 Christian Humpel
Christian Humpel- Laboratory of Psychiatry and Experimental Alzheimer's Research, Department of Psychiatry and Psychotherapy, Medical University of Innsbruck, Innsbruck, Austria
Alzheimer's disease is a severe neurodegenerative disorder of the brain, pathologically characterized by extracellular beta-amyloid plaques, intraneuronal Tau inclusions, inflammation, reactive glial cells, vascular pathology and neuronal cell death. The degradation and clearance of beta-amyloid plaques is an interesting therapeutic approach, and the proteases neprilysin (NEP), insulysin and matrix metalloproteinases (MMP) are of particular interest. The aim of this project was to establish and characterize a simple in vitro model to study the degrading effects of these proteases. Organoytpic brain vibrosections (120 μm thick) were sectioned from adult (9 month old) wildtype and transgenic mice (expressing amyloid precursor protein (APP) harboring the Swedish K670N/M671L, Dutch E693Q, and Iowa D694N mutations; APP_SDI) and cultured for 2 weeks. Plaques were stained by immunohistochemistry for beta-amyloid and Thioflavin S. Our data show that plaques were evident in 2 week old cultures from 9 month old transgenic mice. These plaques were surrounded by reactive GFAP+ astroglia and Iba1+ microglia. Incubation of fresh slices for 2 weeks with 1–0.1–0.01 μg/ml of NEP, insulysin, MMP-2, or MMP-9 showed that NEP, insulysin, and MMP-9 markedly degraded beta-amyloid plaques but only at the highest concentration. Our data provide for the first time a potent and powerful living brain vibrosection model containing a high number of plaques, which allows to rapidly and simply study the degradation and clearance of beta-amyloid plaques in vitro.
Introduction
Sporadic Alzheimer's disease (AD) is a progressive chronic neurodegenerative disorder (at least 97% of all cases are non-genetic) and is characterized by severe beta-amyloid (Aβ) deposition in brain (plaques) and in vessels (cerebral Aβ angiopathy, CAA), Tau pathology, cell death of cholinergic neurons, microglial activation, inflammation and cerebrovascular damage. The causes of AD are yet unknown, but so far the Aβ cascade (Selkoe, 2002) is the most prominent hypothesis and is thought to be the primary event that triggers the pathological cascade in AD. However, there is still an ongoing controversy if Aβ plaques directly cause the disease, or if Aβ plaques are generated secondary to another (e.g., vascular) pathological event.
The amyloid precursor protein (APP) is cleaved by secretases into Aβ peptides (40, 42, or 43 amino acids), and these peptides aggregate under certain conditions and are deposited as Aβ plaques. The plaques consist of a central core of highly aggregated Aβ peptides and a halo around the plaques (Wisniewski et al., 1989). It is well established that the “halo” consists of degenerating nerve fibers and several infiltrating cells, like reactive astrocytes or microglia. The most robust deficits occur in this halo around the plaques within a radius of approximately 20 μm, including local alterations in spine density, neuritic curvature, calcium dysregulation and oxidative stress (Xie et al., 2013). The “core” consists of a dense aggregation of large or several small Aβ depositions, especially the toxic Aβ1–42. Brain capillary vessels penetrate the core and it seems that every plaque is associated with a vessel.
In severe AD, the whole brain is filled with several Aβ plaques. Therapeutic options aim to prevent the deposition of plaques (beta-sheet breaker, secretase inhibitors, Aβ-degrading enzymes) to degrade and clear the plaques (Glabe, 2000; Wang et al., 2006). Immune cells, especially microglia are of special interest, because they (or transformed macrophages) may phagocyte plaques. However, in AD the phagocytic activity of these microglial cells is diminished (D'Andrea et al., 2004; Lee and Landreth, 2010). Thus, strategies explore if endogenous blood cells, especially monocytes, may enter the brain and migrate to plaques; indeed in the AD mouse model this has been demonstrated (Lebson et al., 2010; Hohsfield et al., 2014). Alternatively, it seems likely that infusion of plaque-degrading enzymes may be an attractive strategy to destruct plaques. Indeed, several Aβ-degrading proteases have been identified, including neprilysin (NEP; Iwata et al., 2001; Leissring et al., 2003; Marr et al., 2003), insulin-degrading enzyme (IDE; Qiu et al., 1998; Bennett et al., 2000; Kurochkin, 2001; Farris et al., 2003; Leissring et al., 2003), endothelin-converting enzyme (ECE; Eckman et al., 2001, 2002), angiotensin-converting enzyme (ACE; Hu et al., 2001), plasminogen activators (Van Nostrand and Porter, 1999; Ledesma et al., 2000; Tucker et al., 2000), myelin basic protein (Liao et al., 2009) or different matrix metalloproteinases, such as e.g., MMP-2 (gelatinase A/type IV collagenase) (Roher et al., 1994; Yamada et al., 1995) and MMP-9 (Backstrom et al., 1996; Yin et al., 2006).
However, these proteases have partly a wide range of substrates, are large molecules, and cannot pass the bood-brain barrier (BBB) and find their targets. Different strategies have been tested, especially in vivo in AD mouse models. However, these animal models are all very complex, either because the proteases need to be overexpressed using adeno- or lentiviral vectors (Guan et al., 2009; Liu et al., 2010) or because the enzymes are intracranially injected directly into the brain (Park et al., 2013; Walker et al., 2013) or peripherally applied (e.g., i.v.). Further, studies failed due to the lack of BBB penetration or the short half-life of the enzymes (Davis et al., 2004; Henderson et al., 2014). Thus in order to test a time- and dose specificity and selectivity and a possible combination of these enzymes, we certainly need easy to use in vitro models.
The aim of the present study was to develop a simple in vitro model which contains plaques and can be used to screen for plaque-degenerating enzymatic activity. So far no in vitro models exist, which contain plaques. As we have long-time experience in organotypic brain slices, we aimed to develop an in vitro organotypic vibrosection model which contains plaques and can be used to screen for clearance activities. We report here for the first time, that vibrosections prepared from adult whole brain Alzheimer mice are useful to screen different Aβ-degrading enzymes and this method is easy, fast and potent.
Materials and Methods
APP_SDI Mice and Controls
Wildtype (C57BL/6N) and transgenic APP_SDI (expressing amyloid precursor protein (APP) harboring the Swedish K670N/M671L, Dutch E693Q, and Iowa D694N mutations; C57BL/6-Tg(Thy1-APPSwDutIowa)BWevn/Mmjax) mice were purchased from The Jackson Laboratory and housed at the Medical University of Innsbruck animal facility providing open access to food and water under 12/12 h light-dark cycles. The mice have been generated and extensively characterized by Davis et al. (2004).
