 Alberto Benussi
Alberto Benussi Alessandro Padovani
Alessandro Padovani Barbara Borroni
Barbara Borroni- Centre for Ageing Brain and Neurodegenerative Disorders, Neurology Unit, Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy
Frontotemporal dementia (FTD) is a genetically and pathologically heterogeneous disorder characterized by personality changes, language impairment, and deficits of executive functions associated with frontal and temporal lobe degeneration. Different phenotypes have been defined on the basis of presenting clinical symptoms, i.e., the behavioral variant of FTD, the agrammatic variant of primary progressive aphasia, and the semantic variant of PPA. Some patients have an associated movement disorder, either parkinsonism, as in progressive supranuclear palsy and corticobasal syndrome, or motor neuron disease (FTD–MND). A family history of dementia is found in 40% of cases of FTD and about 10% have a clear autosomal-dominant inheritance. Genetic studies have identified several genes associated with monogenic FTD: microtubule-associated protein tau, progranulin, TAR DNA-binding protein 43, valosin-containing protein, charged multivesicular body protein 2B, fused in sarcoma, and the hexanucleotide repeat expansion in intron 1 of the chromosome 9 open reading frame 72. Patients often present with an extensive phenotypic variability, even among different members of the same kindred carrying an identical disease mutation. The objective of the present work is to review and evaluate available literature data in order to highlight recent advances in clinical, biological, and neuroimaging features of monogenic frontotemporal lobar degeneration and try to identify different mechanisms underlying the extreme phenotypic heterogeneity that characterizes this disease.
Introduction
Frontotemporal dementia (FTD) is the second most common cause of dementia in the presenile age group (<65 years of age), and accounts for 5–15% of all cases of dementia, with a prevalence of 10–15 per 100,000 subjects in the age group of 45–65 years (Bird et al., 2003; Feldman et al., 2003; Borroni et al., 2015b).
Frontotemporal dementia is a genetically and pathologically heterogeneous disorder characterized by personality changes, language impairment, and deficits of executive functions, associated with frontal and temporal lobe degeneration (The Lund and Manchester Groups, 1994; Neary et al., 1998; McKhann et al., 2001).
Current redefined clinical criteria recognize different phenotypes on the basis of presenting clinical symptoms, namely the behavioral variant of FTD (bvFTD), the agrammatic variant of primary progressive aphasia (avPPA), and the semantic variant of PPA (svPPA) (Mesulam, 1982; Gorno-Tempini et al., 2011; Rascovsky et al., 2011). Moreover, some patients have an associated parkinsonism, as in progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS), or motor neuron disease (FTD–MND) (Lomen-Hoerth et al., 2002; Burrell et al., 2011; Armstrong et al., 2013).
A family history of dementia is found in 25–50% of cases of FTD and about 10% have a clear autosomal-dominant inheritance (Seelaar et al., 2008; Rohrer et al., 2009). Genetic studies have identified several genes associated with monogenic FTD: in 1998, mutations in the microtubule-associated protein tau (MAPT) gene on chromosome 17 were identified in families with FTD and parkinsonism (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). In spite of this finding, there have been numerous reports of families with linkage to chromosome 17q21 but without evidence of MAPT pathogenic mutations (Froelich et al., 1997; Kertesz et al., 2000; Rosso et al., 2001; Rademakers et al., 2002).
In 2006, the second FTD-related gene on chromosome 17 was discovered in the progranulin (GRN) gene (Baker et al., 2006; Cruts et al., 2006), accounting for even more cases than mutations in MAPT explaining all the 17q21-linked autosomal-dominant FTD cases not accounted for by MAPT mutations (www.molgen.ua.ac.be/ADMutations/).
Eventually, a genetic linkage to a locus on chromosome 9p was found in many families with FTD and motor neuron disease (Morita et al., 2006; Vance et al., 2006; Luty et al., 2008; Le Ber et al., 2009; Boxer et al., 2011); only recently in 2011, two separate groups identified the defect as being an expanded hexanucleotide repeat in a non-coding region of the chromosome 9 open reading frame 72 (C9orf72) gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011).
Other uncommon mutations have been identified in the valosin-containing protein (VCP) gene, which cause inclusion body myopathy with Paget disease of bone and FTD (Watts et al., 2004), and in the charged multivesicular body protein 2B (CHMP2B) gene, involved in the endosomal–lysosomal pathway (Skibinski et al., 2005). Few reports have also described FTD associated with mutations in the fused in sarcoma (FUS) (Broustal et al., 2010; Van Langenhove et al., 2010), dynactin (DCTN1) (Münch et al., 2005), sequestosome 1 (SQSTM1) (Fecto et al., 2011; Rubino et al., 2012; van der Zee et al., 2014), colony-stimulating factor 1 receptor (CFS1R) (Münch et al., 2005; Guerreiro et al., 2013b), triggering receptor expressed on myeloid cells (TREM2) (Borroni et al., 2013; Guerreiro, 2013), ubiquilin-2 (UBQLN2) (Gellera et al., 2013), and heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) genes (Kim et al., 2013).
Clinical FTD is associated with different types of underlying neuropathology, and the term frontotemporal lobar degeneration (FTLD), characterized by the relative selective degeneration of the frontal and temporal lobes, is used to describe the pathological hallmarks of the disease. Abnormal intracellular accumulation of insoluble proteins have been identified with immunohistochemical techniques and, according to the predominant neuronal inclusions, FTLD cases have been categorized into several types (Mackenzie et al., 2008, 2010). At first, neuronal and glial inclusions of hyperphosphorylated tau protein were identified in FTLD-tau, which represent nearly 30–40% of FTLD cases, and are found in patients carrying MAPT gene mutations, as well as in Pick’s disease, PSP, and corticobasal degeneration (CBD) (Josephs et al., 2006).
Only briefly after the discovery of the GRN gene, TAR DNA-binding protein 43 (TDP-43) inclusions were identified as the predominant hallmark of the originally identified ubiquitin positive inclusions, which were termed FTLD-U (Mackenzie et al., 2006), and subsequently renamed FTLD-TDP (Arai et al., 2006; Neumann et al., 2006). TDP-43 inclusions have also been identified in the majority of amyotrophic lateral sclerosis (ALS) and FTD-MND cases, in particular in patients with mutations of the C9orf72 gene. Four FTLD-TDP subtypes (A–D) have been proposed based on the location of these inclusions (Mackenzie et al., 2011).
Finally in 2009, the ~10% of FTLD cases negative for tau or TDP-43 pathology were characterized by inclusions immunoreactive for FUS, which were then labeled as FTLD-FUS (Munoz et al., 2009; Neumann et al., 2009b; Rademakers et al., 2009). Furthermore, the inclusions positive for FUS were found to also stain for other members of the FET family of DNA/RNA-binding proteins, comprising the TATA-binding protein-associated factor 15 (TAF-15) and Ewing sarcoma protein (EWS), distinguishing FTLD with FUS pathology from ALS with FUS mutations (Neumann et al., 2011).
Patients with monogenic FTD often present with an extensive phenotypic variability, even among different members of the same kindred carrying an identical disease mutation.
The objective of the present work is to review and evaluate available literature data in order to highlight recent advances in clinical, biological, and neuroimaging features of monogenic FTLD and try to identify the hallmarks of different monogenic FTDs.
MAPT Mutations
The MAPT gene on chromosome 17, encoding the tau protein, was the first gene identified in families with FTD and parkinsonism (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). Tau is a natively unfolded protein that is abundantly expressed in the central nervous system where it regulates microtubule assembly and stabilization, being involved in signal transduction and axonal transport (Goedert et al., 1989b). Forty-four pathogenic mutations in the MAPT gene have been identified so far (Cruts et al., 2012), causing the accumulation of hyperphosphorylated tau protein in neurons and glial cells (Hutton et al., 1998).
Alternative mRNA splicing of exons 2, 3, and 10 of the MAPT gene leads to the assembly of six tau isoforms that are expressed in the adult human brain (Neve et al., 1986; Goedert et al., 1989a; Andreadis et al., 1992). The tau isoforms differ from each other by the presence or absence of a 29-aminoacid or 58-aminoacid insert in the amino-terminal half and by the inclusion or not of a 31-aminoacid repeat encoded by exon 10 of MAPT in the carboxy-terminal half of the protein. The inclusion or exclusion of exon 10 results in the production of three isoforms with four repeats each (4R) or three isoforms with three repeats each (3R), respectively (Spillantini and Goedert, 2013). These repeats comprise the microtubule-binding domains of tau, with 4R tau having greater predisposition for microtubule assembly than 3R tau (Goedert and Jakes, 1990).
Mutations can be differentiated into missense mutations in exons 9–13, influencing the normal function of the tau protein in microtubule stabilization, and intronic and some coding mutations modifying the splicing of exon 10 at the mRNA level, resulting in a disproportionate ratio of 3R/4R tau isoforms (Goedert et al., 1989a).
The frequency of MAPT gene mutations is highly variable, ranging from 0–3% in sporadic FTD to 5–20% in familial cases (Houlden et al., 1999; Rizzu et al., 1999; Poorkaj et al., 2001; Binetti et al., 2003; Rademakers et al., 2004; Kaivorinne et al., 2008; Pickering-Brown et al., 2008).
Heterogeneous clinical phenotypes with high penetrance (van Herpen et al., 2003) have been observed in patients with mutations in the MAPT gene, even in families bearing the same pathogenic mutation (Van Swieten et al., 1999; Janssen et al., 2002; Boeve et al., 2005).
Disease onset is also highly variable, ranging between 45 and 65 years, with a mean age of onset of 55 years, nearly 10 years earlier than in patients with GRN mutations (Van Swieten and Spillantini, 2007). The disease may however occasionally present between 20 and 30 years (Van Swieten and Spillantini, 2007; Momeni et al., 2010) or after 70 years in a few mutations (Hayashi et al., 2002). The mean disease duration is 9 years, ranging from 5 to 20 years (Seelaar et al., 2011).
The clinical phenotype of patients with MAPT mutations can be divided into a dementia-dominant type and a parkinsonism-dominant type. The most common presentation, as with GRN mutations, is bvFTD, characterized by behavioral inhibition, impaired social behavior, and obsessive–compulsive disorder (Van Swieten and Spillantini, 2007). As with GRN mutations, patients may subsequently present parkinsonism, which rarely is the exclusive clinical presentation, and more often develops in association with bvFTD (Van Swieten and Spillantini, 2007; Rohrer et al., 2011b). However, some patients have a primary parkinsonian syndrome as in CBS or, more rarely, as in PSP (Dickson et al., 2007; Van Swieten and Spillantini, 2007). Parkinsonian features consist of symmetrical bradykinesia, postural instability and rigidity affecting axial and appendicular musculature, absence of resting tremor, and poor or no responsiveness to levodopa (Ghetti et al., 2015). In patients carrying the N279K mutation, parkinsonism is an early feature and asymmetric resting tremor and levodopa responsiveness have been observed (Tsuboi et al., 2002). Numerous other motor signs and symptoms have been observed, comprising supranuclear gaze palsy, upper and lower motor neuron dysfunction, dystonia, myoclonus, postural and action tremor, apraxia of eyelid opening and closing, dysphagia, and dysarthria (Ghetti et al., 2015).
