 Heinz Jungbluth1,2,3*
Heinz Jungbluth1,2,3* Mathias Gautel3
Mathias Gautel3
- 1Neuromuscular Service, Department of Paediatric Neurology, Evelina Children’s Hospital, St Thomas’ Hospital, London, UK
- 2Department of Basic and Clinical Neuroscience, Institute of Psychiatry, Psychology and Neuroscience (IoPPN), King’s College London, London, UK
- 3Randall Division of Cell and Molecular Biophysics and Cardiovascular Division, King’s College London BHF Centre of Research Excellence, London, UK
Centronuclear myopathies (CNMs) are a genetically heterogeneous group of inherited neuromuscular disorders characterized by clinical features of a congenital myopathy and abundant central nuclei as the most prominent histopathological feature. The most common forms of congenital myopathies with central nuclei have been attributed to X-linked recessive mutations in the MTM1 gene encoding myotubularin (“X-linked myotubular myopathy”), autosomal-dominant mutations in the DNM2 gene encoding dynamin-2 and the BIN1 gene encoding amphiphysin-2 (also named bridging integrator-1, BIN1, or SH3P9), and autosomal-recessive mutations in BIN1, the RYR1 gene encoding the skeletal muscle ryanodine receptor, and the TTN gene encoding titin. Models to study and rescue the affected cellular pathways are now available in yeast, C. elegans, drosophila, zebrafish, mouse, and dog. Defects in membrane trafficking have emerged as a key pathogenic mechanisms, with aberrant T-tubule formation, abnormalities of triadic assembly, and disturbance of the excitation–contraction machinery the main downstream effects studied to date. Abnormal autophagy has recently been recognized as another important collateral of defective membrane trafficking in different genetic forms of CNM, suggesting an intriguing link to primary disorders of defective autophagy with overlapping histopathological features. The following review will provide an overview of clinical, histopathological, and genetic aspects of the CNMs in the context of the key pathogenic mechanism, outline unresolved questions, and indicate promising future lines of enquiry.
Introduction
Centronuclear myopathies (CNMs) are a genetically heterogeneous group of inherited neuromuscular disorders characterized by clinical features of a congenital myopathy and abundant central nuclei as the most prominent histopathological feature [for review, see Jungbluth et al. (2008)]. CNMs are genetically widely heterogeneous and have been attributed to X-linked recessive mutations in MTM1 encoding myotubularin [“X-linked myotubular myopathy (XLMTM)”] (Laporte et al., 1996), autosomal-dominant mutations in DNM2 encoding dynamin-2 (Bitoun et al., 2005) and the BIN1 gene encoding amphiphysin-2 (also named bridging integrator-1, BIN1, or SH3P9) (Bohm et al., 2014), and autosomal-recessive mutations in BIN1 (Nicot et al., 2007), RYR1 encoding the skeletal muscle ryanodine receptor (Wilmshurst et al., 2010), and TTN encoding titin (Ceyhan-Birsoy et al., 2013).
Whilst histopathological abnormalities other than abundant central nuclei are not typically observed in association with MTM1 and BIN1 mutations, the common occurrence of central nuclei, marked variability in fiber size and cores with some of the other genetic backgrounds, in particular recessive RYR1 (Bevilacqua et al., 2011) and TTN mutations (Ceyhan-Birsoy et al., 2013; Chauveau et al., 2014a), have challenged the concept of CNM as a “pure” entity (Romero, 2010) and have suggested a continuum with other congenital myopathies, in particular the core myopathies [for review, see Jungbluth et al. (2011)] and congenital fiber type disproportion (CFTD) (Clarke et al., 2010). The neuromuscular disorder due to dominant mutations in the CCDC78 gene encoding Coiled-Coil Domain-Containing 78 (Majczenko et al., 2012) is another example of a congenital myopathy difficult to classify on histopathological grounds due to the common occurrence of internalized nuclei and cores, containing sarcoplasmic aggregated CCDC78, desmin (DES), actin (ACTA1), and RYR1.
Models to study and rescue the cellular pathways affected in various forms of CNM are now available in yeast (Parrish et al., 2004; Cebollero et al., 2012), C. elegans (Dang et al., 2004; Zou et al., 2009; Neukomm et al., 2011), drosophila (Velichkova et al., 2010), zebrafish (Dowling et al., 2009; Gibbs et al., 2013), mouse (Buj-Bello et al., 2002; Durieux et al., 2010b; Pierson et al., 2012; Fetalvero et al., 2013; Reifler et al., 2014), and dog (Beggs et al., 2010; Bohm et al., 2013). Based on observations in these models, several pathogenic mechanisms have now been suggested, including abnormalities of triads and calcium handling (Al-Qusairi et al., 2009; Dowling et al., 2009; Bohm et al., 2014), as well as defects of the neuromuscular junction (Robb et al., 2011; Dowling et al., 2012), satellite cells (Lawlor et al., 2012), mitochondria, and the DES cytoskeleton (Hnia et al., 2011).
Alterations of the autophagy pathway have recently emerged as a pathogenic mechanism common to different genetic forms of CNM (Al-Qusairi et al., 2013; Fetalvero et al., 2013). Autophagy is a fundamental cellular degradation pathway conserved throughout evolution with important roles in the removal of defective proteins and organelles, defense against infections and adaptation to changing metabolic demands (Mizushima, 2007; Sandri et al., 2013; Wang and Robbins, 2013). Autophagy is physiologically enhanced in neurons and muscle, and in conjunction with the ubiquitin–proteasome pathway, plays a major role in the pathogenesis of muscle atrophy (Sandri, 2013). The autophagy pathway involves several tightly regulated steps, evolving from the initial formation of phagophores to autophagosomes, whose fusion with lysosomes results in the final structures of degradation, autolysosomes [for review, see Mizushima (2007)]. The recent implication of defective autophagy in CNM corresponds to the recognition of its increasing role in a wide range of neuromuscular disorders with both primary and secondary autophagy defects (Merlini and Nishino, 2014). The observation of histopathological features closely resembling CNM in Vici syndrome (McClelland et al., 2010), a severe human multisystem disorders due to recessive mutations affecting the key autophagy regulator epg5 (Cullup et al., 2013), provides additional support for a link between the CNMs and the autophagy pathway.
The majority of the defective proteins implicated in the CNMs to date – myotubularin, dynamin-2, and amphiphysin-2 – are involved in various aspects of membrane trafficking and remodeling relevant to essential cellular processes including endocytosis, intracellular vesicle trafficking, and autophagy [for review, see Cowling et al. (2012)], suggesting a pathogenic “master mechanism” upstream of the more specific downstream pathogenic mechanisms outlined above. However, a link between membrane trafficking and other genes implicated in the CNMs is not immediately obvious, and the communality of clinico-pathological features between MTM1, DNM2, and BIN1-related CNM on one hand and the more recently reported forms due to recessive mutations in RYR1 and TTN on the other hand remains currently unaccounted for on the molecular level.
The following review will give an overview of the key clinical, histopathological, and genetic aspects of the different forms of CNM, outline pathogenic mechanisms where already known, with a particular emphasis on defects in membrane trafficking and autophagy, and summarize unresolved questions and future lines of enquiry. Table 1 summarizes the genes and proteins implicated in the CNMs and outlines their main function(s) where known. Figure 1 illustrates tentative links between the different pathways implicated in the CNMs.
