 Anita Ramanathan
Anita Ramanathan Amy R. Nelson
Amy R. Nelson Abhay P. Sagare
Abhay P. Sagare Berislav V. Zlokovic
Berislav V. Zlokovic- Department of Physiology and Biophysics, Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
Amyloid beta (Aβ) homeostasis in the brain is governed by its production and clearance mechanisms. An imbalance in this homeostasis results in pathological accumulations of cerebral Aβ, a characteristic of Alzheimer’s disease (AD). While Aβ may be cleared by several physiological mechanisms, a major route of Aβ clearance is the vascular-mediated removal of Aβ from the brain across the blood-brain barrier (BBB). Here, we discuss the role of the predominant Aβ clearance protein—low-density lipoprotein receptor-related protein 1 (LRP1)—in the efflux of Aβ from the brain. We also outline the multiple factors that influence the function of LRP1-mediated Aβ clearance, such as its expression, shedding, structural modification and transcriptional regulation by other genes. Finally, we summarize approaches aimed at restoring LRP1-mediated Aβ clearance from the brain.
Introduction
Alzheimer’sdisease (AD) is a neurodegenerative disorder characterized by abnormal elevations of amyloid β (Aβ), neurofibrillary tau tangles and neurovascular dysfunction. Aβ is generated from the transmembranous amyloid precursor protein (APP) after proteolytic cleavage by β- and γ-secretase (Selkoe, 2001) and is proposed to have multiple roles in the brain such as, to name a few, neurotrophic activity, modulation of synaptic plasticity, neurogenesis, metal ion sequestration, antioxidant activity, and calcium homeostasis (del Cárdenas-Aguayo et al., 2014); and also interferes with signal transduction within the neurovascular unit affecting multiple neurovascular functions (Zlokovic, 2011). Thus, regulated clearance of Aβ via receptor-mediated transport potentially allows for controlled regulation of all these Aβ functions at any given time and also prevents accumulation of excess Aβ as a metabolic waste product.
Neurotoxic accumulation of Aβ in the brain has been hypothesized to result from an imbalance in the Aβ homoeostasis i.e., between its production and clearance (Zlokovic, 2011; Musiek and Holtzman, 2015). Recent evidence in humans has suggested that indeed faulty Aβ clearance mechanisms, but not overproduction, contribute to pathological accumulations of cerebral Aβ in late-onset sporadic AD (Mawuenyega et al., 2010). Because impaired clearance of Aβ is now widely identified as a contributing factor towards AD progression, studying Aβ clearance mechanisms, their regulation and devising methods to modulate clearance function is of importance in generating potential AD-related therapeutic interventions (Tarasoff et al., in press).
To prevent Aβ aggregation and deposition, various physiological clearance mechanisms exists to help steer Aβ removal from the brain, namely, transvascular clearance across the blood-brain barrier (BBB; Zlokovic, 2008, 2010, 2011), interstitial fluid (ISF) bulk flow i.e., traditional perivascular clearance (Weller et al., 2008; Hawkes et al., 2012) and glymphatic paravascular clearance (Iliff et al., 2012), cerebrospinal fluid (CSF) absorption (Pollay, 2010) and enzymatic degradation (Farris et al., 2003; Kanemitsu et al., 2003; Hernandez-Guillamon et al., 2015). Aβ removal from brain by transcytosis is typically a concentration-dependent process physiologically driven by a much higher brain-to-plasma concentration gradient of Aβ This receptor-mediated mechanism allows for removal of Aβ from both the mammalian (Zlokovic et al., 2010) and human (Roberts et al., 2014) brain across the largest possible surface area for removal of about 100 cm2 of the capillary endothelium per gram of brain tissue representing the surface of the BBB in vivo (Zlokovic, 2011). Previous studies have demonstrated that the majority of Aβ is cleared by the BBB (~85%) under physiological conditions with a smaller percentage being cleared by ISF bulk flow (Shibata et al., 2000; Zlokovic and Frangione, 2003). Disrupted Aβ transcytosis leads to accumulation of Aβ in the brain causing pathophysiologal changes as shown by multiple previous studies. In this review, we focus on transvascular clearance of Aβ across the BBB mediated by low-density lipoprotein receptor-related protein 1 (LRP1) in association with AD.
Blood-Brain Barrier and LRP1
Maintenance of a toxin-free brain microenvironment is essential for normal neuronal function. The endothelial cells of brain capillaries are compactly connected end-to-end by tight junction proteins forming the BBB, isolating the brain ISF-CSF compartments from the plasma compartment and authorizing entry of molecules from the blood into the brain (Zlokovic, 2008; Abbott et al., 2010). The specialized brain vascular endothelial cells of the BBB, along with pericytes, glial cells and neurons form the neurovascular unit (NVU; Zlokovic, 2011). This arrangement is very different from the highly permeable and lenient systemic capillaries permitting transport of solutes and bigger molecules into the parenchymal tissue space (Mann et al., 1985). At the level of brain capillaries, while oxygen from the blood diffuses freely into the brain, the entry of polar molecules is typically restricted by the BBB (Zlokovic, 2011). However, nutrients can cross the BBB using specific transporters expressed within the brain endothelium (Zlokovic, 2011). The polarized distribution of transporters on the luminal and abluminal endothelial membranes of the BBB allows the brain endothelial cells to control movement of ions, nutrients and other molecules between brain and blood in a highly-regulated manner to match metabolic demands of brain. Although, larger molecules are typically excluded from the brain by the BBB, some proteins and peptides can still cross the BBB slowly if their respective transporters and/or carriers are expressed within the brain endothelium (Zlokovic et al., 1985, 1987; Zlokovic, 1995; Mackic et al., 2002).
At the BBB, the transporter LRP1 plays a pivotal role in maintaining Aβ homeostasis in the central nervous system (CNS). LRP1 is expressed mainly at the abluminal side of the BBB (Zlokovic, 2008; Ueno et al., 2010; Zhao et al., 2015). LRP1 is the primary receptor mediating transport of Aβ across the BBB into circulation, thereby clearing it from the brain. LRP1 is a member of the LDLR family. This cell surface receptor is highly expressed on multiple cell types (Kanekiyo and Bu, 2014). At the neurovascular interface, LRP1 is expressed by vascular endothelial cells forming the BBB, vascular mural cells, namely, pericytes and vascular smooth muscle cells (VSMCs), neurons and astrocytes (Zlokovic et al., 2010; Sagare et al., 2012; Kanekiyo and Bu, 2014).
The distinct structural arrangement of LRP1 enables its multifaceted role as a cargo transporter, a multifunctional scavenger and a signaling receptor involved in endocytosis of its ligands (Lillis et al., 2008). LRP1 recognizes and is involved in the endocytosis of more than 40 different ligands including apolipoprotein E (apoE), APP and Aβ (Zlokovic et al., 2010; Sagare et al., 2012; Kanekiyo and Bu, 2014).
LRP1 physiologically exists in two forms: a cell surface bound form (LRP1) and a truncated soluble form (sLRP1; Figure 1A). The surface-bound LRP1 is composed of two subunits that includes an extracellular α-chain (515 kDa) and a β-chain (85 kDa) containing a short extracellular extension, a single transmembrane domain and a 100 amino acid intracellular domain (Emonard et al., 2014). The characteristic motifs on the cytoplasmic tail of LRP1, namely, two NPXY motifs, one YXXL motif and two di-leucine motifs bind to endocytotic and scaffold adaptors, rapidly trafficking ligands like Aβ into endosomes in a clathrin-dependent manner (Li et al., 2001a; Deane et al., 2004, 2008) transporting them into the vascular lumen. Cell surface levels of LRP1 are controlled by proteolytic cleaving of its ectodomain. Several proteases including beta-secretase 1 (BACE-1), A Disintegrin And Metalloprotease (ADAM)-10, ADAM-12, ADAM-17, membrane type-1 matrix metalloprotease (MT1-MMP) and tissue plasminogen activator (t-PA) have been shown to be involved in shedding of LRP1 (Rozanov et al., 2004; von Arnim et al., 2005; Polavarapu et al., 2007; An et al., 2008; Liu et al., 2009; Selvais et al., 2011). The extracellular domain or ectodomain of LRP1 released after shedding is sLRP1 (Quinn et al., 1997), as described above. The truncated soluble form of LRP1 freely circulates in the plasma and sequesters unbound Aβ in the periphery (Sagare et al., 2007, 2013a). sLRP1 is also detectable in the cerebrospinal fluid (CSF; Qiu et al., 2001; Liu et al., 2009).
