 Satoshi Saito
Satoshi Saito Masafumi Ihara
Masafumi Ihara- 1Department of Regenerative Medicine and Tissue Engineering, National Cerebral and Cardiovascular Center, Suita, Japan
- 2Department of Neurology, National Cerebral and Cardiovascular Center, Suita, Japan
Accumulating evidence has shown a strong relationship between Alzheimer’s disease (AD), cerebral amyloid angiopathy (CAA), and cerebrovascular disease. Cognitive impairment in AD patients can result from cortical microinfarcts associated with CAA, as well as the synaptic and neuronal disturbances caused by cerebral accumulations of β-amyloid (Aβ) and tau proteins. The pathophysiology of AD may lead to a toxic chain of events consisting of Aβ overproduction, impaired Aβ clearance, and brain ischemia. Insufficient removal of Aβ leads to development of CAA and plays a crucial role in sporadic AD cases, implicating promotion of Aβ clearance as an important therapeutic strategy. Aβ is mainly eliminated by three mechanisms: (1) enzymatic/glial degradation, (2) transcytotic delivery, and (3) perivascular drainage (3-“d” mechanisms). Enzymatic degradation may be facilitated by activation of Aβ-degrading enzymes such as neprilysin, angiotensin-converting enzyme, and insulin-degrading enzyme. Transcytotic delivery can be promoted by inhibition of the receptor for advanced glycation end products (RAGE), which mediates transcytotic influx of circulating Aβ into brain. Successful use of the RAGE inhibitor TTP488 in Phase II testing has led to a Phase III clinical trial for AD patients. The perivascular drainage system seems to be driven by motive force generated by cerebral arterial pulsations, suggesting that vasoactive drugs can facilitate Aβ clearance. One of the drugs promoting this system is cilostazol, a selective inhibitor of type 3 phosphodiesterase. The clearance of fluorescent soluble Aβ tracers was significantly enhanced in cilostazol-treated CAA model mice. Given that the balance between Aβ synthesis and clearance determines brain Aβ accumulation, and that Aβ is cleared by several pathways stated above, multi-drugs combination therapy could provide a mainstream cure for sporadic AD.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. AD is pathologically characterized by β-amyloid (Aβ) plaques within the brain parenchyma and Aβ accumulation in blood vessels (cerebral amyloid angiopathy; CAA), as well as by the formation of neurofibrillary tangles and neurodegeneration (Duyckaerts et al., 2009). AD was not previously thought to be closely linked to cerebrovascular disease (CVD), but accumulating lines of evidence have shown a strong relationship between AD and vascular dementia (VaD) (Fotuhi et al., 2009; Kalaria and Ihara, 2013). AD and CVD share common risk factors (Viswanathan et al., 2009; Kalaria et al., 2012), and treatment of vascular risk factors is associated with slower decline in cognitive impairments of AD patients (Deschaintre et al., 2009). The Nun study revealed that CVD plays an important role in determining the presence and severity of the clinical symptoms of AD (Snowdon et al., 1997). Aβ accumulation and other AD changes are also recognized in elderly patients without apparent dementia (Funato et al., 1998; Schneider et al., 2007), which implies a strong relationship between AD neuropathology and the aging processes. Many reports have described that a majority of sporadic dementia patients have a mixture of AD and CVD pathology (Neuropathology Group of Medical Research Council Cognitive Function and Aging Study (MRC CFAS), 2001; Toledo et al., 2013). Hemorrhage, infarctions, and vascular changes are not specific indicators for VaD.
Cerebral amyloid angiopathy often induces lobar hemorrhage and cortical microhemorrhage, which mainly affects the occipital lobe (Charidimou et al., 2012). In addition, imaging technology advances, including 7 T MRI, have identified numerous cortical microinfarcts (CMI), which have been attributed to CAA (Suter et al., 2002; van Veluw et al., 2013; Westover et al., 2013). Cognitive impairment in AD patients may result from hypoperfusion/ischemia and CMIs, as well as synaptic disturbance and neuronal loss caused by Aβ and tau accumulation (Okamoto et al., 2009; Launer et al., 2011; Smith et al., 2012). Small vessel injury is frequent in both AD and VaD. CAA was previously thought to be pathologically different from Binswanger disease, one of the common forms of VaD characterized by arteriolosclerosis and white matter change. However, Binswanger disease and CAA are now often regarded as part of the same spectrum disease; the former labeled type 1 and the latter type 2 small vessel disease (Pantoni, 2010). Both types of arteriopathies make dementia patients vulnerable to hemodynamic fluctuation through impairments in cerebral autoregulation and vascular reactivity (Tanoi et al., 2000; Pimentel-Coelho and Rivest, 2012). Consequently, hypoperfusion induces Aβ overproduction and elimination failure (Zlokovic, 2011; Carare et al., 2013; Elali et al., 2013). Brain ischemia and hypoxia modulates amyloid precursor protein (APP) cleavage enzymes such as β-secretase and γ-secretase, thereby resulting in increased Aβ production (Sun et al., 2006; Guglielmotto et al., 2009; Kitaguchi et al., 2009; Li et al., 2009). Excess Aβ contributes to the impairment of Aβ clearance and CAA (Joachim et al., 1989; Rovelet-Lecrux et al., 2006; Han et al., 2008). Aβ elimination failure could also result from arteriolosclerosis (Weller et al., 2009). Thus, dementia patients with a single simple etiology are scarcely seen, except for juvenile familial AD cases caused by mutations in the APP or presenilin genes, comprising <1% of AD cases (Campion et al., 1999).
In order to explore novel therapies in AD, we must consider the “AD malignant cycle” (Figure 1). In this scheme, cessation of Aβ overproduction is not sufficient to treat patients with sporadic AD, and important components of the cycle, brain ischemia, and CAA should also be noted. Insufficient Aβ clearance seems to be more crucial than Aβ overproduction in sporadic AD patients (Mawuenyega et al., 2010). Even in familial AD cases, the onset of dementia is often delayed until the fifth or sixth decade, suggesting that the aging-associated failure in clearance also plays a part in the pathogenesis of inherited types of the disease (Weller et al., 2008). Therefore, recent work has focused on the failure of Aβ elimination as the most important therapeutic targets and adopted a “neurovascular” approach as a strategy to tackle AD (Vardy et al., 2005; Deane et al., 2008; Carare et al., 2013).
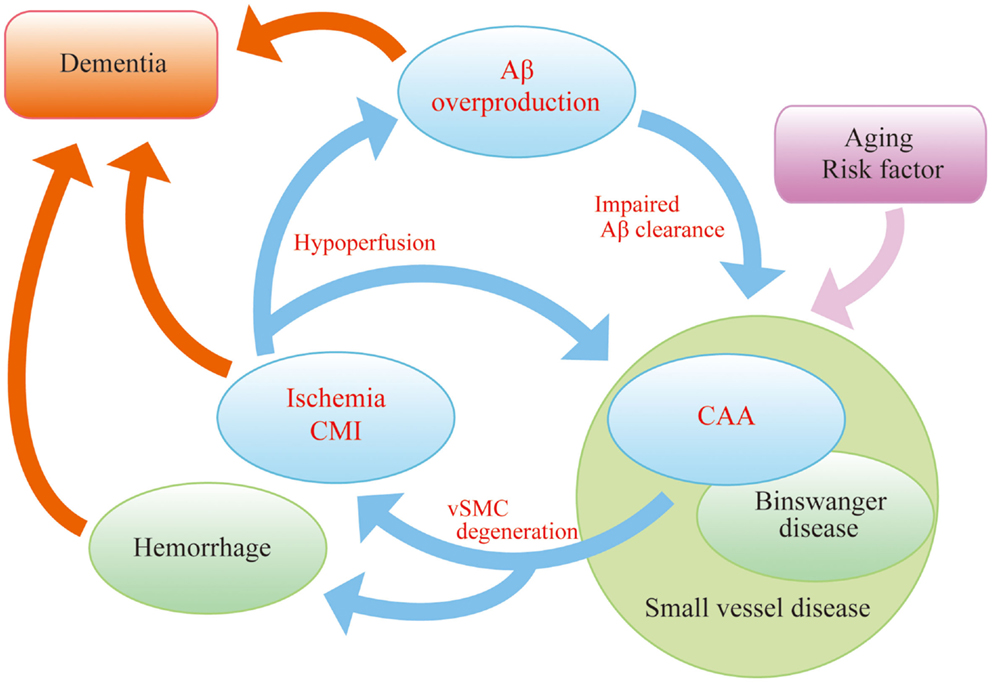
Figure 1. “AD malignant cycle”. Aβ overproduction impairs Aβ elimination leading to vascular smooth muscle cells (vSMC) degeneration, cerebral ischemia, and microinfarcts. Ischemia also induces Aβ overproduction. Such vicious circle consists of core pathology in sporadic AD. Note that cessation of Aβ overproduction is not sufficient to sever the cycle.