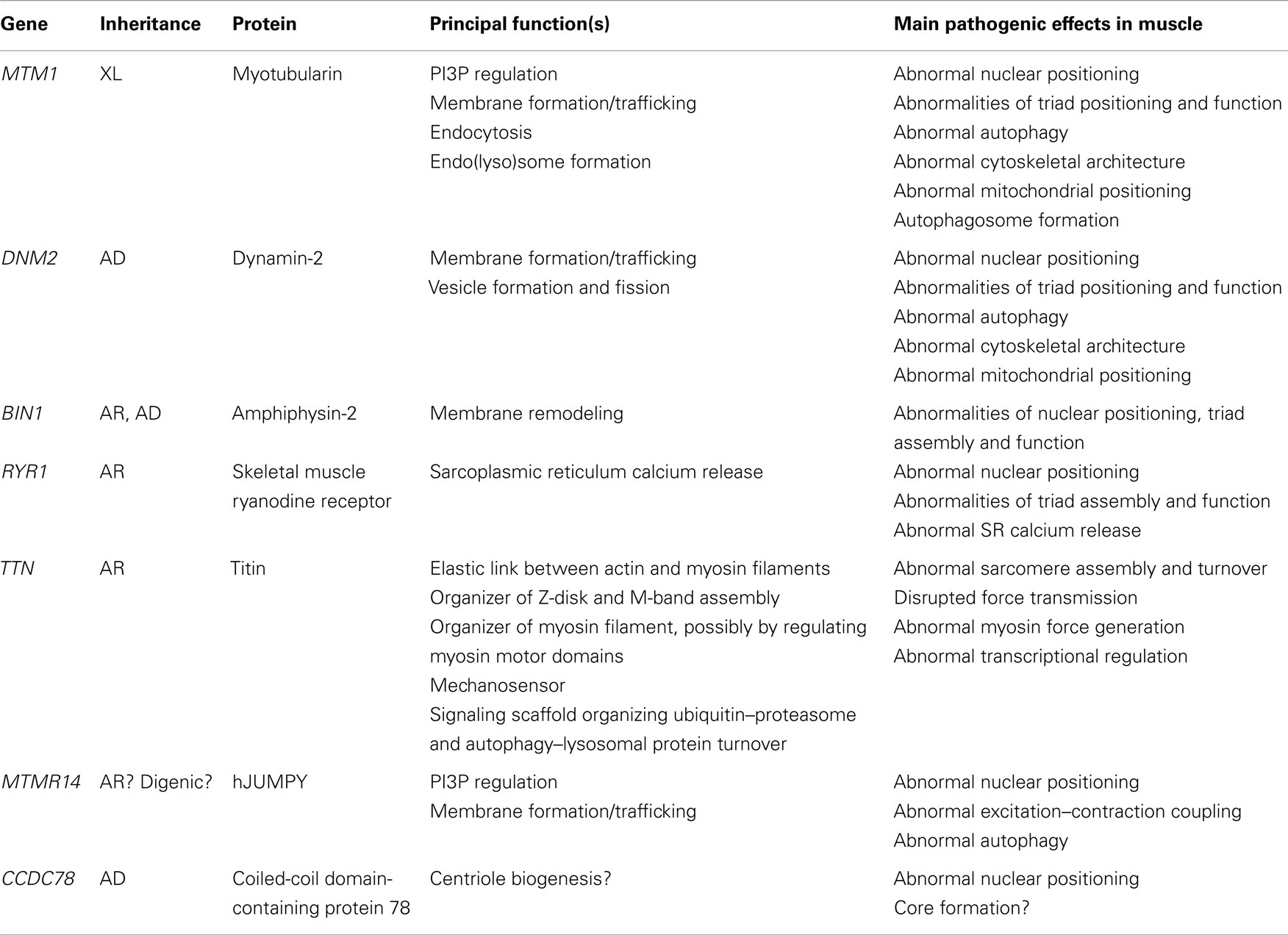
Table 1. Genes and proteins implicated in various forms of centronuclear myopathy (CNM).
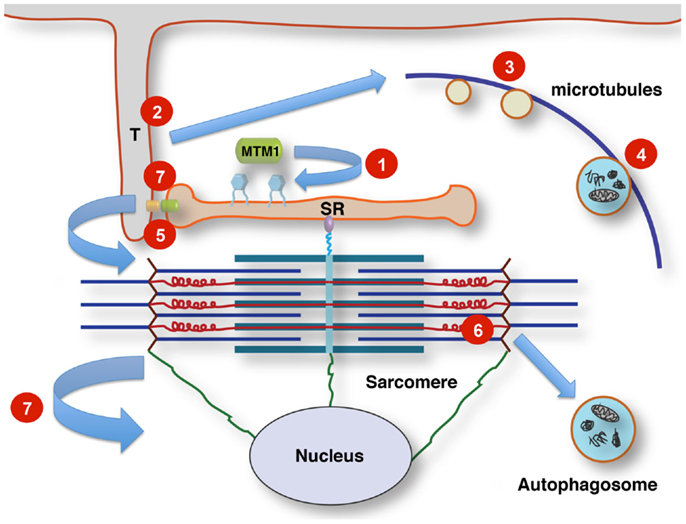
Figure 1. Tentative links between membrane signaling, sarcomere activity, and nuclear positioning. T-tubules (T) link the sarcolemma to the sarcoplasmic reticulum (SR) at the triads by contacts between dihydropyridine receptors (small orange oval) and ryanodine receptors (small green oval). Phosphoinositol phosphates (PIP; blue hexagonal symbols at SR membrane) are turned over by the lipid phosphatase myotubularin, regulating membrane dynamics, and PIP-dependent downstream signaling. This affects multiple pathways, including autophagy. Altered membrane remodeling and microtubular transport will converge on these pathways. Defects affecting Titin’s scaffolding role concerning multiple components of the protein quality control machinery impinge on contractile function, sarcomere turnover and possibly sarcomere-nuclear links. Nuclei and sarcomeres are joined by peripheral cytoskeletal networks, including desmin intermediate filaments and nesprin via “transmembrane actin-associated nuclear links” (Luxton et al., 2011). 1, Mutations affecting lipid phosphatase activity of myotubularin (MTM1 and MTMR14); 2, mutations in components of the membrane remodeling machinery (BIN1); 3, defects in vesicular traffic or microtubule dynamics (DNM2); 4, mutations in the endosomal–autophagosomal–lysosomal pathway (EPG5); 5, defective calcium homeostasis and excitation–contraction coupling (RYR1); 6, defective sarcomeric maintenance and protein quality control (TTN); 7, disrupted nuclear cytoskeleton links, abnormal nuclear positioning and possibly impaired triad function via impaired microtubule/centriole function (CCDC78).
MTM1-Related CNM (“X-Linked Myotubular Myopathy”)
Centronuclear myopathy due to X-linked recessive mutations in the myotubularin (MTM1) gene (also commonly referred to as XLMTM) is a rare congenital myopathy that affects approximately 2/100000 male births per year [for review, see Jungbluth et al. (2008)].
X-linked myotubular myopathy is characterized by a severe phenotype in males with often antenatal onset, profound hypotonia, and weakness at birth with associated severe respiratory and bulbar involvement necessitating invasive ventilation and nasogastric tube feeds. Extraocular muscle involvement is common. The condition is usually fatal within the first year of life but a proportion of more mildly affected males may survive into adolescence or adulthood, sometimes even without requiring constant ventilatory support. Although profound muscle involvement is the most dramatic and earliest feature of myotubularin deficiency, long-term survivors show additional organ manifestations such as hepatic peliosis suggestive of a multisystem disorder (Herman et al., 1999), indicating that myotubularin does play a vital role in tissues other than muscle. Moreover, despite muscle atrophy and weakness, males with XLMTM typically exhibit signs of macrosomia consistent with an overgrowth syndrome (Leguennec et al., 1988; Joseph et al., 1995), suggesting a differential effect of myotubularin deficiency on muscle and other growth pathways. A dilated cardiomyopathy has been reported in two adult brothers with a mild form of XLMTM (Yu et al., 2003), raising the possibility of a cardiac phenotype in long-term survivors that remains to be systematically evaluated. Histopathological features in addition to numerous central nuclei include type 1 predominance and hypotrophy, a region devoid of myofibrils surrounding the central nucleus and necklace fibers (Romero and Bitoun, 2011), but additional sarcomeric disorganization or overt cores are unusual in contrast to the DNM2-, RYR1- and TTN-related forms.
More than 300 MTM1 mutations have been identified to date (Laporte et al., 2000; Herman et al., 2002; Biancalana et al., 2003; Tsai et al., 2005), distributed throughout the entire coding sequence and with only few recurrent substitutions. Genotype–phenotype studies have been limited due to the private nature of many MTM1 mutations, however, not unexpectedly truncating mutations usually give rise to the more severe phenotype whilst non-truncating mutations outside the myotubularin catalytic domain have been associated with milder presentations. Markedly skewed X-inactivation in manifesting females (Jungbluth et al., 2003), as well as complex rearrangements involving the MTM1 locus have also been recently reported (Trump et al., 2011; Amburgey et al., 2013).
Myotubularin defines a family of 14 phosphoinositide phosphatases in mammals [for review, see Laporte et al. (1998, 2001, 2003), Begley and Dixon (2005), Clague and Lorenzo (2005), Robinson and Dixon (2006), and Amoasii et al. (2012)], two of which, MTMR2 and MTMR13, have also been implicated in different forms of Charcot-Marie-Tooth (CMT) disease, CMT4B1 (Berger et al., 2002) and CMT4B2 (Azzedine et al., 2003), respectively. In addition to the catalytic and enzymatically active domain, myotubularin contains four other domains, including a coiled–coiled domain involved in homo- and heterodimer formation. Apart from the recognized interaction with DES, only little is known about interactions with other proteins in skeletal muscle.
Myotubularin dephosphorylates phosphatidylinositol 3- phosphate [PI(3)P] and phosphatidylinositol 3,5-phosphate [PI(3,5)P] [(Blondeau et al., 2000; Taylor et al., 2000; Tronchere et al., 2004); for review, see Tronchere et al. (2003), Robinson and Dixon (2006), and Rohde et al. (2009)], second messengers with a crucial role in membrane trafficking whose production is under the control of specific phosphatidylinositide kinases. In skeletal muscle, the main generator of PI(3)P is the PI3 kinase PIK3C3 (Backer, 2008; Meijer and Klionsky, 2011), a key regulator of a wide range of cellular processes including autophagy, in particular formation and maturation of autophagosomes (Funderburk et al., 2010).