Patients may also develop language impairment, which usually is not a presenting feature (Seelaar et al., 2008; Rohrer et al., 2010b); indeed the P301L mutation was recently observed in a family with impaired single word comprehension suggestive of verbal semantic impairment (Ishizuka et al., 2011), and the V363I and G304S mutations have been reported in cases with avPPA (Munoz et al., 2007; Villa et al., 2011).
As with GRN mutations, there is no clear correlation between the type of mutation and the clinical presentation; however, anecdotal observations suggest that exonic mutations that do not affect the splicing of exon 10 are usually associated with a dementia-predominant phenotype, and that intronic and exonic mutations that affect exon 10 splicing, leading to an overproduction of 4R tau, tend to be associated with a parkinsonism-predominant phenotype (Ghetti et al., 2015).
In some cases, episodic memory impairment has been observed in patients with MAPT mutations, in particular in the R406W mutation, mimicking Alzheimer’s disease (AD) (Tolboom et al., 2010), or in the recently described duplication of the MAPT gene, where the predominant memory impairment was preceded by clinical symptoms characteristic of bvFTD (Rovelet-Lecrux et al., 2010).
Neuroimaging studies have observed heterogeneous patterns of cortical atrophy, but with predominant involvement of the frontal and temporal lobes, with occasional involvement of the parietal lobes (Van Swieten et al., 1999; Janssen et al., 2002; Boeve et al., 2005; Ghetti et al., 2008; Whitwell et al., 2009a,b). The atrophy is particularly prominent in the anteromedial temporal lobes (Spina et al., 2008; Whitwell et al., 2009a,c; Rohrer et al., 2010b), with some studies showing that anteromedial atrophy is a feature of early stages of disease (Boeve et al., 2005). In the majority of individuals, patterns of cortical atrophy are relatively symmetrical (Van Swieten et al., 1999; Ghetti et al., 2008; Rohrer et al., 2011a); however, asymmetry has been observed in some cases (Boeve et al., 2005).
Atrophy of the basal ganglia and of the brainstem have been observed in studies on individual patients, possibly correlating with the parkinsonian features frequently associated with the disease (Arima et al., 2000; Soliveri et al., 2003; Slowinski et al., 2009).
In spite of the fact that temporal lobe atrophy is the most typical finding in patients with MAPT mutations, the pattern of cortical atrophy can differ depending on the specific mutation. Indeed MAPT mutations affecting the splicing of exon 10 are mainly associated with medial temporal atrophy, while mutations in the coding region are primarily associated with lateral temporal lobe atrophy (Whitwell et al., 2009a; Ghetti et al., 2015). These findings, if confirmed in other cohorts of subjects, could help identifying the underlying MAPT mutation.
As in structural imaging, functional imaging with F-18 fluorodeoxyglucose positron emission tomography (FDG-PET) often shows reduced frontal and/or temporal hypometabolism, which resembles the patterns observed in sporadic FTD (Frank et al., 2007).
As in GRN mutations, structural imaging changes can be identified years before expected onset of symptoms in asymptomatic adults at risk of genetic FTD. Rohrer et al. observed differences, between MAPT mutation carriers and non-carriers, in the hippocampus and amygdala 15 years before expected onset, followed by the temporal lobe 10 years before expected onset, and in the insula 5 years before expected onset (Rohrer et al., 2015b).
Functional connectivity in the default mode network (DMN) is altered in MAPT subjects before the occurrence of both atrophy and clinical symptoms, suggesting that changes in functional connectivity are early features of the disease. Asymptomatic MAPT subjects show reduced connectivity in the DMN predominantly between precuneus and lateral temporal lobe, but also medial prefrontal cortex, compared to age-matched controls (Whitwell et al., 2011a). Significantly decreased fractional anisotropy (FA) and increased radial diffusivity in bilateral uncinate fasciculi and reduced FA in the forceps minor have also been observed in asymptomatic MAPT carriers (Dopper et al., 2014).
A preliminary study with the F-18-T807 PET ligand for tau in a patient carrying a MAPT mutation has demonstrated a classic frontotemporal distribution characteristic of inherited tauopathies (Ghetti et al., 2015). These ligands, which characterize tau pathology in vivo (Fodero-Tavoletti et al., 2011; Xia et al., 2013), will undoubtedly improve our future understanding of the pathogenesis of disorders characterized by tau deposition (Villemagne et al., 2015).
GRN Mutations
Since 2006, 69 distinct pathogenic mutations have been identified in the GRN gene, accounting for up to 20% of familial and 5% of sporadic FTD cases (Cruts et al., 2012). Progranulin, which is expressed in many cell types, including neurons and microglia, is a secreted growth factor known for its role in biological processes, including cellular and tissue development, inflammation, wound healing, and cancer, and for its neurotrophic properties. It is proteolytically processed into peptides called granulins, the function of which, in the nervous system, is still largely speculative (Bateman and Bennett, 2009).
Almost all pathogenic mutations cause disease by haploinsufficiency (Baker et al., 2006; Cruts et al., 2006), reducing the levels of progranulin in cerebrospinal fluid, plasma, and serum in symptomatic and asymptomatic GRN mutation carriers (Ghidoni et al., 2008; Finch et al., 2009; Sleegers et al., 2009). Indeed, the measurement of plasmatic progranulin levels is now largely used to screen patients for GRN sequencing (Schofield et al., 2010; Almeida et al., 2013).
A number of factors that influence progranulin expression have been identified in patients carrying GRN mutations. Genetic variants in TMEM106B (Van Deerlin et al., 2010), a gene encoding for the still uncharacterized transmembrane protein 106B, seem to delay the onset of disease in GRN mutation carriers, perhaps increasing the levels of progranulin (Cruchaga et al., 2011; Finch et al., 2011; van der Zee et al., 2011). Granulin expression is also regulated by microRNAs, including miR-29b and miR-107 (Jiao et al., 2010; Wang et al., 2010), and by sortilin (SORT1), a receptor for neurotrophic factors, which mediates progranulin endocytosis and regulates the plasmatic levels of progranulin (Carrasquillo et al., 2010; Hu et al., 2010).
The clinical presentation in patients with GRN mutations is highly variable, and phenotypic heterogeneity can be observed in the same family carrying an identical pathogenic mutation (Beck et al., 2008; Gabryelewicz et al., 2010; Yu et al., 2010; Chen-Plotkin et al., 2011), with an age-dependent and incomplete penetrance (Cruts and Van Broeckhoven, 2008). Age at symptom onset varies significantly, ranging from 35 to 89 years, with an average of 65 years, and a penetrance of 50% by the age of 60 and 90% by the age of 70 years (Gass et al., 2006; van Swieten and Heutink, 2008; Le Ber, 2013).
Within families, there is considerable variation in the age of onset, with a difference of up to 20 years between consecutive generations (Seelaar et al., 2008; van Swieten and Heutink, 2008). Neither age of disease onset nor the duration of the disease seems to have any correlation with the type or location of the GRN mutation.
The initially reported and most common clinical diagnosis in patients carrying GRN mutations is bvFTD, with significant apathy and social withdrawal (Baker et al., 2006; Cruts et al., 2006). In addition, subsequent studies identified, at onset, a clinical presentation characterized by aphasic disorders consistent with avPPA (Snowden et al., 2006; Mesulam et al., 2007; Le Ber et al., 2008), or CBS (Masellis et al., 2006). The aphasic disorder associated with GRN mutations seems nevertheless to be distinct from the avPPA with apraxia of speech observed occasionally in patients with FTLD-tau (Rohrer et al., 2010a,c), or from the logopenic variant of PPA (lvPPA), commonly associated with an atypical presentation of AD pathology (Gorno-Tempini et al., 2011; Rohrer et al., 2012).
Extrapyramidal features have been reported in about 40% of patients with GRN mutations and include CBS with limb apraxia, asymmetrical parkinsonism, and dystonia, although they are rarely a primary symptom and usually emerge after the onset of behavioral or language symptoms (Yu et al., 2010), and only occasionally respond to levodopa (Carecchio et al., 2009; Di Fabio et al., 2010).
In almost 25% of patients hallucinations and delusions can be predominant, frequently causing a misdiagnosis of dementia with Lewy bodies (DLB) (Le Ber et al., 2008; Kelley et al., 2010; Yu et al., 2010).
Furthermore, an AD-like phenotype can be observed in patients with episodic memory deficit, which occurs in 10–30% of cases, or with parietal deficits, such as dyscalculia, visuospatial symptoms, and limb apraxia. This phenotype can be occasionally associated with the c.154delA mutation in GRN (Kelley et al., 2010) or with the genetic variant rs5848, where the minor T allele, located in the 3′-untranslated region of GRN, was found to increase the binding of miR-659 to GRN (Brouwers et al., 2008; Rademakers et al., 2008; Lee et al., 2011; Sheng et al., 2014), consequently reducing serum progranulin levels (Hsiung et al., 2011).
A recent meta-analysis studied the association between the GRN polymorphism rs5848 and risk of neurodegenerative diseases, including FTLD, AD, Parkinson’s disease (PD), and ALS, concluding that rs5848 was associated with an increased risk of AD and PD in the homozygous and recessive models but not with FTLD (Chen et al., 2015).
Since GRN mutations are associated with FTD-TDP pathology, which is frequently observed also in ALS and FTD-ALS, it prompted the search for GRN mutations in patients with associated motor neuron disease. One study identified a missense mutation in a patient with FTD-ALS (p.S120Y) and in a single case of limb onset sporadic ALS (p.T182M) (Schymick et al., 2006), while a later study identified clinical features compatible with MND in 5.4% of patients carrying a GRN mutation; in this cohort, concomitant motor neuron disease was much more common in patients with FTD-TDP pathology not bearing a GRN mutation (26.3%) (Chen-Plotkin et al., 2011).
Recently, it has been shown that a complete GRN deficiency due to a homozygous GRN loss-of-function mutation causes the rare lysosomal storage disease neuronal ceroid lipofuscinosis (Smith et al., 2012). These findings suggest that lysosomal storage disorders and GRN-associated FTLD may share common features (Götzl et al., 2014).
Neuroimaging studies have highlighted that patients carrying a GRN mutation are more likely to show a pronounced asymmetrical atrophy (Huey et al., 2006; Bronner et al., 2007; Beck et al., 2008; Pickering-Brown et al., 2008) affecting either the left or right hemispheres, involving primarily the inferior frontal (Bronner et al., 2007; Beck et al., 2008; Ghetti et al., 2008), temporal and inferior parietal lobes (Mesulam et al., 2007; Pickering-Brown et al., 2008), as well as long intrahemispheric association white matter (WM) tracts (Borroni et al., 2008; Rohrer and Warren, 2011). A prominent feature, which is unusual in FTLD, is the predominance of parietal involvement (Whitwell et al., 2007; Beck et al., 2008; Le Ber et al., 2008), recently confirmed also in presymptomatic mutation carriers (Rohrer et al., 2008, 2015b; Cruchaga et al., 2009). These findings have been also confirmed by voxel-based morphometry (VBM) studies, which detected a widespread and severe pattern of gray matter loss predominantly affecting the frontal, temporal, and parietal lobes (Whitwell et al., 2009b, 2012). Along with cortical involvement, atrophy of deep subcortical structures has been identified in patients carrying GRN mutations, in particular in the caudate nucleus and in the thalamus bilaterally, possibly correlating with the parkinsonian features that are frequently associated since the early stages of the disease (Premi et al., 2014b).