This review mainly focuses on the mechanisms of Aβ elimination and the drug development to facilitate Aβ clearance. The perivascular lymphatic drainage system, one of the Aβ clearance mechanisms, is closely associated with AD and CAA (Carare et al., 2013). In addition, the possibility of drugs enhancing perivascular drainage as well as future strategies for AD and CAA treatment will be reviewed.
Aβ Clearance: 3-d Mechanism
So far, several mechanisms of eliminating Aβ proteins have been identified, which fall into three main categories (3-“d,” Figure 2):
• Enzymatic/glial degradation
• Transcytotic delivery
• Perivascular drainage
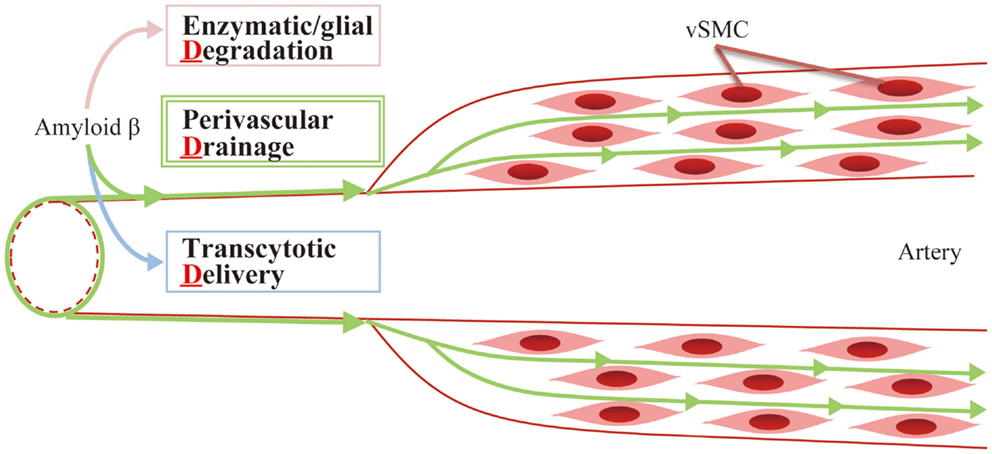
Figure 2. Aβ clearance: 3-d mechanism. Aβ is mainly eliminated by the following mechanisms: (1) enzymatic/glial degradation, (2) transcytotic delivery, and (3) perivascular drainage.
Enzymatic/Glial Degradation
Aβ catabolism is regulated by a series of degrading enzymes as well as glial cells, such as astrocytes and microglia, in the brain parenchyma (Vardy et al., 2005). Among them, neprilysin has received much attention (Iwata et al., 2000). Previous reports have described impaired Aβ degradation in neprilysin-deficient mice (Iwata et al., 2001) and amelioration of Aβ pathology in APP-transgenic mice, when injected with viral vector expressing human neprilysin gene (Marr et al., 2003; Iwata et al., 2004, 2013). Levels of neprilysin mRNA were found to be significantly lower in the hippocampus and middle temporal gyrus of AD brains compared with normal control patients (Yasojima et al., 2001). Decreased neprilysin activity was also associated with CAA (Miners et al., 2006). Thus, the up-regulation of cerebral neprilysin activity could potentially be targeted in the treatment of AD. Indeed, a somatostatin receptor agonist has recently been shown to increase neprilysin activity and decrease Aβ levels in senescence-accelerated mice (Sandoval et al., 2012). However, Meilandt et al. reported that an 11-fold greater neprilysin overexpression failed to reduce pathogenic Aβ oligomers and improve deficits in spatial learning and memory in AD model mice (Meilandt et al., 2009). It was also reported that cerebral Aβ concentration was too low to be degraded by neprilysin (Shibata et al., 2000). The affinity of neprilysin for its physiological substrates (e.g., enkephalins, tachykinins, atrial natriuretic peptide) is within the millimolar range (Hersh and Morihara, 1986), while the levels of Aβ in the brain are normally in the nanomolar range and up to 1 μM/kg even in APP-transgenic mice (Hsiao et al., 1996). Thus, only small concentrations of Aβ will likely bind to neprilysin under physiological and pathological conditions. Many issues should be solved to proceed to drug development of neprilysin activators.
Angiotensin-converting enzyme (ACE) is another Aβ-degrading agent. Captopril, a blood–brain barrier (BBB) penetrating ACE inhibitor, increases Aβ accumulation (Zou et al., 2007), and ACE overexpression in myelomonocytes reduces Aβ deposition in AD model mice (Bernstein et al., 2014). However, brain ACE deficient mice showed no significant alteration in endogenous Aβ levels (Eckman et al., 2006). In addition, two small studies assessing the clinical use of ACE inhibitors, found that they did not deteriorate dementia in AD and amnestic mild cognitive impairment (MCI) patients (Ohrui et al., 2004; Rozzini et al., 2006). Because of such conflicting findings, contributions of ACE to Aβ degradation in the brain per se remain ambiguous.
Insulin-degrading enzyme (IDE) is also known to have Aβ-degrading properties, and hyperinsulinemia in diabetes mellitus competitively inhibits Aβ degradation (Craft and Watson, 2004; Qiu and Folstein, 2006). Indeed, IDE deficient mice demonstrate increased cerebral accumulation of endogenous Aβ with hyperinsulinemia and glucose intolerance (Farris et al., 2003), and IDE overexpression ameliorates Aβ pathology (Leissring et al., 2003), suggesting a link between insulin metabolism and Aβ degradation. However, clinical evidence is still lacking and further studies on the association of IDE with AD pathogenesis may uncover potential treatment targets in AD. Some researchers have labeled AD “type 3 diabetes” (de la Monte and Wands, 2008). If hyperinsulinemia is related to resistance of neuronal cells to insulin, impaired insulin signaling in neurons is thought to lead to neuronal disturbances. A clinical trial assessing intranasal insulin therapy in the treatment of AD and amnestic MCI is anticipated to further elaborate on the relationship between AD and insulin signaling (Craft et al., 2012).
Transcytotic Delivery
The cerebral vasculature originates from large arteries, such as middle cerebral artery and the circle of Willis. These arteries branch into the leptomeningeal arteries, which travel on the surface of the brain in the subarachnoid space. Leptomeningeal arteries further branch into smaller arteries and arterioles consisting of three layers: tunica intima (endothelium), tunica media (smooth muscle cells), and tunica adventitia (mainly collagen fibers). Finally, the terminals of arterioles become capillaries. Capillary lumen and brain parenchyma are separated by the BBB, which prevents the passive exchange of solutes between blood and brain (Iadecola, 2004).
Lipoprotein receptor-related protein-1 (LRP-1), a multifunctional scavenger and signaling receptor, is expressed in neural cells and cerebral microvessels including capillaries, small venules, and arterioles (Wolf et al., 1992; Tooyama et al., 1995; Shibata et al., 2000). LRP-1 has received increasing attention as it mediates transport of Aβ out of the brain across the BBB (Bell and Zlokovic, 2009). Many reports have described the genetic linkage of LRP-1 with AD (Kang et al., 1997; Lambert et al., 1998; Wavrant-DeVrièze et al., 1999) and CAA (Christoforidis et al., 2005). Colocalization of LRP-1 with Aβ was pathologically recognized in senile plaques (Rebeck et al., 1993; Donahue et al., 2006), strengthening the linkage. The relationship is further supported by reduced LRP-1 staining in vessels both in AD patients (Shibata et al., 2000; Donahue et al., 2006) and CAA model mice carrying a vasculotropic Dutch/Iowa mutant form of APP gene (Deane et al., 2004).
Animal experiments have confirmed the importance of transcytosis in the regulation of cerebral Aβ levels. Five hours after microinjection of 125I-labeled Aβ1–40 into the caudate nucleus, 73.8% of labeled tracer had been found in blood across the BBB in young wild-type mice, while 125I-labeled Aβ1–40 in cerebrospinal fluid (CSF) measured 10.7%, and only 15.6% of the dose remained in the brain parenchyma (Shibata et al., 2000). These findings suggest that endothelial transcytosis by LRP-1 and others is probably one of the most prominent pathways in cerebral Aβ clearance, although this study might underestimate other clearance pathways as all the Aβ peptides found in blood are considered to derive from transcytotic delivery.