The fundamental role of myotubularin and its orthologs in PI3P regulation, endocytosis, and endo(lyso)somal function has been documented in drosophila, C. elegans, zebrafish, mouse, and higher mammalian models of myotubularin deficiency [for review, see Cowling et al. (2012)]. Zebrafish morphants following mtm1 morpholino knockdown show abnormal motor behavior and reproduce some of the histopathological features also seen in human XLMTM, associated with increased PI3P levels in muscle (Dowling et al., 2009). In contrast to human XLMTM, the mtm1 knockout mouse develops muscle weakness and atrophy only in the postnatal period, suggesting an effect of myotubularin deficiency on muscle maintenance rather than muscle development (Buj-Bello et al., 2002). Secondary abnormalities of T-tubules, sarcoplasmic reticulum (SR) and the triads (Al-Qusairi et al., 2009; Dowling et al., 2009; Beggs et al., 2010; Toussaint et al., 2011) and, less frequently, abnormalities of intermediate filaments and mitochondrial dynamics (Hnia et al., 2011) have been reported as an important common downstream effect of myotubularin deficiency in zebrafish, mouse, dog, and humans.
Initiation of autophagy, in particular formation of autophagosomes and autophagosome–lysosome fusion, depends on PI3P synthesis (Vergne and Deretic, 2010; Cebollero et al., 2012), and the concerted interaction of autophagy-related (Atg) proteins at the phagophore assembly site (PAS) (Lamb et al., 2013; Ge et al., 2014); considering the important role of myotubularin in regulating PI3P levels in muscle, it is not surprising that alterations of muscle autophagy have now been reported in animal models of XLMTM: In particular, a marked disturbance of autophagy has been reported in zebrafish following double knockdown of the myotubularin family members MTM1 and MTMR14 (Dowling et al., 2010), the latter also known as Jumpy and implicated in very rare digenic forms of CNM (Tosch et al., 2006). Mytotubularin deficiency has also been associated with increased mTORC1 activity, disconnection between starvation and autophagy induction (Fetalvero et al., 2013), increased IGF1R/Akt signaling, upregulation of atrogenes and an increase in autophagy markers in the mtm1 knockout mouse (Al-Qusairi et al., 2013), indicating both up- and downstream effects of murine myotubularin deficiency on the autophagy pathway that are potentially amenable to mTOR inhibition with Rapamycin (Fetalvero et al., 2013) and adeno-associated virus (AAV)-mediated delivery of functional myotubularin (Al-Qusairi et al., 2013). Interestingly, a recent study reporting a muscle-specific conditional knockout of PIK3C3, the phosphatidylinositide 3-kinase critical for PI3P levels in muscle, indicates marked autophagolysosomal abnormalities with histopathological features more suggestive of a muscular dystrophy rather than CNM (Reifler et al., 2014). These observations suggest the autophagy pathway and its upstream regulators as potential therapeutic targets in CNM and, possibly, other forms of neuromuscular disorders.
DNM2-Related CNM
Dominantly inherited DNM2-related CNM is usually much milder than X-linked and recessive forms of CNM although more severe presentations have been reported [for review, see Jungbluth et al. (2008)]. Onset is typically in adolescence or early adulthood, featuring predominant proximal weakness with additional distal involvement particularly in the lower limbs, ptosis with external ophthalmoplegia, and a stable or slowly progressive course. Exertional myalgia may be the presenting feature before the evolution of overt weakness and muscle hypertrophy, occasionally localized, has been observed (Liewluck et al., 2010). Specific dominant intermediate (CMTDIB) and axonal forms of CMT disease (CMT2), respectively, are allelic conditions (Zuchner et al., 2005; Fabrizi et al., 2007). In addition to myopathic changes, EMG and nerve conduction studies may show mild signs of axonal peripheral nerve involvement also in DNM2-related CNM patients (Fischer et al., 2006; Echaniz-Laguna et al., 2007), suggesting a clinical continuum between myopathic and neuropathic manifestations of DNM2 mutations. Other DNM2-mutated patients may feature additional neutropenia (Liewluck et al., 2010) or cataracts (Jungbluth et al., 2010), suggesting a role of dynamin-2 in tissues other than muscle, as well as clinical overlap with multisystem disorders due to primary autophagy defects such as EPG5-related Vici syndrome (Cullup et al., 2013), where cataracts and hematological abnormalities are common. Homozygosity for the DNM2 Phe379Val missense mutation has been recently associated with a congenital lethal syndrome in humans (Koutsopoulos et al., 2013). Histopathological features in addition to centralized nuclei may include type 1 predominance, typical sarcoplasmic radial strands surrounding the central nuclei, increases in connective tissues and cores (Fischer et al., 2006; Schessl et al., 2007; Jeub et al., 2008; Hanisch et al., 2011; Bohm et al., 2012a; Catteruccia et al., 2013).
The DNM2 gene is one of three members of the dynamin family (Praefcke and McMahon, 2004) and ubiquitously expressed, in contrast to DNM1 that is mainly expressed in the brain, and DNM3 expressed in brain and testes. DNM2 encodes a large GTPase protein organized in five functional domains, an N-terminal GTPase domain, a middle domain (MD), a pleckstrin homology (PH) domain, a GTPase effector domain, and a C-terminal proline rich domain (PRD) (McNiven, 2005). Through its PH and PRD domains, dynamin-2 binds to phosphoinositides and SH3 domain proteins such as amphiphysin, respectively. Dominant mutations affecting the dynamin-2 MD have been associated with a mild phenotype of CNM (Bitoun et al., 2005), whilst more severe presentations with neonatal onset have been attributed to heterozygous de novo dominant mutations affecting the PH domain (Bitoun et al., 2007; Jungbluth et al., 2010). The dynamin-2 PH domain is also predominantly affected by DNM2 mutations causing primary neuropathic phenotypes. A recurrent DNM2 mutation (c.1393C > T; p.Arg465Trp) has been identified in a number of unrelated autosomal-dominant pedigrees with a mild form of CNM.
Dynamins are involved in membrane fission, and the role of various isoforms including dynamin-2 in clathrin-dependent and independent endocytosis, vesicle formation and processing (Jones et al., 1998; Praefcke and McMahon, 2004; Durieux et al., 2010a) has been documented in various models of dynamin deficiency. Additional roles have been proposed in the microtubule network, actin cytoskeleton assembly (Gu et al., 2010), and centrosome cohesion (Thompson et al., 2004), the latter of potential relevance for the nuclear abnormalities observed in DNM2-related CNM.
Murine models of the common human DNM2 R465W dominant CNM mutation do replicate aspects of the human phenotype, and not unexpected considering the close links between endocytic and autophagic pathways, show variable abnormalities of autophagy: Durieux et al. (2010b, 2012) demonstrated a slowly progressive myopathy with upregulation of genes involved in ubiquitin–proteosome (UPS) and autophagy pathways in a heterozygous knock-in mouse model of the common CMT-associated heterozygous DNM2 mutation R465W. Mice homozygous for the R465W mutation showed a severe phenotype similar to what has been observed in other mouse models of dysregulated autophagy (Durieux et al., 2012), characterized by increased glycogen storage, hepatomegaly, hypoglycemia, and early lethality. The same mice showed microscopic evidence of delayed autophagosome maturation and of reduced autophagic flux on in vitro studies. Another mouse model generated by intramuscular AAV injection of mutant R465W–DNM2 generated histopathological abnormalities and T-tubule defects similar to those observed in humans and animal models of other forms of CNM, suggesting a muscle maintenance defect as the principal abnormality also in DNM2-related CNM. An intriguing and potentially therapeutically exploitable link between DNM2- and MTM1-related pathways has been recently indicated by demonstrating rescue of the XLMTM phenotype through dynamin-2 reduction in mice (Cowling et al., 2014).
BIN1-Related CNM
Autosomal-recessive BIN1-related CNM has only been reported in a small number of families associated with a mild to moderate phenotype characterized by early-childhood onset, extraocular muscle involvement and slowly progressive muscle weakness, and atrophy (Nicot et al., 2007; Claeys et al., 2010). However, more severe, early-onset lethal (Nicot et al., 2007) and rapidly progressive presentations due to homozygous BIN1 mutations affecting splicing have been reported (Bohm et al., 2010, 2013). Dominant inheritance of BIN1 mutations has also been recently recognized (Bohm et al., 2014). In addition to central nuclei, type 1 fiber-type predominance may be an additional feature, but sarcomeric disorganization and core-like areas are uncommon [for review, see Jungbluth et al. (2008)].
BIN1 encodes amphiphysin-2, a protein belonging to the BAR (Bin/Amphiphysin/Rvs) domain-containing family of proteins (Peter et al., 2004) involved in various key cellular processes including membrane recycling and endocyotosis [for review, see Prokic et al. (2014)]. Corresponding to other proteins implicated in the CNMs, BIN1 also contains a phosphoinositide-binding domain and is involved in T-tubule formation. Mutations affecting the BIN1 BAR domain impair membrane tubulation and result in structural abnormalities (Wu et al., 2014). BIN1 is ubiquitously expressed but subject to tissue-specific alternative splicing, whereas amphiphysin 1, the other member of the amphiphysin family, is mainly expressed in brain. BIN1 downregulation has been associated with cancer progression and cardiac disease, whereas BIN1 overexpression has been linked to an increased risk for late-onset Alzheimer disease [for review, see Prokic et al. (2014)].