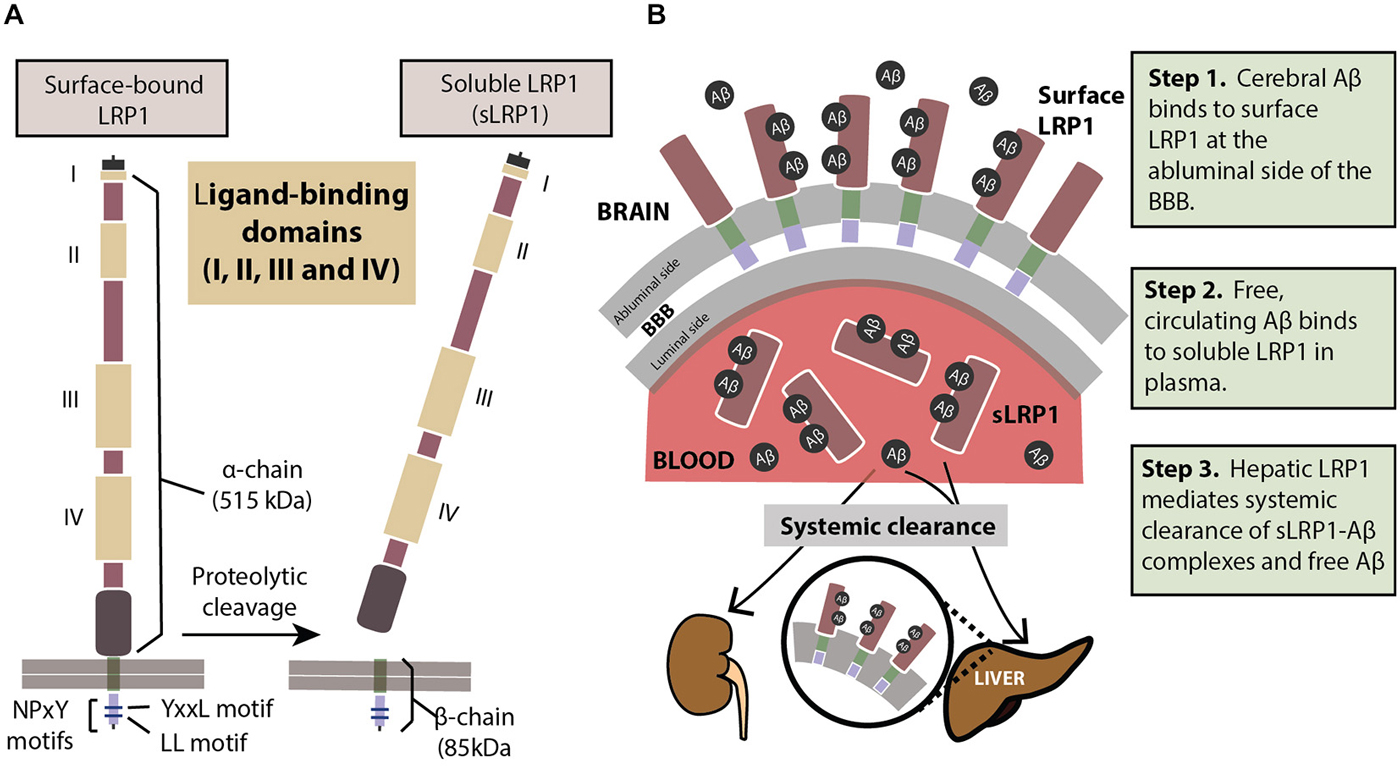
Figure 1. (A) The structure of LRP1. The extracellular domain consists of a heavy α-chain (515 kDa) with four ligand-binding domains. The light chain or the β-chain (85 kDa) extends into the intracellular compartment where it has two characteristic NPxY motifs involved in endocytosis and cell signaling. Proteolytic cleavage of surface-bound LRP1 yields the soluble sLRP1. (B) The three steps of LRP1-mediated Aβ clearance from brain (step 1), from blood (step 2) and from the body (step 3). For description of these three steps please see panel (B). See “Role of LRP1 in Aβ Clearance” Section for a more detailed explanation.
The endocytotic efficiency of LRP1 is dynamically regulated by post-translational phosphorylation of serine, tyrosine and threonine residues on its cytoplasmic tail (Bu et al., 1998; van der Geer, 2002), which also enables its downstream cell signaling functions. Phosphorylation on serine and tyrosine residues by protein kinase Cα (PKCα) enables the LRP1 intracellular domain to bind to adaptor protein Disabled-1 (Dab1) decreasing endocytosis by 25% (Ranganathan et al., 2004), whereas, phosphorylation on serine residues by protein kinase A (PKA) increases LRP1 endocytosis (Li et al., 2001b). Distinct from this, phosphorylation of tyrosine by Src activates the Ras signaling pathway rather than endocytosis (Barnes et al., 2003).
Interestingly, our recent study (Zhao et al., 2015) indicates that phosphatidylinositol binding clathrin assembly protein (PICALM; Dreyling et al., 1996; Tebar et al., 1999), a highly-validated risk factor for AD confirmed in several genome-wide association studies (Harold et al., 2009; Lambert et al., 2009; Carrasquillo et al., 2010, 2015; Chen et al., 2012; Tanzi, 2012; Liu et al., 2013; Morgen et al., 2014) binds to the intracellular tail of LRP1 at the YXXL domain and regulates endocytsosis of LRP1-Aβ complexes. Aβ binding to LRP1 presumably elicits a conformational change in its cytoplasmic tail, enabling PICALM-regulated endosomal transcytosis of Aβ (Zhao et al., 2015).
Role of LRP1 in Aβ Clearance
In order to prevent pathological accumulations of Aβ in the brain, Aβ clearance from the cerebral milieu into periphery and out of the system is of prime importance. The multi-site role of the key Aβ-binding receptor, LRP1, helps eliminate systemic Aβ in a three-step serial clearance mechanism (Figure 1B; Zlokovic et al., 2010; Sagare et al., 2012). Step 1 (BBB). At the site of the BBB, surface LRP1 is expressed on the endothelial membrane facing the brain (abluminal). This membrane-bound LRP1 binds cerebral Aβ and rapidly initiates its clearance into the blood (luminal) side (Shibata et al., 2000; Deane et al., 2004, 2008; Ito et al., 2006; Bell et al., 2007; Sagare et al., 2007). Radiolabelled and unlabeled Aβ administered intracerebrally has appeared intact in plasma, indicating Aβ efflux from the brain by cerebrovascular LRP1 (Shiiki et al., 2004; Bell et al., 2007). Step 2 (Plasma). sLRP1 in the periphery contributes to Aβ clearance by sequestering free Aβ in circulation (Sagare et al., 2007, 2013a). Coimmunoprecipitation of sLRP1-bound Aβ in neurologically normal humans has indicated that circulating sLRP1 can sequester 70–90% of plasma Aβ (Sagare et al., 2007), thereby driving the Aβ gradient in favor of efflux across the BBB. This endogenous peripheral Aβ “sink” created by sLRP1 promotes Aβ clearance from the brain into circulation. Step 3 (Liver). LRP1 was first identified in the liver (Herz et al., 1988), and at the hepatic site, is responsible for binding to and clearing Aβ from the system (Tamaki et al., 2006; Sagare et al., 2011b; Sutcliffe et al., 2011; Sehgal et al., 2012). In addition to the liver, sLRP1-Aβ complexes are also removed by the kidney and spleen (Sagare et al., 2007).
Recent Techniques to Study Aβ Clearance from the Brain
For many years, methods involving intracerebral injection of radiolabelled Aβ and studying its efflux from the brain into the plasma and/or CSF has been a validated method to predict Aβ clearance. Over the recent years, there has been an increased interest in exploring the brain ISF pool for Aβ levels, its metabolic dynamics and clearance rates. To this effect, an analytical technique—microdialysis (Figure 2)—which has been popularly used to study neurotransmitters, has recently been recruited to study soluble Aβ in the brain of awake APP overexpressing mice (Cirrito et al., 2003; Chefer et al., 2009; Sagare et al., 2013b; Zhao et al., 2015).
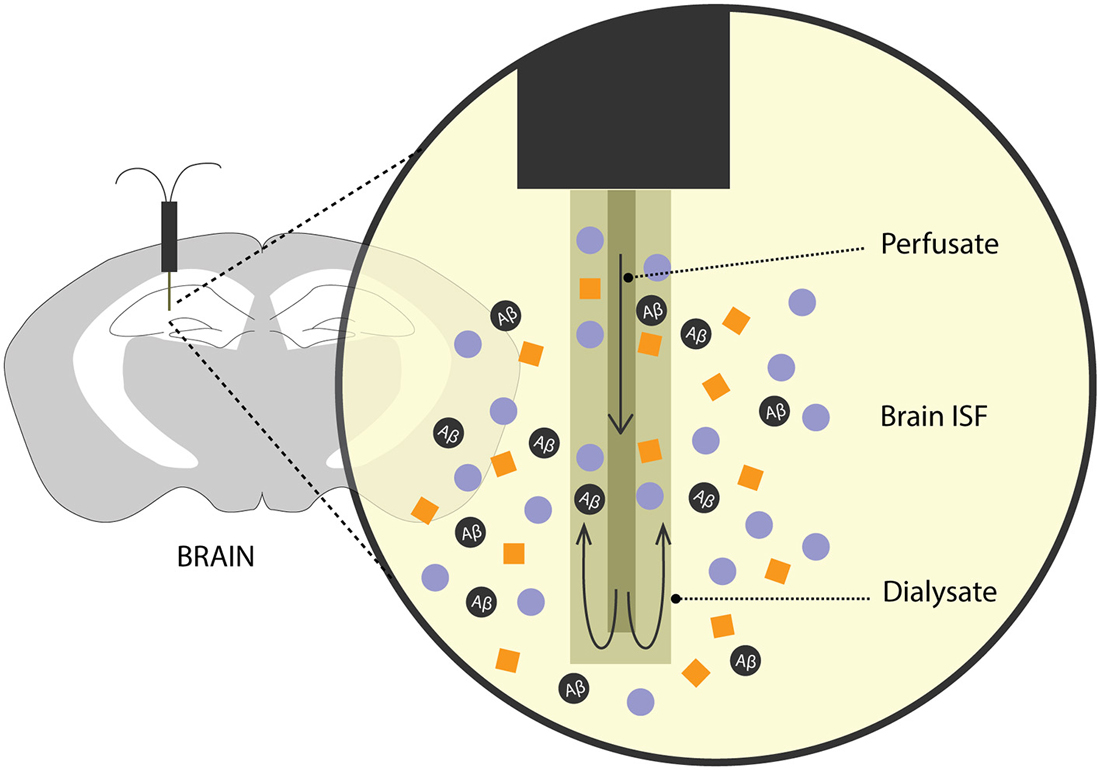
Figure 2. Brain microdialysis. A microdialysis probe is surgically implanted into a specific brain region of interest. Aβ and other solutes from the brain ISF freely enter the semipermeable membrane of the microdialysis probe. Fractions of microdialysates obtained over a period of time are then used to quantify Aβ levels reflecting steady-state levels and/or clearance of Aβ from the brain after inhibition of Aβ production.