LRP-1 binds to Aβ directly (Deane et al., 2004), but also binds indirectly via its ligands including α2-macroglobulin, receptor-associated protein, and apolipoprotein E (ApoE) (Narita et al., 1997; Bu, 2009; Kanekiyo and Bu, 2009). ApoE is the main chaperone of Aβ in central nervous system (Holtzman et al., 2012; Zolezzi et al., 2014). To date, three isoforms of ApoE have been described (ε2, ε3, and ε4), and the ApoE ε4 variant is considered to be one of the most relevant risk factors for AD and CAA (Premkumar et al., 1996; Zolezzi et al., 2014). ApoE immunoreactivity is common in amyloid plaques, suggesting that ApoE interacts with Aβ directly in AD brains and could strongly influence the rate of Aβ removal (Namba et al., 1991; Holtzman et al., 2012). Several authors have proposed ApoE as therapeutic target for Aβ clearance (Cramer et al., 2012; Zolezzi and Inestrosa, 2014). Cramer et al. reported that bexarotene, a retinoid X receptor agonist, stimulated the ApoE-dependent Aβ clearance through the actions of liver X receptors and peroxisome proliferator-activated nuclear receptor gamma in AD model mice (Cramer et al., 2012). As a result, cognitive deficits improved with reduced burden of Aβ plaque. However, some conflicting reports have been also documented (Fitz et al., 2013; Price et al., 2013; Tesseur et al., 2013; Veeraraghavalu et al., 2013). Further analysis and experimentation should be performed.
Receptor for advanced glycation end products (RAGE), an immunoglobulin supergene family member, is also known to be a key molecule in Aβ transcytosis (Yan et al., 2012). Strong staining for RAGE has been reported in the vessels of AD patients (Yan et al., 1996; Donahue et al., 2006) and has been shown to mediate influx of circulating Aβ into brain across the BBB (Deane et al., 2003). In addition, RAGE contributes to Aβ-related synaptic dysfunction and microglial activation (Yan et al., 1996; Origlia et al., 2008, 2010). These findings suggest that RAGE could be a therapeutic target in AD and CAA. Indeed, a RAGE inhibitor ameliorated cerebral Aβ burden and normalized cognitive performance in APP-transgenic mice (Deane et al., 2012). The phase III 18 month clinical trial of the RAGE inhibitor TTP488 is being planned for mild to moderate AD patients (The U.S. National Institutes of Health, 2014); positive results in phase II testing have been reported (Burstein et al., 2014).
Perivascular Drainage
The central nervous system is devoid of conventional lymphatic vessels, unlike other organs containing networks of lymphatic vessels, which process various substances, such as wastes, fluid, proteins, and cells from tissues to lymph nodes. However, the lymphatic perivascular drainage system in the brain performs the main function assigned to systemic lymphatic vessels. Analysis of the lymphatic perivascular drainage system dates back as far as the nineteenth century, where it was shown that Indian ink injected into cisterna magna drained to the cervical lymph nodes (Schwalbe, 1869; Weller et al., 2010).
The detail of perivascular drainage system has been examined mainly by intracranial injection of various tracers, including 125I-labeled albumin (Bradbury et al., 1981; Szentistványi et al., 1984; Yamada et al., 1991), Indian ink (Zhang et al., 1992), and various fluorescent tracers (Carare et al., 2008). Recently, this drainage pathway was also confirmed by multi-photon imaging (Arbel-Ornath et al., 2013).
Fluorescent tracers, injected to the striatum, spread diffusely through the extracellular spaces of the brain parenchyma and enter the walls of blood vessels almost immediately. Confocal microscopy showed tracers colocalize with laminin in the basement membranes of capillary walls. Injected tracers were cleared from the basement membranes in the walls of capillaries and arteries, while some tracers were taken up by smooth muscle cells and perivascular macrophages (Zhang et al., 1992; Carare et al., 2008). Studies using radiolabeled tracers showed that drainage of interstitial fluid (ISF) and solutes continues along tunica media and the tunica adventitia of leptomeningeal and major cerebral arteries, through the base of the skull to the deep cervical lymph nodes (Szentistványi et al., 1984; Weller et al., 2010). Tissue soluble Aβ was detected by enzyme immunoassay in meningeal arteries and intracranial arteries but not in extracranial vessels (Shinkai et al., 1995). The clearance system leading to cervical lymph nodes was confirmed by subsequent injection into the inferior colliculus (Ball et al., 2010). Theoretical models have indicated that arterial pulsations could be the motive force behind ISF and solutes being driven centrifugally from the brain by reflection waves that follow each cardiac pulse wave (Schley et al., 2006).
This drainage route closely corresponds with the distribution of Aβ in the basement membranes of capillary and artery walls in CAA (Weller et al., 1998), which implies that the congestion of drainage pathway is associated with the pathogenesis of CAA. The fact that CAA was accelerated in the brains of immunized AD patients and that the CSF Aβ concentration was decreased both in AD and CAA patients may result from an impaired perivascular drainage pathway (Nicoll et al., 2004; Patton et al., 2006; Verbeek et al., 2009). Consistent with this, perivascular drainage of solutes is impaired in the aging mouse brain and in the presence of CAA (Hawkes et al., 2011). The fact that cerebral Aβ clearance was delayed after photothrombosis within individual vessels or middle cerebral artery occlusion (Garcia-Alloza et al., 2011), and after bilateral common carotid artery stenosis (Okamoto et al., 2012), further supports the notion that brain ischemia and impaired arterial pulsation could be an exacerbation factor of CAA. Consistent with the experimental data is the clinical finding that arterial stiffness, indicated by pulse wave velocity, has been associated with Aβ deposition in the brains of non-demented elderly adults (Hughes et al., 2013). Therefore, vasoactive drugs could have potential in the improvement of lymphatic congestion and facilitation of Aβ clearance in the brain.
Convincing Effects of Phosphodiesterase Inhibitor
Among varieties of vasoactive drugs, cilostazol, a selective inhibitor of type 3 phosphodiesterase (PDE), is likely to be a promising agent for AD and CAA (Figure 3). PDE3 can hydrolyze both cAMP and cGMP, while increasing cAMP level is a major pharmacological effect of cilostazol (Ikeda, 1999). PDE3 is widely expressed in central nervous system and up-regulated in Aβ-positive vessels, especially in vascular smooth muscle cells (vSMC) (Maki et al., 2014), suggesting the possibility that PDE3 inhibition could be therapeutic for CAA. Cilostazol possesses multiple effects, such as increasing pulse rate (Shinohara et al., 2010) and arterial elasticity (Han et al., 2013), prolonging pulse duration time (Aruna and Naidu, 2007), and dilating cerebral vessels (Tanaka et al., 1989; Birk et al., 2004a,b); such vasoactive actions may promote efficiency of perivascular drainage. In support of this, clearance of fluorescent soluble Aβ tracers is significantly enhanced in cilostazol-treated CAA model mice, thereby resulting in maintenance of vascular integrity, amelioration of Aβ deposits (Figure 4), and prevention of cognitive decline (Maki et al., 2014). Memory-preserving activity of cilostazol has been demonstrated in aged wild-type mice (Yanai et al., 2014) and a rat model of chronic cerebral hypoperfusion (Watanabe et al., 2006), suggesting that cilostazol could be a potential disease modifying therapy of AD and other dementing disorders.
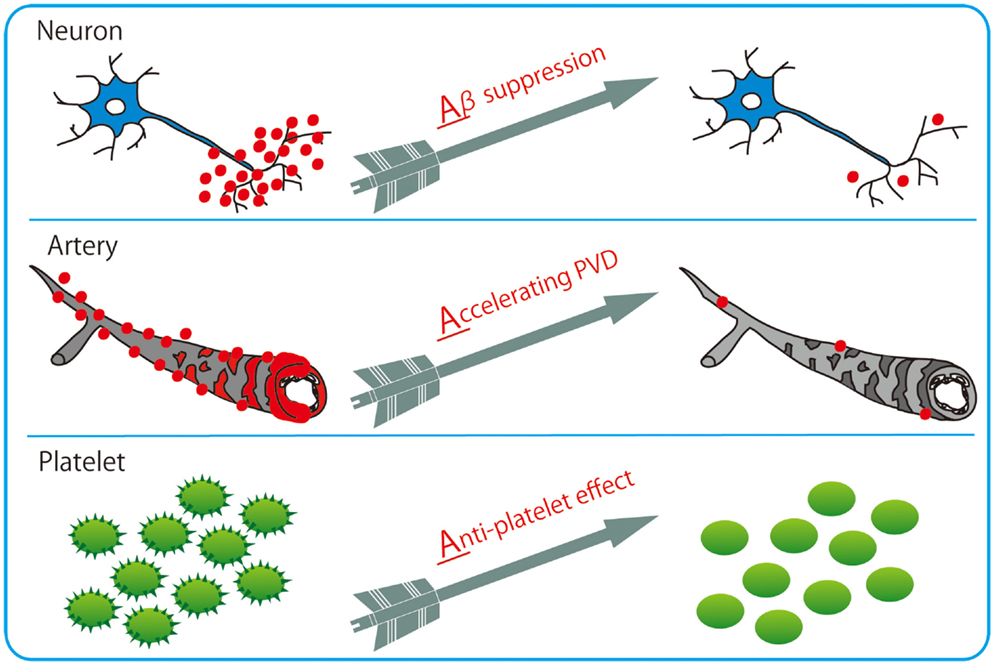
Figure 3. Cilostazol with 3 Arrows: triple effects toward potential resolution of dementia. Cilostazol, a selective inhibitor of PDE3, has pleiotropic capabilities of suppressing Aβ production in neurons, enhancing Aβ clearance through perivascular drainage system, and inhibiting platelet aggregation (anti-platelet effects).