Organotypic Vibrosections of Adult Mice
For vibrosection cultures 9 month old adult mice were used. All experiments conformed to Austrian guidelines on the ethical use of animals and all efforts were made to minimize the number of animals used and their suffering. For all experiments a minimum of n = 6 slices was analyzed. Usually 2 slices per group including a control were incubated, which was repeated at least in 3 independent experiments. Vibrosections were performed as described in detail under sterile conditions (Ullrich et al., 2011). The animals were rapidly sacrificed, the brains dissected and sagittally cut. The brains were glued (Glue Loctite) onto the chuck of a water cooled vibratome Leica VT1000A (UV sterilized and under a Laminar Flow), and triggered close to a commercial shave racer. Under aseptic conditions, 120 μm vibrosections were cut and collected in sterile medium. The organotypic vibrosections were carefully placed onto a sterile 0.4 μm pore membrane (Millipore HTTP02500), which was then placed into a 0.4 μm membrane insert (Millipore PICM03050) within a 6-well plate. Vibrosections were cultured in 6-well plates (Greiner) at 37°C and 5% CO2 with 1.2 ml/well of the following culture medium: 50% MEM/HEPES (Gibco), 25% heat inactivated horse serum (Gibco/Lifetech, Austria), 25% Hanks' solution (Gibco), 2 mM NaHCO3 (Merck, Austria), 6.5 mg/ml glucose (Merck, Germany), 2 mM glutamine (Merck, Germany), pH 7.2. Vibrosections were incubated for 2 weeks where they attach to the membranes. In order to study if slices develop plaques, vibrosections from 4 month old APP_SDI Tg mice (which display no plaques at this stage) were incubated for up to 5 months.
Recombinant proteases were dissolved in 100 μl sterile medium, aliquoted and stored frozen at −20°C; at the day of use, aliquots were diluted in sterile medium and not filtered. Vibrosections were cultured with or without 1, 0.1, or 0.01 μg/ml recombinant human neprilysin/CD10 (R&D, Nr. 1182-ZNC-010), human insulysin/IDE (R&D systems, nr. 2496-ZN-010), human matrix-metalloproteinase-2 (MMP-2; Peprotech Nr. 420-02) or human matrix-metalloproteinase-9 (Sigma, nr. M8945). At the end of the experiment, vibrosections were fixed for 3 h at 4°C in 4% paraformaldehyde (PAF)/10 mM phosphate-buffered saline (PBS) and then stored at 4°C in PBS until use.
Immunohistochemistry
Immunohistochemistry was performed as previously described under free-floating conditions (Ullrich et al., 2011). The vibrosections were washed with PBS and incubated in PBS/0.1% Triton (T-PBS) for 30 min at 20°C while shaking. To quench endogenous peroxidase, sections were treated with PBS/1%H2O2/5% methanol. After incubation, the sections were then blocked in T-PBS/20% horse serum (GIBCO Invitrogen)/0.2% BSA (SERVA) for 30 min at 20°C shaking. Following blocking with mouse IgG blocking reagent (Vector MKB-2213), brain sections were incubated with primary antibody [beta-amyloid, 1–16 (6E10) Covance SIG-39300; glial fibriallary acid protein GFAP Millipore AB5541; microglial Iba1 Wako 019-19741] in T-PBS/0.2% BSA overnight at 20°C. The sections were then washed and incubated with the corresponding biotinylated secondary (Aβ-mouse; GFAP-chicken; Iba1-rabbit) antibody (1:200, Vector Laboratories) in T-PBS/0.2% BSA for 1 h at 20°C shaking. Following secondary antibody incubation, sections were rinsed with PBS and incubated in avidin-biotin complex solution (Elite ABC kit, Vector Laboratories) for 1 h at 20°C shaking. Finally, the sections were washed with 50 mM Tris-buffered saline (TBS) and then incubated in 0.5 mg/ml 3,3′-diaminobenzidine (DAB, Sigma)/TBS/0.003% H2O2 at 20°C in the dark until a signal was detected. Once DAB staining was visible, the reaction was stopped by adding TBS to the sections. The brain sections were rinsed with TBS, inserted into a 6-well plate (slice down to well surface) directly onto a drop of Vectashield (Vector), cover-slipped and then evaluated under an inverse microscope (Leica DM IRB). Some sections were stained with Thioflavin S (Sigma) to label plaques. Some sections were stained for fluorescence using Alexa-488 (Aβ) and using Alexa-546 (GFAP).
Western Blot
Western blot analysis was performed as previously described by us (Hohsfield et al., 2014). Slices were incubated for 2 weeks, and all slices from 3 wells were taken and pooled in an Eppendorf tube, then dissolved in 100 μl ice-cold PBS containing a protease inhibitor cocktail (P-8340, Sigma), homogenized using an ultrasonic device (Hielscher Ultrasonic Processor, Germany) and then centrifuged at 16,000 × g for 60 min at 4°C. The supernatant was collected (=soluble extract) and the pellet was dissolved in 50 μl 70% formic acid, vortexed and neutralized with 150 μl 7N NaOH (=insoluble extract). Then, 20 μl of the extracts were loaded onto 10% Bis-Tris SDS-polyacrylamide gels, and separated for 25 min at 200 V and then electrotransferred to nylon-PVDF Immobilon-PSQ membranes for 90 min at 30 V in 20% methanol blotting buffer. The Western Breeze Chromogenic System was used for the detection of specific proteins in cortical extracts. Briefly, blots were blocked for 30 min in blocking buffer, incubated with primary antibodies against beta-amyloid (1:1000) at 4°C overnight, washed, and then incubated in alkaline phosphatase conjugated anti-mouse IgG for 30 min. After washing, bound antibodies were detected using an enhanced chemiluminescence (ECL) system. As a control aggregated beta-amyloid standards (Calbiochem Aβ1–42, PP69, Mw = 4417 kDa) were loaded. Aggregation was performed as described by Ryan et al. (2010). Briefly, 250 μg recombinant Aβ1–42was dissolved in 250 μl 5% acidic acid (226 μM), 10 min sonicated in a water bath, then diluted to 100 μM in PBS + 0.005% sodiumdodecylsulfate (SDS), then incubated overnight at 4°C, and further diluted in PBS to 11 μM, and incubated for 2 weeks at 4°C. Then the aggregates were further incubated for 2 weeks at 37°C with or without 1 μg/ml MMP-9.