Furthermore, cerebellar atrophy can be observed in some cases (Nuytemans et al., 2007; Benussi et al., 2008).
Subcortical WM involvement, ranging from T2-weighted MRI hyperintensities (Nuytemans et al., 2007; Le Ber et al., 2008; Kelley et al., 2009) to widespread WM lesions may be linked to progranulin pathological processes in a subset of GRN mutation carriers (Caroppo et al., 2014).
Annualized rates of whole brain atrophy, and consequently of ventricular expansion, are greatest in GRN, followed by sporadic FTD, C9ORF72, and lastly MAPT (Whitwell et al., 2015), in which similar rates of hippocampal atrophy have been observed (Whitwell et al., 2011b).
Using single photon emission computed tomography (SPECT), hypoperfusion of the hippocampus and that of the cingulate gyrus have been observed, denoting the association of these regions with the pathogenesis of GRN mutations (Le Ber et al., 2008).
Recent studies have highlighted that structural imaging and cognitive changes can be identified years before expected onset of symptoms in asymptomatic adults at risk of genetic FTD. In the study by Rohrer et al., in the GRN group, differences were observed between carriers and non-carriers in the insula at 15 years before expected onset, then in the temporal and parietal lobes at 10 years before expected onset, with the earliest subcortical area affected being the striatum at 5 years before expected onset. GRN mutation carriers also showed significantly greater asymmetry than non-carriers at 5 years before expected onset (Rohrer et al., 2015b).
In functional connectivity studies, Premi et al. observed in asymptomatic carriers of the GRN Thr272fs mutation, decreased brain connectivity within the left frontoparietal network and increased connectivity in the executive network compared with healthy controls (Premi et al., 2013b; 2014a). Furthermore, the TMEM106B at-risk polymorphism (T/T) was associated with decreased connectivity within the ventral salience network (i.e., middle frontal gyrus) and the left frontoparietal network (i.e., left precuneus) (Premi et al., 2013a). Further studies have confirmed the patterns of brain atrophy and WM tract abnormalities in frontal-parietal circuits and within the uncinate fasciculus, supporting the hypothesis that WM damage is an early feature in the FTD disease process and can be detected at least a decade before the estimated symptom onset in asymptomatic mutation carriers (Dopper et al., 2014; Pievani et al., 2014).
C9orf72 Mutations
The strong association between FTD and ALS has become increasingly apparent due to a rapidly expanding body of clinical, epidemiological, genetic, and molecular evidence (Gros-Louis et al., 2006) and, in 2006, a locus was mapped on chromosome 9p21 in FTD-ALS families (Vance et al., 2006). Until 2011, despite numerous efforts, the disease mutation remained elusive (Momeni et al., 2006; Morita et al., 2006; Valdmanis et al., 2007; Luty et al., 2008; Le Ber et al., 2009; Boxer et al., 2011; Pearson et al., 2011). Eventually, DeJesus-Hernandez and colleagues observed the non-Mendelian inheritance of a GGGGCC hexanucleotide repeat, located in a non-coding region of the C9orf72 gene, in the large VSM-20 family (Vancouver, San Francisco and Mayo family 20) affected by FTD-ALS, with the intuition that the repeat expansion, which was identified in the parents but not in the affected children by using fluorescent PCR, was too large to be amplified by the PCR method and could only be identified by repeated-primed PCR assay and Southern blot analysis (DeJesus-Hernandez et al., 2011). With the use of next-generation sequencing, another group independently recognized the repeat expansion in a Welsh family affected by FTD-ALS (Renton et al., 2011).
Until recently, C9orf72 was a completely uncharacterized protein with unknown function. The pathogenesis underlying the C9orf72 mutation still remains unclear, but likely includes haploinsufficiency through loss of gene expression (Gijselinck et al., 2012; Cruts et al., 2013; van der Zee et al., 2013), gain-of-function mechanisms with secondary RNA toxicity caused by sequestration of RNA-binding proteins (DeJesus-Hernandez et al., 2011; Lagier-Tourenne et al., 2013), or both (Rohrer et al., 2015a).
Two different protein isoforms of the C9orf72 gene are predicted to be generated from a total of three different transcripts variants, depending on the localization of the GGGGCC repeat, either in the promoter or in the intron 1 of the gene (DeJesus-Hernandez et al., 2011). All three different transcripts have been found to be reduced in blood (Mori et al., 2013a; Davidson et al., 2014), cortex, cerebellum, and spinal cord (Belzil et al., 2013; Donnelly et al., 2013). This reduced expression possibly reflects, to a limited extent, the hypermethylation of a CpG island upstream of the repeat (Xi et al., 2013; Belzil et al., 2014; Liu et al., 2014), and the methylation of lysine residues on H3 and H4 histones, which bind the GGGGCC repeats and suppress transcription (Xi et al., 2013; Davidson et al., 2014; Russ et al., 2014), acting as a possible innate protective mechanism (Liu et al., 2014; McMillan et al., 2015; Xi et al., 2015).
As previously mentioned, the neuropathological hallmarks, associated with C9orf72 mutations, are the TDP-43 immunoreactive inclusions, i.e., subtypes A and B (Mahoney et al., 2012; Lashley et al., 2014), which have been observed in many cerebral structures, including the extramotor cerebral cortex, hippocampus, basal ganglia, substantia nigra, and lower motor neurons of the brainstem and spinal cord (Rademakers et al., 2012). In addition to the TDP-43 pathology, a distinctive and singular feature is the presence of abundant TDP-43 negative, p62-positive inclusions, with a unique star-like morphology, in the cerebellum, hippocampus, and frontal cortex (Al-Sarraj et al., 2011; Simon-Sanchez et al., 2012; Snowden et al., 2012; Troakes et al., 2012; Mori et al., 2013b).
Recently, Mori and colleagues tried to characterize these inclusions, speculating that they may consist of dipeptide repeat (DPR) proteins which are the result of the bidirectional translation of the non-coding GGGGCC expansion repeat through a mechanism of non-ATG-initiated translation, termed RAN translation (Mori et al., 2013c). Indeed, the researchers demonstrated that the GGGGCC expansion is bidirectionally translated into five distinct DPR proteins (Mori et al., 2013c), which have been observed in high load in neocortical regions, hippocampus, cerebellum and, to some extent, in motor neurons (Mackenzie et al., 2013). Moreover, the recent observation, in a Belgian C9orf72 carrier, of neuropathological aggregates of a DPR core surrounded by TDP-43 inclusions, suggested that DPR aggregation due to RAN translation could be the initial pathological event that possibly precedes and potentially triggers TDP-43 accumulation (Mackenzie et al., 2013; Mori et al., 2013c; van der Zee and Van Broeckhoven, 2014).
These findings have led to the suggestion that C9orf72 expansion cases should be reclassified, from a neuropathological prospective, as FTLD-DPR, since identified cases can fit into one of two FTLD-TDP subtypes (A or B), or might even lack TDP-43 pathology (Snowden et al., 2012; Mori et al., 2013c).
Despite these new findings implicating both loss-of-function and gain-of-function mechanism with new insights into the roles of sense (GGGGCC) and antisense (GGCCCC) repeat RNA and DPR proteins, the relative contribution to these mechanisms and the molecular pathways that lead to neurodegeneration are yet to be elucidated (Rohrer et al., 2015a).
The number of repeat units of the C9orf72 expansion in affected patients can range from at least several hundred to thousands of copies, whereas unaffected healthy controls harbor 2–24 repeat units (DeJesus-Hernandez et al., 2011; Beck et al., 2013; van Blitterswijk et al., 2013). So far, there is no clear evidence for intergenerational anticipation, although some studies have identified earlier disease onset in subsequent generations (Stewart et al., 2012; Benussi et al., 2014).
Whilst it is now rather clear that expansions of several hundred copies are definitely pathogenic, it is not known which is the smallest hexanucleotide expansion that can confer a risk of developing the disease. Indeed, in vitro studies have demonstrated an inverse correlation between repeat size and gene expression starting at just 9 repeats, with a reduction of up to 50% in expression for 24 expansion repeats (van der Zee et al., 2013).
Furthermore, as in other non-coding repeat expansion disorders, evidence for somatic instability of the C9orf72 repeat has been observed (DeJesus-Hernandez et al., 2011), resulting in a range of mutation sizes in particular tissues with variation between tissues in the same individual. This possibly suggest that intermediate repeats detected in blood-sample-derived DNA might be associated with massive expansions in the brain (Rohrer et al., 2015a), making it even harder to determine correlations between genetics, phenotype, and neuropathology (Lavedan et al., 1993; Matsuura et al., 2004). Indeed, a recent study tried to estimate, with Southern blotting techniques and densitometry, the repeat size of the most abundant GGGGCC expansion species in patients with ALS and FTD-ALS. Comparing different tissues, substantial variation in repeat sizes between samples from the cerebellum, frontal cortex, and blood have been identified. In particular, longer repeat sizes in the cerebellum seem to be associated with a survival disadvantage, indicating that expansion size does affect disease severity (Majounie et al., 2012).
As with GRN mutations, TMEM106B has been identified as a genetic modifier also in C9orf72 cases, demonstrating a significantly later age at death and age at onset for the rs1990622 major allele (T) carriers (Gallagher et al., 2014).
Since the discovery of the C9orf72 mutation, testing of patients’ cohorts worldwide has found an average frequency of the pathogenic expansion in 37% of familial ALS, 6% of sporadic ALS, 21% of familial FTD, and 6% of sporadic FTD. Epidemiological studies have all established that the C9orf72 mutation is the major genetic cause of familial ALS (more frequent than superoxidose dismutase 1 mutations), and comparable in frequency to GRN mutations in familial FTD cases (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Chiò et al., 2012; Hsiung et al., 2012; Majounie et al., 2012; Cruts et al., 2013; van der Zee et al., 2013).
Age at disease onset is widely variable, ranging from 27 to 83 years (mean 50 years), with about 50% patients affected by age 60 and with a penetrance reaching 90% at age 70. Disease duration is also variable, ranging from 1 to 22 years, with the development of ALS as a strong negative prognostic factor (Boeve et al., 2012; Chiò et al., 2012; Cooper-Knock et al., 2012; Gijselinck et al., 2012; Hsiung et al., 2012; Stewart et al., 2012).