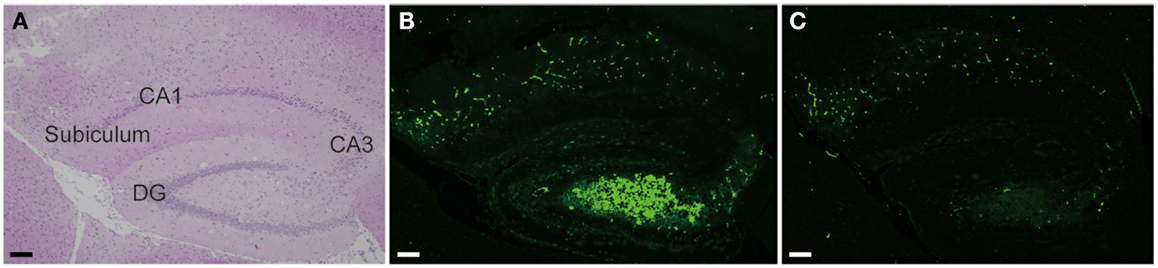
Figure 4. Cilostazol reduced Aβ deposition. Hippocampal images obtained from 17-month-old homozygous Tg-SwDI mice, a model of CAA, treated with vehicle (A,B) or cilostazol (C) for 15 months show that cilostazol treatment reduced levels of Aβ deposits in the hippocampus compared with vehicle treatment. Scale bars indicate 100 μm. (A) HE staining. (B,C) Thioflavin-S staining.
Recently, Nedergaad et al. suggested the “glymphatic pathway,” consisting of para-arterial CSF influx route, para-venous ISF efflux route, and convective bulk fluid flux (Iliff and Nedergaard, 2013; Nedergaard, 2013), as another clearance system in central nervous system. Aβ proteins may be cleared through this perivascular pathway, as well as the perivascular drainage system (Iliff et al., 2012), although the relationship to CAA pathogenesis remains to be clarified as Aβ does not accumulate in the venous system. Cerebral arterial pulsation with a vasoactive agent dobutamine drives perivascular CSF–ISF exchange (Iliff et al., 2013). Further investigation is required to determine whether other vasoactive drugs such as cilostazol could have a potential to facilitate paravascular clearance.
Many inhibitors of other PDE subtypes have been reported to produce cognitive enhancement (Reneerkens et al., 2009) and have been associated with neuronal cAMP signaling activation. Rolipram, a PDE4 inhibitor, reverses the decrease in cAMP regulatory element-binding protein (CREB) phosphorylation, which results in persistent improvement in synaptic function in AD model mice (Gong et al., 2004). Sildenafil, a PDE5 inhibitor, decreases Aβ levels in extracts of cerebral cortex and improves associative and spatial memory in AD model mice (Puzzo et al., 2009). Caffeine is a non-specific PDE inhibitor (Yoshimura, 2005), and its beneficial effects have been clarified in many clinical AD studies (Eskelinen et al., 2009; Eskelinen and Kivipelto, 2010). Caffeine stimulates cAMP-dependent protein kinase A signaling and increases CREB phosphorylation in AD model mice (Arendash et al., 2006; Zeitlin et al., 2011). Protein kinase A activation then suppresses the expression of Aβ-synthesizing enzymes such as β- and γ-secretase, leading to reduced Aβ production (Arendash et al., 2009). Cilostazol also reduces Aβ production in vitro (Lee et al., 2012, 2014; Maki et al., 2014), and suppresses Aβ-induced tauopathy and tau phosphorylation in vitro (Lee et al., 2012, 2014). However, as only a minor fraction of cilostazol passes through BBB (Akiyama et al., 1985), it remains to be elucidated whether these positive effects of cilostazol do occur in AD patients.
Cilostazol has a wide variety of pleiotropic effects capable of inducing neurogenesis (Lee et al., 2009; Tanaka et al., 2010), promoting oligodendrocyte precursor cell differentiation (Miyamoto et al., 2013), preventing lipid peroxidation (Hiramatsu et al., 2010; Kurtoglu et al., 2014), enhancing cholesterol efflux from macrophages (Nakaya et al., 2010), ameliorating insulin resistance (Wada et al., 2013), reducing inflammatory burden (Otsuki et al., 2001; Tsai et al., 2008; Hattori et al., 2009), and improving systemic lymphatic function by inducing proliferation and stabilization of lymphatic endothelial cells (Kimura et al., 2014). In a clinical setting, cilostazol is currently used as an anti-platelet drug (Gotoh et al., 2000; Shinohara et al., 2010), and may be used to prevent ischemic events in patients with CAA. Major manifestations of CAA include lobar hemorrhage and cortical microhemorrhage, as well as CMI. As most CAA patients are elderly (Zhang-Nunes et al., 2006), this necessitates the use of anti-platelet drugs with little risk of hemorrhage (Charidimou et al., 2012). The second Cilostazol Stroke Prevention Study (CSPS2) for patients with cerebral infarction showed that the hemorrhagic stroke was significantly less frequent in cilostazol treatment than with aspirin (Shinohara et al., 2010; Uchiyama et al., 2014). The prevention of cerebral hemorrhage may be explained by reproducible experimental evidence showing that cilostazol inhibits expression of matrix metalloproteinase-9 and protects vascular endothelial cells (Ishiguro et al., 2010; Hase et al., 2012; Kasahara et al., 2012). Endothelial protection with cilostazol mediates increase in nitric oxide, which dilates blood vessels (Oyama et al., 2011), leading to increased cerebral blood flow (Mochizuki et al., 2001; Matsumoto et al., 2011; Sakurai et al., 2013). These results suggest that cilostazol could be suitable for patients with both AD and CVD, the most common type of dementia in the elderly.
Favorable effects have already been described in observational clinical studies, which demonstrated the efficacy of cilostazol in patients with MCI (Taguchi et al., 2013), donepezil-treated patients with clinically probable AD (Arai and Takahashi, 2009; Ihara et al., 2014), and AD with CVD (Sakurai et al., 2013). Randomized placebo-controlled clinical trials are being planned for patients with MCI.
Future Strategy for AD and CAA Treatment
Aging inevitably increases the amount of Aβ burden in the brain, implying a strong relationship between impaired Aβ metabolism and age (Funato et al., 1998). Since heterogeneity and multimorbidity are common in the elderly (Barnett et al., 2012), dementia likely originates from a combination of different pathological substrates. As the population ages, the distribution of AD shifts to older ages in developed countries (Hebert et al., 2013), resulting in an increasing number of demented patients with numerous complicated etiologies. Given that the balance between Aβ synthesis and clearance determines brain Aβ accumulation, and that Aβ is cleared by several pathways stated above, multi-drugs combination therapy would likely be necessary for sporadic AD with complicated etiologies. Combination therapy has already been applied to various diseases, such as hypertension, diabetes mellitus, and malignant tumors. The ultimate goal will be to develop a sovereign remedy of AD, and we hope that the recent rapid advances in drug development will enable us to delay the onset or modify the progression of cognitive impairment with multi-targeting therapies. Further investigation from various viewpoints will thus be essential for the development of novel treatment for AD and CAA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akiyama, H., Kudo, S., and Shimizu, T. (1985). The absorption, distribution and excretion of a new antithrombotic and vasodilating agent, cilostazol, in rat, rabbit, dog and man. Arzneimittelforschung 35, 1124–1132.