The essential role of amphiphysins and their orthologs in endocytosis, membrane remodeling and recycling has been documented in drosophila and C. elegans models of amphiphysin deficiency [for review, see Cowling et al. (2012)]. A recent Bin1-deficient zebrafish model of BIN1-related CNM reproduces the histopathological features of the human phenotype, and indicates abnormal calcium release resulting from aberrant triad formation as an important pathogenic mechanism downstream of the principal membrane remodeling abnormality (Smith et al., 2014). The T-tubule and triadic abnormalities observed in the Bin1-deficient zebrafish model are similar to those observed in MTM1- and DNM2-related CNM (Toussaint et al., 2011), indicating a shared pathogenic mechanism due to implication of the defective proteins in the same pathway. BIN1-deficient mice show early lethality (Muller et al., 2003), but murine skeletal muscle has not yet been thoroughly analyzed. A recent mouse model of Bin1 depletion in the heart shows abnormalities of T-tubule folding resulting in free diffusion of local extracellular calcium and potassium ions, prolonged action-potential duration and increased susceptibility to ventricular arrhythmias (Hong et al., 2014).
BIN1 deficiency has not yet been associated with defects in the autophagy pathway, however, it is of note that structurally related BAR domain-containing proteins such as SH3P2 translocate to the PAS following autophagy induction and appear to play a role in autophagosome formation (Zhuang et al., 2013).
RYR1-Related CNM
Recessive mutations in RYR1 are another cause of congenital myopathies with central nuclei (Wilmshurst et al., 2010). RYR1 mutations are one of the most common causes of inherited neuromuscular disorders, ranging from the malignant hyperthermia susceptibility (MHS) trait without any associated weakness to various congenital myopathies, including mainly dominantly inherited Central Core Disease (CCD) as well as mainly recessively inherited Multi-minicore Disease (MmD) [for review, see Jungbluth et al. (2011)], CFTD (Clarke et al., 2010), and CNM (Wilmshurst et al., 2010). The genetics of RYR1-related myopathies are not infrequently complex, occasionally with two clearly pathogenic RYR1 mutations occurring on the same allele or running independently in the same family, possibly accounting for the wide phenotypical variability and variable penetrance (Klein et al., 2012). There is substantial clinical and pathological overlap between MmD, CFTD, and CNM due to recessive RYR1 mutations, and it appears appropriate to view these conditions as part of a recessive RYR1-related continuum rather than completely distinct entities.
Clinically, RYR1-related CNM is of intermediate severity compared to other genetic forms, with facial weakness, external ophthalmoplegia, predominantly proximal muscle involvement but less pronounced bulbar or respiratory impairment (Wilmshurst et al., 2010). There is however, a more severe end of the spectrum, with some profoundly affected males showing clinical presentations indistinguishable from the XLMTM phenotype. Patients with RYR1-related CNM show a marked tendency to improve over time, even following an initially severe presentation, a feature also in other recessive RYR1-related myopathies (Bohm et al., 2012b) that remains currently unexplained.
On the pathological level, central and multiple internalized nuclei are often the principal histopathological feature when muscle biopsy is performed early in life (Jungbluth et al., 2007), but other histopathological features typically associated with recessive RYR1-related myopathies – marked type 1 predominance or uniformity, fiber type disproportion and cores – may evolve over time (Bevilacqua et al., 2011).
In contrast to dominantly inherited MHS and CCD where most features can be explained by abnormal calcium release from the mutant RyR1 channel [for review, see Treves et al. (2005)], the pathogenesis of recessive RYR1-related myopathies is currently only partly understood and probably more complex. Recessive RYR1 genotypes, often featuring compound heterozygosity for RYR1 missense and truncating mutations, result in reduced expression of the RyR1 protein and secondary reduction of the DHPR receptor, its principal ligand (Wilmshurst et al., 2010; Bevilacqua et al., 2011; Zhou et al., 2013). Additional upregulation of the IP3R receptor may be found in some cases, an observation currently of uncertain significance (Zhou et al., 2013). Whilst the concomitant reduction of RyR1 and DHPR and the resulting excitation–contraction (EC) coupling defect are likely to explain the weakness, muscle atrophy as well as histopathological features of fiber-type disproportion and centralized nuclei are not readily explained by alterations of calcium release in recessive RYR1-related myopathies.
In contrast to other genetic forms of CNM, defects in membrane trafficking and autophagy have not been implicated in recessive RYR1-related CNM. However, it is of note that marked autophagy abnormalities have been observed in mice following induced reduction of the DHPR receptor (Pietri-Rouxel et al., 2010), a secondary feature also in recessive RYR1-related myopathies. In addition, the recent implication of annexin-1 and annexin-5, members of the annexin family of proteins that bind to phospholipid membranes in a calcium-dependent manner, in autophagosome maturation (Ghislat and Knecht, 2012) suggests a potential link between disturbed calcium homeostasis and autophagy regulation that may warrant further investigation. Effect of altered calcium release on autophagic pathways have been previously considered but with conflicting conclusions (East and Campanella, 2013).
TTN-Related CNM
Recessive mutations in TTN encoding the giant sarcomeric ruler protein titin have recently been identified by next generation sequencing in five individuals selected from a cohort of 29 unrelated and genetically unresolved patients with a clinico-pathological diagnosis of CNM (Ceyhan-Birsoy et al., 2013). TTN mutations have recently also been indicated as one of the most common identifiable genetic causes of dilated cardiomyopathy (Herman et al., 2012), and have been implicated in a wide range of neuromuscular disorders, including late-onset tibial muscular dystrophy, limb girdle muscular dystrophy type 2J (LGMD2J), hereditary myopathy with early respiratory failure (HMERF), and early-onset myopathy with fatal cardiomyopathy [reviewed recently in Chauveau et al. (2014b)]. Although the functional relevance and clear genotype–phenotype correlations have been established for many unequivocally pathogenic TTN mutations, it is also important to bear in mind that truncating TTN variants are exceedingly common [over 6000 in the December 2014 release of the 1000 genomes database, and ca. 3% of controls reported in Herman et al. (2012)] and that pathogenicity is not always immediately evident even if those variants are truncating. This might be due to tissue-specific and developmentally regulated exon usage, especially in the extensively differentially spliced I-band region of titin (Bang et al., 2001), making some truncating variants penetrant in only a small subset of titin isoforms. But also, truncating mutations near the C-terminus appear, on the whole, to be recessive without an adult phenotype (Carmignac et al., 2007; Ceyhan-Birsoy et al., 2013; Chauveau et al., 2014a). The reasons for the recessive inheritance of truncating variants in constitutively expressed exons remains to be understood.
Clinical features of the five patients with TTN-related CNM in the study by Ceyhan-Birsoy et al. (2013) were characterized by early-childhood onset, generalized weakness, and respiratory impairment, but without evidence of cardiac involvement at the time of the last follow-up in childhood or late adolescence (5–19 years). In contrast to other genetic forms of CNM, extraocular muscles were spared and in one case CK levels were increased >1000 IU/l. Histopathological features included increases in connective tissue, fiber type disproportion, and type 1 predominance and hypotrophy. In contrast to MTM1-related CNM, but corresponding to findings in the RYR1-related form, central and internalized nuclei were typically multiple rather than single. Similar observations were made in the seminal paper on the recessive truncating TTN-linked early-onset Salih myopathy (Carmignac et al., 2007) and in four families with compound heterozygous TTN variants in Autosomal-Recessive Multi-minicore Disease with Heart Disease (AR MmD-HD) (Chauveau et al., 2014a). Patients with AR MmD-HD show marked centronucleation with additional morphological changes, notably the formation of protein aggregates and Z-disk streaming that show ultrastructural similarities to those found in myofibrillar myopathy. Patients also display various cardiac phenotypes, from left-ventricular non-compaction to septal defects (ASD and VSD) and dilated cardiomyopathy, in some instances requiring transplantation (Chauveau et al., 2014a). Additional findings of core-like areas on oxidative stain and myofibrillar disruption on EM, in particular Z-disk streaming and sarcomere disruption, suggest that TTN-related CNM and AR MmD-HD may be part of a TTN-related histopathological spectrum rather than a pure entity, again corresponding to observations in the RYR1-related form.
The pathogenesis of TTN-related CNM and in particular its association with pathways affected in other forms of CNM, if any, remains currently uncertain. Most mutations identified in TTN-related CNM give rise to significant C-terminal truncations, with or without the expression of disruptive missense variants, resulting in secondary reduction of interacting proteins such as nebulin and calpain-3 that may contribute to the phenotype. Calpain-3 is required for the normal recruitment of RyR1 receptors to the triad, a function that, if disturbed, may give rise to similar abnormalities of triad assembly and EC coupling as seen in other genetic forms of CNM. A common feature of TTN-linked AR MmD-HD and CNM is, however, the disruption of titin M-band linked interactions; of these, three are possibly mechanistically related to pathways linked to the “classical” CNM variants. Firstly, M-band titin links the sarcomere to the SR via its interactions with the giant protein obscurin (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2003; Fukuzawa et al., 2008) and thus contributes to the organized integration of the EC-coupling machinery of T-tubules, junctional SR, and sarcomeres. Intriguingly, obscurin knockout mice also develop a myopathic phenotype with centralized nuclei and disordered SR (Lange et al., 2009). Secondly, the M-band associated kinase domain of titin is linked to the control of protein turnover via the autophagy cargo adaptors Nbr1 and SQSTM1 [reviewed in Gautel (2011)]. Lastly, mutations in the C-terminus of titin are linked to secondary calpain-3 deficiency also in the case of adult titinopathies (Udd, 2012), likely due to the abrogation of a calpain-3 binding site near the C-terminus of titin (Charton et al., 2010). While the connections between titin mutations, protein turnover, and abnormal nuclear positioning in titin-associated CNM-like myopathies are currently unclear, accumulating evidence suggests that protein turnover via autophagy and calpain-mediated turnover converge on M-band titin and that these connections are concerted with physical links to the SR and triad systems. If such links exist, it seems plausible that the ablation or functional disruption of titin-linked autophagy functions in M-band titinopathies (Chauveau et al., 2014a) result in partial phenotypic overlap with membrane-associated components of the autophagy machinery.