Clinical phenotype is highly heterogeneous and variable even within families affected by the same mutation. The most common clinical presentations are FTD (Boeve et al., 2012; Dobson-Stone et al., 2012; Englund et al., 2012; Hsiung et al., 2012; Mahoney et al., 2012; Sha et al., 2012; Snowden et al., 2012; Devenney et al., 2014), ALS (Boeve et al., 2012; Byrne et al., 2012; Chiò et al., 2012; Cooper-Knock et al., 2012; Stewart et al., 2012; Coon et al., 2013; Montuschi et al., 2015), or the combination of both. The most frequent FTD subtype is bvFTD, with avPPA observed less frequently, although other phenotypes have been described, and even cases with classical bvFTD or ALS have extensive phenotypic heterogeneity. bvFTD associated with the C9orf72 mutation may present with classical features, as apathy, disinhibition, socially inappropriate behavior, abnormal eating behavior, and loss of empathy (Rascovsky et al., 2011). Nevertheless, behavioral changes with features of psychosis, including hallucinations and delusions, are not rarely observed in these patients (Arighi et al., 2012; Boeve et al., 2012; Dobson-Stone et al., 2012; Englund et al., 2012; Mahoney et al., 2012; Snowden et al., 2012; Galimberti et al., 2013; Kertesz et al., 2013; Devenney et al., 2014), and can even predominate at disease onset and lead to an initial diagnosis of schizophrenia (Galimberti et al., 2014a), bipolar disorder (Galimberti et al., 2014b), obsessive–compulsive disorder (Calvo et al., 2012), depressive pseudodementia (Bieniek et al., 2014), or DLB (Robinson et al., 2014).
Mutations in C9orf72 have been rarely observed in patients with PPA (Renton et al., 2011; Dobson-Stone et al., 2012; Englund et al., 2012; Mahoney et al., 2012; Sha et al., 2012; Simon-Sanchez et al., 2012; Snowden et al., 2012; Van Langenhove et al., 2013; Saint-Aubert et al., 2014).
As in GRN and MAPT mutations, deficits in episodic memory at onset, mimicking AD have been reported also in patients with C9orf72 mutations (Wojtas et al., 2012; Cacace et al., 2013; Harms et al., 2013; Murray et al., 2013; Pletnikova et al., 2014). Moreover, extrapyramidal movement disorder (usually an akinetic-rigid syndrome) (Murray et al., 2011; Boeve et al., 2012; Cooper-Knock et al., 2012; Floris et al., 2012; Hsiung et al., 2012; Simon-Sanchez et al., 2012) and cerebellar signs have also been reported (Hsiung et al., 2012). Most patients, during the course of the disease, eventually develop some abnormalities of behavior, language, and motor function (Boeve, 2011; Byrne et al., 2012; Cooper-Knock et al., 2012; Hsiung et al., 2012; Stewart et al., 2012).
Structural neuroimaging in patients with C9orf72 expansions does not reveal a characteristic pattern of atrophy, unlike GRN or MAPT mutations. A prevalently symmetrical, bilateral, cortical, and subcortical atrophy, predominantly affecting the frontotemporal regions, has been observed (Boeve et al., 2012; Dobson-Stone et al., 2012; Englund et al., 2012; Mahoney et al., 2012; Sha et al., 2012; Snowden et al., 2012; Whitwell et al., 2012; Devenney et al., 2014).
Neuroimaging studies using VBM analysis have observed extensive atrophy of the frontal, insular, temporal, and parietal cortical and subcortical regions, including prominent atrophy of the thalamus and cerebellum (Mahoney et al., 2012; Whitwell et al., 2012; Irwin et al., 2013).
Concordant with gray matter atrophy, WM atrophy of long intrahemispheric, commissural, and corticospinal tracts has been observed with diffusion tractography (Mahoney et al., 2012).
As with structural neuroimaging, functional neuroimaging using FDG-PET and SPECT has shown frontal, temporal, and parietal cortical and subcortical alterations (Boeve et al., 2012; Adeli et al., 2014; Suárez-Calvet et al., 2014). Seed-based intrinsic connectivity analyses in C9orf72 carriers versus non-carriers showed connectivity reductions in the salience and sensorimotor networks, with severity of behavioral symptom correlating with diminished salience network connectivity and increased DMN connectivity (Lee et al., 2014).
As in GRN and MAPT mutations, presymptomatic imaging signatures can be identified in asymptomatic adults at risk of genetic FTD. Rohrer et al. observed differences, between C9orf72 mutation carriers and non-carriers, in subcortical areas, including the thalamus, the insula, and posterior cortical areas 25 years before expected onset, followed by the frontal and temporal lobes 20 years before expected onset, and in the cerebellum 10 years before expected onset (Rohrer et al., 2015b).
VCP Mutations
Missense mutations in the VCP gene, located on chromosome 9p13.3, were originally reported in 2004 in North American families with a specific autosomal dominant multisystem syndrome called inclusion body myopathy with Paget disease of bone and/or FTD (IBMPFD) (Watts et al., 2004), with neuropathology consistent with type D TDP-43 pathology (van der Zee et al., 2009).
Valosin-containing protein, also known as p97, is a member of the ATPases associated with various cellular activities (AAA) family of proteins and is involved in multiple cellular processes, including protein degradation via both proteasome and autophagy pathways, in membrane fusion, transcriptional activation, apoptosis, and molecular chaperoning (Nalbandian et al., 2011; Yamanaka et al., 2012). The role of mutant VCP in the pathogenesis of IBMPFD or FTD-ALS remains elusive.
So far, 19 pathogenic mutations in VCP have been described (Cruts et al., 2012). The frequency of VCP mutations in FTD is about 1.6%, making it a rare but significant cause of FTD (van der Zee et al., 2009). Nearly 80% of VCP mutation carriers have a positive family history with a wide variability in phenotypic presentation, even within the same family.
The disease usually begins at the age of 40 with musculoskeletal symptoms not dissimilar to limb-girdle muscular dystrophy, affecting more than 80% patients. Moreover, nearly 60% patients develop adult-onset Paget disease of bone (Watts et al., 2004). FTD usually develops later on in the disease course (age 55–60 years), affecting about 30% of patients, with a classical bvFTD phenotype (Kovach et al., 2001; Guyant-Marechal et al., 2006; Kimonis et al., 2008); occasional psychiatric disorders have been reported (Jacquin et al., 2013). However, although rare, VCP mutations have been found in patients with a dementia-only phenotype (van der Zee et al., 2009). Early language deficits and semantic impairment have been rarely described (Kim et al., 2011).
VCP mutations have been identified also in patients with an MND/ALS phenotype, occasionally associated with FTD (Johnson et al., 2010; Miller et al., 2012; Hirano et al., 2015); these findings are supported by the pyramidal tract dysfunction which has been observed in some cases (Kumar et al., 2010).
The term multisystem proteinopathy has been recently proposed to replace the term IBMPFD to describe this phenotype (Benatar et al., 2013).
MRI findings range widely, from cases showing no signs of frontal lobe atrophy to patients with prominent, asymmetric, focal atrophy in the anterior and temporal lobes (Gidaro et al., 2008; Djamshidian et al., 2009; Stojkovic et al., 2009; Kim et al., 2011; Surampalli et al., 2015). FDG-PET imaging has revealed severe glucose hypometabolism in the bilateral frontotemporoparietal areas (Guyant-Marechal et al., 2006; Stojkovic et al., 2009; Kim et al., 2011).
TARDBP Mutations
Transactive response DNA-binding protein 43 (TDP-43) is a ubiquitously expressed and highly conserved ribonucleoprotein, encoded by the TARDBP gene on chromosome 1, with a putative role in the regulation of transcriptional activity, of messenger RNA splicing, exon skipping, and microRNA biogenesis of several genes. It has been proposed that, in neurodegenerative disorders, TDP-43 can be redistributed from the nucleus to the cytoplasm and sequestered into inclusions that are mainly composed of abnormally phosphorylated and C-terminally truncated TDP-43 fragments (Buratti and Baralle, 2008; Polymenidou et al., 2011; Tollervey et al., 2011).
The identification of mutations within the TARDBP gene as causative for both familial and sporadic ALS provided additional proofs for the pathogenic role of TDP-43 in neurodegeneration (Kabashi et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008).
Mutations in the TARDBP gene were initially described in 2008 in patients with ALS, both in familial and sporadic cases (Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008). So far, mutations in the TARDBP gene account for 2–3% of ALS patients (Gitcho et al., 2008; Kabashi et al., 2008; Kühnlein et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008; Daoud et al., 2009).
Although previous studies failed to identify a significant role for TARDBP mutations in FTD (Rollinson et al., 2007; Gallone et al., 2009; Gijselinck et al., 2009; Schumacher et al., 2009), recently it has been reported that TARDBP mutations might be causative of FTD with MDN. These uncommon cases present with a heterogeneous phenotype, almost always associated with a significant motor neuron involvement, including FTD-MND (Benajiba et al., 2009; Chiò et al., 2010), FTD-MND with extrapyramidal symptoms (Borghero et al., 2011; Chiò et al., 2011; Quadri et al., 2011; Mosca et al., 2012), and MND with supranuclear palsy (Kovacs et al., 2009; Borghero et al., 2011). Pure FTD cases without motor neuron involvement carrying a TARDBP mutation are even more exceptional. Indeed, less than 16 cases have been reported so far (Borroni et al., 2009, 2010; Gitcho et al., 2009; Kovacs et al., 2009; Gelpi et al., 2014; Synofzik et al., 2014; Moreno et al., 2015), with only four cases having neuropathological confirmation (Gitcho et al., 2009; Kovacs et al., 2009; Gelpi et al., 2014; Moreno et al., 2015). The most common phenotype that was described was bvFTD followed by PPA, in particular with svPPA, with a heterogeneous age of onset, ranging from 29 to 77 years.
Neuroimaging studies, achieved only in case reports and small series, show predominant temporal lobe involvement (Borroni et al., 2009, 2010; Gitcho et al., 2009; Kovacs et al., 2009; Gelpi et al., 2014; Synofzik et al., 2014; Moreno et al., 2015).
TDP-43 inclusions, as previously outlined, have been observed also in non-TARDBP-associated genetic forms of FTLD, including GRN, VCP, and C9orf72 mutations, suggesting that dysregulation of TDP-43 might be a pivotal downstream pathogenic mechanism involved in neurodegeneration in each of these disorders (Rademakers et al., 2012).
CHMP2B Mutations
The CHMP2B gene, located on chromosome 3p11.2, encodes the charged multivesicular body protein 2B, which has been identified in 2005 in a large Danish family with an autosomal form of FTLD (Skibinski et al., 2005), first described in 1987 (Gydesen et al., 1987). The average age of onset in this family was 55 years, ranging from 46 to 65 years. Since 2005, researchers identified CHMP2B missense variants in FTD and FTD-ALS cases, representing less than 1% of familial forms of FTLD, although the pathogenicity of these variants remain to be elucidated (Parkinson et al., 2006; Cox et al., 2010).
The phenotypic expression of the originally described family comprises bvFTD that may be associated with late parkinsonism, dystonia, myoclonus, and pyramidal symptoms (Skibinski et al., 2005).