Arai, H., and Takahashi, T. (2009). A combination therapy of donepezil and cilostazol for patients with moderate Alzheimer disease: pilot follow-up study. Am. J. Geriatr. Psychiatry 17, 353–354. doi:10.1097/JGP.0b013e31819431ea
Arbel-Ornath, M., Hudry, E., Eikermann-Haerter, K., Hou, S., Gregory, J. L., Zhao, L., et al. (2013). Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 126, 353–364. doi:10.1007/s00401-013-1145-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arendash, G. W., Mori, T., Cao, C., Mamcarz, M., Runfeldt, M., Dickson, A., et al. (2009). Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J. Alzheimers Dis. 17, 661–680. doi:10.3233/JAD-2009-1087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arendash, G. W., Schleif, W., Rezai-Zadeh, K., Jackson, E. K., Zacharia, L. C., Cracchiolo, J. R., et al. (2006). Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 142, 941–952. doi:10.1016/j.neuroscience.2006.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aruna, D., and Naidu, M. U. (2007). Pharmacodynamic interaction studies of Ginkgo biloba with cilostazol and clopidogrel in healthy human subjects. Br. J. Clin. Pharmacol. 63, 333–338. doi:10.1111/j.1365-2125.2006.02759.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ball, K. K., Cruz, N. F., Mrak, R. E., and Dienel, G. A. (2010). Trafficking of glucose, lactate, and amyloid-beta from the inferior colliculus through perivascular routes. J. Cereb. Blood Flow Metab. 30, 162–176. doi:10.1038/jcbfm.2009.206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barnett, K., Mercer, S. W., Norbury, M., Watt, G., Wyke, S., and Guthrie, B. (2012). Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet 380, 37–43. doi:10.1016/S0140-6736(12)60240-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118, 103–113. doi:10.1007/s00401-009-0522-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bernstein, K. E., Koronyo, Y., Salumbides, B. C., Sheyn, J., Pelissier, L., Lopes, D. H., et al. (2014). Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Invest. 124, 1000–1012. doi:10.1172/JCI66541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Birk, S., Edvinsson, L., Olesen, J., and Kruuse, C. (2004a). Analysis of the effects of phosphodiesterase type 3 and 4 inhibitors in cerebral arteries. Eur. J. Pharmacol. 489, 93–100. doi:10.1016/j.ejphar.2004.02.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Birk, S., Kruuse, C., Petersen, K. A., Jonassen, O., Tfelt-Hansen, P., and Olesen, J. (2004b). The phosphodiesterase 3 inhibitor cilostazol dilates large cerebral arteries in humans without affecting regional cerebral blood flow. J. Cereb. Blood Flow Metab. 24, 1352–1358. doi:10.1097/01.WCB.0000143536.22131.D7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bradbury, M. W., Cserr, H. F., and Westrop, R. J. (1981). Drainage of cerebral interstitial fluid into deep cervical lymph of the rabbit. Am. J. Physiol. 240, F329–F336.
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi:10.1038/nrn2620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burstein, A. H., Grimes, I., Galasko, D. R., Aisen, P. S., Sabbagh, M., and Mjalli, A. M. (2014). Effect of TTP488 in patients with mild to moderate Alzheimer’s disease. BMC Neurol. 14:12. doi:10.1186/1471-2377-14-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Campion, D., Dumanchin, C., Hannequin, D., Dubois, B., Belliard, S., Puel, M., et al. (1999). Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 65, 664–670. doi:10.1086/302553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carare, R. O., Bernardes-Silva, M., Newman, T. A., Page, A. M., Nicoll, J. A., Perry, V. H., et al. (2008). Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 34, 131–144. doi:10.1111/j.1365-2990.2007.00926.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carare, R. O., Hawkes, C. A., Jeffrey, M., Kalaria, R. N., and Weller, R. O. (2013). Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol. Appl. Neurobiol. 39, 593–611. doi:10.1111/nan.12042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Charidimou, A., Gang, Q., and Werring, D. J. (2012). Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J. Neurol. Neurosurg. Psychiatr. 83, 124–137. doi:10.1136/jnnp-2011-301308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christoforidis, M., Schober, R., and Krohn, K. (2005). Genetic-morphologic association study: association between the low density lipoprotein-receptor related protein (LRP) and cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 31, 11–19. doi:10.1111/j.1365-2990.2004.00614.x
Craft, S., Baker, L. D., Montine, T. J., Minoshima, S., Watson, G. S., Claxton, A., et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69, 29–38. doi:10.1001/archneurol.2011.233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Craft, S., and Watson, G. S. (2004). Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 3, 169–178. doi:10.1016/S1474-4422(04)00681-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi:10.1126/science.1217697
de la Monte, S. M., and Wands, J. R. (2008). Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2, 1101–1113. doi:10.1177/193229680800200619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deane, R., Du Yan, S., Submamaryan, R. K., Larue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi:10.1038/nm890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deane, R., Sagare, A., and Zlokovic, B. V. (2008). The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr. Pharm. Des. 14, 1601–1605. doi:10.2174/138161208784705487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deane, R., Singh, I., Sagare, A. P., Bell, R. D., Ross, N. T., Larue, B., et al. (2012). A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest. 122, 1377–1392. doi:10.1172/JCI58642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43, 333–344. doi:10.1016/j.neuron.2004.07.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deschaintre, Y., Richard, F., Leys, D., and Pasquier, F. (2009). Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology 73, 674–680. doi:10.1212/WNL.0b013e3181b59bf3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A., Silverberg, G. D., Miller, M. C., et al. (2006). RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 112, 405–415. doi:10.1007/s00401-006-0115-3
Duyckaerts, C., Delatour, B., and Potier, M. C. (2009). Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36. doi:10.1007/s00401-009-0532-1
Eckman, E. A., Adams, S. K., Troendle, F. J., Stodola, B. A., Kahn, M. A., Fauq, A. H., et al. (2006). Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J. Biol. Chem. 281, 30471–30478. doi:10.1074/jbc.M605827200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elali, A., Thériault, P., Préfontaine, P., and Rivest, S. (2013). Mild chronic cerebral hypoperfusion induces neurovascular dysfunction, triggering peripheral beta-amyloid brain entry and aggregation. Acta Neuropathol Commun 1, 75. doi:10.1186/2051-5960-1-75
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eskelinen, M. H., and Kivipelto, M. (2010). Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimers Dis. 20(Suppl. 1), S167–S174. doi:10.3233/JAD-2010-1404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eskelinen, M. H., Ngandu, T., Tuomilehto, J., Soininen, H., and Kivipelto, M. (2009). Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J. Alzheimers Dis. 16, 85–91. doi:10.3233/JAD-2009-0920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100, 4162–4167. doi:10.1073/pnas.0230450100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fitz, N. F., Cronican, A. A., Lefterov, I., and Koldamova, R. (2013). Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924–c. doi:10.1126/science.1235809
Fotuhi, M., Hachinski, V., and Whitehouse, P. J. (2009). Changing perspectives regarding late-life dementia. Nat. Rev. Neurol. 5, 649–658. doi:10.1038/nrneurol.2009.175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Funato, H., Yoshimura, M., Kusui, K., Tamaoka, A., Ishikawa, K., Ohkoshi, N., et al. (1998). Quantitation of amyloid beta-protein (A beta) in the cortex during aging and in Alzheimer’s disease. Am. J. Pathol. 152, 1633–1640.
Garcia-Alloza, M., Gregory, J., Kuchibhotla, K. V., Fine, S., Wei, Y., Ayata, C., et al. (2011). Cerebrovascular lesions induce transient β-amyloid deposition. Brain 134, 3697–3707. doi:10.1093/brain/awr300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gong, B., Vitolo, O. V., Trinchese, F., Liu, S., Shelanski, M., and Arancio, O. (2004). Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Invest. 114, 1624–1634. doi:10.1172/JCI22831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gotoh, F., Tohgi, H., Hirai, S., Terashi, A., Fukuuchi, Y., Otomo, E., et al. (2000). Cilostazol stroke prevention study: a placebo-controlled double-blind trial for secondary prevention of cerebral infarction. J. Stroke Cerebrovasc. Dis. 9, 147–157. doi:10.1053/jscd.2000.7216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guglielmotto, M., Aragno, M., Autelli, R., Giliberto, L., Novo, E., Colombatto, S., et al. (2009). The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J. Neurochem. 108, 1045–1056. doi:10.1111/j.1471-4159.2008.05858.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, B. H., Zhou, M. L., Abousaleh, F., Brendza, R. P., Dietrich, H. H., Koenigsknecht-Talboo, J., et al. (2008). Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J. Neurosci. 28, 13542–13550. doi:10.1523/JNEUROSCI.4686-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, S. W., Lee, S. S., Kim, S. H., Lee, J. H., Kim, G. S., Kim, O. J., et al. (2013). Effect of cilostazol in acute lacunar infarction based on pulsatility index of transcranial Doppler (ECLIPse): a multicenter, randomized, double-blind, placebo-controlled trial. Eur. Neurol. 69, 33–40. doi:10.1159/000338247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hase, Y., Okamoto, Y., Fujita, Y., Kitamura, A., Nakabayashi, H., Ito, H., et al. (2012). Cilostazol, a phosphodiesterase inhibitor, prevents no-reflow and hemorrhage in mice with focal cerebral ischemia. Exp. Neurol. 233, 523–533. doi:10.1016/j.expneurol.2011.11.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hattori, Y., Suzuki, K., Tomizawa, A., Hirama, N., Okayasu, T., Hattori, S., et al. (2009). Cilostazol inhibits cytokine-induced nuclear factor-kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Cardiovasc. Res. 81, 133–139. doi:10.1093/cvr/cvn226
Hawkes, C. A., Hartig, W., Kacza, J., Schliebs, R., Weller, R. O., Nicoll, J. A., et al. (2011). Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 121, 431–443. doi:10.1007/s00401-011-0801-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hebert, L. E., Weuve, J., Scherr, P. A., and Evans, D. A. (2013). Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783. doi:10.1212/WNL.0b013e31828726f5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hersh, L. B., and Morihara, K. (1986). Comparison of the subsite specificity of the mammalian neutral endopeptidase 24.11 (enkephalinase) to the bacterial neutral endopeptidase thermolysin. J. Biol. Chem. 261, 6433–6437.
