushima, K. (2002b). Type IV collagen prevents amyloid beta-protein fibril formation. Life Sci. 70, 1555–1564. doi:10.1016/S0024-3205(01)01528-4
Larson, E. B., Shadlen, M. F., Wang, L., McCormick, W. C., Bowen, J. D., Teri, L., et al. (2004). Survival after initial diagnosis of Alzheimer disease. Ann. Intern. Med. 140, 501–509. doi:10.7326/0003-4819-140-7-200404060-00008
Launer, L. J., Ross, G. W., Petrovitch, H., Masaki, K., Foley, D., White, L. R., et al. (2000). Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol. Aging 21, 49–55. doi:10.1016/S0197-4580(00)00096-8
McKee, K. K., Capizzi, S., and Yurchenco, P. D. (2009). Scaffold-forming and adhesive contributions of synthetic laminin-binding proteins to basement membrane assembly. J. Biol. Chem. 284, 8984–8994. doi:10.1074/jbc.M809719200
Miao, J., Xu, F., Davis, J., Otte-Holler, I., Verbeek, M. M., and Van Nostrand, W. E. (2005). Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am. J. Pathol. 167, 505–515. doi:10.1016/S0002-9440(10)62993-8
Miner, J. H. (1999). Renal basement membrane components. Kidney Int. 56, 2016–2024. doi:10.1046/j.1523-1755.1999.00785.x
Miner, J. H., and Yurchenco, P. D. (2004). Laminin functions in tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 20, 255–284. doi:10.1146/annurev.cellbio.20.010403.094555
Miyakawa, T., Shimoji, A., Kuramoto, R., and Higuchi, Y. (1982). The relationship between senile plaques and cerebral blood vessels in Alzheimer’s disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Arch. B Cell Pathol. 40, 121–129. doi:10.1007/BF02932857
Natte, R., Maat-Schieman, M. L., Haan, J., Bornebroek, M., Roos, R. A., and van Duinen, S. G. (2001). Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann. Neurol. 50, 765–772. doi:10.1002/ana.10040
Noonan, D. M., and Hassell, J. R. (1993). Perlecan, the large low-density proteoglycan of basement membranes: structure and variant forms. Kidney Int. 43, 53–60. doi:10.1038/ki.1993.10
Ohrui, T., Matsui, T., Yamaya, M., Arai, H., Ebihara, S., Maruyama, M., et al. (2004). Angiotensin-converting enzyme inhibitors and incidence of Alzheimer’s disease in Japan. J. Am. Geriatr. Soc. 52, 649–650. doi:10.1111/j.1532-5415.2004.52178_7.x
Patton, R. L., Kalback, W. M., Esh, C. L., Kokjohn, T. A., Van Vickle, G. D., Luehrs, D. C., et al. (2006). Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am. J. Pathol. 169, 1048–1063.
Perlmutter, L. S. (1994). Microvascular pathology and vascular basement membrane components in Alzheimer’s disease. Mol. Neurobiol. 9, 33–40. doi:10.1007/BF02816103
Perlmutter, L. S., Myers, M. A., and Barron, E. (1994). Vascular basement membrane components and the lesions of Alzheimer’s disease: light and electron microscopic analyses. Microsc. Res. Tech. 28, 204–215. doi:10.1002/jemt.1070280305
Perris, R. (1997). The extracellular matrix in neural crest-cell migration. Trends Neurosci. 20, 23–31. doi:10.1016/S0166-2236(96)10063-1
Pfeifer, L. A., White, L. R., Ross, G. W., Petrovitch, H., and Launer, L. J. (2002a). Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58, 1629–1634. doi:10.1212/WNL.58.11.1629
Pfeifer, M., Boncristiano, S., Bondolfi, L., Stalder, A., Deller, T., Staufenbiel, M., et al. (2002b). Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 298, 1379. doi:10.1126/science.1078259
Rasi, K., Hurskainen, M., Kallio, M., Staven, S., Sormunen, R., Heape, A. M., et al. (2010). Lack of collagen XV impairs peripheral nerve maturation and, when combined with laminin-411 deficiency, leads to basement membrane abnormalities and sensorimotor dysfunction. J. Neurosci. 30, 14490–14501. doi:10.1523/JNEUROSCI.2644-10.2010
Roher, A. E., Kuo, Y. M., Esh, C., Knebel, C., Weiss, N., Kalback, W., et al. (2003). Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol. Med. 9, 112–122.