Evaluation of Plaques
Vibrosections at the cortical level were photographed with the Leica inverse microscope at a 10× magnification under a red filter. The exposure time was always 23 ms with the bright light set at the lowest level. The software Openlab was used at a Mac computer connected to the microscope. Pictures were saved as JPG files and the analysis was performed using Image J. The pictures were transformed to a 8-bit grayscale image. The calibration was set at 0.470 (distance in pixels), 1.00 (known distance), 1.0 (pixel aspect ratio) and μm (unit in lenght) and global was activated. The picture was transformed into a binary image and the threshold was adapted to 30–40. The number of particles was counted setting the size to 100–8000 μm2. The number of plaques was counted in a defined circle in 2.5 mm2 area. Examples of plaque evaluations are given in the Figures 4E–G.
Statistical Analysis
Statistical analysis was performed by a Kruskal–Wallis test and Dunn's post-hoc test, where p < 0.05 represents significance.
Results
Plaques in the 9 Month Old APP_SDI Mice
Organotypic vibrosections were cultured for 2 weeks and the plaques were stained by immunohistochemistry for Aβ or Thioflavin S. In cultured wildtype mice vibrosections (9 month old, 120 μm, 2 weeks) no plaques were visible (Figure 1A). In cultured APP_SDI mice vibrosections a high number of plaques were stained by Aβ immunostaining (Figure 1B) or Thioflavin S dye (Figure 1D). As a control the primary antibody was omitted in transgenic vibrosections and no plaques were found (Figure 1C). It became evident, that reactive astrocytes expressing GFAP were markedly enhanced in transgenic (Figure 2B) but not wildtype (Figure 2A) vibrosections, which centered around the Aβ plaques (Figures 2E–G). Similarly, microglial Iba1 staining was markedly increased in transgenic versus wildtype vibrosections (Figures 2C,D).

Figure 1. Beta-amyloid plaques in the amyloid-precursor protein_SDI mouse model vibrosections. Organotypic vibrosections (120 μm thick) of 9 month old wildtype (Wt, A) or transgenic (Tg, B–D) mice were prepared and cultured for 2 weeks, then fixed and immunohistochemically stained for beta-amyloid (A–C) or Thioflavin S (D). As a control Tg sections were stained without primary antibody (C). Note a dense number of plaques in 2 week old cultured vibrosections of 9 month old APP_SDI mice (B). Scale bar in A = 200 μm (A–C) and 120 μm (D).
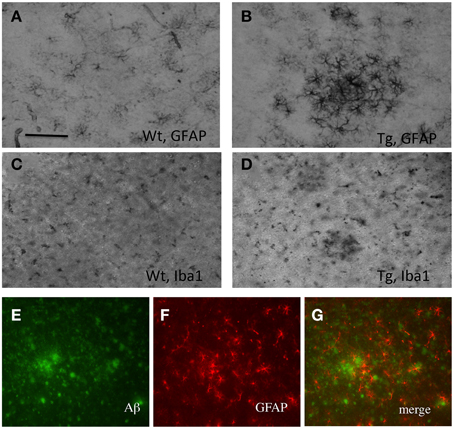
Figure 2. Characterization of plaques in vibrosections of 9 month old amyloid-precursor protein_SDI transgenic mouse model. Organotypic vibrosections of 9 month old wildtype (Wt; A,C) or transgenic (Tg; B,D–G) mice were prepared (120 μm thick) and cultured for 2 weeks, then fixed and immunohistochemically stained for astroglial glial-fibrillary acidic protein (GFAP; A,B,F) or microglial Iba1 (C,D). Astroglial GFAP (F) was co-stained with beta-amyloid (E), showing reactive astroglia around plaques (G). Scale bar in A = 150 μm (A–D) and 100 μm (E–G).
Development of Plaques
In order to study if slices develop plaques, vibrosections from 4 month old APP_SDI Tg mice (which display no plaques at this stage) were incubated for up to 5 months and then stained for Aβ plaques. Slices incubated for 1 or 2 months did not show any Aβ staining (Figures 3A,B). However, after 3 months and more pronounced after 4 and 5 months of incubation several intracellular Aβ-like depositions were seen, but no extracellular plaques were found (Figures 3C–E).

Figure 3. Vibrosections from 4 month old APP_SDI mice (having no plaques at this stage) were incubated for 1 (A), 2 (B), or 5 (C–E) months, then fixed and stained for Aβ. No staining was visible after 1 or 2 months of incubation, however, after 3 and more pronounced after 4 and 5 months several Aβ+ intracellular depositions were seen. The 2 pictures in the last row (D,E) show high magnifications taken from panel (C). However, at any stage no extracellular plaques were found. Scale bar in A = 200 μm (A–C) and 12 μm (D,E).
Clearance of Plaques
When 9 month old vibrosections of APP_SDI mice were cultured for 2 weeks with 1 μg/ml recombinant neprilysin (Figure 4D) or insulysin or MMP-9, a significant reduction of the number of plaques was observed (Figure 5) compared to controls (Figure 4A). The incubation with 1 μg/ml MMP-2 was not successful (Figure 5). No effect on plaque clearance was seen when the vibrosections were incubated with 100 or 10 ng/ml of all tested enzymes (Figures 4B,C, 5).
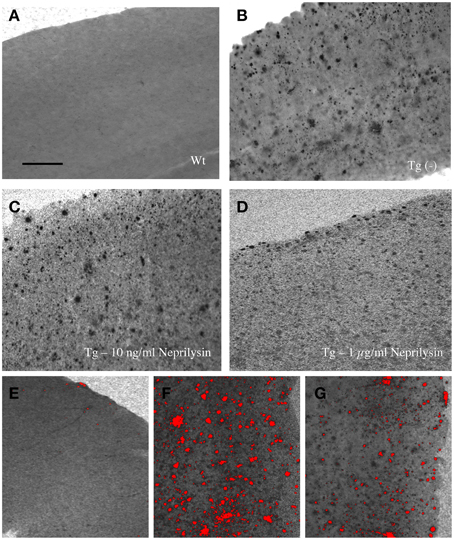
Figure 4. Effects of neprilysin on beta-amyloid (Aβ) plaques in 9 month old amyloid-precursor protein_SDI mouse model vibrosections and computer-assisted analysis (E-G). Organotypic vibrosections of 9 month old wildtype (Wt, A,E) or transgenic (Tg, B–D,F,G) mice were prepared (120 μm thick) and cultured for 2 weeks without (A,B) or with 10 ng/ml (C) or 1 μg/ml (D) neprilysin, then fixed and immunohistochemically stained for beta-amyloid (A–G). Note the decreased number of plaques in vibrosections incubated with 1 μg/ml recombinant neprilysin for 2 weeks (D). The number of plaques was counted using computer-assisted imaging (E–G), showing no plaques in Wt vibrosections (E), a high number of plaques in Tg vibrosections (F) and a reduced number of plaques in the neprilysin treated vibrosections (G). Scale bar in A = 200 μm (A–G).