Hiramatsu, M., Takiguchi, O., Nishiyama, A., and Mori, H. (2010). Cilostazol prevents amyloid beta peptide(25-35)-induced memory impairment and oxidative stress in mice. Br. J. Pharmacol. 161, 1899–1912. doi:10.1111/j.1476-5381.2010.01014.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holtzman, D. M., Herz, J., and Bu, G. (2012). Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2, a006312. doi:10.1101/cshperspect.a006312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. doi:10.1126/science.274.5284.99
Hughes, T. M., Kuller, L. H., Barinas-Mitchell, E. J., Mackey, R. H., Mcdade, E. M., Klunk, W. E., et al. (2013). Pulse wave velocity is associated with β-amyloid deposition in the brains of very elderly adults. Neurology 81, 1711–1718. doi:10.1212/01.wnl.0000435301.64776.37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi:10.1038/nrn1387
Ihara, M., Nishino, M., Taguchi, A., Yamamoto, Y., Hattori, Y., Saito, S., et al. (2014). Cilostazol add-on therapy in patients with mild dementia receiving donepezil: a retrospective study. PLoS ONE 9:e89516. doi:10.1371/journal.pone.0089516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ikeda, Y. (1999). Antiplatelet therapy using cilostazol, a specific PDE3 inhibitor. Thromb. Haemost. 82, 435–438.
Iliff, J. J., and Nedergaard, M. (2013). Is there a cerebral lymphatic system? Stroke 44, S93–S95. doi:10.1161/STROKEAHA.112.678698
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4, 147ra111. doi:10.1126/scitranslmed.3003748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iliff, J. J., Wang, M., Zeppenfeld, D. M., Venkataraman, A., Plog, B. A., Liao, Y., et al. (2013). Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 33, 18190–18199. doi:10.1523/JNEUROSCI.1592-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ishiguro, M., Mishiro, K., Fujiwara, Y., Chen, H., Izuta, H., Tsuruma, K., et al. (2010). Phosphodiesterase-III inhibitor prevents hemorrhagic transformation induced by focal cerebral ischemia in mice treated with tPA. PLoS ONE 5:e15178. doi:10.1371/journal.pone.0015178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Mizukami, H., Shirotani, K., Takaki, Y., Muramatsu, S., Lu, B., et al. (2004). Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain. J. Neurosci. 24, 991–998. doi:10.1523/JNEUROSCI.4792-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Sekiguchi, M., Hattori, Y., Takahashi, A., Asai, M., Ji, B., et al. (2013). Global brain delivery of neprilysin gene by intravascular administration of AAV vector in mice. Sci. Rep. 3, 1472. doi:10.1038/srep01472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550–1552. doi:10.1126/science.1059946
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., et al. (2000). Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150. doi:10.1038/72237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joachim, C. L., Mori, H., and Selkoe, D. J. (1989). Amyloid beta-protein deposition in tissues other than brain in Alzheimer’s disease. Nature 341, 226–230. doi:10.1038/341226a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalaria, R. N., Akinyemi, R., and Ihara, M. (2012). Does vascular pathology contribute to Alzheimer changes? J. Neurol. Sci. 322, 141–147. doi:10.1016/j.jns.2012.07.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalaria, R. N., and Ihara, M. (2013). Dementia: vascular and neurodegenerative pathways-will they meet? Nat. Rev. Neurol. 9, 487–488. doi:10.1038/nrneurol.2013.164
Kanekiyo, T., and Bu, G. (2009). Receptor-associated protein interacts with amyloid-beta peptide and promotes its cellular uptake. J. Biol. Chem. 284, 33352–33359. doi:10.1074/jbc.M109.015032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, D. E., Saitoh, T., Chen, X., Xia, Y., Masliah, E., Hansen, L. A., et al. (1997). Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer’s disease. Neurology 49, 56–61. doi:10.1212/WNL.49.1.56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kasahara, Y., Nakagomi, T., Matsuyama, T., Stern, D., and Taguchi, A. (2012). Cilostazol reduces the risk of hemorrhagic infarction after administration of tissue-type plasminogen activator in a murine stroke model. Stroke 43, 499–506. doi:10.1161/STROKEAHA.111.635417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kimura, T., Hamazaki, T. S., Sugaya, M., Fukuda, S., Chan, T., Tamura-Nakano, M., et al. (2014). Cilostazol improves lymphatic function by inducing proliferation and stabilization of lymphatic endothelial cells. J. Dermatol. Sci. 74, 150–158. doi:10.1016/j.jdermsci.2014.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kitaguchi, H., Tomimoto, H., Ihara, M., Shibata, M., Uemura, K., Kalaria, R. N., et al. (2009). Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. 1294, 202–210. doi:10.1016/j.brainres.2009.07.078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurtoglu, T., Basoglu, H., Ozkisacik, E. A., Cetin, N. K., Tataroglu, C., Yenisey, C., et al. (2014). Effects of cilostazol on oxidative stress, systemic cytokine release, and spinal cord injury in a rat model of transient aortic occlusion. Ann. Vasc. Surg. 28, 479–488. doi:10.1016/j.avsg.2013.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lambert, J. C., Wavrant-De Vrièze, F., Amouyel, P., and Chartier-Harlin, M. C. (1998). Association at LRP gene locus with sporadic late-onset Alzheimer’s disease. Lancet 351, 1787–1788. doi:10.1016/S0140-6736(05)78749-3
Launer, L. J., Hughes, T. M., and White, L. R. (2011). Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia aging study autopsy study. Ann. Neurol. 70, 774–780. doi:10.1002/ana.22520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. R., Park, S. Y., Kim, H. Y., Shin, H. K., Lee, W. S., Rhim, B. Y., et al. (2012). Protection by cilostazol against amyloid-β(1-40)-induced suppression of viability and neurite elongation through activation of CK2α in HT22 mouse hippocampal cells. J. Neurosci. Res. 90, 1566–1576. doi:10.1002/jnr.23037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. R., Shin, H. K., Park, S. Y., Kim, H. Y., Lee, W. S., Rhim, B. Y., et al. (2014). Attenuation of β-amyloid-induced tauopathy via activation of CK2α/SIRT1: targeting for cilostazol. J. Neurosci. Res. 92, 206–217. doi:10.1002/jnr.23310
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J. H., Shin, H. K., Park, S. Y., Kim, C. D., Lee, W. S., and Hong, K. W. (2009). Cilostazol preserves CA1 hippocampus and enhances generation of immature neuroblasts in dentate gyrus after transient forebrain ischemia in rats. Exp. Neurol. 215, 87–94. doi:10.1016/j.expneurol.2008.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. doi:10.1016/S0896-6273(03)00787-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, L., Zhang, X., Yang, D., Luo, G., Chen, S., and Le, W. (2009). Hypoxia increases Abeta generation by altering beta- and gamma-cleavage of APP. Neurobiol. Aging 30, 1091–1098. doi:10.1016/j.neurobiolaging.2007.10.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maki, T., Okamoto, Y., Carare, R., Hase, Y., Hattori, Y., Hawkes, C., et al. (2014). Phosphodiesterase III inhibitor promotes drainage of cerebrovascular β-amyloid. Ann Clin Transl Neurol. 1, 519–533. doi:10.1002/acn3.79
Marr, R. A., Rockenstein, E., Mukherjee, A., Kindy, M. S., Hersh, L. B., Gage, F. H., et al. (2003). Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23, 1992–1996.