In freely moving rodents, cerebral microdialysis enables continuous, time-lapsed readouts of Aβ levels in the brain ISF. Briefly, the microdialysis method entails implanting a small microdialysis probe (~ 200 μm in diameter) into a discrete brain region. Each probe has a characteristic recovery rate for the analyte of interest, here, Aβ, that is optimized in vitro. The probe comprises of a semipermeable membrane of a limited porosity, (in this case, 35 kDa) and is perfused with artificial CSF. Cerebral molecules up to 35 kDa in size can freely enter the membrane and are recovered through the microdialysate. Fractions of microdialysates can be collected over different periods of times to represent a neurochemical “snapshot” of the cerebral milieu around the probe along a certain time period. Aβ in the microdialysate is then quantified using biochemical assays like enzyme-linked immunosorbent assay (ELISA). In addition to obtaining steady-state measurements of Aβ, its efflux kinetics like half-life can be studied by administering a potent γ-secretase inhibitor, compound E which inhibits the production of soluble Aβ. The ISF Aβ levels from the microdialysate, upon compound E injection, now represent its clearance from the brain. This method has been used extensively in AD mouse models to obtain valuable information on genetic factors that affect clearance Aβ (Cirrito et al., 2003; Farris et al., 2007; Sagare et al., 2013b; Zhao et al., 2015). Intracerebral microdialysis has also been used to obtain human ISF Aβ concentrations in patients undergoing invasive intracranial monitoring (Brody et al., 2008).
A pioneering method was recently developed to measure rates of Aβ synthesis and clearance in human subjects. After intravenously infusing a stable isotope-labeled amino acid (13C6-leucine; Bateman et al., 2006; Mawuenyega et al., 2010) CSF and plasma were sampled over a 36-h period. The fractional synthesis rate (FSR) and fractional clearance rate (FCR) of in vivo Aβ was then quantified using high-resolution tandem mass spectrometry. This method was instrumental in demonstrating that diminished Aβ clearance, and not overproduction, contributes towards cerebral Aβ accumulation in late-onset AD (Bateman et al., 2006; Mawuenyega et al., 2010).
Factors Regulating LRP1-Mediated Aβ Clearance
The function of the BBB-localized LRP1 in actively removing Aβ from the brain is regulated by several factors. Fluctuations in LRP1 expression levels and structural modifications directly affect Aβ clearance. LRP1 expression is further regulated by several vascular genes in brain endothelium and VSMCs, and genetic risk factors for late-onset AD that can all modulate LRP1-mediated Aβ clearance (Figure 3).
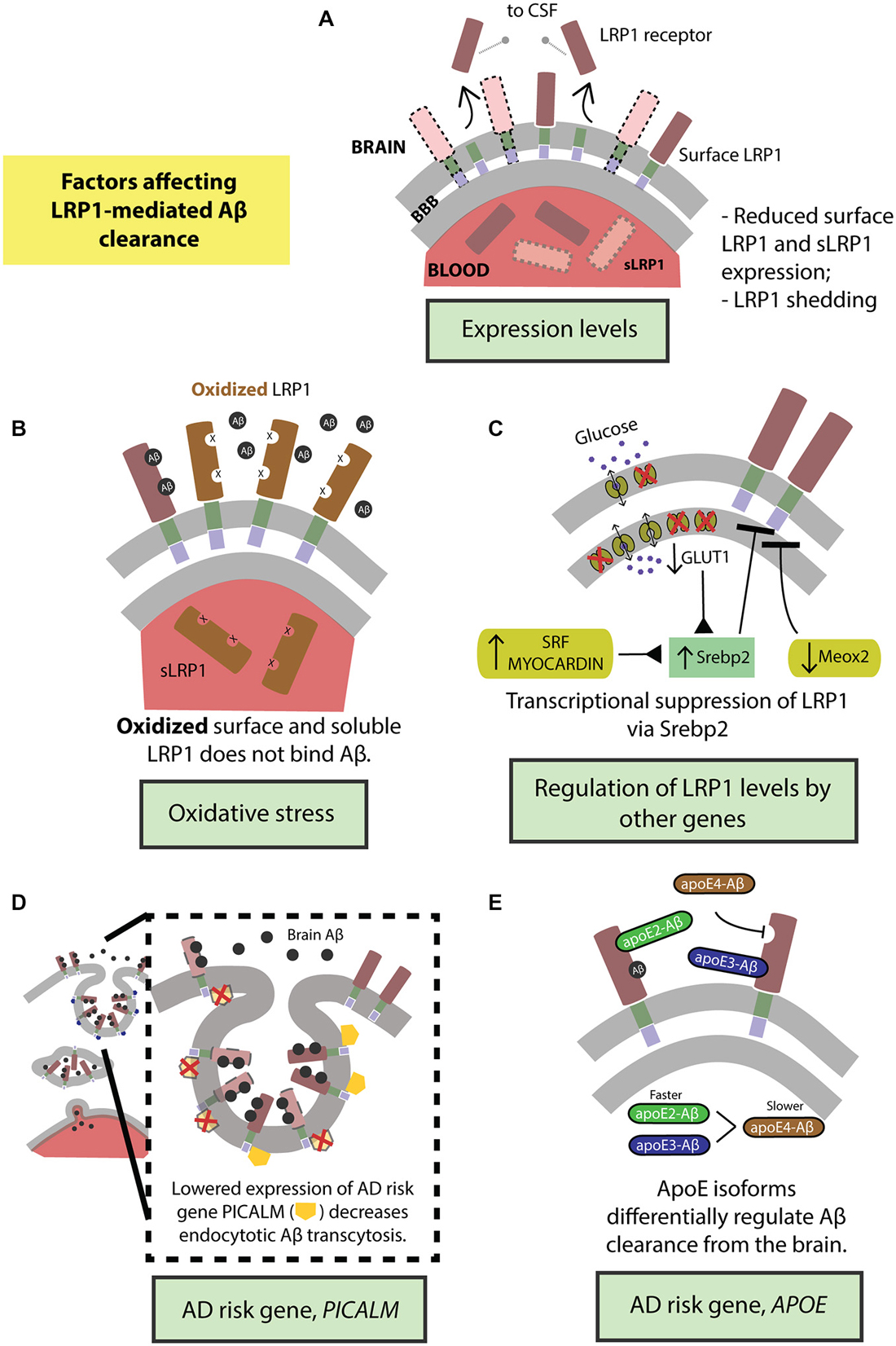
Figure 3. Factors regulating transvascular LRP1-mediated Aβ clearance. (A) Age-dependent reduction in LRP1 levels and its shedding and (B) oxidation of LRP1. LRP1 expression and Aβ clearance are further regulated by (C) transcriptional suppression by Srebp2 and SLC2A1 (encoding glucose transporter, Glut1) in brain endothelial cells and SRF and MYOCD in vascular smooth muscle cells (VSMCs), as well as by MEOX2 in brain endothelial cells. Moreover, well-known highly replicated Alzheimer’s disease (AD) genetic risk factors including PICALM (D) and APOE4 gene (E) influence endocytosis and transcytosis of Aβ-LRP1 complexes across brain endothelium of the blood-brain barrier. See “Factors Regulating LRP1-Mediated Aβ Clearance” Section for a more detailed explanation.
Expression Levels
LRP1 expression levels are significantly reduced in brain endothelial cells in normally aging and AD humans and animal models (Kang et al., 2000; Shibata et al., 2000; Deane et al., 2004; Donahue et al., 2006) leading to higher levels of Aβ in the brain (Figure 3A). Additionally, LRP1 levels are also diminished on VSMCs in AD patients (Bell et al., 2009). Importantly, there is a significant negative correlation between the expression of LRP1 on microvessels and Aβ accumulation in cerebrovasculature and brain parenchyma (Shibata et al., 2000; Donahue et al., 2006). Also, sLRP1 fraction bound to Aβ40 and Aβ42 is significantly reduced in Mild Cognitive Impairment (MCI) and AD subjects that corresponds to elevated levels of free Aβ40 and Aβ42 in plasma (Figure 3A; Sagare et al., 2011a). This free plasma Aβ can cross the BBB and re-enter the brain in a concentration-dependent manner mediated by the receptor for advanced glycation end products (RAGE), the major Aβ influx receptor at the BBB transporting Aβ from blood into the brain (Deane et al., 2003; Ujiie et al., 2003; Zlokovic, 2008). Interestingly, RAGE expression levels are increased in AD endothelium that is associated with cerebrovascular and brain accumulation of Aβ (Yan et al., 1996; Deane et al., 2003, 2012; Silverberg et al., 2010). Therefore, the accumulation of Aβ in the brain can be attributed to the cumulative effects of reduced expression of surface-bound LRP1 and sLRP1, causing higher cerebral and plasma Aβ levels respectively, and increased expression of RAGE, increasing the re-entry of Aβ into the brain.
Receptor Shedding
Lipoprotein receptors on the BBB are susceptible to ectodomain shedding which alters endocytotic transport and clearance of molecules, including Aβ, from the brain. Treating human brain endothelial cells with Aβ causes shedding of sLRP1 (Bachmeier et al., 2014; Figure 3A). Similarly, intracranial infusion of Aβ into mouse brain results in sLRP1 shedding (Bachmeier et al., 2014). In both the in vitro and in vivo experiments, the amount of sLRP1 shedding was reduced in the presence of ApoE2 or ApoE3 but not ApoE4 (Bachmeier et al., 2014). Furthermore, sLRP1 levels are increased in CSF from aged and AD subjects due to shedding (Qiu et al., 2001) caused by ADAM10 and ADAM17 (Liu et al., 2009). The dysregulation of shedded sLRP1, in part due to the presence of Aβ, could impair Aβ clearance from the brain and contribute to the pathogenesis of AD.