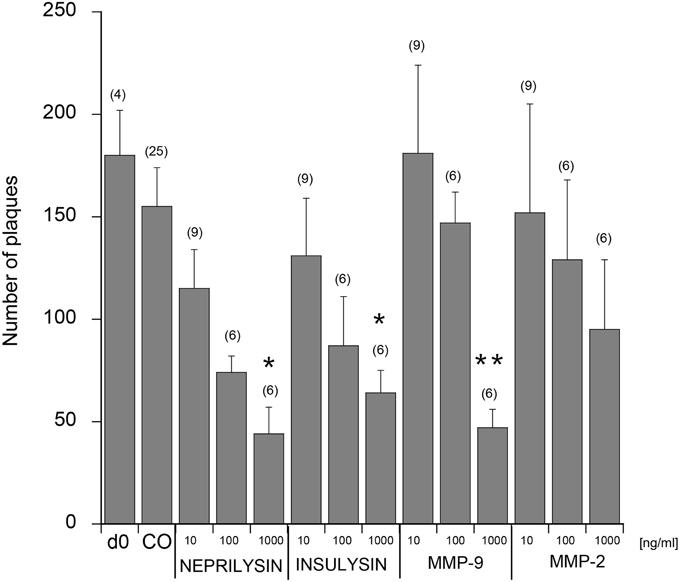
Figure 5. Quantitative analysis of beta-amyloid (Aβ) plaques in the cortical vibrosections. Vibrosections of 9 month old adult amyloid-precursor protein_SDI mouse model were cultured for 2 weeks without (CO) or with 10, 100, or 1000 ng/ml neprilysin, insulysin, matrix metalloproteinase (MMP) −2 or −9. Slices were then fixed and immunohistochemically stained for plaques with a beta-amyloid antibody. As a comparison the number of plaques is given from brain sections at day zero (d0). The number of beta-amyloid plaques was evaluated using computer-assisted microscopy. Values are given as mean ± SEM plaques per 2.5 mm2; the values in parenthesis give the number of analyzed slices. Statistical analysis was performed by a Kruskal–Wallis test and Dunn's post-hoc test (*p < 0.05; **p < 0.01).
Western Blot Analysis
The Aβ standards were detectable as a small peptide at approximately 4 kDa with a detection limit as low as 10 ng per lane (Figure 6). When aggregated Aβ was loaded several aggregated forms (mainly 8–12–16 kDa) were visible, however, also larger smear-like aggregates were found (Figure 6). No Aβ-immunoreactivity was seen in soluble aggregates or soluble slice extracts (Figure 6). When insoluble slice extracts were analyzed by Western Blot, no staining was seen in Wt mice, however, a strong staining was seen for 4–16 kDa Aβ forms and a strong larger smear (Figure 6). However, no clear difference was seen between MMP-9 treated or untreated vibrosections (Figure 6).
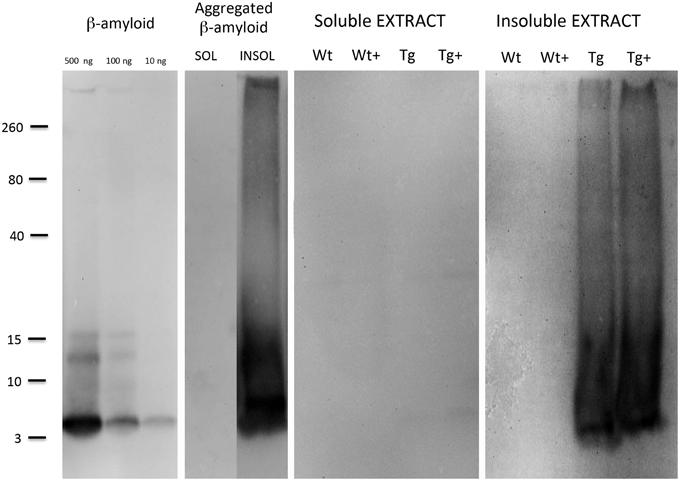
Figure 6. Western Blot analysis of brain slice extracts. Beta-amyloid (10–100–500 ng per lane), or aggregated beta-amyloid (2w 4°C + 2w 37°C) or slice extracts (either soluble or insoluble extracts) were loaded onto gels, separated for 25 min, blotted with 20% methanol and analyzed for beta-amyloid by enhanced chemiluminescence. Size markers are given on the left side as kDa. Slices were prepared from 9 month old wildtype (Wt) or APP_SDI transgenic (Tg) mice and incubated for 2 weeks with or without 1 μg/ml MMP-9 (Wt+ or Tg+).
Discussion
The organotypic brain slice and vibrosection model is well-established and serves as a validated tool to study neurodegeneration or neuroprotection (Stoppini et al., 1991; Gähwiler et al., 1997; Schatz et al., 1999; Weis et al., 2001; Humpel and Weis, 2002; Ullrich et al., 2011). Usually brain slices are derived from young postnatal brains, however, such slices are not useful, because plaques develop usually between 4 and 6 months of age in tg mice. Previously, we have developed a model from postnatal wildtype rats (Marksteiner and Humpel, 2008), however, such “Aβ-like depositions” are not fully comparable to the Aβ plaques-derived from adult brains. In the present study we developed for the first time an organotypic vibrosection model from adult AD mice, and demonstrate that this model shows Aβ plaques and is useful to screen for Aβ-degrading enzymes.
Adult Brain Slices
Usually most brain slices are derived from postnatal animals, as it is difficult to properly prepare and maintain adult slices. Culturing of adult brain slices is very tricky and has been reported by only a few groups with several limitations, especially the short life time and reduced viability of the slices. Finley et al. (2004) cultured juvenile young adult (postnatal P20-21 days) hippocampal slices and reported maintained neuronal architecture and synaptic activity. Kim et al. (2013) established adult organotypic hippocampal slices in serum-free culture medium for 30 days and found well preserved astrocytes, neural processes and synapses. Su et al. (2011) cultured adult hippocampal slices which led to a time-dependent reproducible cell death. They reported that slices lost 50% of their cells after 6 days in vitro, while brain-derived neurotrophic factor supplementation promoted cell survival. Wilhelmi et al. (2002) reported the culturing of adult hippocampal slices in a cerebrospinal fluid-like medium, which markedly prolonged culture times up to 5–6 days. Anyhow, the need of special supplemented adult slice (serum-free) medium needs to be further optimized. So far, none of the published conditions for adult slices are accepted and established.