Neuropathological examination revealed ubiquitin/p62-positive, TDP-43/FUS negative neuronal inclusions, termed FTLD-U (Holm et al., 2007, 2009).
FUS Mutations
The genetic heterogeneity of FTD is further emphasized by the occurrence of rare mutations in the FUS gene, located on chromosome 16, which was first identified as a fusion oncogene in human liposarcomas (Crozat et al., 1993; Rabbitts et al., 1993).
FUS, as TAF-15 and EWS, belongs to the FET family of DNA/RNA-binding proteins, which are highly conserved, ubiquitously expressed, multifunctional binding proteins (Tan and Manley, 2009) that can bind to a large number of overlapping RNA targets (Hoell et al., 2011). Although the precise physiological function of FUS has not been fully elucidated, growing evidence suggests that the protein is involved in various cellular processes, including cell proliferation, DNA repair, transcription regulation, and multiple levels of RNA and microRNA processing (Lagier-Tourenne et al., 2010; Deng et al., 2014).
In 2009, FUS gene mutations were identified as the primary disease-associated gene for familial (Kwiatkowski et al., 2009; Vance et al., 2009; Doi et al., 2010) and sporadic ALS (Belzil et al., 2009). This mutation accounted for nearly 3% of familial ALS cases, characterized by FUS-positive, TDP-43 negative inclusions (Kwiatkowski et al., 2009; Vance et al., 2009), consequently termed ALS-FUS. Eventually, FUS was found to be the most characteristic marker of the remaining tau-negative and/or TDP-43 negative FTLD cases, encompassing three closely related but distinct clinicopathological entities: atypical FTLD-U (aFTLD-U), neuronal intermediate filament inclusion disease (NIFID), and basophilic inclusion body disease (BIBD) (Munoz et al., 2009; Neumann et al., 2009a,b; Urwin et al., 2010).
FUS mutations in patients with clinical FTLD are rare, and only four FUS mutations have so far been identified in patients with FTD-MND/ALS or in their families (Ticozzi et al., 2009; Blair et al., 2010; Broustal et al., 2010; Yan et al., 2010). Moreover, two cases with a pure FTD phenotype without motor neuron involvement have also been reported (Van Langenhove et al., 2010; Huey et al., 2012). Nevertheless, no autopsy data are yet available to confirm FUS pathology in any patient with FTLD and FUS mutations.
In contrast to ALS with FUS pathology, which is mostly caused by FUS mutations, FTLD-FUS tends to be sporadic, with none yet associated with a mutation in the FUS gene (Rademakers et al., 2012).
Different pathogenic mechanisms in ALS-FUS and FTLD-FUS also arise from a recent study in which, in ALS-FUS, no co-accumulation of other FET-proteins into FUS-positive inclusions was found, in remarkable contrast to FTLD-FUS, in which TAF-15 and EWS were found to co-accumulate in FUS-positive cytoplasmic inclusions (Neumann et al., 2011).
Structural neuroimaging studies show predominant volume loss in the anterior temporal lobe, amygdala, anterior hippocampus, anterior insula, and anterior cingulate cortex (Lee et al., 2012). Bilateral anterior striatal atrophy is particularly prominent in the head of the caudate nucleus, which appears to be significantly greater in FTLD-FUS compared with FTLD-TDP and FTLD-tau, suggesting that severe caudate atrophy may be a useful clinical feature to predict FTLD-FUS pathology (Josephs et al., 2010; Lee et al., 2012).
Other Genetic Causes: DCTN1 and TREM2
There are few reports of DCTN1 mutations in patients with FDT and MND/ALS (Münch et al., 2005; Newsway et al., 2010), variably associated with parkinsonism, hypoventilation, dysautonomia, weight loss, and behavioral disturbances configuring the recently described Perry syndrome, which is associated with TDP-43 pathology (Farrer et al., 2009; Ohshima et al., 2010). Notwithstanding, no mutations were found in large FTD or MND/ALS cohorts (Seelaar et al., 2008; Vilariño-Güell et al., 2009).
Homozygous loss-of-function mutations in TREM2 on chromosome 6p21, encoding the triggering receptor expressed on myeloid cells 2 protein, have been initially associated with an autosomal recessive form of early onset dementia presenting with bone cysts and consequent fractures called polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, also known as Nasu-Hakola disease (Paloneva et al., 2000, 2002).
In 2013, two studies identified a rare heterozygous variant (R47H) on the second exon of TREM2, showing an approximately three- to fivefold increased risk in late onset AD (Guerreiro et al., 2013c; Jonsson et al., 2013). Furthermore, homozygous and compound heterozygous TREM2 exon 2 mutations were identified in rare families presenting with autosomal recessive bvFTD without any clinical or radiological evidence of bone involvement (Giraldo et al., 2013; Guerreiro, 2013; Guerreiro et al., 2013a; Lattante et al., 2013; Rayaprolu et al., 2013; Le Ber et al., 2014). Patients typically have very young age at onset (20–50 years) and, in addition to behavioral changes, they present with early parietal and hippocampal deficits, extrapyramidal features, and seizures. Neuroimaging frequently shows frontotemporal atrophy and extensive WM abnormalities (Guerreiro, 2013; Le Ber et al., 2014).
Recently, Borroni and colleagues evaluated the frequency of heterozygous TREM2 mutations as risk factor in FTD patients, confirming that the Q33X, R47H, and T66M mutations were significantly associated with the disease and absent in healthy subjects (Borroni et al., 2013), in line with further reports (Cuyvers et al., 2013).
Conclusion
The striking clinical, genetic, and pathological heterogeneity are undisputable features of FTLD. The clinical phenotypes range from social to language impairment, variably associated with MND/ALS or extrapyramidal symptoms (see Table 1). In the past 9 years, considerable progress has been made toward unraveling the genetic causes of FTLD, with apparently all common FTD-related genes discovered, comprising C9orf72, MAPT, GRN, TARDBP, VCP, CHMP2B, and FUS, currently identifying 50–60% of the familial forms of FTD, although rare mutations in other genes could explain a small number of the remaining families (Rademakers et al., 2012; Le Ber, 2013).
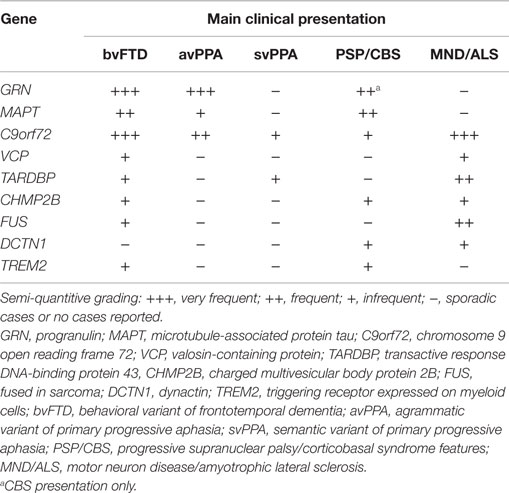
Table 1. Main clinical presentation in monogenic FTLD.
Neuropathology is variably associated with two predominant hallmarks, namely FTLD-tau (found in MAPT mutations) and FTLD-TDP (found in GRN, C9orf72, TARDPB, and VCP mutations), although it is occasionally associated with FTLD-FUS (found in FUS mutations only in MND/ALS), FTLD-U (found in CHMP2B mutations) and the newly identified FTLD-DPR in C9orf72 mutations.
Why different genes linked to different molecular pathways are responsible for a neurodegenerative process involving mainly the frontotemporal lobes is still a matter for debate, and the shared mechanisms that lead to predominant frontal and temporal degeneration remain to be identified.
As previously outlined, frequently there is poor correspondence between the type of mutation or neuropathological feature and the clinical phenotype; indeed, neuropathology is predicable only in known genetic defects and few clinical or neuroanatomical features have a specific molecular association (Rohrer et al., 2011a). Both clinically and anatomically, different genetic mutations show substantial overlap between diseases. However, certain distinctive clinical and neuroimaging features emerge and might help in driving neurology practice in molecular diagnostics. Patients with C9orf72 repeat expansion usually have an early disease onset and a family history of FTD (mainly bvFTD with disinhibition or svPPA), which is variably associated with MND/ALS. MAPT mutation carriers often present with a very early age of onset bvFTD, associated with parkinsonism, which is a common feature later in the disease course in all types of genetic FTLD. GRN mutations carriers frequently present with bvFTD, avPPA, and concomitant CBS at onset. TARDBP sequencing should be performed in familial or sporadic C9orf72 negative FTD-MND/ALS cases. The association of inclusion body myopathy and Paget’s disease are indicative of VCP mutations (Rohrer and Warren, 2011; Borroni and Padovani, 2013; Le Ber, 2013; Van Langenhove et al., 2013).
In addition to these distinct clinical features, from a neuroimaging perspective, strikingly asymmetric interhemispheric atrophy with frontoparietal involvement is indicative of GRN mutations, while relative symmetric atrophy with particular involvement of the anterior temporal lobes is indicative of MAPT mutations (Rohrer et al., 2010b; Whitwell et al., 2012). On the other hand, C9orf72 mutation carriers tend to have a more generalized and symmetrical atrophy (Rohrer et al., 2010b; Whitwell et al., 2012; Van Langenhove et al., 2013) (see Table 2).
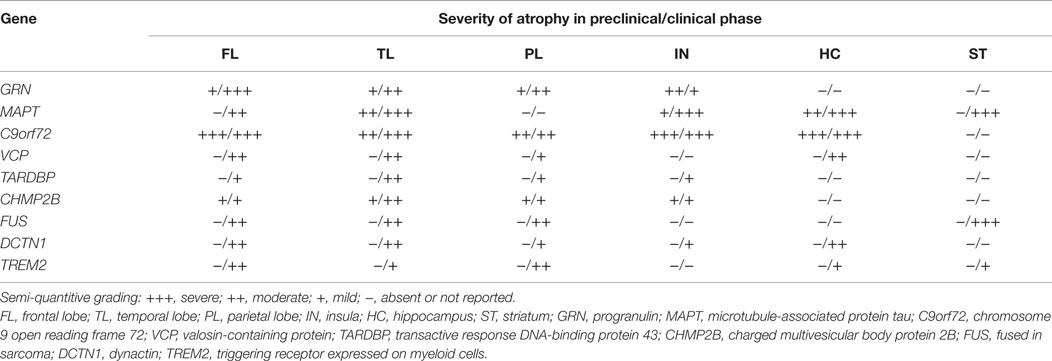
Table 2. Preclinical and clinical neuroanatomical correlates in monogenic FTLD.
Numerous attempts have been made to identify an in vivo biomarker that can accurately predict the underlying neuropathology and consequently point out which gene should be analyzed, but a clear-cut association between clinical or neuroradiological features and characteristic histological inclusions has not been yet established (Kim et al., 2007; Josephs et al., 2008; McMillan et al., 2013). Plasma progranulin levels have been shown to be reliable predictors of GRN mutations, and should be performed before GRN sequencing (Finch et al., 2009; Ghidoni et al., 2012). Furthermore, the recently identified cerebrospinal phospho-tau181 to total-tau ratio appears to predict FTLD-TDP pathology with high accuracy, thus leading to the search of genes associated with TDP-43 neuropathology (Hu et al., 2013; Grossman et al., 2014; Borroni et al., 2015a).