Oxidation
There is an increase in oxidative stress in the brain of aged and AD subjects (Moosmann and Behl, 2002; Chen and Zhong, 2014). This is evidenced by increased biomarker levels in the blood that reflect oxidative stress in the brain (Beal, 2005; Torres et al., 2011). Oligomeric Aβ induces oxidative stress that is believed to contribute to AD pathologies (Drake et al., 2003; Boyd-Kimball et al., 2006; Clementi et al., 2006). Importantly, LRP1 is oxidized in AD hippocampus and does not bind Aβ leading to increased Aβ deposition (Figure 3B; Drake et al., 2003). In addition to surface LRP1, in AD patients and animal models, sLRP1 binding to Aβ is disrupted by oxidation (Figure 3B; Sagare et al., 2012). Oxidized sLRP1, which does not bind to Aβ, is associated with elevated levels of free Aβ40 and Aβ42 in the plasma (Sagare et al., 2011a). Our previous studies have shown a significant positive correlation between CSF tau/Aβ42 ratios and oxidized sLRP plasma levels (Sagare et al., 2011a). Therefore, oxidation of both LRP1 and sLRP1 negatively impacts Aβ clearance from the brain. Moreover, there is likely a vicious cycle since Aβ induces LRP1 oxidation rendering the receptor less capable of clearing/reducing Aβ levels in the brain.
Regulation of LRP1 Expression by Genes in Vascular Cells
The expression of LRP1 is negatively regulated by its only known transcriptional suppressor; the sterol regulatory element binding protein (SREBP2; Llorente-Cortés et al., 2006, 2007). Recent studies have evidenced how Aβ clearance mechanisms in the CNS are indirectly altered by vascular- and metabolism-related genes via SREBP2-mediated regulation of LRP1 (Figure 3C).
LRP1 and GLUT1. The brain, which exclusively depends on the vasculature for essential metabolites, receives its supply of glucose across the BBB through the glucose transporter (GLUT1; encoded by SLC2A1). In AD, there is a reduction in this GLUT1 on cerebral microvessels (Mooradian et al., 1997). Diminished uptake of glucose, as studied by positron emission tomography (PET) using a glucose analog, 18F-2-fluoro-2-deoxy-D-glucose (FDG), is reported to be a forerunner to brain atrophy (Hunt et al., 2007), and has been observed in individuals with a genetic risk for AD, with a positive familial AD history, as well as those with none or mild cognitive deficits to eventually go on to develop AD (Hunt et al., 2007; Herholz, 2010).
Our group has recently demonstrated that GLUT1 deficiency in a transgenic AD mouse model overexpressing human APP Swedish mutant, APPsw/0, accelerated amyloid load and aggravated Aβ accumulation (Winkler et al., 2015). Incidentally, GLUT1 heterozygous mice (Slc2a1+/−) also expressed lower levels of LRP1 with respect to controls (Slc2a1+/+); the trend in diminished LRP1 expression further exacerbating with the addition of APP phenotype in Slc2a1+/−APPSw/0 (Winkler et al., 2015). Re-expression and silencing of Slc2a1 correspondingly boosted and decreased LRP1 levels, explaining the reversible nature of the GLUT1-LRP1 relationship; while suppressing LRP1 levels did not affect GLUT1 expression, indicating that GLUT1 acts upstream to LRP1. In deciphering the molecular mechanism behind this relationship, it was observed that Slc2a1 deficiency upregulates the SREBP2 transcription factor, in turn downregulating LRP1 expression (Figure 3C; Winkler et al., 2015).
Reductions in glucose transporters observed in AD (Kalaria and Harik, 1989; Horwood and Davies, 1994; Simpson et al., 1994; Mooradian et al., 1997), extends its effects beyond apparent hypometabolism to essentially affect Aβ clearance mechanisms by regulating LRP1 levels in the cerebral endothelia.
LRP1 and vascular-related genes. The expression levels of several vascular-related genes are altered in AD. For example, transcriptome profiling of human brain endothelial cells has indicated that the expression of mesenchyme homeobox gene 2 (MEOX2), a regulator of vascular differentiation and remodeling, is reduced in AD (Wu et al., 2005). The downregulation of MEOX2, and consequently the encoded protein, growth arrest-specific homeobox (GAX), is associated with altered angiogenesis, cerebral hypoperfusion and accumulation of brain Aβ (Wu et al., 2005). Unsurprisingly, MEOX2 expression affects Aβ homeostasis by regulating LRP1 expression. Low levels of MEOX2, as studied in vivo and in vitro models, leads to diminished LRP1 levels at the BBB by promoting its proteosomal degradation (Figure 3C; Wu et al., 2005). On the other hand, two other interrelated transcriptional factors constituting the critical regulators of VSMCs differentiation, namely, serum response factor (SRF) and myocardin (MYOCD) are, in turn, upregulated in AD (Chow et al., 2007; Bell et al., 2009). The overexpression profile of SRF/MYOCD initiates a hypercontractile phenotype in the cerebral arteries through increased expression of SRF/MYOCD-regulated contractile proteins, thereby resulting in cerebral hypoperfusion, diminished neurovascular coupling and cerebral amyloid angiopathy (CAA; Chow et al., 2007). Cerebral VSMCs from AD patients with CAA exhibit, along with overexpressed SRF/MYOCD, an accumulation of Aβ and significantly lower levels of LRP in comparison with age-matched healthy controls (Bell et al., 2009). Following this, it was then observed that SRF/MYOCD overexpression in VSMCs transcriptionally regulates LRP1 levels by transactivation of SREBP2, diminishing LRP1 surface expression and affecting Aβ efflux from the brain (Figure 3C; Bell et al., 2009).
Regulation of LRP1-Mediated Aβ Endocytosis and Clearance by AD Risk Genes
PICALM. While the extracellular domain of LRP1 binds a diverse array of ligands, the intracellular cytoplasmic domain is actively involved in ligand endocytosis (Krieger and Herz, 1994; Reekmans et al., 2010). Recently, a gene crucial for endocytotic internalization of receptors—PICALM (Sorkin and von Zastrow, 2009; Treusch et al., 2011) encoding phosphatidylinositol binding clathrin assembly protein (Dreyling et al., 1996; Tebar et al., 1999), is a highly-validated risk factor for AD and has been confirmed in several genome-wide association studies (Harold et al., 2009; Lambert et al., 2009; Carrasquillo et al., 2010, 2015; Chen et al., 2012; Tanzi, 2012; Liu et al., 2013; Morgen et al., 2014). Interestingly, in our most recent work (Zhao et al., 2015), we observed that Picalm haploinsufficiency imparts diminished clearance of cerebral Aβ and accelerated amyloid pathology. Because the capillaries lining the BBB prolifically express PICALM (Baig et al., 2010; Parikh et al., 2014), we investigated whether this high-risk AD gene regulated the internalization of the main Aβ-clearance receptor, LRP1, thereby affecting Aβ clearance. Using primary human brain endothelial cells, we observed that fluorescently-labeled Aβ40-LRP1 complex rapidly colocalizes with PICALM, and remains associated with it for several minutes after exposure, indicating a downstream mechanism of PICALM-regulated clathrin-dependent endocytosis of Aβ40-LRP1 (Figure 3D). Further delineating the endocytotic fate Aβ40-LRP1, proximity ligation studies with several downstream players from endosomal pathway revealed that PICALM-driven Aβ40-LRP1 internalization is shunted away from lysosomal degradation and instead directed towards a transcytotic clearance pathway, shuttling Aβ from the brain into circulation (Figure 3D; Zhao et al., 2015). This seminal finding marks the first of its kind, mechanistically answering the integral question on the relation between the AD risk factor PICALM, amyloid load and LRP1 function.
The binding of PICALM to the intracellular tail of LRP1 at the YXXL domain is specific for Aβ as the ligand, and did not occur with other LRP1 ligands, like, apoE and α2-macroglobulin (A2M; Zhao et al., 2015). Aβ binding to LRP1 presumably elicits a conformational change in its cytoplasmic tail, enabling PICALM-regulated endosomal Aβ transcytosis. PICALM is downregulated in AD (Zhao et al., 2015); these reductions potentially contribute to an exacerbation in disease pathology by hindering LRP1-mediated Aβ transport, further tipping the Aβ balance in the brain (Figure 3D; Zlokovic et al., 2010; Sagare et al., 2012).
Importantly, single-nucleotide polymorphisms (SNPs) in PICALM, located upstream of the gene coding region but not in the coding region, have been identified to influence AD risk (Harold et al., 2009; Lambert et al., 2009; Carrasquillo et al., 2010, 2015; Chen et al., 2012; Tanzi, 2012; Liu et al., 2013; Morgen et al., 2014). It has been reported that some AD-associated SNPs influence PICALM expression (Raj et al., 2012). We recently studied the highly validated rs3851179 PICALM variants whose rs3851179A allele is associated with a lower AD risk than the rs3851179G allele (Lambert et al., 2009, 2013) using inducible pluripotent stem cell (iPSC)-derived endothelial cells. These studies revealed that the protective rs3851179A allele significantly increased PICALM expression and, more importantly, Aβ clearance, reiterating the essential role of PICALM in Aβ clearance (Zhao et al., 2015).