Development of Plaques in Vibrosections
In our first approach we wanted to test if plaques develop in organotypic vibrosections. Thus, we cultured brain sections from 4 month old transgenic mice. At this stage no plaques are seen in the brain. We hypothesized that culturing of such slices for up to 5 months may induce Aβ plaque deposition, because 9 month old tg mice contain a large plaque load. Our data shows that up to 2 months of culturing, no Aβ-immunostaining was evident, however, after 3–5 months of culturing a strong intracellular Aβ immunoreactivity was found. At any stage we never observed extracellular Aβ plaque depositions in the vibrosections. Thus, this model may not be useful to screen for the effects of Aβ-degrading enzymes, but may rather be very useful to test the processes of Aβ processing, release and extracellular deposition to plaques. Thus, further experiments are necessary to test the conditions and factors, which may induce an extracellular plaque deposition in vitro similar as seen in the vivo situation.
Plaques in Advanced AD Stages
In order to study plaques in vitro and to investigate Aβ-degrading enzymes, we developed an in vitro vibrosection model from 9 month old tg mice, which contain a high plaque load at this stage in the whole brain (Davis et al., 2004). Our data clearly show that culturing of 120 μm adult vibrosections for 2 weeks maintains many Aβ plaques. These plaques in vitro are typically of the same pattern and size as the plaques seen in vivo, showing strong Aβ depositions. The number of plaques did not increase during culturing for 2 weeks. Using Western blots we confirmed that the vibrosections contained aggregated insoluble Aβ at this stage. Further we show, that these plaques are surrounded by reactive healthy GFAP+ astrocytes. Similarly, also reactive Iba1+ microglia have been found in these slices. This is a typical cellular pattern, that reactive glial cells surround the plaques. However, we cannot exclude that additional traumatic effects at both sides of the vibrosection occurs due to slicing, which may influence the plaque composition.
Degradation and Clearance of Plaques with Neprilysin
Neprilysin is a 85–110 kDa plasma membrane glycoprotein of the neutral zinc metalloendo-peptidase family expressed by neurons and cerebrovascular smooth muscle cells. Neprilysin seems to be the most potent Aβ-degrading enzyme (Iwata et al., 2001) and several studies demonstrated its effect. In fact, several cleavage sites of NEP within the Aβ sequence have been identified (Grimm et al., 2013). However, it is worth mentioning that NEP cleaves monomeric Aβ1–40 more efficiently than Aβ1–42; in vitro NEP degraded 27% of monomeric Aβ1–42, but cleaved 73% of Aβ1–40 monomers (Grimm et al., 2013). It has been shown that intracerebral infusion of a recombinant soluble neprilysin from insect cells into AD mice improved memory and reduced Aβ accumulation in the brain (Park et al., 2013). Further an intravenous infusion of a NEP fusion protein resulted in dose-dependent clearance of Aβ, but did not affect brain Aβ, however, an intracranial infusion was successful (Walker et al., 2013). Liu et al. (2010) produced a secreted form of NEP by adeno-associated transduction and showed that plasma NEP decreased plasma Aβ by 30% and cleared brain Aβ by 28–55% within 3 months. In contrast, overexpression of NEP on the surface of leucocytes (by lentiviral induction) and periphal infusion reduced soluble Aβ in brain by 30% and lowered CAA Aβ by 50–60% (Guan et al., 2009). In contrast, another group fused NEP to albumin to increase plasma half-life time and showed that repated i.v. infusion for 4 months in Tg2576 mice (and rats and monkeys) cleared plasma Aβ but not brain or CSF Aβ (Henderson et al., 2014). Some studies were conducted also in vitro, however, their conclusions are limited due to the simplicity of the model. Miners et al. (2011) showed that NEP overexpression in primary human adult cerebrovascular smooth muscle cells counteracted Aβ-induced toxicity. In cultured astrocytes, ketamine (an NMDA antagonist) and MK-801 decreased NEP expression via de-phosphorylation of p38MAPK, suggesting that these signal pathways are linked to Aβ degradation (Yamamoto et al., 2013). All in all, the in vivo data are partly not conclusive, although NEP seems to have the most prominent Aβ-degrading activity. Our present data indeed show that recombinant NEP degrades Aβ plaques, but only at the highest concentration of 1 μg/ml. However, we cannot exclude that the added proteases underwent autodegradation or that the enzymes added to the medium did not efficiently diffuse into the slice. In this case it would be helpful to determine the catalytic activity in slice extracts under optimized conditions.
Insulysin, MMPs, and Other Enzymes
Yan et al. (2006) reported that MMP-9 was very potent to degrade soluble fibrillar Aβ, but this ability was not shared by other enzymes examined, including ECE, IDE and neprilysin. In fact, MMP-9 was also the most potent enzyme to degrade Aβ in our hands. MMP-9 is a zinc dependent metalloprotease and initially synthesized as the inactive proenzyme pro-MMP-9, which is cleaved into an active form upon release by other proteases. MMP-9 is expressed in many cells in the brain, neurons, astroglia, microglia, vascular cells and is increased in the brain of AD patients (Dzwonek et al., 2004; Nalivaeva et al., 2008). MMP-9 is expressed by reactive astrocytes around Aβ plaques, and it is suggested that these cells may contribute in plaque clearance (Yan et al., 2006; Yin et al., 2006).
Our data show, however, in contrast to Yan et al. (2006) that also neprilysin and IDE was very potent to degrade Aβ plaques. This must not be necessarily a contrast, because, the cleavage site of the enzymes must be considered. Studies examining different fragments of Aβ report on the importance of 2 critical regions for aggregation at positions 17–21 and 30–35 (see Yan et al., 2006). The suggested cleavage site of MMP-9 is Phe20-Ala21 and Ala30-Ile31 (Yan et al., 2006). However, the plaques in the different AD mouse models are heterogenous, because the mutation sites differ within the Aβ sequence. In our APP_SDI mouse model 3 mutations have been inserted (Swedish K670N/M671L); Dutch E22Q (E693Q) and Iowa D23N (D694N). Thus, the effects of the different proteases can vary in the different AD mouse models used.