Rare Congenital Myopathies with Central Nuclei
Congenital myopathies with features of CNM with or without additional histopathological abnormalities due to uncommon genetic backgrounds have been observed in isolated families.
Tosch and colleagues reported single heterozygous missense variants in hJUMPY (also known as MTMR14, a member of the myotubularin family) in two sporadic cases with features of CNM and uncertain inheritance (Tosch et al., 2006). Although both variants were demonstrated to reduce the enzymatic activity of hJUMPY, identification of an additional DNM2 mutation in one patient suggests that a second mutation may be required for full manifestation of clinical features; this is also in keeping with the observation of a more severe phenotype in the MTM1–MTMR14 zebrafish double knockout compared to knockout of each single gene (Dowling et al., 2010).
Autosomal-dominant mutations in CCDC78 have also recently been identified in a single family characterized by core-like areas and increased internalized nuclei (Majczenko et al., 2012); CCDC78 encodes a skeletal muscle protein enriched in the perinuclear region and at the sarcolemma and possibly triad (Majczenko et al., 2012), suggesting a possible link with a pathogenic mechanism, abnormal triad assembly, and resulting disturbance of EC coupling, common to other forms of CNM. CCDC78 plays a key role in centriole biogenesis (Klos Dehring et al., 2013); the impaired function in CNM4 and the link to potential triad malfunction or abnormal nuclear positioning via impaired microtubule function is currently elusive.
Conclusion and Outlook
Recent years have seen substantial advances in our understanding of the CNMs, in particular those due to mutations in MTM1, DNM2, and BIN1, encoding proteins intricately linked in various aspects of phosphoinositide metabolism and membrane trafficking, with aberrant T-tubule formation, abnormalities of triad assembly and disturbance of the EC machinery as the most important downstream effects studied to date. Abnormal autophagy has recently been recognized as another important pathogenic mechanism in different genetic forms of CNM, suggesting an intriguing link to primary disorders of defective autophagy with overlapping histopathological features. These findings have illustrated the role of defective pathways common to several genetic forms of CNM that may be potentially amenable to therapeutic intervention. It remains currently uncertain if the proteins encoded by genes more recently implicated in the CNMs, in particular RYR1 and TTN, are involved with the same pathways or linked with altogether different mechanisms. The functional links between the genetic mechanisms implicated in CNM are tentative at the moment, and it has to be seen whether all myopathies clinically classified as CNM indeed join into a common pathomechanistic pathway. Although the mechanisms outlined above may at least partially explain the muscle weakness and atrophy observed in different forms of CNM, other aspects such as the consistent abnormality of nuclear positioning remain currently unaccounted for. The molecular machinery involved in nuclear positioning is currently only partially understood [for review, see Osorio and Gomes (2014)], but emerging evidence suggests that normal positioning of the nucleus is a prerequisite for its normal functioning (Metzger et al., 2012). Recent work has already suggested a link between N-WASP and BIN1-related nuclear positioning and triad organization (Falcone et al., 2014). Further investigation of the CNMs as a paradigm of disorders with nuclear positioning as the most prominent pathological hallmark will advance our understanding of the intricate interaction between the nucleus, microtubules and the actomyosin cytoskeleton (Luxton et al., 2011; Cadot et al., 2012), and delineate the importance of the interplay of these structures for cellular function in health and disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Grant support from the Myotubular Trust/Sparks (Grant Ref. 12KCL01-MT) is gratefully acknowledged. Mathias Gautel holds the British Heart Foundation Chair of Molecular Cardiology.
References
Al-Qusairi, L., Prokic, I., Amoasii, L., Kretz, C., Messaddeq, N., Mandel, J. L., et al. (2013). Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. FASEB J. 27, 3384–3394. doi:10.1096/fj.12-220947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Al-Qusairi, L., Weiss, N., Toussaint, A., Berbey, C., Messaddeq, N., Kretz, C., et al. (2009). T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc. Natl. Acad. Sci. U. S. A. 106, 18763–18768. doi:10.1073/pnas.0900705106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amburgey, K., Lawlor, M. W., Del Gaudio, D., Cheng, Y. W., Fitzpatrick, C., Minor, A., et al. (2013). Large duplication in MTM1 associated with myotubular myopathy. Neuromuscul. Disord. 23, 214–218. doi:10.1016/j.nmd.2012.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amoasii, L., Hnia, K., and Laporte, J. (2012). Myotubularin phosphoinositide phosphatases in human diseases. Curr. Top. Microbiol. Immunol. 362, 209–233. doi:10.1007/978-94-007-5025-8_10
Azzedine, H., Bolino, A., Taieb, T., Birouk, N., Di Duca, M., Bouhouche, A., et al. (2003). Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am. J. Hum. Genet. 72, 1141–1153. doi:10.1086/375034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Backer, J. M. (2008). The regulation and function of class III PI3Ks: novel roles for Vps34. Biochem. J. 410, 1–17. doi:10.1042/BJ20071427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bagnato, P., Barone, V., Giacomello, E., Rossi, D., and Sorrentino, V. (2003). Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. 160, 245–253. doi:10.1083/jcb.200208109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bang, M. L., Centner, T., Fornoff, F., Geach, A. J., Gotthardt, M., McNabb, M., et al. (2001). The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 89, 1065–1072. doi:10.1161/hh2301.100981
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beggs, A. H., Bohm, J., Snead, E., Kozlowski, M., Maurer, M., Minor, K., et al. (2010). MTM1 mutation associated with X-linked myotubular myopathy in labrador retrievers. Proc. Natl. Acad. Sci. U. S. A. 107, 14697–14702. doi:10.1073/pnas.1003677107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Begley, M. J., and Dixon, J. E. (2005). The structure and regulation of myotubularin phosphatases. Curr. Opin. Struct. Biol. 15, 614–620. doi:10.1016/j.sbi.2005.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berger, P., Bonneick, S., Willi, S., Wymann, M., and Suter, U. (2002). Loss of phosphatase activity in myotubularin-related protein 2 is associated with Charcot-Marie-Tooth disease type 4B1. Hum. Mol. Genet. 11, 1569–1579. doi:10.1093/hmg/11.13.1569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bevilacqua, J. A., Monnier, N., Bitoun, M., Eymard, B., Ferreiro, A., Monges, S., et al. (2011). Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol. Appl. Neurobiol. 37, 271–284. doi:10.1111/j.1365-2990.2010.01149.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biancalana, V., Caron, O., Gallati, S., Baas, F., Kress, W., Novelli, G., et al. (2003). Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum. Genet. 112, 135–142. doi:10.1007/s00439-002-0869-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bitoun, M., Bevilacqua, J. A., Prudhon, B., Maugenre, S., Taratuto, A. L., Monges, S., et al. (2007). Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann. Neurol. 62, 666–670. doi:10.1002/ana.21235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bitoun, M., Maugenre, S., Jeannet, P. Y., Lacene, E., Ferrer, X., Laforet, P., et al. (2005). Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 37, 1207–1209. doi:10.1038/ng1657
Blondeau, F., Laporte, J., Bodin, S., Superti-Furga, G., Payrastre, B., and Mandel, J. L. (2000). Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum. Mol. Genet. 9, 2223–2229. doi:10.1093/oxfordjournals.hmg.a018913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohm, J., Biancalana, V., Dechene, E. T., Bitoun, M., Pierson, C. R., Schaefer, E., et al. (2012a). Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum. Mutat. 33, 949–959. doi:10.1002/humu.22067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohm, J., Leshinsky-Silver, E., Vassilopoulos, S., Le Gras, S., Lerman-Sagie, T., Ginzberg, M., et al. (2012b). Samaritan myopathy, an ultimately benign congenital myopathy, is caused by a RYR1 mutation. Acta Neuropathol. 124, 575–581. doi:10.1007/s00401-012-1007-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohm, J., Biancalana, V., Malfatti, E., Dondaine, N., Koch, C., Vasli, N., et al. (2014). Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain 137, 3160–3170. doi:10.1093/brain/awu272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohm, J., Vasli, N., Maurer, M., Cowling, B., Shelton, G. D., Kress, W., et al. (2013). Altered splicing of the BIN1 muscle-specific exon in humans and dogs with highly progressive centronuclear myopathy. PLoS Genet. 