ApoE. ApoE is a 34 kDa glycoprotein produced mainly by the glial cells in brain and liver in the periphery (Huang and Mahley, 2014). ApoE plays an important role in lipid metabolism (Mahley, 1988). In humans, the apoE exists in three isoforms, apoE2, apoE3 and apoE4 which differ from each other by either one or two amino acids at position 112 and 158 (Huang and Mahley, 2014). Several genome wide association studies in the past two decades identified APOE4 as a major genetic risk factor for AD (Corder et al., 1993; Saunders et al., 1993; Tanzi, 2012). Recent studies suggest that apoE4 contributes to vascular and neuronal dysfunction via both Aβ-dependent and Aβ-independent pathways (Deane et al., 2008; Bell et al., 2012; Hudry et al., 2013; Huang and Mahley, 2014; Casey et al., 2015; Halliday et al., 2015). ApoE interacts with Aβ and plays an important role in its metabolism and AD pathogenesis (Wisniewski and Frangione, 1992; Holtzman et al., 2000; DeMattos et al., 2004; Bell et al., 2007; Deane et al., 2008; Jiang et al., 2008; Castellano et al., 2011; Zlokovic, 2013; Tai et al., 2014). Previous work from our group has shown that lipidation status of apoE can affect its binding to Aβ and clearance across the BBB (Martel et al., 1997; Bell et al., 2007; Deane et al., 2008). Our studies have also shown that rapid BBB clearance of Aβ complexed to apoE2 and apoE3 occurs mainly by LRP1 (Figure 3E), by contrast, apoE4-Aβ complexes are removed by slower very low density lipoprotein receptor (VLDLR)-mediated internalization and transcytosis (Bell et al., 2007; Deane et al., 2008).
Restoration of LRP1
In addressing the reduced expression of LRP1 on AD capillaries, a potential method of selectively targeting LRP1 for its restoration in aging or in diseased state is via the delivery of gene transfer vectors. Viral-mediated gene transfer methods, especially adeno-associated viral (AAV) systems have proven to be effective both in different peripheral cell types as well as in the CNS (Davidson et al., 2000; Mingozzi and High, 2011), are safe and more commonly used for targeted gene therapy. In general, gene therapy directed towards neurological disorders remains challenging due to the restricted entry of vectors into the brain authorized by the BBB endothelial lining. However, targeting a receptor like LRP1 is more achievable due to its favorable position on the endothelial membrane, making it directly accessible to therapeutic interventions that may be administered intravenously. In animal models, vascular endothelial cells have been successfully transduced with AAV-2 vector system by using peptides with a high affinity for cerebral vasculature, specific for normal and diseased states identified by in vivo phage panning (Chen et al., 2009). Recently, AAV-9, a serotype less affected by human neutralizing antibodies, has been developed for a highly efficient transduction of endothelial cells (Varadi et al., 2012). Using these recently-developed gene transfer techniques offering a vascular-directed biodistribution, it is conceivable to use AAV-based targeting vectors to deliver LRP1 whole cDNAs or a part of its domains to restore reduced LRP1 expression at the BBB in AD.
Additionally, another candidate site for restoration or selective enhancement of LRP1 by gene therapy is the liver (Sagare et al., 2012). Restoring normal LRP1 levels in hepatocytes by use of the highly successful liver-directed AAV-based gene transfer methods (Mingozzi and High, 2011; Wang et al., 2011) can systemically “vacuum” out the peripheral Aβ and, in turn, promote the removal of brain Aβ by driving the Aβ gradient.
Moreover, Aβ clearance therapy is feasible at the peripheral “sink” constituent of Aβ homeostasis (Zlokovic et al., 2010; Sagare et al., 2011b, 2012, 2013a). In fact, circulating sLRP1-Aβ in plasma serves as an early biomarker for mild cognitive impairment preceding AD-type dementia (Sagare et al., 2011a). Soluble LRP1 and/or its wild-type recombinant cluster IV, WT-LRPIV, bind to free Aβ, preventing it from reentering the brain via the RAGE receptor, and therapeutically enabling reductions in Aβ pathology (Sagare et al., 2007). In order to limit the binding of the recombinant LRPIV to only the neurotoxic Aβ while excluding other LRP-binding ligands, a mutant of LRPIV was recently developed, exhibiting higher binding affinity to Aβ (Sagare et al., 2013a). This mutant, LRPIV-D3674 effectively replaced oxidized sLRP1, is 25–27% more effective than WT-LRPIV in clearing brain endogenous Aβ, and within 3 months of treatment, significantly reduced Aβ levels in hippocampus and cortex of APPsw/0 mice (Sagare et al., 2013a). LRPIV-D3674 is thus capable of regulating Aβ levels in the periphery, and forms the rationale for an efficient Aβ clearance therapy. Restoring reduced or oxidized sLRP1, that contributes to ~70% of bound Aβ (Sagare et al., 2007), can restore the natural peripheral “sink” and salvage the irregular Aβ homeostasis in AD.
In developing these LRP1-targeted efforts, it is, however, important to exert caution in Aβ specificity and the safety of the therapies as LRP1 has a multi-functional role as an endocytotic and a cell signaling receptor in several other systemic mechanisms.
Conclusion
AD is a growing epidemic. There are currently 5.2 million Americans suffering from this disease and the number of individuals affected is projected to triple by the year 2050 (Sano et al., 2013). Failure in Aβ clearance is highly recognized in relation to AD pathogenesis in sporadic or late-onset AD, the most common form of AD occurring in over 95% of patients (Tanzi, 2012). Here, we have briefly reviewed the transvascular Aβ clearance from the brain mediated by LRP1.
Because the cerebrovasculature highly influences Aβ homeostasis it would be beneficial to develop therapeutic strategies in AD including both, the neuronal and vascular components of the CNS. In addition to the advantage that the vascular system is easily accessible from the periphery for pharmacological and genetic manipulations, vascular damage patterns typically precede neuronal injury, providing an opportunistic window for treatment—a concept proposed in the vascular two-hit hypothesis (Zlokovic, 2011). According to the vascular two-hit hypothesis, an initial vascular insult to the brain (hit 1) elicited by hypoxia, hypoperfusion or a disrupted BBB precedes observed amyloid pathology in AD. Accumulated Aβ (hit 2), predominantly an effect of faulty Aβ clearance in late-onset AD, now triggers a pathological cascade of neuronal injury, cognitive decline and AD-dementia.
LRP1 plays a regulatory role in both, the hit 1 (before Aβ accumulation) as well as the hit 2 (amyloid pathology) phases of the AD disease progression, making it an invaluable target in AD clearance therapy (Zlokovic et al., 2010). Currently, there are no drugs that can prevent or reverse AD. Targeted LRP1-based therapies in restoration/enhancement of surface LRP1 at the BBB or on hepatocytes, or the peripheral application of LRPIV Aβ-binding clusters are currently being explored, and show potential in serving as Aβ clearance therapies (Sagare et al., 2012, 2013a). Due to the numerous factors like oxidative stress, receptor shedding, and the influence of other genes, among others, that regulate LRP1 expression and function, thereby affecting Aβ clearance, there is a compelling need to adapt a multifaceted approach in addressing therapeutic interventions. Pharmacological interventions may be used in conjunction with gene therapy; for example, administration of withanolides and withanosides reverse AD pathology in APP/PS1 mice by enhancing LRP1 in the brain microvessels and liver (Sehgal et al., 2012), olive-oil-derived oleocanthal enhances cerebral Aβ clearance by upregulating LRP1 at the BBB (Abuznait et al., 2013). Moreover, changes in lifestyle can have a meaningful impact as preventive measures in AD. An antioxidant-rich diet, such as the recently investigated Mediterranean-DASH (Dietary Approaches to Stop Hypertension) Intervention for Neurodegenerative Delay (MIND) diet has shown potential in reducing AD risk (Morris et al., 2015), presumably by rousing the body’s natural anti-oxidant defense mechanism. Incorporating physical activity into one’s lifestyle serves as a pro-angiogenic factor having beneficial effects on cognition. Additionally, another promising compound performing as a vasculoprotectant, antioxidant and an anti-inflammatory agent is the activated protein C (APC; Griffin et al., 2002, 2015).
AD-associated genes regulating LRP1 function, like APOE, SLC2A1 and PICALM, too extend potential as targets for AD therapy in alleviating AD pathology and/or amend Aβ imbalance. Future studies focused on genetically engineered mice possessing variants in these AD risk genes can generate more knowledge on the role of LRP1-mediate transvacular clearance of Aβ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This manuscript is supported in part by the National Institutes of Health grants AG039452, AG23084, and NS34467 and Zilkha Senior Scholar Support to BVZ.