In addition, a number of enzymes or chemicals have been reported to degrade plaques, however, often their effects are not well characterized, regarding either dosage or specificity of the substrate or selectivity to find the targets. Such a new enzyme is e.g., nardilysin (a N-arginine dibasic convertase; NRDc, initially identified as a metalloendopeptidase of the M16 family), which has been reported to prevent amyloid plaque formation by enhancing alpha-secretase in an AD mouse model (Ohno et al., 2014). Finally, several other enzymes or substrates may induce Aβ degradation/clearance in the brain and may become a potent therapeutic target, such as e.g., BACE2, nattokinase or neprilysin-2 (Nalivaeva et al., 2008; Hsu et al., 2009; Hafez et al., 2010; Abdul-Hay et al., 2012). Further, different substrates (e.g., estrogens or BRI2 protein) may indirectly have Aβ-degrading activity via activation of Aβ-degrading enzymes (Kilger et al., 2011; Merlo and Sortino, 2012).
Advantage of the Adult AD In Vitro Model
Many of the reported enzymes have been shown to degrade soluble Aβ, but not all of them have been convincingly shown to degrade Aβ fibrils or compact plaques. The problems and limits are clearly that the effects of these enzymes cannot be easily studied in vivo in mouse models. The enzymes are large molecules, do not enter the BBB, have a wide range of substrates and side effects on peripheral organs or blood cells, when administered into the vein. Intraventricular infusions break up the BBB resulting in influx of blood-derived substrates and enzymes. Further, the stability of the applied enzymes is a problem, and only small amounts may enter the plaque site. An interesting experimental approach has been reported by Yan et al. (2006), who prepared 5 μm thin snap frozen cryostat sections from saline perfused AD mice (APP/PS1), incubated them in 70 nM protease (MMP-9, ECE, IDE, NEP) at 37°C for 5 days and stained them with Thioflavin S and observed degradation of plaques by MMP-9 only. This is an interesting model, however, since the slices have been fresh frozen, they are no longer considered as “living.” So far good in vitro models are missing and our novel model may provide for the first time a compact system to demonstrate clearance of plaques directly in a living mouse brain model allowing to study time- and dose-dependent effects.
Limit of the Model
This in vitro AD model has definitively several limitations. The most severe limitation is the low neuronal viability of the adult slices. Although it was out of focus to characterize the neuronal viability, we have preliminary data that the very sensitive dopaminergic and cholinergic neurons did not survive well in our adult wt or tg vibrosections even when incubated with growth factors. In fact, more work is needed to study and enhance the survival of neurons in adult slices, and the culture conditions must be adapted and optimized. Further, we cannot exclude that the enzymes (at highest concentration) may damage the vibrosections, especially the neurons. Cell death assays including e.g., propidiumiodine stainings, neuN immunostainings, and MAP-2 Western Blots may help to further characterize the adult slice model. So, far this present model may be limited but very powerful to study plaque development and degradation using Aβ-degrading enzymes.
Another problem, although not limited to the slice culture conditions, is the lack of Tau pathology, the second important pathological condition in AD. In most APP overexpressing models only Aβ plaques occur and no Tau pathology is evident. Alternatively, a triple transgenic mouse model (3xTg-AD, B6; 129-Psen1tm1MpmTg(APPSwe, tauB301L)1Lfa/J) could be useful which contains βA as well as Tau pathology. The problem is that the plaques and Tau pathology occur only at very lates age, approximately at 15–20 months, and it is not very convenient to use such old mice for cultures. Last not least the transgenic AD mouse model reflects more a genetic model of AD, which accounts for only <5% of all AD cases. As more than 95% are sporadic AD and the causes for AD are not known, animal models for sporadic AD have not been described to date.
Clear or Prevent?
The application of Aβ-degrading enzymes is a very interesting therapeutic option to degrade and clear plaques in severe AD patients. The problems of delivery, stability and selectivity must be resolved and thus this approach could become a potent technology to clear plaques at an end-stage. However, we also need therapeutic options to prevent plaques. Thus strategies need to be developed to apply such Aβ-degrading enzymes at a very early stage of the disease, such as in mild cognitive impairment. In fact, PET analysis shows that some individuals display already plaques at early age without having any cognitive symptoms. Such therapeutic approaches with proteases, however, must be safe, easy and specific. Organotypic vibrosections may help to understand and establish such therapeutic options with Aβ-degrading enzymes.
In conclusion our data provide for the first time a potent and powerful living brain vibrosection model containing a high number of plaques, which allows to rapidly and simply study the degradation and clearance of Aβ plaques in vitro.
Author Contributions
CH designed and analyzed the data and wrote the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Karin Albrecht for excellent technical help. I thank Mag. Nina Daschil for help in establishing adult organotypic brain slices and animal care. This study was supported by the Austrian Science Funds Sonderforschungsbereich F4405-B19 and by FWF P24734-B24.
References
Abdul-Hay, S. O., Sahara, T., McBride, M., Kang, D., and Leissring, M. A. (2012). Identification of BACE2 as an avid ß-amyloid-degrading protease. Mol. Neurodegener. 7:46. doi: 10.1186/1750-1326-7-46
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Backstrom, J. R., Lim, G. P., Cullen, M. J., and Tökés, Z. A. (1996). Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-beta peptide (1-40). J. Neurosci. 16, 7910–7919.