Matsumoto, S., Shimodozono, M., Miyata, R., and Kawahira, K. (2011). Effect of cilostazol administration on cerebral hemodynamics and rehabilitation outcomes in poststroke patients. Int. J. Neurosci. 121, 271–278. doi:10.3109/00207454.2010.551431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774. doi:10.1126/science.1197623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meilandt, W. J., Cisse, M., Ho, K., Wu, T., Esposito, L. A., Scearce-Levie, K., et al. (2009). Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J. Neurosci. 29, 1977–1986. doi:10.1523/JNEUROSCI.2984-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miners, J. S., Van Helmond, Z., Chalmers, K., Wilcock, G., Love, S., and Kehoe, P. G. (2006). Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 65, 1012–1021. doi:10.1097/01.jnen.0000240463.87886.9a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyamoto, N., Pham, L. D., Hayakawa, K., Matsuzaki, T., Seo, J. H., Magnain, C., et al. (2013). Age-related decline in oligodendrogenesis retards white matter repair in mice. Stroke 44, 2573–2578. doi:10.1161/STROKEAHA.113.001530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mochizuki, Y., Oishi, M., and Mizutani, T. (2001). Effects of cilostazol on cerebral blood flow, P300, and serum lipid levels in the chronic stage of cerebral infarction. J. Stroke Cerebrovasc. Dis. 10, 63–69. doi:10.1053/jscd.2001.24657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakaya, K., Ayaori, M., Uto-Kondo, H., Hisada, T., Ogura, M., Yakushiji, E., et al. (2010). Cilostazol enhances macrophage reverse cholesterol transport in vitro and in vivo. Atherosclerosis 213, 135–141. doi:10.1016/j.atherosclerosis.2010.07.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Namba, Y., Tomonaga, M., Kawasaki, H., Otomo, E., and Ikeda, K. (1991). Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 541, 163–166. doi:10.1016/0006-8993(91)91092-F
Narita, M., Holtzman, D. M., Schwartz, A. L., and Bu, G. (1997). Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J. Neurochem. 69, 1904–1911. doi:10.1046/j.1471-4159.1997.69051904.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nedergaard, M. (2013). Neuroscience. Garbage truck of the brain. Science 340, 1529–1530. doi:10.1126/science.1240514
Neuropathology Group of Medical Research Council Cognitive Function and Aging Study (MRC CFAS). (2001). Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet 357, 169–175. doi:10.1016/S0140-6736(00)03589-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicoll, J. A., Yamada, M., Frackowiak, J., Mazur-Kolecka, B., and Weller, R. O. (2004). Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer’s disease. Pro-CAA position statement. Neurobiol. Aging 25, 589–597. doi:10.1016/j.neurobiolaging.2004.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohrui, T., Tomita, N., Sato-Nakagawa, T., Matsui, T., Maruyama, M., Niwa, K., et al. (2004). Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology 63, 1324–1325. doi:10.1212/01.WNL.0000140705.23869.E9
Okamoto, Y., Ihara, M., Fujita, Y., Ito, H., Takahashi, R., and Tomimoto, H. (2009). Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport 20, 990–996. doi:10.1097/WNR.0b013e32832d2e6a
Okamoto, Y., Yamamoto, T., Kalaria, R. N., Senzaki, H., Maki, T., Hase, Y., et al. (2012). Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 123, 381–394. doi:10.1007/s00401-011-0925-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Origlia, N., Bonadonna, C., Rosellini, A., Leznik, E., Arancio, O., Yan, S. S., et al. (2010). Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. 30, 11414–11425. doi:10.1523/JNEUROSCI.2127-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Origlia, N., Righi, M., Capsoni, S., Cattaneo, A., Fang, F., Stern, D. M., et al. (2008). Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J. Neurosci. 28, 3521–3530. doi:10.1523/JNEUROSCI.0204-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Otsuki, M., Saito, H., Xu, X., Sumitani, S., Kouhara, H., Kurabayashi, M., et al. (2001). Cilostazol represses vascular cell adhesion molecule-1 gene transcription via inhibiting NF-kappaB binding to its recognition sequence. Atherosclerosis 158, 121–128. doi:10.1016/S0021-9150(01)00431-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oyama, N., Yagita, Y., Kawamura, M., Sugiyama, Y., Terasaki, Y., Omura-Matsuoka, E., et al. (2011). Cilostazol, not aspirin, reduces ischemic brain injury via endothelial protection in spontaneously hypertensive rats. Stroke 42, 2571–2577. doi:10.1161/STROKEAHA.110.609834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pantoni, L. (2010). Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701. doi:10.1016/S1474-4422(10)70104-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patton, R. L., Kalback, W. M., Esh, C. L., Kokjohn, T. A., Van Vickle, G. D., Luehrs, D. C., et al. (2006). Amyloid-beta peptide remnants in an-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am. J. Pathol. 169, 1048–1063. doi:10.2353/ajpath.2006.060269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pimentel-Coelho, P. M., and Rivest, S. (2012). The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer’s disease. Eur. J. Neurosci. 35, 1917–1937. doi:10.1111/j.1460-9568.2012.08126.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Premkumar, D. R., Cohen, D. L., Hedera, P., Friedland, R. P., and Kalaria, R. N. (1996). Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am. J. Pathol. 148, 2083–2095.
Price, A. R., Xu, G., Siemienski, Z. B., Smithson, L. A., Borchelt, D. R., Golde, T. E., et al. (2013). Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924–d. doi:10.1126/science.1234089
Puzzo, D., Staniszewski, A., Deng, S. X., Privitera, L., Leznik, E., Liu, S., et al. (2009). Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J. Neurosci. 29, 8075–8086. doi:10.1523/JNEUROSCI.0864-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, W. Q., and Folstein, M. F. (2006). Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol. Aging 27, 190–198. doi:10.1016/j.neurobiolaging.2005.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rebeck, G. W., Reiter, J. S., Strickland, D. K., and Hyman, B. T. (1993). Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 11, 575–580. doi:10.1016/0896-6273(93)90070-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reneerkens, O. A., Rutten, K., Steinbusch, H. W., Blokland, A., and Prickaerts, J. (2009). Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology (Berl.) 202, 419–443. doi:10.1007/s00213-008-1273-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerrière, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi:10.1038/ng1718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rozzini, L., Chilovi, B. V., Bertoletti, E., Conti, M., Del Rio, I., Trabucchi, M., et al. (2006). Angiotensin converting enzyme (ACE) inhibitors modulate the rate of progression of amnestic mild cognitive impairment. Int. J. Geriatr. Psychiatry 21, 550–555. doi:10.1002/gps.1523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakurai, H., Hanyu, H., Sato, T., Kume, K., Hirao, K., Kanetaka, H., et al. (2013). Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: a pilot study. Geriatr. Gerontol. Int. 13, 90–97. doi:10.1111/j.1447-0594.2012.00866.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sandoval, K. E., Farr, S. A., Banks, W. A., Crider, A. M., Morley, J. E., and Witt, K. A. (2012). Somatostatin receptor subtype-4 agonist NNC 26-9100 decreases extracellular and intracellular Abeta(1-42) trimers. Eur. J. Pharmacol. 683, 116–124. doi:10.1016/j.ejphar.2012.03.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schley, D., Carare-Nnadi, R., Please, C. P., Perry, V. H., and Weller, R. O. (2006). Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J. Theor. Biol. 238, 962–974. doi:10.1016/j.jtbi.2005.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, J. A., Arvanitakis, Z., Bang, W., and Bennett, D. A. (2007). Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. doi:10.1212/01.wnl.0000271090.28148.24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwalbe, G. (1869). Der arachnoidalraum ein lymphraum und sein zusammenhang mit dem perichoroidalraum. Zentralb Med Wiss 7, 465–467.
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi:10.1172/JCI10498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shinkai, Y., Yoshimura, M., Ito, Y., Odaka, A., Suzuki, N., Yanagisawa, K., et al. (1995). Amyloid beta-proteins 1-40 and 1-42(43) in the soluble fraction of extra- and intracranial blood vessels. Ann. Neurol. 38, 421–428. doi:10.1002/ana.410380312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shinohara, Y., Katayama, Y., Uchiyama, S., Yamaguchi, T., Handa, S., Matsuoka, K., et al. (2010). Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol. 9, 959–968. doi:10.1016/S1474-4422(10)70198-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, E. E., Schneider, J. A., Wardlaw, J. M., and Greenberg, S. M. (2012). Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 11, 272–282. doi:10.1016/S1474-4422(11)70307-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Snowdon, D. A., Greiner, L. H., Mortimer, J. A., Riley, K. P., Greiner, P. A., and Markesbery, W. R. (1997). Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277, 813–817.
Sun, X., He, G., Qing, H., Zhou, W., Dobie, F., Cai, F., et al. (2006). Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A 103, 18727–18732. doi:10.1073/pnas.0606298103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suter, O. C., Sunthorn, T., Kraftsik, R., Straubel, J., Darekar, P., Khalili, K., et al. (2002). Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 33, 1986–1992. doi:10.1161/01.STR.0000024523.82311.77
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szentistványi, I., Patlak, C. S., Ellis, R. A., and Cserr, H. F. (1984). Drainage of interstitial fluid from different regions of rat brain. Am. J. Physiol. 246, F835–F844.
Taguchi, A., Takata, Y., Ihara, M., Kasahara, Y., Tsuji, M., Nishino, M., et al. (2013). Cilostazol improves cognitive function in patients with mild cognitive impairment: a retrospective analysis. Psychogeriatrics 13, 164–169. doi:10.1111/psyg.12021
Tanaka, K., Gotoh, F., Fukuuchi, Y., Amano, T., Uematsu, D., Kawamura, J., et al. (1989). Effects of a selective inhibitor of cyclic AMP phosphodiesterase on the pial microcirculation in feline cerebral ischemia. Stroke 20, 668–673. doi:10.1161/01.STR.20.5.668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanaka, Y., Tanaka, R., Liu, M., Hattori, N., and Urabe, T. (2010). Cilostazol attenuates ischemic brain injury and enhances neurogenesis in the subventricular zone of adult mice after transient focal cerebral ischemia. Neuroscience 171, 1367–1376. doi:10.1016/j.neuroscience.2010.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanoi, Y., Okeda, R., and Budka, H. (2000). Binswanger’s encephalopathy: serial sections and morphometry of the cerebral arteries. Acta Neuropathol. 100, 347–355. doi:10.1007/s004010000203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tesseur, I., Lo, A. C., Roberfroid, A., Dietvorst, S., Van Broeck, B., Borgers, M., et al. (2013). Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924–e. doi:10.1126/science.1233937
The U.S. National Institutes of Health. (2014). Evaluation of the Efficacy and Safety of TTP488 in Patients with Mild Alzheimer’s Disease. Available at: http://clinicaltrial.gov/ct2/show/NCT02080364?term=TTP488&rank=2
Toledo, J. B., Arnold, S. E., Raible, K., Brettschneider, J., Xie, S. X., Grossman, M., et al. (2013). Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the national Alzheimer’s coordinating centre. Brain 136, 2697–2706.