Considering the vast clinical, genetic, and pathological heterogeneity associated with this disease, the development of accurate in vivo biomarkers is mandatory for guiding genetic screening, for defining distinctive therapeutic approaches and helping in patients’ selection in future clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adeli, A., Savica, R., Lowe, V. J., Vemuri, P., Knopman, D. S., DeJesus-Hernandez, M., et al. (2014). The GGGGCC repeat expansion in C9ORF72 in a case with discordant clinical and FDG-PET findings: PET trumps syndrome. Neurocase 20, 110–120. doi:10.1080/13554794.2012.732090
Almeida, M. R., Baldeiras, I., Ribeiro, M. H., Santiago, B., Machado, C., Massano, J., et al. (2013). Progranulin peripheral levels as a screening tool for the identification of subjects with progranulin mutations in a Portuguese cohort. Neurodegener. Dis. 13, 214–223. doi:10.1159/000352022
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi:10.1007/s00401-011-0911-2
Andreadis, A., Brown, W. M., and Kosik, K. S. (1992). Structure and novel exons of the human tau gene. Biochemistry 31, 10626–10633. doi:10.1021/bi00158a027
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi:10.1016/j.bbrc.2006.10.093
Arighi, A., Fumagalli, G. G., Jacini, F., Fenoglio, C., Ghezzi, L., Pietroboni, A. M., et al. (2012). Early onset behavioral variant frontotemporal dementia due to the C9ORF72 hexanucleotide repeat expansion: psychiatric clinical presentations. J. Alzheimers Dis. 31, 447–452. doi:10.3233/JAD-2012-120523
Arima, K., Kowalska, A., Hasegawa, M., Mukoyama, M., Watanabe, R., Kawai, M., et al. (2000). Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology 54, 1787–1795. doi:10.1212/wnl.54.9.1787
Armstrong, M. J., Borroni, B., Litvan, I., Lang, A. E., Bak, T. H., Bhatia, K. P., et al. (2013). Criteria for the diagnosis of corticobasal degeneration. Neurology 80, 496–503. doi:10.1212/WNL.0b013e31827f0fd1
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi:10.1038/nature05016
Bateman, A., and Bennett, H. P. J. (2009). The granulin gene family: from cancer to dementia. Bioessays 31, 1245–1254. doi:10.1002/bies.200900086
Beck, J., Poulter, M., Hensman, D., Rohrer, J. D., Mahoney, C. J., Adamson, G., et al. (2013). Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 92, 345–353. doi:10.1016/j.ajhg.2013.01.011
Beck, J., Rohrer, J. D., Campbell, T., Morrison, K. E., Goodall, E. F., Warrington, E. K., et al. (2008). A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 131, 706–720. doi:10.1093/brain/awm320
Belzil, V. V., Bauer, P. O., Gendron, T. F., Murray, M. E., Dickson, D., and Petrucelli, L. (2014). Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Res. 1584, 15–21. doi:10.1016/j.brainres.2014.02.015
Belzil, V. V., Bauer, P. O., Prudencio, M., Gendron, T. F., Stetler, C. T., Yan, I. K., et al. (2013). Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 126, 895–905. doi:10.1007/s00401-013-1199-1
Belzil, V. V., Valdmanis, P. N., Dion, P. A., Daoud, H., Kabashi, E., Noreau, A., et al. (2009). Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 73, 1176–1179. doi:10.1212/WNL.0b013e3181bbfeef
Benajiba, L., Le Ber, I., Camuzat, A., Lacoste, M., Thomas-Antérion, C., Couratier, P., et al. (2009). TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 65, 470–473. doi:10.1002/ana.21612
Benatar, M., Wuu, J., Fernandez, C., Weihl, C. C., Katzen, H., Steele, J., et al. (2013). Motor neuron involvement in multisystem proteinopathy: implications for ALS. Neurology 80, 1874–1880. doi:10.1212/WNL.0b013e3182929fc3
Benussi, L., Binetti, G., Sina, E., Gigola, L., Bettecken, T., Meitinger, T., et al. (2008). A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol. Aging 29, 427–435. doi:10.1016/j.neurobiolaging.2006.10.028
Benussi, L., Rossi, G., Glionna, M., Tonoli, E., Piccoli, E., Fostinelli, S., et al. (2014). C9ORF72 hexanucleotide repeat number in frontotemporal lobar degeneration: a genotype-phenotype correlation study. J. Alzheimers Dis. 38, 799–808. doi:10.3233/JAD-131028
Bieniek, K. F., van Blitterswijk, M., Baker, M. C., Petrucelli, L., Rademakers, R., and Dickson, D. W. (2014). Expanded C9ORF72 hexanucleotide repeat in depressive pseudodementia. JAMA Neurol. 71, 775–781. doi:10.1001/jamaneurol.2013.6368
Binetti, G., Nicosia, F., Benussi, L., Ghidoni, R., Feudatari, E., Barbiero, L., et al. (2003). Prevalence of TAU mutations in an Italian clinical series of familial frontotemporal patients. Neurosci. Lett. 338, 85–87. doi:10.1016/S0304-3940(02)01330-7
Bird, T., Knopman, D., VanSwieten, J., Rosso, S., Feldman, H., Tanabe, H., et al. (2003). Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann. Neurol. 54, S29–S31. doi:10.1002/ana.10572
Blair, I. P., Williams, K. L., Warraich, S. T., Durnall, J. C., Thoeng, A. D., Manavis, J., et al. (2010). FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 81, 639–645. doi:10.1136/jnnp.2009.194399
Boeve, B. F. (2011). Progressive supranuclear palsy. Parkinsonism Relat. Disord. 18, S192–S194. doi:10.1016/S1353-8020(11)70060-8
Boeve, B. F., Boylan, K. B., Graff-Radford, N. R., DeJesus-Hernandez, M., Knopman, D. S., Pedraza, O., et al. (2012). Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 135, 765–783. doi:10.1093/brain/aws004
Boeve, B. F., Tremont-Lukats, I. W., Waclawik, A. J., Murrell, J. R., Hermann, B., Jack, C. R., et al. (2005). Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain 128, 752–772. doi:10.1093/brain/awh356
Borghero, G., Floris, G., Cannas, A., Marrosu, M. G., Murru, M. R., Costantino, E., et al. (2011). A patient carrying a homozygous p.A382T TARDBP missense mutation shows a syndrome including ALS, extrapyramidal symptoms, and FTD. Neurobiol. Aging 32, .e1–.e5. doi:10.1016/j.neurobiolaging.2011.06.009
Borroni, B., Alberici, A., Premi, E., Archetti, S., Garibotto, V., Agosti, C., et al. (2008). Brain magnetic resonance imaging structural changes in a pedigree of asymptomatic progranulin mutation carriers. Rejuvenation Res. 11, 585–595. doi:10.1089/rej.2007.0623
Borroni, B., Archetti, S., Del Bo, R., Papetti, A., Buratti, E., Bonvicini, C., et al. (2010). TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 13, 509–517. doi:10.1089/rej.2010.1017
Borroni, B., Benussi, A., Archetti, S., Galimberti, D., Parnetti, L., Nacmias, B., et al. (2015a). Csf p-tau181/tau ratio as biomarker for TDP pathology in frontotemporal dementia. Amyotroph. Lateral Scler. Frontotemporal Degener. 16, 86–91. doi:10.3109/21678421.2014.971812
Borroni, B., Turrone, R., Galimberti, D., Nacmias, B., Alberici, A., Benussi, A., et al. (2015b). Italian frontotemporal dementia network (FTD group-SINDEM): sharing clinical and diagnostic procedures in frontotemporal dementia in Italy. Neurol. Sci. 36, 751–757. doi:10.1007/s10072-014-2033-9
Borroni, B., Bonvicini, C., Alberici, A., Buratti, E., Agosti, C., Archetti, S., et al. (2009). Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 30, E974–E983. doi:10.1002/humu.21100
Borroni, B., Ferrari, F., Galimberti, D., Nacmias, B., Barone, C., Bagnoli, S., et al. (2013). Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 35, .e7–.e10. doi:10.1016/j.neurobiolaging.2013.09.017
Borroni, B., and Padovani, A. (2013). Dementia: a new algorithm for molecular diagnostics in FTLD. Nat. Rev. Neurol. 9, 241–242. doi:10.1038/nrneurol.2013.72
Boxer, A. L., Mackenzie, I. R., Boeve, B. F., Baker, M., Seeley, W. W., Crook, R., et al. (2011). Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J. Neurol. Neurosurg. Psychiatry 82, 196–203. doi:10.1136/jnnp.2009.204081
Bronner, I. F., Rizzu, P., Seelaar, H., van Mil, S. E., Anar, B., Azmani, A., et al. (2007). Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur. J. Hum. Genet. 15, 369–374. doi:10.1038/sj.ejhg.5201772
Broustal, O., Camuzat, A., Guillot-Noël, L., Guy, N., Millecamps, S., Deffond, D., et al. (2010). FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers Dis. 22, 765–769. doi:10.3233/JAD-2010-100837
Brouwers, N., Sleegers, K., Engelborghs, S., Maurer-Stroh, S., Gijselinck, I., van der Zee, J., et al. (2008). Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71, 656–664. doi:10.1212/01.wnl.0000319688.89790.7a
Buratti, E., and Baralle, F. E. (2008). Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 13:867–878. doi:10.2741/2727
Burrell, J. R., Kiernan, M. C., Vucic, S., and Hodges, J. R. (2011). Motor neuron dysfunction in frontotemporal dementia. Brain 134, 2582–2594. doi:10.1093/brain/awr195
Byrne, S., Elamin, M., Bede, P., Shatunov, A., Walsh, C., Corr, B., et al. (2012). Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 11, 232–240. doi:10.1016/S1474-4422(12)70014-5
Cacace, R., Van Cauwenberghe, C., Bettens, K., Gijselinck, I., van der Zee, J., Engelborghs, S., et al. (2013). C9orf72 G4C2 repeat expansions in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 34, .e1–.e7. doi:10.1016/j.neurobiolaging.2012.12.019
Calvo, A., Moglia, C., Canosa, A., Cistaro, A., Valentini, C., Carrara, G., et al. (2012). Amyotrophic lateral sclerosis/frontotemporal dementia with predominant manifestations of obsessive-compulsive disorder associated to GGGGCC expansion of the c9orf72 gene. J. Neurol. 259, 2723–2725. doi:10.1007/s00415-012-6640-1
Carecchio, M., Fenoglio, C., De Riz, M., Guidi, I., Comi, C., Cortini, F., et al. (2009). Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic mild cognitive impairment converted to Alzheimer’s disease. J. Neurol. Sci. 287, 291–293. doi:10.1016/j.jns.2009.07.011
Caroppo, P., Le Ber, I., Camuzat, A., Clot, F., Naccache, L., Lamari, F., et al. (2014). Extensive white matter involvement in patients with frontotemporal lobar degeneration. JAMA Neurol. 71, 1562. doi:10.1001/jamaneurol.2014.1316
Carrasquillo, M. M., Nicholson, A. M., Finch, N., Gibbs, J. R., Baker, M., Rutherford, N. J., et al. (2010). Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am. J. Hum. Genet. 87, 890–897. doi:10.1016/j.ajhg.2010.11.002
Chen, Y., Li, S., Su, L., Sheng, J., Lv, W., Chen, G., et al. (2015). Association of progranulin polymorphism rs5848 with neurodegenerative diseases: a meta-analysis. J. Neurol. 262, 814–822. doi:10.1007/s00415-014-7630-2
Chen-Plotkin, A. S., Martinez-Lage, M., Sleiman, P. M. A., Hu, W., Greene, R., Wood, E. M., et al. (2011). Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch. Neurol. 68, 488–497. doi:10.1001/archneurol.2011.53