Bennett, R. G., Duckworth, W. C., and Hamel, F. G. (2000). Degradation of amylin by insulin-degrading enzyme. J. Biol. Chem. 275, 36621–36625. doi: 10.1074/jbc.M006170200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
D'Andrea, M. R., Cole, G. M., and Ard, M. D. (2004). The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol. Aging 25, 675–683. doi: 10.1016/j.neurobiolaging.2003.12.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, J., Xu, F., Deane, R., Romanov, G., Previti, M. L., Zeigler, K., et al. (2004). Early-onset and robust cerebral microvascular accumulation of beta-amyloid-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of beta-amyloid-protein precursor. J. Biol. Chem. 279, 20296–20306. doi: 10.1074/jbc.M312946200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dzwonek, J., Rylski, M., and Kaczmarek, L. (2004). Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Lett. 567, 129–135. doi: 10.1016/j.febslet.2004.03.070
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eckman, E. A., Reed, D. K., and Eckman, C. B. (2001). Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J. Biol. Chem. 276, 24540–24548. doi: 10.1074/jbc.M007579200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eckman, E. A., Watson, M., Marlow, L., Sambamurti, K., and Eckman, C. B. (2002). Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 278, 2081–2084. doi: 10.1074/jbc.C200642200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finley, M., Fairman, D., Liu, D., Li, P., Wood, A., and Cho, S. (2004). Functional validation of adult hippocampal organotypic cultures as an in vitro model of brain injury. Brain Res. 1001, 125–132. doi: 10.1016/j.brainres.2003.12.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gähwiler, B. H., Capogna, M., Debanne, D., McKinney, R. A., and Thompson, S. M. (1997). Organotypic slice cultures: a technique has come of age. Trends Neurosci. 20, 471–477. doi: 10.1016/S0166-2236(97)01122-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Glabe, C. (2000). Does Alzheimer disease tilt the scales of amyloid degradation versus accumulation? Nat. Med. 6, 133–134. doi: 10.1038/72215
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grimm, M. O., Mett, J., Stahlmann, C. P., Haupenthal, V. J., Zimmer, V. C., and Hartmann, T. (2013). Neprilysin and Aβ clearance: impact of the APP intracellular domain in NEP regulation and implications in Alzheimer's disease. Front. Aging Neurosci. 5:98. doi: 10.3389/fnagi.2013.00098
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guan, H., Liu, Y., Police, S., Kim, M. H., Oddo, S., LaFerla, F. M., et al. (2009). Peripherally expressed neprilysin reduces brain amyloid burden: a novel approach for treating Alzheimer's disease. J. Neurosci. Res. 87, 1462–1473. doi: 10.1002/jnr.21944
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hafez, D., Huang, J. Y., Huynh, A. M., Valtierra, S., Rockenstein, E., Bruno, A. M., et al. (2010). Neprilysin-2 is an important β-amyloid degrading enzyme. Am. J. Pathol. 178, 306–312. doi: 10.1016/j.ajpath.2010.11.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Henderson, S., Andersson, C., Janson, J., Goldschmidt, T. J., Appelkvist, P., Bogstedt, A., et al. (2014). Sustained peripheral depletion of amyloid-beta with a novel form of neprilysin does not affect central levels of amyloid-beta. Brain 137(Pt 2), 553–564. doi: 10.1093/brain/awt308
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hohsfield, L. A., Ehrlich, D., and Humpel, C. (2014). Intravenous infusion of NGF-secreting monocytes supports the survival of cholinergic neurons in the nucleus basalis of Meynert in hypercholesterolemia Brown Norway rats. J. Neurosci. Res. 92, 298–306. doi: 10.1002/jnr.23309
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hsu, R. L., Lee, K. T., Wang, J. H., Lee, L. Y., and Chen, R. P. (2009). Amyloid-degrading ability of nattokinase from Bacillus subtilis natto. J. Agric. Food Chem. 57, 503–508. doi: 10.1021/jf803072r
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hu, J., Igarashi, A., Kamata, M., and Nakagawa, H. (2001). Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide: retards Amyloid beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J. Biol. Chem. 276, 47863–47868. doi: 10.1074/jbc.M104068200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Humpel, C., and Weis, C. (2002). Nerve growth factor and cholinergic CNS neurons studied in organotypic brain slices: implications in Alzheimer's disease? J. Neural Transm. Suppl. 62, 253–263. doi: 10.1007/978-3-7091-6139-5_23
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwata, N., Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., et al. (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kilger, E., Buehler, A., Woelfing, H., Kumar, S., Kaeser, S. A., Nagarathinam, A., et al. (2011). BRI2 protein regulates β-amyloid degradation by increasing levels of secreted insulin-degrading enzyme (IDE). J. Biol. Chem. 286, 37446–37457. doi: 10.1074/jbc.M111.288373
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, H., Kim, E., Park, M., Lee, E., and Namkoong, K. (2013). Organotypic hippocampal slice culture from the adult mouse brain: a versatile tool for translational neuropsychopharmacology. Prog. Neuropsychopharmacol. Biol. Psychiatry 41, 36–43. doi: 10.1016/j.pnpbp.2012.11.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kurochkin, I. V. (2001). Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem. Sci. 26, 421–425. doi: 10.1016/S0968-0004(01)01876-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lebson, L., Nash, K., Herber, D., Carty, N., Lee, D. C., Li, Q., et al. (2010). Trafficking CD11b-positive blood cells deliver therapeutic genes to the brain of amyloid-depositing transgenic mice. J. Neurosci. 30, 9651–9658. doi: 10.1523/JNEUROSCI.0329-10.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ledesma, M. D., Da Silva, J. S., Crassaerts, K., Delacourte, A., De Strooper, B., and Dotti, C. G. (2000). Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep. 1, 530–535. doi: 10.1093/embo-reports/kvd107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, C. Y., and Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117, 949–960. doi: 10.1007/s00702-010-0433-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. doi: 10.1016/S0896-6273(03)00787-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liao, M. C., Ahmed, M., Smith, S. O., and Van Nostrand, W. E. (2009). Degradation of amyloid beta protein by purified myelin basic protein. J. Biol. Chem. 284, 28917–28925. doi: 10.1074/jbc.M109.050856
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, Y., Studzinski, C., Murphy, M. P., Klein, R. L., and Hersh, L. B. (2010). Circulating neprilysin clears brain amyloid. Mol. Cell. Neurosci. 45, 101–107 doi: 10.1016/j.mcn.2010.05.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marksteiner, J., and Humpel, C. (2008). Beta-amyloid expression, release and extracellular deposition in aged rat brain slices. Mol. Psychiatry 13, 939–952. doi: 10.1038/sj.mp.4002072
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marr, R. A., Rockenstein, E., Mukherjee, A., Kindy, M. S., Hersh, L. B., Gage, F. H., et al. (2003). Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23, 1992–1996.