9:e1003430. doi:10.1371/journal.pgen.1003430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohm, J., Yis, U., Ortac, R., Cakmakci, H., Kurul, S. H., Dirik, E., et al. (2010). Case report of intrafamilial variability in autosomal recessive centronuclear myopathy associated to a novel BIN1 stop mutation. Orphanet J. Rare Dis. 5, 35. doi:10.1186/1750-1172-5-35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buj-Bello, A., Laugel, V., Messaddeq, N., Zahreddine, H., Laporte, J., Pellissier, J. F., et al. (2002). The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. U. S. A. 99, 15060–15065. doi:10.1073/pnas.212498399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cadot, B., Gache, V., Vasyutina, E., Falcone, S., Birchmeier, C., and Gomes, E. R. (2012). Nuclear movement during myotube formation is microtubule and dynein dependent and is regulated by Cdc42, Par6 and Par3. EMBO Rep. 13, 741–749. doi:10.1038/embor.2012.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carmignac, V., Salih, M. A., Quijano-Roy, S., Marchand, S., Al Rayess, M. M., Mukhtar, M. M., et al. (2007). C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann. Neurol. 61, 340–351. doi:10.1002/ana.21089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catteruccia, M., Fattori, F., Codemo, V., Ruggiero, L., Maggi, L., Tasca, G., et al. (2013). Centronuclear myopathy related to dynamin 2 mutations: clinical, morphological, muscle imaging and genetic features of an Italian cohort. Neuromuscul. Disord. 23, 229–238. doi:10.1016/j.nmd.2012.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cebollero, E., Van Der Vaart, A., Zhao, M., Rieter, E., Klionsky, D. J., Helms, J. B., et al. (2012). Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr. Biol. 22, 1545–1553. doi:10.1016/j.cub.2012.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ceyhan-Birsoy, O., Agrawal, P. B., Hidalgo, C., Schmitz-Abe, K., Dechene, E. T., Swanson, L. C., et al. (2013). Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 81, 1205–1214. doi:10.1212/WNL.0b013e3182a6ca62
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Charton, K., Daniele, N., Vihola, A., Roudaut, C., Gicquel, E., Monjaret, F., et al. (2010). Removal of the calpain 3 protease reverses the myopathology in a mouse model for titinopathies. Hum. Mol. Genet. 19, 4608–4624. doi:10.1093/hmg/ddq388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chauveau, C., Bonnemann, C., Julien, C., Kho, A. L., Marks, H., Talim, B., et al. (2014a). Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum. Mol. Genet. 23, 980–991. doi:10.1093/hmg/ddt494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chauveau, C., Rowell, J., and Ferreiro, A. (2014b). A rising titan: TTN review and mutation update. Hum. Mutat. 35, 1046–1059. doi:10.1002/humu.22611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Claeys, K. G., Maisonobe, T., Bohm, J., Laporte, J., Hezode, M., Romero, N. B., et al. (2010). Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology 74, 519–521. doi:10.1212/WNL.0b013e3181cef7f9
Clague, M. J., and Lorenzo, O. (2005). The myotubularin family of lipid phosphatases. Traffic 6, 1063–1069. doi:10.1111/j.1600-0854.2005.00338.x
Clarke, N. F., Waddell, L. B., Cooper, S. T., Perry, M., Smith, R. L., Kornberg, A. J., et al. (2010). Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum. Mutat. 31, E1544–E1550. doi:10.1002/humu.21278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cowling, B. S., Chevremont, T., Prokic, I., Kretz, C., Ferry, A., Coirault, C., et al. (2014). Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Invest. 124, 1350–1363. doi:10.1172/JCI71206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cowling, B. S., Toussaint, A., Muller, J., and Laporte, J. (2012). Defective membrane remodeling in neuromuscular diseases: insights from animal models. PLoS Genet. 8:e1002595. doi:10.1371/journal.pgen.1002595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cullup, T., Kho, A. L., Dionisi-Vici, C., Brandmeier, B., Smith, F., Urry, Z., et al. (2013). Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 45, 83–87. doi:10.1038/ng.2497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dang, H., Li, Z., Skolnik, E. Y., and Fares, H. (2004). Disease-related myotubularins function in endocytic traffic in Caenorhabditis elegans. Mol. Biol. Cell 15, 189–196. doi:10.1091/mbc.E03-08-0605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dowling, J. J., Joubert, R., Low, S. E., Durban, A. N., Messaddeq, N., Li, X., et al. (2012). Myotubular myopathy and the neuromuscular junction: a novel therapeutic approach from mouse models. Dis. Model Mech. 5, 852–859. doi:10.1242/dmm.009746
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dowling, J. J., Low, S. E., Busta, A. S., and Feldman, E. L. (2010). Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum. Mol. Genet. 19, 2668–2681. doi:10.1093/hmg/ddq153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dowling, J. J., Vreede, A. P., Low, S. E., Gibbs, E. M., Kuwada, J. Y., Bonnemann, C. G., et al. (2009). Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 5:e1000372. doi:10.1371/journal.pgen.1000372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Durieux, A. C., Prudhon, B., Guicheney, P., and Bitoun, M. (2010a). Dynamin 2 and human diseases. J. Mol. Med. 88, 339–350. doi:10.1007/s00109-009-0587-4
Durieux, A. C., Vignaud, A., Prudhon, B., Viou, M. T., Beuvin, M., Vassilopoulos, S., et al. (2010b). A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 19, 4820–4836. doi:10.1093/hmg/ddq413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Durieux, A. C., Vassilopoulos, S., Laine, J., Fraysse, B., Brinas, L., Prudhon, B., et al. (2012). A centronuclear myopathy – dynamin 2 mutation impairs autophagy in mice. Traffic 13, 869–879. doi:10.1111/j.1600-0854.2012.01348.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
East, D. A., and Campanella, M. (2013). Ca2+ in quality control: an unresolved riddle critical to autophagy and mitophagy. Autophagy 9, 1710–1719. doi:10.4161/auto.25367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Echaniz-Laguna, A., Nicot, A. S., Carre, S., Franques, J., Tranchant, C., Dondaine, N., et al. (2007). Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul. Disord. 17, 955–959. doi:10.1016/j.nmd.2007.06.467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fabrizi, G. M., Ferrarini, M., Cavallaro, T., Cabrini, I., Cerini, R., Bertolasi, L., et al. (2007). Two novel mutations in dynamin-2 cause axonal Charcot-Marie-Tooth disease. Neurology 69, 291–295. doi:10.1212/01.wnl.0000265820.51075.61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falcone, S., Roman, W., Hnia, K., Gache, V., Didier, N., Laine, J., et al. (2014). N-WASP is required for Amphiphysin-2/BIN1-dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Mol. Med. 6, 1455–1475. doi:10.15252/emmm.201404436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fetalvero, K. M., Yu, Y., Goetschkes, M., Liang, G., Valdez, R. A., Gould, T., et al. (2013). Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol. Cell. Biol. 33, 98–110. doi:10.1128/MCB.01075-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischer, D., Herasse, M., Bitoun, M., Barragan-Campos, H. M., Chiras, J., Laforet, P., et al. (2006). Characterization of the muscle involvement in dynamin 2-related centronuclear myopathy. Brain 129, 1463–1469. doi:10.1093/brain/awl071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fukuzawa, A., Lange, S., Holt, M. R., Vihola, A., Carmignac, V., Ferreiro, A., et al. (2008). Interactions with titin and myomesin target obscurin and its small homologue, obscurin-like 1, to the sarcomeric M-band: implications for hereditary myopathies. J. Cell. Sci. 121, 1841–1851. doi:10.1242/jcs.028019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Funderburk, S. F., Wang, Q. J., and Yue, Z. (2010). The Beclin 1-VPS34 complex – at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362. doi:10.1016/j.tcb.2010.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gautel, M. (2011). Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch. 462, 119–134. doi:10.1007/s00424-011-0946-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ge, L., Baskaran, S., Schekman, R., and Hurley, J. H. (2014). The protein-vesicle network of autophagy. Curr. Opin. Cell Biol. 29C, 18–24. doi:10.1016/j.ceb.2014.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghislat, G., and Knecht, E. (2012). New Ca(2+)-dependent regulators of autophagosome maturation. Commun. Integr. Biol. 5, 308–311. doi:10.4161/cib.20076