References
Abbott, N. J., Patabendige, A. A. K., Dolman, D. E. M., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Abuznait, A. H., Qosa, H., Busnena, B. A., El Sayed, K. A., and Kaddoumi, A. (2013). Olive-oil-derived oleocanthal enhances β-amyloid clearance as a potential neuroprotective mechanism against Alzheimer’s disease: in vitro and in vivo studies. ACS Chem. Neurosci. 4, 973–982. doi: 10.1021/cn400024q
An, J., Zhang, C., Polavarapu, R., Zhang, X., Zhang, X., and Yepes, M. (2008). Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein induce Akt phosphorylation in the ischemic brain. Blood 112, 2787–2794. doi: 10.1182/blood-2008-02-141630
Bachmeier, C., Shackleton, B., Ojo, J., Paris, D., Mullan, M., and Crawford, F. (2014). Apolipoprotein E isoform-specific effects on lipoprotein receptor processing. Neuromolecular Med. 16, 686–696. doi: 10.1007/s12017-014-8318-6
Baig, S., Joseph, S. A., Tayler, H., Abraham, R., Owen, M. J., Williams, J., et al. (2010). Distribution and expression of picalm in Alzheimer disease. J. Neuropathol. Exp. Neurol. 69, 1071–1077. doi: 10.1097/NEN.0b013e3181f52e01
Barnes, H., Ackermann, E. J., and van der Geer, P. (2003). v-Src induces Shc binding to tyrosine 63 in the cytoplasmic domain of the LDL receptor-related protein 1. Oncogene 22, 3589–3597. doi: 10.1038/sj.onc.1206504
Bateman, R. J., Munsell, L. Y., Morris, J. C., Swarm, R., Yarasheski, K. E., and Holtzman, D. M. (2006). Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861. doi: 10.1038/nm1438
Beal, M. F. (2005). Oxidative damage as an early marker of Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 26, 585–586. doi: 10.1016/j.neurobiolaging.2004.09.022
Bell, R. D., Deane, R., Chow, N., Long, X., Sagare, A., Singh, I., et al. (2009). SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat. Cell Biol. 11, 143–153. doi: 10.1038/ncb1819
Bell, R. D., Sagare, A. P., Friedman, A. E., Bedi, G. S., Holtzman, D. M., Deane, R., et al. (2007). Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 27, 909–918. doi: 10.1038/sj.jcbfm.9600419
Bell, R. D., Winkler, E. A., Singh, I., Sagare, A. P., Deane, R., Wu, Z., et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. doi: 10.1038/nature11087
Boyd-Kimball, D., Poon, H. F., Lynn, B. C., Cai, J., Pierce, W. M., Klein, J. B., et al. (2006). Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1–42): implications for Alzheimer’s disease. Neurobiol. Aging 27, 1239–1249. doi: 10.1016/j.neurobiolaging.2005.07.001
Brody, D. L., Magnoni, S., Schwetye, K. E., Spinner, M. L., Esparza, T. J., Stocchetti, N., et al. (2008). Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321, 1221–1224. doi: 10.1126/science.1161591
Bu, G., Sun, Y., Schwartz, A. L., and Holtzman, D. M. (1998). Nerve growth factor induces rapid increases in functional cell surface low density lipoprotein receptor-related protein. J. Biol. Chem. 273, 13359–13365. doi: 10.1074/jbc.273.21.13359
Carrasquillo, M. M., Belbin, O., Hunter, T. A., Ma, L., Bisceglio, G. D., Zou, F., et al. (2010). Replication of CLU, CR1 and PICALM associations with alzheimer disease. Arch. Neurol. 67, 961–964. doi: 10.1001/archneurol.2010.147
Carrasquillo, M. M., Crook, J. E., Pedraza, O., Thomas, C. S., Pankratz, V. S., Allen, M., et al. (2015). Late-onset Alzheimer’s risk variants in memory decline, incident mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 36, 60–67. doi: 10.1016/j.neurobiolaging.2014.07.042
Casey, C. S., Atagi, Y., Yamazaki, Y., Shinohara, M., Tachibana, M., Fu, Y., et al. (2015). Apolipoprotein E inhibits cerebrovascular pericyte mobility through a RhoA-mediated pathway. J. Biol. Chem. 290, 14208–14217. doi: 10.1074/jbc.M114.625251
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Chefer, V. I., Thompson, A. C., Zapata, A., and Shippenberg, T. S. (2009). Overview of brain Microdialysis. Curr. Protoc. Neurosci. Chapter 7:Unit7.1. doi: 10.1002/0471142301.ns0701s47
Chen, Y. H., Chang, M., and Davidson, B. L. (2009). Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat. Med. 15, 1215–1218. doi: 10.1038/nm.2025
Chen, L. H., Kao, P. Y. P., Fan, Y. H., Ho, D. T. Y., Chan, C. S. Y., Yik, P. Y., et al. (2012). Polymorphisms of CR1, CLU and PICALM confer susceptibility of Alzheimer’s disease in a southern Chinese population. Neurobiol. Aging 33, 210.e1–210.e7. doi: 10.1016/j.neurobiolaging.2011.09.016
Chen, Z., and Zhong, C. (2014). Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 30, 271–281. doi: 10.1007/s12264-013-1423-y
Chow, N., Bell, R. D., Deane, R., Streb, J. W., Chen, J., Brooks, A., et al. (2007). Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc. Natl. Acad. Sci. U S A 104, 823–828. doi: 10.1073/pnas.0608251104
Cirrito, J. R., May, P. C., O’Dell, M. A., Taylor, J. W., Parsadanian, M., Cramer, J. W., et al. (2003). In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J. Neurosci. 23, 8844–8853.
Clementi, M. E., Pezzotti, M., Orsini, F., Sampaolese, B., Mezzogori, D., Grassi, C., et al. (2006). Alzheimer’s amyloid beta-peptide (1–42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem. Biophys. Res. Commun. 342, 206–213. doi: 10.1016/j.bbrc.2006.01.137
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Davidson, B. L., Stein, C. S., Heth, J. A., Martins, I., Kotin, R. M., Derksen, T. A., et al. (2000). Recombinant adeno-associated virus type 2, 4 and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc. Natl. Acad. Sci. U S A 97, 3428–3432. doi: 10.1073/pnas.97.7.3428
Deane, R., Du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi: 10.1038/nm890
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/JCI36663
Deane, R., Singh, I., Sagare, A. P., Bell, R. D., Ross, N. T., LaRue, B., et al. (2012). A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Invest. 122, 1377–1392. doi: 10.1172/JCI58642
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43, 333–344. doi: 10.1016/j.neuron.2004.07.017
del Cárdenas-Aguayo, M. C., del Silva-Lucero, M. C., Cortes-Ortiz, M., Jiménez-Ramos, B., Gómez-Virgilio, L., Ramírez-Rodríguez, G., et al. (2014). “Physiological role of amyloid beta in neural cells: the cellular trophic activity,” in Neurochemistry, ed. T. Heinbockel (InTech), ISBN: 978-953-51-1237-2. Available at: http://www.intechopen.com/books/neurochemistry/physiological-role-of-amyloid-beta-in-neural-cells-the-cellular-trophic-activity
DeMattos, R. B., Cirrito, J. R., Parsadanian, M., May, P. C., O’Dell, M. A., Taylor, J. W., et al. (2004). ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41, 193–202. doi: 10.1016/s0896-6273(03)00850-x
Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A., Silverberg, G. D., Miller, M. C., et al. (2006). RAGE, LRP-1 and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 112, 405–415. doi: 10.1007/s00401-006-0115-3
Drake, J., Link, C. D., and Butterfield, D. A. (2003). Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol. Aging 24, 415–420. doi: 10.1016/s0197-4580(02)00225-7
Dreyling, M. H., Martinez-Climent, J. A., Zheng, M., Mao, J., Rowley, J. D., and Bohlander, S. K. (1996). The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc. Natl. Acad. Sci. U S A 93, 4804–4809. doi: 10.1073/pnas.93.10.4804
Emonard, H., Théret, L., Bennasroune, A. H., and Dedieu, S. (2014). Regulation of LRP-1 expression: make the point. Pathol. Biol. (Paris) 62, 84–90. doi: 10.1016/j.patbio.2014.02.002
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U S A 100, 4162–4167. doi: 10.1073/pnas.0230450100
Farris, W., Schütz, S. G., Cirrito, J. R., Shankar, G. M., Sun, X., George, A., et al. (2007). Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. 171, 241–251. doi: 10.2353/ajpath.2007.070105
Griffin, J. H., Zlokovic, B., and Fernández, J. A. (2002). Activated protein C: potential therapy for severe sepsis, thrombosis, and stroke. Semin. Hematol. 39, 197–205. doi: 10.1053/shem.2002.34093
Griffin, J. H., Zlokovic, B. V., and Mosnier, L. O. (2015). Activated protein C: biased for translation. Blood 125, 2898–2907. doi: 10.1182/blood-2015-02-355974
Halliday, M. R., Rege, S. V., Ma, Q., Zhao, Z., Miller, C. A., Winkler, E. A., et al. (2015). Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. doi: 10.1038/jcbfm.2015.44 [Epub ahead of print].
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
Hawkes, C. A., Sullivan, P. M., Hands, S., Weller, R. O., Nicoll, J. A. R., and Carare, R. O. (2012). Disruption of arterial perivascular drainage of amyloid-β from the brains of mice expressing the human APOE ε4 allele. PLoS One 7:e41636. doi: 10.1371/journal.pone.0041636
Herholz, K. (2010). Cerebral glucose metabolism in preclinical and prodromal Alzheimer’s disease. Expert Rev. Neurother. 10, 1667–1673. doi: 10.1586/ern.10.136
Hernandez-Guillamon, M., Mawhirt, S., Blais, S., Montaner, J., Neubert, T. A., Rostagno, A., et al. (2015). Sequential Abeta degradation by the matrix metalloproteases MMP-2 and MMP-9. J. Biol. Chem. 290, 15078–15091. doi: 10.1074/jbc.m114.610931
Herz, J., Hamann, U., Rogne, S., Myklebost, O., Gausepohl, H., and Stanley, K. K. (1988). Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J. 7, 4119–4127.