Merlo, S., and Sortino, M. A. (2012). Estrogen activates matrix metalloproteinases-2 and -9 to increase beta amyloid degradation. Mol. Cell. Neurosci. 49, 423–429. doi: 10.1016/j.mcn.2012.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miners, J., Kehoe, P., and Love, S. (2011). Neprilysin protects against cerebral amyloid angiopathy and beta-amyloid-induced degeneration of cerebrovascular smooth muscle cells. Brain Pathol. 21, 594–605. doi: 10.1111/j.1750-3639.2011.00486.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nalivaeva, N. N., Fisk, L. R., Belyaev, N. D., and Turner, A. J. (2008). Amyloid-degrading enzymes as therapeutic targets in Alzheimer's disease. Curr. Alzheimer Res. 5, 212–224. doi: 10.2174/156720508783954785
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ohno, M., Hiraoka, Y., Nishi, K., Saijo, S., Matsuoka, T., Tomimoto, H., et al. (2014). Nardilysin prevents amyloid plaque formation by enhancing gamma-secretase activity in an Alzheimer's disease mouse model. Neurobiol. Aging 35, 213–222. doi: 10.1016/j.neurobiolaging.2013.07.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, M., Lee, J. K., Ahn, J., Jin, H. K., Park, J. S., and Bae, J. S. (2013). Recombinant soluble neprilysin reduces amyloid-beta accumulation and improves memory impairment in Alzheimer's disease mice. Brain Res. 1529, 113–124. doi: 10.1016/j.brainres.2013.05.045
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., et al. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 273, 32730–32738. doi: 10.1074/jbc.273.49.32730
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roher, A. E., Kasunic, T. C., Woods, A. S., Cotter, R. J., Ball, M. J., and Fridman, R. (1994). Proteolysis of A beta peptide from Alzheimer disease brain by gelatinase A. Biochem. Biophys. Res. Commun. 205, 1755–1761. doi: 10.1006/bbrc.1994.2872
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ryan, D., Narrow, W. C. F. H., and Bowers, W. J. (2010). An improved method for generating consistent soluble amyloid-beta oligomer preparations for in vitro neurotoxicity studies. J. Neurosci. Methods 190, 171–179. doi: 10.1016/j.jneumeth.2010.05.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schatz, S., Kaufmann, W. A., Saria, A., and Humpel, C. (1999). Dopamine neurons in a simple GDNF-treated meso-striatal organotypic co-culture model. Exp. Brain Res. 127, 270–278. doi: 10.1007/s002210050796
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stoppini, L., Buchs, P. A., and Muller, D. (1991). A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182. doi: 10.1016/0165-0270(91)90128-M
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Su, T., Paradiso, B., Long, Y. S., Liao, W. P., and Simonato, M. (2011). Evaluation of cell damagae in organotypic hippocampal slice culture from adult mouse: a potential model system to study neuroprotection. Brain Res. 1385, 68–76. doi: 10.1016/j.brainres.2011.01.115
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tucker, H. M., Kihiko, M., Caldwell, J. N., Wright, S., Kawarabayashi, T., Price, D., et al. (2000). The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 20, 3937–3946.
Ullrich, C., Daschil, N., and Humpel, C. (2011). Organotypic vibrosections: novel whole sagittal brain cultures. J. Neurosci. Methods 201, 131–141. doi: 10.1016/j.jneumeth.2011.07.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Nostrand, W. E., and Porter, M. (1999). Plasmin cleavage of the amyloid beta-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 38, 11570–11576. doi: 10.1021/bi990610f
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walker, J., Pacoma, R., Ou, W., Alves, J., Mason, D. E., Peters, E. C., et al. (2013). Enhanced proteolytic clearance of plasma Amyloi-beta by peripherally administered neprilysin does not result in reduced levels of brain Amyloid-beta in mice. J. Neurosci. 33, 2457–2464. doi: 10.1523/JNEUROSCI.3407-12.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, D. S., Dickson, D. W., and Malter, J. S. (2006). beta-Amyloid degradation and Alzheimer's disease. J. Biomed. Biotechnol. 2006:58406. doi: 10.1155/JBB/2006/58406
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weis, C., Marksteiner, J., and Humpel, C. (2001). Nerve growth factor and glial cell line-derived neurotrophic factor restore the cholinergic phenotype in organotypic brain slices of the basal nucleus of Meynert. Neuroscience 102, 129–138. doi: 10.1016/S0306-4522(00)00452-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wilhelmi, E., Schöder, U. H., Benabdallah, A., Sieg, F., Breder, J., and Reymann, K. G. (2002). Organotypic brain-slice cultures from adult rats: approaches for a prolonged culture time. Altern. Lab. Anim. 30, 275–283. doi: 10.1371/journal.pone.0045017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wisniewski, H. M., Wen, G. Y., and Kim, K. S. (1989). Comparison of four staining methods on the detection of neuritic plaques. Acta Neuropathol. 78, 22–27. doi: 10.1007/BF00687398
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xie, H., Guan, J., Borrelli, L. A., Xu, J., Serrano-Pozo, A., and Bacskai, B. J. (2013). Mitochondrial alterations near amyloid plaques in an Alzheimer's disease mouse model. J. Neurosci. 33, 17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamada, T., Miyazaki, K., Koshikawa, N., Takahashi, M., Akatsu, H., and Yamamoto, T. (1995). Selective localization of gelatinase A, an enzyme degrading beta-amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol. 89, 199–203. doi: 10.1007/BF00309334
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamamoto, N., Arima, H., Kasahara, R., Taniura, H., Hirate, H., Sugiura, T., et al. (2013). Ketamine reduces amyloid beta-protein degradation by suppressing neprilysin expression in primary cultured astrocytes. Neurosci. Lett. 545, 54–58. doi: 10.1016/j.neulet.2013.04.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yan, P., Hu, X., Yin, K., Bateman, R. J., Cirrito, J. R., Xiao, Q., et al. (2006). Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J. Biol. Chem. 281, 24566–24574. doi: 10.1074/jbc.M602440200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yin, K. J., Yan, P., Hu, X., Xiao, Q., Pan, X., Bateman, R., et al. (2006). Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci. 26, 10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer, beta-amyloid plaques, clearance, in vitro model, organotypic brain slices, vibrosections
Citation: Humpel C (2015) Organotypic vibrosections from whole brain adult Alzheimer mice (overexpressing amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as a model to study clearance of beta-amyloid plaques. Front. Aging Neurosci. 7:47. doi: 10.3389/fnagi.2015.00047
Received: 16 December 2014; Paper pending published: 04 February 2015;
Accepted: 24 March 2015; Published: 09 April 2015.
Edited by:
Roxana Octavia Carare, University of Southampton, UKReviewed by:
Uwe-Karsten Hanisch, University of Leipzig, GermanyAndreas Vlachos, Goethe-University Frankfurt, Germany
Copyright © 2015 Humpel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Humpel, Laboratory of Psychiatry and Experimental Alzheimer's Research, Department of Psychiatry and Psychotherapy, Medical University of Innsbruck, Anichstr. 35, A-6020 Innsbruck, AustriaY2hyaXN0aWFuLmh1bXBlbEBpLW1lZC5hYy5hdA==