Schley, D., Carare-Nnadi, R., Please, C. P., Perry, V. H., and Weller, R. O. (2006). Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J. Theor. Biol. 238, 962–974. doi:10.1016/j.jtbi.2005.07.005
Scholz, W. (1938). Studien zur pathologie der Hirgefässe. II. Die drusige entartung der Hirnarterien und capillaren. Z. Ges. Neurol. Psychiatr. 162, 694–715. doi:10.1007/BF02890989
Shah, N. S., Vidal, J. S., Masaki, K., Petrovitch, H., Ross, G. W., Tilley, C., et al. (2012). Midlife blood pressure, plasma beta-amyloid, and the risk for Alzheimer disease: the Honolulu Asia Aging Study. Hypertension 59, 780–786. doi:10.1161/HYPERTENSIONAHA.111.178962
Shimizu, H., Ghazizadeh, M., Sato, S., Oguro, T., and Kawanami, O. (2009). Interaction between beta-amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer’s disease. J. Clin. Neurosci. 16, 277–282. doi:10.1016/j.jocn.2008.04.009
Shin, H. K., Jones, P. B., Garcia-Alloza, M., Borrelli, L., Greenberg, S. M., Bacskai, B. J., et al. (2007). Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 130(Pt 9), 2310–2319. doi:10.1093/brain/awm156
Sink, K. M., Leng, X., Williamson, J., Kritchevsky, S. B., Yaffe, K., Kuller, L., et al. (2009). Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension: results from the Cardiovascular Health Study. Arch. Intern. Med. 169, 1195–1202. doi:10.1001/archinternmed.2009.175
Sixt, M., Engelhardt, B., Pausch, F., Hallmann, R., Wendler, O., and Sorokin, L. M. (2001). Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J. Cell Biol. 153, 933–946. doi:10.1083/jcb.153.5.933
Sorokin, L. (2010). The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 10, 712–723. doi:10.1038/nri2852
Suzuki, N., Iwatsubo, T., Odaka, A., Ishibashi, Y., Kitada, C., and Ihara, Y. (1994). High tissue content of soluble beta 1-40 is linked to cerebral amyloid angiopathy. Am J Pathol 145(2):452–460.
Szentistvanyi, I., Patlak, C. S., Ellis, R. A., and Cserr, H. F. (1984). Drainage of interstitial fluid from different regions of rat brain. Am. J. Physiol. 246(6 Pt 2), F835–F844.
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006296. doi:10.1101/cshperspect.a006296
Teeling, J. L., Carare, R. O., Glennie, M. J., and Perry, V. H. (2012). Intracerebral immune complex formation induces inflammation in the brain that depends on Fc receptor interaction. Acta Neuropathol. 124, 479–490. doi:10.1007/s00401-012-0995-3
Thyboll, J., Kortesmaa, J., Cao, R., Soininen, R., Wang, L., Iivanainen, A., et al. (2002). Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol. Cell. Biol. 22, 1194–1202. doi:10.1128/MCB.22.4.1194-1202.2002
Tian, J., Shi, J., Smallman, R., Iwatsubo, T., and Mann, D. M. (2006). Relationships in Alzheimer’s disease between the extent of Abeta deposition in cerebral blood vessel walls, as cerebral amyloid angiopathy, and the amount of cerebrovascular smooth muscle cells and collagen. Neuropathol. Appl. Neurobiol. 32, 332–340. doi:10.1111/j.1365-2990.2006.00732.x
Timpl, R. (1996). Macromolecular organization of basement membranes. Curr. Opin. Cell Biol. 8, 618–624. doi:10.1016/S0955-0674(96)80102-5
Timpl, R., and Brown, J. C. (1996). Supramolecular assembly of basement membranes. Bioessays 18, 123–132. doi:10.1002/bies.950180208
Vinters, H. V. (1987). Cerebral amyloid angiopathy. A critical review. Stroke 18, 311–324. doi:10.1161/01.STR.18.2.311
Vinters, H. V., Wang, Z. Z., and Secor, D. L. (1996). Brain parenchymal and microvascular amyloid in Alzheimer’s disease. Brain Pathol. 6, 179–195. doi:10.1111/j.1750-3639.1996.tb00799.x