Holtzman, D. M., Bales, K. R., Tenkova, T., Fagan, A. M., Parsadanian, M., Sartorius, L. J., et al. (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 97, 2892–2897. doi: 10.1073/pnas.050004797
Horwood, N., and Davies, D. C. (1994). Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch. 425, 69–72. doi: 10.1007/bf00193951
Huang, Y., and Mahley, R. W. (2014). Apolipoprotein E: structure and function in lipid metabolism, neurobiology and Alzheimer’s diseases. Neurobiol. Dis. 72(Pt. A), 3–12. doi: 10.1016/j.nbd.2014.08.025
Hudry, E., Dashkoff, J., Roe, A. D., Takeda, S., Koffie, R. M., Hashimoto, T., et al. (2013). Gene transfer of human Apoe Isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 5:212ra161. doi: 10.1126/scitranslmed.3007000
Hunt, A., Schönknecht, P., Henze, M., Seidl, U., Haberkorn, U., and Schröder, J. (2007). Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res. 155, 147–154. doi: 10.1016/j.pscychresns.2006.12.003
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Ito, S., Ohtsuki, S., and Terasaki, T. (2006). Functional characterization of the brain-to-blood efflux clearance of human amyloid-beta peptide (1–40) across the rat blood-brain barrier. Neurosci. Res. 56, 246–252. doi: 10.1016/j.neures.2006.07.006
Jiang, Q., Lee, C. Y. D., Mandrekar, S., Wilkinson, B., Cramer, P., Zelcer, N., et al. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693. doi: 10.1016/j.neuron.2008.04.010
Kalaria, R. N., and Harik, S. I. (1989). Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J. Neurochem. 53, 1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x
Kanekiyo, T., and Bu, G. (2014). The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 6:93. doi: 10.3389/fnagi.2014.00093
Kanemitsu, H., Tomiyama, T., and Mori, H. (2003). Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci. Lett. 350, 113–116. doi: 10.1016/s0304-3940(03)00898-x
Kang, D. E., Pietrzik, C. U., Baum, L., Chevallier, N., Merriam, D. E., Kounnas, M. Z., et al. (2000). Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Invest. 106, 1159–1166. doi: 10.1172/jci11013
Krieger, M., and Herz, J. (1994). Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL Receptor-Related Protein (LRP). Annu. Rev. Biochem. 63, 601–637. doi: 10.1146/annurev.bi.63.070194.003125
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Li, Y., Lu, W., Marzolo, M. P., and Bu, G. (2001a). Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J. Biol. Chem. 276, 18000–18006. doi: 10.1074/jbc.m101589200
Li, Y., van Kerkhof, P., Marzolo, M. P., Strous, G. J., and Bu, G. (2001b). Identification of a major cyclic AMP-dependent protein kinase A phosphorylation site within the cytoplasmic tail of the low-density lipoprotein receptor-related protein: implication for receptor-mediated endocytosis. Mol. Cell. Biol. 21, 1185–1195. doi: 10.1128/mcb.21.4.1185-1195.2001
Lillis, A. P., Van Duyn, L. B., Murphy-Ullrich, J. E., and Strickland, D. K. (2008). LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918. doi: 10.1152/physrev.00033.2007
Liu, G., Zhang, S., Cai, Z., Ma, G., Zhang, L., Jiang, Y., et al. (2013). PICALM gene rs3851179 polymorphism contributes to Alzheimer’s disease in an Asian population. Neuromolecular Med. 15, 384–388. doi: 10.1007/s12017-013-8225-2
Liu, Q., Zhang, J., Tran, H., Verbeek, M. M., Reiss, K., Estus, S., et al. (2009). LRP1 shedding in human brain: roles of ADAM10 and ADAM17. Mol. Neurodegener. 4:17. doi: 10.1186/1750-1326-4-17
Llorente-Cortés, V., Costales, P., Bernués, J., Camino-Lopez, S., and Badimon, L. (2006). Sterol regulatory element-binding protein-2 negatively regulates low density lipoprotein receptor-related protein transcription. J. Mol. Biol. 359, 950–960. doi: 10.1016/j.jmb.2006.04.008
Llorente-Cortés, V., Royo, T., Otero-Viñas, M., Berrozpe, M., and Badimon, L. (2007). Sterol regulatory element binding proteins downregulate LDL receptor-related protein (LRP1) expression and LRP1-mediated aggregated LDL uptake by human macrophages. Cardiovasc. Res. 74, 526–536. doi: 10.1016/j.cardiores.2007.02.020
Mackic, J. B., Bading, J., Ghiso, J., Walker, L., Wisniewski, T., Frangione, B., et al. (2002). Circulating amyloid-β peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer’s disease lesions. Vascul. Pharmacol. 38, 303–313. doi: 10.1016/s1537-1891(02)00198-2
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630. doi: 10.1126/science.3283935
Mann, G. E., Zlokovic, B. V., and Yudilevich, D. L. (1985). Evidence for a lactate transport system in the sarcolemmal membrane of the perfused rabbit heart: kinetics of unidirectional influx, carrier specificity and effects of glucagon. Biochim. Biophys. Acta 819, 241–248. doi: 10.1016/0005-2736(85)90179-8
Martel, C. L., Mackic, J. B., Matsubara, E., Governale, S., Miguel, C., Miao, W., et al. (1997). Isoform-specific effects of apolipoproteins E2, E3 and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer’s amyloid beta. J. Neurochem. 69, 1995–2004. doi: 10.1046/j.1471-4159.1997.69051995.x
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774. doi: 10.1126/science.1197623
Mingozzi, F., and High, K. A. (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 12, 341–355. doi: 10.1038/nrg2988
Mooradian, A. D., Chung, H. C., and Shah, G. N. (1997). GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol. Aging 18, 469–474. doi: 10.1016/s0197-4580(97)00111-5
Moosmann, B., and Behl, C. (2002). Antioxidants as treatment for neurodegenerative disorders. Expert Opin. Investig. Drugs 11, 1407–1435. doi: 10.1517/13543784.11.10.1407
Morgen, K., Ramirez, A., Frölich, L., Tost, H., Plichta, M. M., Kölsch, H., et al. (2014). Genetic interaction of PICALM and APOE is associated with brain atrophy and cognitive impairment in Alzheimer’s disease. Alzheimers Dement. 10, S269–S276. doi: 10.1016/j.jalz.2013.11.001
Morris, M. C., Tangney, C. C., Wang, Y., Sacks, F. M., Bennett, D. A., and Aggarwal, N. T. (2015). MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. doi: 10.1016/j.jalz.2014.11.009 [Epub ahead of print].
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 18, 800–806. doi: 10.1038/nn.4018
Parikh, I., Fardo, D. W., and Estus, S. (2014). Genetics of PICALM expression and Alzheimer’s disease. PLoS One 9:e91242. doi: 10.1371/journal.pone.0091242
Polavarapu, R., Gongora, M. C., Yi, H., Ranganthan, S., Lawrence, D. A., Strickland, D., et al. (2007). Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood 109, 3270–3278. doi: 10.1182/blood-2006-08-043125
Pollay, M. (2010). The function and structure of the cerebrospinal fluid outflow system. Cerebrospinal Fluid Res. 7:9. doi: 10.1186/1743-8454-7-9
Qiu, Z., Strickland, D. K., Hyman, B. T., and Rebeck, G. W. (2001). Elevation of LDL receptor-related protein levels via ligand interactions in Alzheimer disease and in vitro. J. Neuropathol. Exp. Neurol. 60, 430–440.
Quinn, K. A., Grimsley, P. G., Dai, Y. P., Tapner, M., Chesterman, C. N., and Owensby, D. A. (1997). Soluble low density lipoprotein receptor-related protein (LRP) circulates in human plasma. J. Biol. Chem. 272, 23946–23951. doi: 10.1074/jbc.272.38.23946
Raj, T., Shulman, J. M., Keenan, B. T., Chibnik, L. B., Evans, D. A., Bennett, D. A., et al. (2012). Alzheimer disease susceptibility loci: evidence for a protein network under natural selection. Am. J. Hum. Genet. 90, 720–726. doi: 10.1016/j.ajhg.2012.02.022
Ranganathan, S., Liu, C.-X., Migliorini, M. M., Von Arnim, C. A. F., Peltan, I. D., Mikhailenko, I., et al. (2004). Serine and threonine phosphorylation of the low density lipoprotein receptor-related protein by protein kinase Calpha regulates endocytosis and association with adaptor molecules. J. Biol. Chem. 279, 40536–40544. doi: 10.1074/jbc.m407592200
Reekmans, S. M., Pflanzner, T., Gordts, P. L. S. M., Isbert, S., Zimmermann, P., Annaert, W., et al. (2010). Inactivation of the proximal NPXY motif impairs early steps in LRP1 biosynthesis. Cell. Mol. Life Sci. 67, 135–145. doi: 10.1007/s00018-009-0171-7
Roberts, K. F., Elbert, D. L., Kasten, T. P., Patterson, B. W., Sigurdson, W. C., Connors, R. E., et al. (2014). Amyloid-β efflux from the central nervous system into the plasma. Ann. Neurol. 76, 837–844. doi: 10.1002/ana.24270
Rozanov, D. V., Hahn-Dantona, E., Strickland, D. K., and Strongin, A. Y. (2004). The low density lipoprotein receptor-related protein LRP is regulated by membrane type-1 matrix metalloproteinase (MT1-MMP) proteolysis in malignant cells. J. Biol. Chem. 279, 4260–4268. doi: 10.1074/jbc.m311569200
Sagare, A. P., Bell, R. D., Srivastava, A., Sengillo, J. D., Singh, I., Nishida, Y., et al. (2013a). A lipoprotein receptor cluster IV mutant preferentially binds amyloid-β and regulates its clearance from the mouse brain. J. Biol. Chem. 288, 15154–15166. doi: 10.1074/jbc.m112.439570
Sagare, A. P., Bell, R. D., Zhao, Z., Ma, Q., Winkler, E. A., Ramanathan, A., et al. (2013b). Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 4:2932. doi: 10.1038/ncomms3932
Sagare, A., Deane, R., Bell, R. D., Johnson, B., Hamm, K., Pendu, R., et al. (2007). Clearance of amyloid-beta by circulating lipoprotein receptors. Nat. Med. 13, 1029–1031. doi: 10.1038/nm1635
Sagare, A. P., Deane, R., Zetterberg, H., Wallin, A., Blennow, K., and Zlokovic, B. V. (2011a). Impaired lipoprotein receptor-mediated peripheral binding of plasma amyloid-β is an early biomarker for mild cognitive impairment preceding Alzheimer’s disease. J. Alzheimers Dis. 24, 25–34. doi: 10.3233/JAD-2010-101248
Sagare, A. P., Winkler, E. A., Bell, R. D., Deane, R., and Zlokovic, B. V. (2011b). From the liver to the blood-brain barrier: an interconnected system regulating brain amyloid-β levels. J. Neurosci. Res. 89, 967–968. doi: 10.1002/jnr.22670
Sagare, A. P., Deane, R., and Zlokovic, B. V. (2012). Low-density lipoprotein receptor-related protein 1: a physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacol. Ther. 136, 94–105. doi: 10.1016/j.pharmthera.2012.07.008
Sano, M., Dahlman, K., Sewell, M., and Zhu, C. W. (2013). “The economics of caring for individuals with Alzheimer’s disease,” in Caregiving for Alzheimer’s Disease and Related Disorders Caregiving: Research • Practice • Policy, eds S. H. Zarit and R. C. Talley (New York: Springer), 71–90. Available at: http://link.springer.com/chapter/10.1007/978-1-4614-5335-2_5. [accessed May 31, 2015].