Gibbs, E. M., Clarke, N. F., Rose, K., Oates, E. C., Webster, R., Feldman, E. L., et al. (2013). Neuromuscular junction abnormalities in DNM2-related centronuclear myopathy. J. Mol. Med. 91, 727–737. doi:10.1007/s00109-013-0994-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gu, C., Yaddanapudi, S., Weins, A., Osborn, T., Reiser, J., Pollak, M., et al. (2010). Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J. 29, 3593–3606. doi:10.1038/emboj.2010.249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanisch, F., Muller, T., Dietz, A., Bitoun, M., Kress, W., Weis, J., et al. (2011). Phenotype variability and histopathological findings in centronuclear myopathy due to DNM2 mutations. J. Neurol. 258, 1085–1090. doi:10.1007/s00415-010-5889-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herman, D. S., Lam, L., Taylor, M. R., Wang, L., Teekakirikul, P., Christodoulou, D., et al. (2012). Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 366, 619–628. doi:10.1056/NEJMoa1110186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herman, G. E., Finegold, M., Zhao, W., De Gouyon, B., and Metzenberg, A. (1999). Medical complications in long-term survivors with X-linked myotubular myopathy. J. Pediatr. 134, 206–214. doi:10.1016/S0022-3476(99)70417-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herman, G. E., Kopacz, K., Zhao, W., Mills, P. L., Metzenberg, A., and Das, S. (2002). Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum. Mutat. 19, 114–121. doi:10.1002/humu.10033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hnia, K., Tronchere, H., Tomczak, K. K., Amoasii, L., Schultz, P., Beggs, A. H., et al. (2011). Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J. Clin. Invest. 121, 70–85. doi:10.1172/JCI44021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, T., Yang, H., Zhang, S. S., Cho, H. C., Kalashnikova, M., Sun, B., et al. (2014). Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 20, 624–632. doi:10.1038/nm.3543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeub, M., Bitoun, M., Guicheney, P., Kappes-Horn, K., Strach, K., Druschky, K. F., et al. (2008). Dynamin 2-related centronuclear myopathy: clinical, histological and genetic aspects of further patients and review of the literature. Clin. Neuropathol. 27, 430–438. doi:10.5414/NPP27430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, S. M., Howell, K. E., Henley, J. R., Cao, H., and McNiven, M. A. (1998). Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science 279, 573–577. doi:10.1126/science.279.5350.573
Joseph, M., Pai, G. S., Holden, K. R., and Herman, G. (1995). X-linked myotubular myopathy: clinical observations in ten additional cases. Am. J. Med. Genet. 59, 168–173. doi:10.1002/ajmg.1320590211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jungbluth, H., Cullup, T., Lillis, S., Zhou, H., Abbs, S., Sewry, C., et al. (2010). Centronuclear myopathy with cataracts due to a novel dynamin 2 (DNM2) mutation. Neuromuscul. Disord. 20, 49–52. doi:10.1016/j.nmd.2009.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jungbluth, H., Sewry, C. A., Buj-Bello, A., Kristiansen, M., Orstavik, K. H., Kelsey, A., et al. (2003). Early and severe presentation of X-linked myotubular myopathy in a girl with skewed X-inactivation. Neuromuscul. Disord. 13, 55–59. doi:10.1016/S0960-8966(02)00194-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jungbluth, H., Sewry, C. A., and Muntoni, F. (2011). Core myopathies. Semin. Pediatr. Neurol. 18, 239–249. doi:10.1016/j.spen.2011.10.005
Jungbluth, H., Wallgren-Pettersson, C., and Laporte, J. (2008). Centronuclear (myotubular) myopathy. Orphanet J. Rare Dis. 3, 26. doi:10.1186/1750-1172-3-26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jungbluth, H., Zhou, H., Sewry, C. A., Robb, S., Treves, S., Bitoun, M., et al. (2007). Centronuclear myopathy due to a de novo dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul. Disord. 17, 338–345. doi:10.1016/j.nmd.2007.01.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klein, A., Lillis, S., Munteanu, I., Scoto, M., Zhou, H., Quinlivan, R., et al. (2012). Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum. Mutat. 33, 981–988. doi:10.1002/humu.22056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klos Dehring, D. A., Vladar, E. K., Werner, M. E., Mitchell, J. W., Hwang, P., and Mitchell, B. J. (2013). Deuterosome-mediated centriole biogenesis. Dev. Cell 27, 103–112. doi:10.1016/j.devcel.2013.08.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kontrogianni-Konstantopoulos, A., Jones, E. M., Van Rossum, D. B., and Bloch, R. J. (2003). Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell 14, 1138–1148. doi:10.1091/mbc.E02-07-0411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koutsopoulos, O. S., Kretz, C., Weller, C. M., Roux, A., Mojzisova, H., Bohm, J., et al. (2013). Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur. J. Hum. Genet. 21, 637–642. doi:10.1038/ejhg.2012.226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lamb, C. A., Yoshimori, T., and Tooze, S. A. (2013). The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774. doi:10.1038/nrm3696
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lange, S., Ouyang, K., Meyer, G., Cui, L., Cheng, H., Lieber, R. L., et al. (2009). Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell. Sci. 122, 2640–2650. doi:10.1242/jcs.046193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laporte, J., Bedez, F., Bolino, A., and Mandel, J. L. (2003). Myotubularins, a large disease-associated family of cooperating catalytically active and inactive phosphoinositides phosphatases. Hum. Mol. Genet. 2, R285–R292. doi:10.1093/hmg/ddg273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laporte, J., Biancalana, V., Tanner, S. M., Kress, W., Schneider, V., Wallgren-Pettersson, C., et al. (2000). MTM1 mutations in X-linked myotubular myopathy. Hum. Mutat. 15, 393–409. doi:10.1002/(SICI)1098-1004(200005)15:5<393::AID-HUMU1>3.0.CO;2-R
Laporte, J., Blondeau, F., Buj-Bello, A., and Mandel, J. L. (2001). The myotubularin family: from genetic disease to phosphoinositide metabolism. Trends Genet. 17, 221–228. doi:10.1016/S0168-9525(01)02245-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laporte, J., Blondeau, F., Buj-Bello, A., Tentler, D., Kretz, C., Dahl, N., et al. (1998). Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum. Mol. Genet. 7, 1703–1712. doi:10.1093/hmg/7.11.1703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laporte, J., Hu, L. J., Kretz, C., Mandel, J. L., Kioschis, P., Coy, J. F., et al. (1996). A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 13, 175–182. doi:10.1038/ng0696-175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lawlor, M. W., Alexander, M. S., Viola, M. G., Meng, H., Joubert, R., Gupta, V., et al. (2012). Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am. J. Pathol. 181, 961–968. doi:10.1016/j.ajpath.2012.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leguennec, J. C., Bernier, J. P., and Lamarche, J. (1988). High stature in neonatal myotubular myopathy. Acta Paediatr. Scand. 77, 610–611. doi:10.1111/j.1651-2227.1988.tb10714.x
Liewluck, T., Lovell, T. L., Bite, A. V., and Engel, A. G. (2010). Sporadic centronuclear myopathy with muscle pseudohypertrophy, neutropenia, and necklace fibers due to a DNM2 mutation. Neuromuscul. Disord. 20, 801–804. doi:10.1016/j.nmd.2010.07.273
Luxton, G. W., Gomes, E. R., Folker, E. S., Worman, H. J., and Gundersen, G. G. (2011). TAN lines: a novel nuclear envelope structure involved in nuclear positioning. Nucleus 2, 173–181. doi:10.1073/pnas.1000824108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Majczenko, K., Davidson, A. E., Camelo-Piragua, S., Agrawal, P. B., Manfready, R. A., Li, X., et al. (2012). Dominant mutation of CCDC78 in a unique congenital myopathy with prominent internal nuclei and atypical cores. Am. J. Hum. Genet. 91, 365–371. doi:10.1016/j.ajhg.2012.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McClelland, V., Cullup, T., Bodi, I., Ruddy, D., Buj-Bello, A., Biancalana, V., et al. (2010). Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am. J. Med. Genet. A 152A, 741–747. doi:10.1002/ajmg.a.33296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meijer, A. J., and Klionsky, D. J. (2011). Vps34 is a phosphatidylinositol 3-kinase, not a phosphoinositide 3-kinase. Autophagy 7, 563–564. doi:10.4161/auto.7.6.14873
Merlini, L., and Nishino, I. (2014). 201st ENMC international workshop: autophagy in muscular dystrophies – translational approach, 1-3 November 2013, Bussum, The Netherlands. Neuromuscul. Disord. 24, 546–561. doi:10.1016/j.nmd.2014.03.009
Metzger, T., Gache, V., Xu, M., Cadot, B., Folker, E. S., Richardson, B. E., et al. (2012). MAP and kinesin-dependent nuclear positioning is required for skeletal muscle function. Nature 484, 120–124. doi:10.1038/nature10914
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mizushima, N. (2007). Autophagy: process and function. Genes Dev. 21, 2861–2873. doi:10.1101/gad.1599207
Muller, A. J., Baker, J. F., Duhadaway, J. B., Ge, K., Farmer, G., Donover, P. S., et al. (2003). Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol. Cell. Biol. 23, 4295–4306. doi:10.1128/MCB.23.12.4295-4306.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neukomm, L. J., Nicot, A. S., Kinchen, J. M., Almendinger, J., Pinto, S. M., Zeng, S., et al. (2011). The phosphoinositide phosphatase MTM-1 regulates apoptotic cell corpse clearance through CED-5-CED-12 in C. elegans. Development 138, 2003–2014. doi:10.1242/dev.060012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicot, A. S., Toussaint, A., Tosch, V., Kretz, C., Wallgren-Pettersson, C., Iwarsson, E., et al. (2007). Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 39, 1134–1139. doi:10.1038/ng2086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osorio, D. S., and Gomes, E. R. (2014). Connecting the nucleus to the cytoskeleton for nuclear positioning and cell migration. Adv. Exp. Med. Biol. 773, 505–520. doi:10.1007/978-1-4899-8032-8_23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parrish, W. R., Stefan, C. J., and Emr, S. D. (2004). Essential role for the myotubularin-related phosphatase Ymr1p and the synaptojanin-like phosphatases Sjl2p and Sjl3p in regulation of phosphatidylinositol 3-phosphate in yeast. Mol. Biol. Cell 15, 3567–3579. doi:10.1091/mbc.E04-03-0209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peter, B. J., Kent, H. M., Mills, I. G., Vallis, Y., Butler, P. J., Evans, P. R., et al. (2004). BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303, 495–499. doi:10.1126/science.1092586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pierson, C. R., Dulin-Smith, A. N., Durban, A. N., Marshall, M. L., Marshall, J. T., Snyder, A. D., et al. (2012). Modeling the human MTM1 p.R69C mutation in murine Mtm1 results in exon 4 skipping and a less severe myotubular myopathy phenotype. Hum. Mol. Genet. 21, 811–825. doi:10.1093/hmg/ddr512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pietri-Rouxel, F., Gentil, C., Vassilopoulos, S., Baas, D., Mouisel, E., Ferry, A., et al. (2010). DHPR alpha1S subunit controls skeletal muscle mass and morphogenesis. EMBO J. 29, 643–654. doi:10.1038/emboj.2009.366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Praefcke, G. J., and McMahon, H. T. (2004). The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 5, 133–147. doi:10.1038/nrm1313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prokic, I., Cowling, B. S., and Laporte, J. (2014). Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. 92, 453–463. doi:10.1007/s00109-014-1138-1