Chiò, A., Borghero, G., Pugliatti, M., Ticca, A., Calvo, A., Moglia, C., et al. (2011). Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch. Neurol. 68, 594–598. doi:10.1001/archneurol.2010.352
Chiò, A., Borghero, G., Restagno, G., Mora, G., Drepper, C., Traynor, B. J., et al. (2012). Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135, 784–793. doi:10.1093/brain/awr366
Chiò, A., Calvo, A., Moglia, C., Restagno, G., Ossola, I., Brunetti, M., et al. (2010). Amyotrophic lateral sclerosis-frontotemporal lobar dementia in 3 families with p.Ala382Thr TARDBP mutations. Arch. Neurol. 67, 1002–1009. doi:10.1001/archneurol.2010.173
Coon, E. A., Daube, J. R., DeJesus-Hernandez, M., Adeli, A., Savica, R., Parisi, J. E., et al. (2013). Clinical and electrophysiologic variability in amyotrophic lateral sclerosis within a kindred harboring the C9ORF72 repeat expansion. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 132–137. doi:10.3109/17482968.2012.724075
Cooper-Knock, J., Hewitt, C., Highley, J. R., Brockington, A., Milano, A., Man, S., et al. (2012). Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135, 751–764. doi:10.1093/brain/awr365
Cox, L. E., Ferraiuolo, L., Goodall, E. F., Heath, P. R., Higginbottom, A., Mortiboys, H., et al. (2010). Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 5:e9872. doi:10.1371/journal.pone.0009872
Crozat, A., Aman, P., Mandahl, N., and Ron, D. (1993). Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363, 640–644. doi:10.1038/363640a0
Cruchaga, C., Fernandez-Seara, M. A., Seijo-Martinez, M., Samaranch, L., Lorenzo, E., Hinrichs, A., et al. (2009). Cortical atrophy and language network reorganization associated with a novel progranulin mutation. Cereb. Cortex 19, 1751–1760. doi:10.1093/cercor/bhn202
Cruchaga, C., Graff, C., Chiang, H.-H., Wang, J., Hinrichs, A. L., Spiegel, N., et al. (2011). Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol. 68, 581–586. doi:10.1001/archneurol.2010.350
Cruts, M., Gijselinck, I., van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. doi:10.1038/nature05017
Cruts, M., Gijselinck, I., Van Langenhove, T., van der Zee, J., and Van Broeckhoven, C. (2013). Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 36, 450–459. doi:10.1016/j.tins.2013.04.010
Cruts, M., Theuns, J., and Van Broeckhoven, C. (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344. doi:10.1002/humu.22117
Cruts, M., and Van Broeckhoven, C. (2008). Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 24, 186–194. doi:10.1016/j.tig.2008.01.004
Cuyvers, E., Bettens, K., Philtjens, S., Van Langenhove, T., Gijselinck, I., van der Zee, J., et al. (2013). Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 35, 1–9. doi:10.1016/j.neurobiolaging.2013.09.009
Daoud, H., Valdmanis, P. N., Kabashi, E., Dion, P., Dupre, N., Camu, W., et al. (2009). Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 46, 112–114. doi:10.1136/jmg.2008.062463
Davidson, Y. S., Barker, H., Robinson, A. C., Thompson, J. C., Harris, J., Troakes, C., et al. (2014). Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2, 70. doi:10.1186/2051-5960-2-70
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi:10.1016/j.neuron.2011.09.011
Deng, H., Gao, K., and Jankovic, J. (2014). The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 10, 337–348. doi:10.1038/nrneurol.2014.78
Devenney, E., Hornberger, M., Irish, M., Mioshi, E., Burrell, J., Tan, R., et al. (2014). Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol. 71, 331–339. doi:10.1001/jamaneurol.2013.6002
Di Fabio, R., Tessa, A., Simons, E. J., Santorelli, F. M., Casali, C., Serrao, M., et al. (2010). Familial frontotemporal dementia with parkinsonism associated with the progranulin c.C1021T (p.Q341X) mutation. Parkinsonism Relat. Disord. 16, 484–485. doi:10.1016/j.parkreldis.2010.05.001
Dickson, D. W., Rademakers, R., and Hutton, M. L. (2007). Progressive supranuclear palsy: pathology and genetics. Brain Pathol. 17, 74–82. doi:10.1111/j.1750-3639.2007.00054.x
Djamshidian, A., Schaefer, J., Haubenberger, D., Stogmann, E., Zimprich, F., Auff, E., et al. (2009). A novel mutation in the VCP gene (G157R) in a german family with inclusion-body myopathy with paget disease of bone and frontotemporal dementia. Muscle Nerve 39, 389–391. doi:10.1002/mus.21225
Dobson-Stone, C., Hallupp, M., Bartley, L., Shepherd, C. E., Halliday, G. M., Schofield, P. R., et al. (2012). C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 79, 995–1001. doi:10.1212/wnl.0b013e3182684634
Doi, H., Koyano, S., Suzuki, Y., Nukina, N., and Kuroiwa, Y. (2010). The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 66, 131–133. doi:10.1016/j.neures.2009.10.004
Donnelly, C. J., Zhang, P.-W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi:10.1016/j.neuron.2013.10.015
Dopper, E. G. P., Rombouts, S. A. R. B., Jiskoot, L. C., Heijer den, T., de Graaf, J. R. A., de Koning, I., et al. (2014). Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology 83, e19–26. doi:10.1212/WNL.0000000000000583
Englund, E., Gustafson, L., Passant, U., Majounie, E., Renton, A. E., Traynor, B. J., et al. (2012). Familial Lund frontotemporal dementia caused by C9ORF72 hexanucleotide expansion. Neurobiol. Aging 33, .e13–.e16. doi:10.1016/j.neurobiolaging.2012.02.019
Farrer, M. J., Hulihan, M. M., Kachergus, J. M., Dächsel, J. C., Stoessl, A. J., Grantier, L. L., et al. (2009). DCTN1 mutations in Perry syndrome. Nat. Genet. 41, 163–165. doi:10.1038/ng.293
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi:10.1001/archneurol.2011.250
Feldman, H., Levy, A. R., Hsiung, G.-Y., Peters, K. R., Donald, A., Black, S. E., et al. (2003). A Canadian cohort study of cognitive impairment and related dementias (ACCORD): study methods and baseline results. Neuroepidemiology 22, 265–274. doi:10.1016/j.jalz.2011.03.006
Finch, N., Baker, M., Crook, R., Swanson, K., Kuntz, K., Surtees, R., et al. (2009). Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132, 583–591. doi:10.1093/brain/awn352
Finch, N., Carrasquillo, M. M., Baker, M., Rutherford, N. J., Coppola, G., DeJesus-Hernandez, M., et al. (2011). TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76, 467–474. doi:10.1212/WNL.0b013e31820a0e3b
Floris, G., Borghero, G., Cannas, A., Stefano, F., Costantino, E., Murru, M. R., et al. (2012). Frontotemporal dementia with psychosis, parkinsonism, visuo-spatial dysfunction, upper motor neuron involvement associated to expansion of C9ORF72: a peculiar phenotype? J. Neurol. 259, 1749–1751. doi:10.1007/s00415-012-6444-3
Fodero-Tavoletti, M. T., Okamura, N., Furumoto, S., Mulligan, R. S., Connor, A. R., McLean, C. A., et al. (2011). 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 134, 1089–1100. doi:10.1093/brain/awr038
Frank, A. R., Wszolek, Z. K., Jack, C. R., and Boeve, B. F. (2007). Distinctive MRI findings in pallidopontonigral degeneration (PPND). Neurology 68, 620–621. doi:10.1212/01.wnl.0000254614.39759.3d
Froelich, S., Basun, H., Forsell, C., Lilius, L., Axelman, K., Andreadis, A., et al. (1997). Mapping of a disease locus for familial rapidly progressive frontotemporal dementia to chromosome 17q12-21. Am. J. Med. Genet. 74, 380–385. doi:10.1002/(SICI)1096-8628(19970725)74:4<380::AID-AJMG8>3.0.CO;2-T
Gabryelewicz, T., Masellis, M., Berdynski, M., Bilbao, J. M., Rogaeva, E., St George-Hyslop, P., et al. (2010). Intra-familial clinical heterogeneity due to FTLD-U with TDP-43 proteinopathy caused by a novel deletion in progranulin gene (PGRN). J. Alzheimers Dis. 22, 1123–1133. doi:10.3233/JAD-2010-101413
Galimberti, D., Fenoglio, C., Serpente, M., Villa, C., Bonsi, R., Arighi, A., et al. (2013). Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: late-onset psychotic clinical presentation. Biol. Psychiatry 74, 384–391. doi:10.1016/j.biopsych.2013.01.031
Galimberti, D., Reif, A., Dell’Osso, B., Kittel-Schneider, S., Leonhard, C., Herr, A., et al. (2014a). C9ORF72 hexanucleotide repeat expansion is a rare cause of schizophrenia. Neurobiol. Aging 35, .e7–.e10. doi:10.1016/j.neurobiolaging.2013.12.004
Galimberti, D., Reif, A., Dell’Osso, B., Palazzo, C., Villa, C., Fenoglio, C., et al. (2014b). C9ORF72 hexanucleotide repeat expansion as a rare cause of bipolar disorder. Bipolar Disord. 16, 448–449. doi:10.1111/bdi.12169
Gallagher, M. D., Suh, E., Grossman, M., Elman, L., McCluskey, L., van Swieten, J. C., et al. (2014). TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 127, 407–418. doi:10.1007/s00401-013-1239-x
Gallone, S., Giordana, M. T., Scarpini, E., Rainero, I., Rubino, E., Fenoglio, P., et al. (2009). Absence of TARDBP gene mutations in an italian series of patients with frontotemporal lobar degeneration. Dement. Geriatr. Cogn. Disord. 28, 239–243. doi:10.1159/000241876
Gass, J., Cannon, A., Mackenzie, I. R., Boeve, B., Baker, M., Adamson, J., et al. (2006). Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 15, 2988–3001. doi:10.1093/hmg/ddl241
Gellera, C., Tiloca, C., Del Bo, R., Corrado, L., Pensato, V., Agostini, J., et al. (2013). Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 84, 183–187. doi:10.1136/jnnp-2012-303433
Gelpi, E., van der Zee, J., Turon Estrada, A., Van Broeckhoven, C., and Sánchez-Valle, R. (2014). TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol. Appl. Neurobiol. 40, 225–230. doi:10.1111/nan.12063
Ghetti, B., Oblak, A. L., Boeve, B. F., Johnson, K. A., Dickerson, B. C., and Goedert, M. (2015). Invited review: frontotemporal dementia caused by microtubule-associated protein taugene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46. doi:10.1111/nan.12213
Ghetti, B., Spina, S., Murrell, J. R., Huey, E. D., Pietrini, P., Sweeney, B., et al. (2008). In vivo and postmortem clinicoanatomical correlations in frontotemporal dementia and parkinsonism linked to chromosome 17. Neurodegener. Dis. 5, 215–217. doi:10.1159/000113706
Ghidoni, R., Benussi, L., Glionna, M., Franzoni, M., and Binetti, G. (2008). Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71, 1235–1239. doi:10.1212/01.wnl.0000325058.10218.fc
Ghidoni, R., Paterlini, A., and Benussi, L. (2012). Circulating progranulin as a biomarker for neurodegenerative diseases. Am. J. Neurodegener. Dis. 1, 180–190.