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., et al. (1993). Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472. doi: 10.1212/wnl.43.8.1467
Sehgal, N., Gupta, A., Valli, R. K., Joshi, S. D., Mills, J. T., Hamel, E., et al. (2012). Withania somnifera reverses Alzheimer’s disease pathology by enhancing low-density lipoprotein receptor-related protein in liver. Proc. Natl. Acad. Sci. U S A 109, 3510–3515. doi: 10.1073/pnas.1112209109
Selvais, C., D’Auria, L., Tyteca, D., Perrot, G., Lemoine, P., Troeberg, L., et al. (2011). Cell cholesterol modulates metalloproteinase-dependent shedding of low-density lipoprotein receptor-related protein-1 (LRP-1) and clearance function. FASEB J. 25, 2770–2781. doi: 10.1096/fj.10-169508
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/jci10498
Shiiki, T., Ohtsuki, S., Kurihara, A., Naganuma, H., Nishimura, K., Tachikawa, M., et al. (2004). Brain insulin impairs amyloid-beta(1–40) clearance from the brain. J. Neurosci. 24, 9632–9637. doi: 10.1523/jneurosci.2236-04.2004
Silverberg, G. D., Miller, M. C., Messier, A. A., Majmudar, S., Machan, J. T., Donahue, J. E., et al. (2010). Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J. Neuropathol. Exp. Neurol. 69, 98–108. doi: 10.1097/NEN.0b013e3181c8ad2f
Simpson, I. A., Chundu, K. R., Davies-Hill, T., Honer, W. G., and Davies, P. (1994). Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 35, 546–551. doi: 10.1002/ana.410350507
Sorkin, A., and von Zastrow, M. (2009). Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 10, 609–622. doi: 10.1038/nrm2748
Sutcliffe, J. G., Hedlund, P. B., Thomas, E. A., Bloom, F. E., and Hilbush, B. S. (2011). Peripheral reduction of β-amyloid is sufficient to reduce brain β-amyloid: implications for Alzheimer’s disease. J. Neurosci. Res. 89, 808–814. doi: 10.1002/jnr.22603
Tai, L. M., Mehra, S., Shete, V., Estus, S., Rebeck, G. W., Bu, G., et al. (2014). Soluble apoE/Aβ complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol. Neurodegener. 9:2. doi: 10.1186/1750-1326-9-2
Tamaki, C., Ohtsuki, S., Iwatsubo, T., Hashimoto, T., Yamada, K., Yabuki, C., et al. (2006). Major involvement of low-density lipoprotein receptor-related protein 1 in the clearance of plasma free amyloid beta-peptide by the liver. Pharm. Res. 23, 1407–1416. doi: 10.1007/s11095-006-0208-7
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006296. doi: 10.1101/cshperspect.a006296
Tarasoff, J., Carare, R., Osorio, R., Glodzil, L., Butler, T., Fieremans, E., et al. (in press). Clearance systems in the brain and Alzheimer disease. Nat. Rev. Neurol.
Tebar, F., Bohlander, S. K., and Sorkin, A. (1999). Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin and the impact of overexpression on clathrin-mediated traffic. Mol. Biol. Cell 10, 2687–2702. doi: 10.1091/mbc.10.8.2687
Torres, L. L., Quaglio, N. B., de Souza, G. T., Garcia, R. T., Dati, L. M. M., Moreira, W. L., et al. (2011). Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 26, 59–68. doi: 10.3233/JAD-2011-110284
Treusch, S., Hamamichi, S., Goodman, J. L., Matlack, K. E. S., Chung, C. Y., Baru, V., et al. (2011). Functional links between Aβ toxicity, endocytic trafficking and Alzheimer’s disease risk factors in yeast. Science 334, 1241–1245. doi: 10.1126/science.1213210
Ueno, M., Nakagawa, T., Wu, B., Onodera, M., Huang, C.-L., Kusaka, T., et al. (2010). Transporters in the brain endothelial barrier. Curr. Med. Chem. 17, 1125–1138. doi: 10.2174/092986710790827816
Ujiie, M., Dickstein, D. L., Carlow, D. A., and Jefferies, W. A. (2003). Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 10, 463–470. doi: 10.1080/mic.10.6.463.470
van der Geer, P. (2002). Phosphorylation of LRP1: regulation of transport and signal transduction. Trends Cardiovasc. Med. 12, 160–165. doi: 10.1016/s1050-1738(02)00154-8
Varadi, K., Michelfelder, S., Korff, T., Hecker, M., Trepel, M., Katus, H. A., et al. (2012). Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene Ther. 19, 800–809. doi: 10.1038/gt.2011.143
von Arnim, C. A. F., Kinoshita, A., Peltan, I. D., Tangredi, M. M., Herl, L., Lee, B. M., et al. (2005). The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J. Biol. Chem. 280, 17777–17785. doi: 10.1074/jbc.m414248200
Wang, L., Bell, P., Lin, J., Calcedo, R., Tarantal, A. F., and Wilson, J. M. (2011). AAV8-mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta). Mol. Ther. 19, 2012–2020. doi: 10.1038/mt.2011.151
Weller, R. O., Subash, M., Preston, S. D., Mazanti, I., and Carare, R. O. (2008). Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 18, 253–266. doi: 10.1111/j.1750-3639.2008.00133.x
Winkler, E. A., Nishida, Y., Sagare, A. P., Rege, S. V., Bell, R. D., Perlmutter, D., et al. (2015). GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 18, 521–530. doi: 10.1038/nn.3966
Wisniewski, T., and Frangione, B. (1992). Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 135, 235–238. doi: 10.1016/0304-3940(92)90444-c
Wu, Z., Guo, H., Chow, N., Sallstrom, J., Bell, R. D., Deane, R., et al. (2005). Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 11, 959–965. doi: 10.1038/nm1287
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. doi: 10.1038/382685a0
Zhao, Z., Sagare, A. P., Ma, Q., Halliday, M. R., Kong, P., Kisler, K., et al. (2015). Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 18, 978–987. doi: 10.1038/nn.4025
Zlokovic, B. V. (1995). Cerebrovascular permeability to peptides: manipulations of transport systems at the blood-brain barrier. Pharm. Res. 12, 1395–1406. doi: 10.1023/A:1016254514167
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Zlokovic, B. V. (2010). Neurodegeneration and the neurovascular unit. Nat. Med. 16, 1370–1371. doi: 10.1038/nm1210-1370
Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12, 723–738. doi: 10.1038/nrn3114
Zlokovic, B. V. (2013). Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol. 70, 440–444. doi: 10.1001/jamaneurol.2013.2152
Zlokovic, B. V., Begley, D. J., and Chain-Eliash, D. G. (1985). Blood-brain barrier permeability to leucine-enkephalin,d-Alanine2-d-leucine5-enkephalin and their N-terminal amino acid (tyrosine). Brain Res. 336, 125–132. doi: 10.1016/0006-8993(85)90423-8
Zlokovic, B. V., Deane, R., Sagare, A. P., Bell, R. D., and Winkler, E. A. (2010). Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer’s amyloid β-peptide elimination from the brain. J. Neurochem. 115, 1077–1089. doi: 10.1111/j.1471-4159.2010.07002.x
Zlokovic, B. V., and Frangione, B. (2003). “Transport-clearance hypothesis for Alzheimer’s disease and potential therapeutic implications,” in Aβ Metabolism in Alzheimer’s Disease, ed. T. C. Saido (Georgetown, TX: Landes Bioscience), 114–122.
Keywords: amyloid β clearance, blood-brain barrier, lipoprotein receptor-related protein 1 (LRP1), PICALM, Alzheimer’s disease (AD)
Citation: Ramanathan A, Nelson AR, Sagare AP and Zlokovic BV (2015) Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 7:136. doi: 10.3389/fnagi.2015.00136
Received: 02 June 2015; Accepted: 02 July 2015;
Published: 15 July 2015.
Edited by:
Roxana Octavia Carare, University of Southampton, UKReviewed by:
Ignacio Torres-Aleman, Cajal Institute, SpainRobert Marr, Rosalind Franklin University of Medicine and Science, USA
Stéphane Dedieu, CNRS UMR 7369 MEDYC and Reims University, France
Copyright © 2015 Ramanathan, Nelson, Sagare and Zlokovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Berislav V. Zlokovic, Department of Physiology and Biophysics, Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, 1501 San Pablo Street, Los Angeles, CA 90089, USA,emxva292aWNAdXNjLmVkdQ==