Tooyama, I., Kawamata, T., Akiyama, H., Kimura, H., Moestrup, S. K., Gliemann, J., et al. (1995). Subcellular localization of the low density lipoprotein receptor-related protein (alpha 2-macroglobulin receptor) in human brain. Brain Res. 691, 235–238. doi:10.1016/0006-8993(95)00735-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, C. S., Lin, F. Y., Chen, Y. H., Yang, T. L., Wang, H. J., Huang, G. S., et al. (2008). Cilostazol attenuates MCP-1 and MMP-9 expression in vivo in LPS-administrated balloon-injured rabbit aorta and in vitro in LPS-treated monocytic THP-1 cells. J. Cell. Biochem. 103, 54–66. doi:10.1002/jcb.21388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uchiyama, S., Shinohara, Y., Katayama, Y., Yamaguchi, T., Handa, S., Matsuoka, K., et al. (2014). Benefit of cilostazol in patients with high risk of bleeding: subanalysis of cilostazol stroke prevention study 2. Cerebrovasc. Dis. 37, 296–303. doi:10.1159/000360811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Veluw, S. J., Zwanenburg, J. J., Engelen-Lee, J., Spliet, W. G., Hendrikse, J., Luijten, P. R., et al. (2013). In vivo detection of cerebral cortical microinfarcts with high-resolution 7T MRI. J. Cereb. Blood Flow Metab. 33, 322–329. doi:10.1038/jcbfm.2012.196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vardy, E. R., Catto, A. J., and Hooper, N. M. (2005). Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimer’s disease. Trends Mol. Med. 11, 464–472. doi:10.1016/j.molmed.2005.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veeraraghavalu, K., Zhang, C., Miller, S., Hefendehl, J. K., Rajapaksha, T. W., Ulrich, J., et al. (2013). Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340, 924–f. doi:10.1126/science.1235505
Verbeek, M. M., Kremer, B. P., Rikkert, M. O., Van Domburg, P. H., Skehan, M. E., and Greenberg, S. M. (2009). Cerebrospinal fluid amyloid beta(40) is decreased in scerebral amyloid angiopathy. Ann. Neurol. 66, 245–249. doi:10.1002/ana.21694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Viswanathan, A., Rocca, W. A., and Tzourio, C. (2009). Vascular risk factors and dementia: how to move forward? Neurology 72, 368–374. doi:10.1212/01.wnl.0000341271.90478.8e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wada, T., Onogi, Y., Kimura, Y., Nakano, T., Fusanobori, H., Ishii, Y., et al. (2013). Cilostazol ameliorates systemic insulin resistance in diabetic db/db mice by suppressing chronic inflammation in adipose tissue via modulation of both adipocyte and macrophage functions. Eur. J. Pharmacol. 707, 120–129. doi:10.1016/j.ejphar.2013.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, T., Zhang, N., Liu, M., Tanaka, R., Mizuno, Y., and Urabe, T. (2006). Cilostazol protects against brain white matter damage and cognitive impairment in a rat model of chronic cerebral hypoperfusion. Stroke 37, 1539–1545. doi:10.1161/01.STR.0000221783.08037.a9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wavrant-DeVrièze, F., Lambert, J. C., Stas, L., Crook, R., Cottel, D., Pasquier, F., et al. (1999). Association between coding variability in the LRP gene and the risk of late-onset Alzheimer’s disease. Hum. Genet. 104, 432–434. doi:10.1007/s004390050980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Djuanda, E., Yow, H. Y., and Carare, R. O. (2009). Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 117, 1–14. doi:10.1007/s00401-008-0457-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Galea, I., Carare, R. O., and Minagar, A. (2010). Pathophysiology of the lymphatic drainage of the central nervous system: implications for pathogenesis and therapy of multiple sclerosis. Pathophysiology 17, 295–306. doi:10.1016/j.pathophys.2009.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Massey, A., Newman, T. A., Hutchings, M., Kuo, Y. M., and Roher, A. E. (1998). Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am. J. Pathol. 153, 725–733. doi:10.1016/S0002-9440(10)65616-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. doi:10.1111/j.1750-3639.2008.00133.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westover, M. B., Bianchi, M. T., Yang, C., Schneider, J. A., and Greenberg, S. M. (2013). Estimating cerebral microinfarct burden from autopsy samples. Neurology 80, 1365–1369. doi:10.1212/WNL.0b013e31828c2f52
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolf, B. B., Lopes, M. B., Vandenberg, S. R., and Gonias, S. L. (1992). Characterization and immunohistochemical localization of alpha 2-macroglobulin receptor (low-density lipoprotein receptor-related protein) in human brain. Am. J. Pathol. 141, 37–42.
Yamada, S., Depasquale, M., Patlak, C. S., and Cserr, H. F. (1991). Albumin outflow into deep cervical lymph from different regions of rabbit brain. Am. J. Physiol. 261, H1197–H1204.
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. doi:10.1038/382685a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, S. S., Chen, D., Yan, S., Guo, L., Du, H., and Chen, J. X. (2012). RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer’s disease. Front Biosci (Schol Ed) 4:240–250. doi:10.2741/265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yanai, S., Semba, Y., Ito, H., and Endo, S. (2014). Cilostazol improves hippocampus-dependent long-term memory in mice. Psychopharmacology (Berl.) 231, 2681–2693. doi:10.1007/s00213-014-3442-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yasojima, K., Akiyama, H., Mcgeer, E. G., and Mcgeer, P. L. (2001). Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci. Lett. 297, 97–100. doi:10.1016/S0304-3940(00)01675-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoshimura, H. (2005). The potential of caffeine for functional modification from cortical synapses to neuron networks in the brain. Curr. Neuropharmacol. 3, 309–316. doi:10.2174/157015905774322543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeitlin, R., Patel, S., Burgess, S., Arendash, G. W., and Echeverria, V. (2011). Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer’s transgenic mice. Brain Res. 1417, 127–136. doi:10.1016/j.brainres.2011.08.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, E. T., Richards, H. K., Kida, S., and Weller, R. O. (1992). Directional and compartmentalised drainage of interstitial fluid and cerebrospinal fluid from the rat brain. Acta Neuropathol. 83, 233–239. doi:10.1007/BF00296784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang-Nunes, S. X., Maat-Schieman, M. L., Van Duinen, S. G., Roos, R. A., Frosch, M. P., and Greenberg, S. M. (2006). The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 16, 30–39. doi:10.1111/j.1750-3639.2006.tb00559.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12, 723–738. doi:10.1038/nrn3114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zolezzi, J. M., Bastías-Candia, S., Santos, M. J., and Inestrosa, N. C. (2014). Alzheimer’s disease: relevant molecular and physiopathological events affecting amyloid-β brain balance and the putative role of PPARs. Front. Aging Neurosci. 6:176. doi:10.3389/fnagi.2014.00176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zolezzi, J. M., and Inestrosa, N. C. (2014). Brain metabolite clearance: impact on Alzheimer’s disease. Metab. Brain Dis. 29, 553–561. doi:10.1007/s11011-014-9527-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zou, K., Yamaguchi, H., Akatsu, H., Sakamoto, T., Ko, M., Mizoguchi, K., et al. (2007). Angiotensin-converting enzyme converts amyloid beta-protein 1-42 (Abeta(1-42)) to Abeta(1-40), and its inhibition enhances brain Abeta deposition. J. Neurosci. 27, 8628–8635. doi:10.1523/JNEUROSCI.1549-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer’s disease, cerebral amyloid angiopathy, treatment, perivascular drainage, cilostazol
Citation: Saito S and Ihara M (2014) New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 6:290. doi: 10.3389/fnagi.2014.00290
Received: 14 July 2014; Paper pending published: 31 August 2014;
Accepted: 01 October 2014; Published online: 20 October 2014.
Edited by:
Roxana Octavia Carare, University of Southampton, UKReviewed by:
Nibaldo C. Inestrosa, Pontifical Catholic University of Chile, ChileValentina Echeverria Moran, Bay Pines VA Medical Center, USA
Copyright: © 2014 Saito and Ihara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masafumi Ihara, Department of Neurology, National Cerebral and Cardiovascular Center, 5-7-1 Fujishiro-Dai, Osaka, Suita 565-8565, Japan e-mail:aWhhcmFAbmN2Yy5nby5qcA==