Vogels, O. J., Broere, C. A., ter Laak, H. J., ten Donkelaar, H. J., Nieuwenhuys, R., and Schulte, B. P. (1990). Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol. Aging 11, 3–13. doi:10.1016/0197-4580(90)90056-6
Wang, P., and Olbricht, W. L. (2011). Fluid mechanics in the perivascular space. J. Theor. Biol. 274, 52–57. doi:10.1016/j.jtbi.2011.01.014
Weller, R. O., Massey, A., Newman, T. A., Hutchings, M., Kuo, Y. M., and Roher, A. E. (1998). Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am. J. Pathol. 153, 725–733. doi:10.1016/S0002-9440(10)65616-7
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. doi:10.1111/j.1750-3639.2008.00133.x
Whitehouse, P. J., Price, D. L., Struble, R. G., Clark, A. W., Coyle, J. T., and Delon, M. R. (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215, 1237–1239. doi:10.1126/science.7058341
Wilcock, D. M., Rojiani, A., Rosenthal, A., Subbarao, S., Freeman, M. J., Gordon, M. N., et al. (2004). Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflammation 1, 24. doi:10.1186/1742-2094-1-24
Wisniewski, H. M., and Wegiel, J. (1994). Beta-amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol. 87, 233–241. doi:10.1007/BF00296738
Wisniewski, H. M., Wegiel, J., and Kotula, L. (1996). Review. David Oppenheimer Memorial Lecture 1995: some neuropathological aspects of Alzheimer’s disease and its relevance to other disciplines. Neuropathol. Appl. Neurobiol. 22, 3–11. doi:10.1111/j.1365-2990.1996.tb00839.x
Wu, C., Ivars, F., Anderson, P., Hallmann, R., Vestweber, D., Nilsson, P., et al. (2009). Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat. Med. 15, 519–527. doi:10.1038/nm.1957
Wyss-Coray, T., Lin, C., Sanan, D. A., Mucke, L., and Masliah, E. (2000). Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 156, 139–150. doi:10.1016/S0002-9440(10)64713-X
Yousif, L. F., Di Russo, J., and Sorokin, L. (2013). Laminin isoforms in endothelial and perivascular basement membranes. Cell Adh. Migr. 7, 101–110. doi:10.4161/cam.22680
Yurchenco, P. D., and Schittny, J. C. (1990). Molecular architecture of basement membranes. FASEB J. 4, 1577–1590.
Zarow, C., Barron, E., Chui, H. C., and Perlmutter, L. S. (1997). Vascular basement membrane pathology and Alzheimer’s disease. Ann. N. Y. Acad. Sci. 826, 147–160. doi:10.1111/j.1749-6632.1997.tb48467.x
Zhang, E. T., Richards, H. K., Kida, S., and Weller, R. O. (1992). Directional and compartmentalised drainage of interstitial fluid and cerebros- pinal fluid from the rat brain. Acta Neuropathol. 83, 233–239. doi:10.1007/BF00296784
Keywords: Alzheimer’s disease, cerebral amyloid angiopathy, perivascular drainage, basement membrane, β-amyloid
Citation: Morris AWJ, Carare RO, Schreiber S and Hawkes CA (2014) The cerebrovascular basement membrane: role in the clearance of β-amyloid and cerebral amyloid angiopathy. Front. Aging Neurosci. 6:251. doi: 10.3389/fnagi.2014.00251
Received: 29 July 2014; Accepted: 05 September 2014;
Published online: 19 September 2014.
Edited by:
Emil C. Toescu, Birmingham University, UKReviewed by:
Bogdan O. Popescu, Colentina Clinical Hospital, RomaniaChristoph Kleinschnitz, University of Würzburg, Germany
Copyright: © 2014 Morris, Carare, Schreiber and Hawkes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheryl A. Hawkes, Clinical and Experimental Sciences, University of Southampton, LD67 (MP806), Southampton General Hospital, Hampshire, Southampton SO16 6YD, UK e-mail:Yy5oYXdrZXNAc290b24uYWMudWs=