Reifler, A., Li, X., Archambeau, A. J., McDade, J. R., Sabha, N., Michele, D. E., et al. (2014). Conditional knockout of pik3c3 causes a murine muscular dystrophy. Am. J. Pathol. 184, 1819–1830. doi:10.1016/j.ajpath.2014.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robb, S. A., Sewry, C. A., Dowling, J. J., Feng, L., Cullup, T., Lillis, S., et al. (2011). Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul. Disord. 21, 379–386. doi:10.1016/j.nmd.2011.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robinson, F. L., and Dixon, J. E. (2006). Myotubularin phosphatases: policing 3-phosphoinositides. Trends Cell Biol. 16, 403–412. doi:10.1016/j.tcb.2006.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rohde, H. M., Tronchere, H., Payrastre, B., and Laporte, J. (2009). Detection of myotubularin phosphatases activity on phosphoinositides in vitro and ex vivo. Methods Mol. Biol. 462, 265–278. doi:10.1007/978-1-60327-115-8_18
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Romero, N. B. (2010). Centronuclear myopathies: a widening concept. Neuromuscul. Disord. 20, 223–228. doi:10.1016/j.nmd.2010.01.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Romero, N. B., and Bitoun, M. (2011). Centronuclear myopathies. Semin. Pediatr. Neurol. 18, 250–256. doi:10.1016/j.spen.2011.10.006
Sandri, M. (2013). Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 45, 2121–2129. doi:10.1016/j.biocel.2013.04.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sandri, M., Coletto, L., Grumati, P., and Bonaldo, P. (2013). Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell. Sci. 126, 5325–5333. doi:10.1242/jcs.114041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schessl, J., Medne, L., Hu, Y., Zou, Y., Brown, M. J., Huse, J. T., et al. (2007). MRI in DNM2-related centronuclear myopathy: evidence for highly selective muscle involvement. Neuromuscul. Disord. 17, 28–32. doi:10.1016/j.nmd.2006.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, L. L., Gupta, V. A., and Beggs, A. H. (2014). Bridging integrator 1 (Bin1) deficiency in zebrafish results in centronuclear myopathy. Hum. Mol. Genet. 23, 3566–3578. doi:10.1093/hmg/ddu067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, G. S., Maehama, T., and Dixon, J. E. (2000). Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. U. S. A. 97, 8910–8915. doi:10.1073/pnas.160255697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thompson, H. M., Cao, H., Chen, J., Euteneuer, U., and McNiven, M. A. (2004). Dynamin 2 binds gamma-tubulin and participates in centrosome cohesion. Nat. Cell Biol. 6, 335–342. doi:10.1038/ncb1112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tosch, V., Rohde, H. M., Tronchere, H., Zanoteli, E., Monroy, N., Kretz, C., et al. (2006). A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum. Mol. Genet. 15, 3098–3106. doi:10.1093/hmg/ddl250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toussaint, A., Cowling, B. S., Hnia, K., Mohr, M., Oldfors, A., Schwab, Y., et al. (2011). Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. 121, 253–266. doi:10.1007/s00401-010-0754-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Treves, S., Anderson, A. A., Ducreux, S., Divet, A., Bleunven, C., Grasso, C., et al. (2005). Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul. Disord. 15, 577–587. doi:10.1016/j.nmd.2005.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tronchere, H., Buj-Bello, A., Mandel, J. L., and Payrastre, B. (2003). Implication of phosphoinositide phosphatases in genetic diseases: the case of myotubularin. Cell. Mol. Life Sci. 60, 2084–2099. doi:10.1007/s00018-003-3062-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tronchere, H., Laporte, J., Pendaries, C., Chaussade, C., Liaubet, L., Pirola, L., et al. (2004). Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin in mammalian cells. J. Biol. Chem. 279, 7304–7312. doi:10.1074/jbc.M311071200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trump, N., Cullup, T., Verheij, J. B., Manzur, A., Muntoni, F., Abbs, S., et al. (2011). X-linked myotubular myopathy due to a complex rearrangement involving a duplication of MTM1 exon 10. Neuromuscul. Disord. 22, 384–388. doi:10.1016/j.nmd.2011.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, T. C., Horinouchi, H., Noguchi, S., Minami, N., Murayama, K., Hayashi, Y. K., et al. (2005). Characterization of MTM1 mutations in 31 Japanese families with myotubular myopathy, including a patient carrying 240 kb deletion in Xq28 without male hypogenitalism. Neuromuscul. Disord. 15, 245–252. doi:10.1016/j.nmd.2004.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Udd, B. (2012). Distal myopathies – new genetic entities expand diagnostic challenge. Neuromuscul. Disord. 22, 5–12. doi:10.1016/j.nmd.2011.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Velichkova, M., Juan, J., Kadandale, P., Jean, S., Ribeiro, I., Raman, V., et al. (2010). Drosophila Mtm and class II PI3K coregulate a PI(3)P pool with cortical and endolysosomal functions. J. Cell Biol. 190, 407–425. doi:10.1083/jcb.200911020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vergne, I., and Deretic, V. (2010). The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 584, 1313–1318. doi:10.1016/j.febslet.2010.02.054
Wang, X., and Robbins, J. (2013). Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell. Cardiol. 71, 16–24. doi:10.1016/j.yjmcc.2013.11.006
Wilmshurst, J. M., Lillis, S., Zhou, H., Pillay, K., Henderson, H., Kress, W., et al. (2010). RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol. 68, 717–726. doi:10.1002/ana.22119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, T., Shi, Z., and Baumgart, T. (2014). Mutations in BIN1 associated with centronuclear myopathy disrupt membrane remodeling by affecting protein density and oligomerization. PLoS ONE 9:e93060. doi:10.1371/journal.pone.0093060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, S., Manson, J., White, S., Bourne, A., Waddy, H., Davis, M., et al. (2003). X-linked myotubular myopathy in a family with three adult survivors. Clin. Genet. 64, 148–152. doi:10.1034/j.1399-0004.2003.00118.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, H., Rokach, O., Feng, L., Munteanu, I., Mamchaoui, K., Wilmshurst, J. M., et al. (2013). RyR1 deficiency in congenital myopathies disrupts excitation-contraction coupling. Hum. Mutat. 34, 986–996. doi:10.1002/humu.22326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhuang, X., Wang, H., Lam, S. K., Gao, C., Wang, X., Cai, Y., et al. (2013). A BAR-domain protein SH3P2, which binds to phosphatidylinositol 3-phosphate and ATG8, regulates autophagosome formation in Arabidopsis. Plant Cell 25, 4596–4615. doi:10.1105/tpc.113.118307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zou, W., Lu, Q., Zhao, D., Li, W., Mapes, J., Xie, Y., et al. (2009). Caenorhabditis elegans myotubularin MTM-1 negatively regulates the engulfment of apoptotic cells. PLoS Genet. 5:e1000679. doi:10.1371/journal.pgen.1000679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zuchner, S., Noureddine, M., Kennerson, M., Verhoeven, K., Claeys, K., De Jonghe, P., et al. (2005). Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat. Genet. 37, 289–294. doi:10.1038/ng1514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: centronuclear myopathy, myotubular myopathy, MTM1 myotubularin gene, DNM2 dynamin-2 gene, BIN1 bridging integrator-1/amphiphysin-2 gene, RYR1 ryanodine receptor-1 gene, TTN titin gene, autophagy
Citation: Jungbluth H and Gautel M (2014) Pathogenic mechanisms in centronuclear myopathies. Front. Aging Neurosci. 6:339. doi: 10.3389/fnagi.2014.00339
Received: 22 October 2014; Paper pending published: 06 November 2014;
Accepted: 02 December 2014; Published online: 19 December 2014.
Edited by:
Luciano Merlini, Istituto Ortopedico Rizzoli IRCCS, ItalyReviewed by:
Jocelyn Laporte, INSERM, FranceDenise Alessandra Cassandrini, IRCCS Fondazione Stella Maris, Italy
Copyright: © 2014 Jungbluth and Gautel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heinz Jungbluth, Children’s Neuroscience Centre, St Thomas’ Hospital, Floor 01 – Staircase D South Wing, London SE1 7EH, UK e-mail:aGVpbnouanVuZ2JsdXRoQGdzdHQubmhzLnVr