Gidaro, T., Modoni, A., Sabatelli, M., Tasca, G., Broccolini, A., and Mirabella, M. (2008). An Italian family with inclusion-body myopathy and frontotemporal dementia due to mutation in the VCP gene. Muscle Nerve 37, 111–114. doi:10.1002/mus.20890
Gijselinck, I., Sleegers, K., Engelborghs, S., Robberecht, W., Martin, J.-J., Vandenberghe, R., et al. (2009). Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol. Aging 30, 1329–1331. doi:10.1016/j.neurobiolaging.2007.11.002
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi:10.1016/S1474-4422(11)70261-7
Giraldo, M., Lopera, F., Siniard, A. L., Corneveaux, J. J., Schrauwen, I., Carvajal, J., et al. (2013). Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer’s disease. Neurobiol. Aging 34, .e11–.e18. doi:10.1016/j.neurobiolaging.2013.02.016
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi:10.1002/ana.21344
Gitcho, M. A., Bigio, E. H., Mishra, M., Johnson, N., Weintraub, S., Mesulam, M., et al. (2009). TARDBP 3’-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 118, 633–645. doi:10.1007/s00401-009-0571-7
Goedert, M., and Jakes, R. (1990). Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230.
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989a). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. doi:10.1016/0896-6273(89)90210-9
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989b). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399. doi:10.1002/chin.200613246
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Mendez, M., Cappa, S. F., Ogar, J. M., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 75, 1006–1014. doi:10.1212/WNL.0b013e31821103e6
Götzl, J. K., Mori, K., Damme, M., Fellerer, K., Tahirovic, S., Kleinberger, G., et al. (2014). Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 127, 845–860. doi:10.1007/s00401-014-1262-6
Gros-Louis, F., Gaspar, C., and Rouleau, G. A. (2006). Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 956–972. doi:10.1016/j.bbadis.2006.01.004
Grossman, M., Elman, L., McCluskey, L., McMillan, C. T., Boller, A., Powers, J., et al. (2014). Phosphorylated tau as a candidate biomarker for amyotrophic lateral sclerosis. JAMA Neurol. 71, 442. doi:10.1001/jamaneurol.2013.6064
Guerreiro, R., Bilgic, B., Guven, G., Bras, J., Rohrer, J., Lohmann, E., et al. (2013a). Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol. Aging 34, .e1–.e5. doi:10.1016/j.neurobiolaging.2013.06.005
Guerreiro, R., Kara, E., Le Ber, I., Bras, J., Rohrer, J. D., Taipa, R., et al. (2013b). Genetic analysis of inherited leukodystrophies: genotype-phenotype correlations in the CSF1R gene. JAMA Neurol. 70, 875–882. doi:10.1001/jamaneurol.2013.698
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013c). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi:10.1056/NEJMoa1211851
Guerreiro, R. J. (2013). Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia–like syndrome without bone involvement. JAMA Neurol. 70, 78. doi:10.1001/jamaneurol.2013.579
Guyant-Marechal, L., Laquerriere, A., Duyckaerts, C., Dumanchin, C., Bou, J., Dugny, F., et al. (2006). Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology 67, 644–651. doi:10.1212/01.wnl.0000225184.14578.d3
Gydesen, S., Hagen, S., Klinken, L., Abelskov, J., and Sørensen, S. A. (1987). Neuropsychiatric studies in a family with presenile dementia different from Alzheimer and pick disease. Acta Psychiatr. Scand. 76, 276–284. doi:10.1111/j.1600-0447.1987.tb02896.x
Harms, M., Benitez, B. A., Cairns, N., Cooper, B., Cooper, P., Mayo, K., et al. (2013). C9orf72 hexanucleotide repeat expansions in clinical alzheimer disease. JAMA Neurol. 70, 736. doi:10.1001/2013.jamaneurol.537
Hayashi, S., Toyoshima, Y., Hasegawa, M., Umeda, Y., Wakabayashi, K., Tokiguchi, S., et al. (2002). Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann. Neurol. 51, 525–530. doi:10.1002/ana.10163
Hirano, M., Nakamura, Y., Saigoh, K., Sakamoto, H., Ueno, S., Isono, C., et al. (2015). VCP gene analyses in Japanese patients with sporadic amyotrophic lateral sclerosis identify a new mutation. Neurobiol. Aging 36, .e1–.e6. doi:10.1016/j.neurobiolaging.2014.10.012
Hoell, J. I., Larsson, E., Runge, S., Nusbaum, J. D., Duggimpudi, S., Farazi, T. A., et al. (2011). RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 18, 1428–1431. doi:10.1038/nsmb.2163
Holm, I. E., Englund, E., Mackenzie, I. R. A., Johannsen, P., and Isaacs, A. M. (2007). A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J. Neuropathol. Exp. Neurol. 66, 884–891. doi:10.1097/nen.0b013e3181567f02
Holm, I. E., Isaacs, A. M., and Mackenzie, I. R. A. (2009). Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol. 118, 719–720. doi:10.1007/s00401-009-0593-1
Houlden, H., Baker, M., Adamson, J., Grover, A., Waring, S., Dickson, D., et al. (1999). Frequency of tau mutations in three series of non-Alzheimer’s degenerative dementia. Ann. Neurol. 46, 243–248. doi:10.1002/1531-8249(199908)46:2<243::AID-ANA14>3.0.CO;2-L
Hsiung, G. Y. R., DeJesus-Hernandez, M., Feldman, H. H., Sengdy, P., Bouchard-Kerr, P., Dwosh, E., et al. (2012). Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135, 709–722. doi:10.1093/brain/awr354
Hsiung, G.-Y. R., Fok, A., Feldman, H. H., Rademakers, R., and Mackenzie, I. R. A. (2011). rs5848 polymorphism and serum progranulin level. J. Neurol. Sci. 300, 28–32. doi:10.1016/j.jns.2010.10.009
Hu, F., Padukkavidana, T., Vægter, C. B., Brady, O. A., Zheng, Y., Mackenzie, I. R., et al. (2010). Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667. doi:10.1016/j.neuron.2010.09.034
Hu, W. T., Watts, K., Grossman, M., Glass, J., Lah, J. J., Hales, C., et al. (2013). Reduced CSF p-Tau181 to Tau ratio is a biomarker for FTLD-TDP. Neurology 81, 1945–1952. doi:10.1212/01.wnl.0000436625.63650.27
Huey, E. D., Ferrari, R., Moreno, J. H., Jensen, C., Morris, C. M., Potocnik, F., et al. (2012). FUS and TDP43 genetic variability in FTD and CBS. Neurobiol. Aging 33, .e9–.e17. doi:10.1016/j.neurobiolaging.2011.08.004
Huey, E. D., Grafman, J., Wassermann, E. M., Pietrini, P., Tierney, M. C., Ghetti, B., et al. (2006). Characteristics of frontotemporal dementia patients with a Progranulinmutation. Ann. Neurol. 60, 374–380. doi:10.1002/ana.20969
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi:10.1038/31508
Irwin, D. J., McMillan, C. T., Brettschneider, J., Libon, D. J., Powers, J., Rascovsky, K., et al. (2013). Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 84, 163–169. doi:10.1136/jnnp-2012-303507
Ishizuka, T., Nakamura, M., Ichiba, M., and Sano, A. (2011). Familial semantic dementia with P301L mutation in the tau gene. Dement. Geriatr. Cogn. Disord. 31, 334–340. doi:10.1159/000328412
Jacquin, A., Rouaud, O., Soichot, P., Béjot, Y., Dygai-Cochet, I., Sarazin, M., et al. (2013). Psychiatric presentation of frontotemporal dementia associated with inclusion body myopathy due to the VCP mutation (R155H) in a French family. Case Rep. Neurol. 5, 187–194. doi:10.1159/000356481
Janssen, J. C., Warrington, E. K., Morris, H. R., Lantos, P., Brown, J., Revesz, T., et al. (2002). Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 58, 1161–1168. doi:10.1212/WNL.58.8.1161
Jiao, J., Herl, L. D., Farese, R. V., and Gao, F.-B. (2010). MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS ONE 5:e10551. doi:10.1371/journal.pone.0010551
Johnson, J. O., Mandrioli, J., Benatar, M., Van Deerlin, V. M., Trojanowski, J. Q., Gibbs, J. R., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi:10.1016/j.neuron.2010.11.036
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi:10.1056/NEJMoa1211103
Josephs, K. A., Petersen, R. C., Knopman, D. S., Boeve, B. F., Whitwell, J. L., Duffy, J. R., et al. (2006). Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 66, 41–48. doi:10.1212/01.wnl.0000191307.69661.c3
Josephs, K. A., Whitwell, J. L., Dickson, D. W., Boeve, B. F., Knopman, D. S., Petersen, R. C., et al. (2008). Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol. Aging 29, 280–289. doi:10.1016/j.neurobiolaging.2006.09.019
Josephs, K. A., Whitwell, J. L., Parisi, J. E., Petersen, R. C., Boeve, B. F., Jack, C. R. Jr., et al. (2010). Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur. J. Neurol. 17, 969–975. doi:10.1111/j.1468-1331.2010.02975.x
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Velde, C. V., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi:10.1038/ng.132
Kaivorinne, A.-L., Krüger, J., Kuivaniemi, K., Tuominen, H., Moilanen, V., Majamaa, K., et al. (2008). Role of MAPT mutations and haplotype in frontotemporal lobar degeneration in Northern Finland. BMC Neurol. 8:48. doi:10.1186/1471-2377-8-48
Kelley, B. J., Haidar, W., Boeve, B. F., Baker, M., Graff-Radford, N. R., Krefft, T., et al. (2009). Prominent phenotypic variability associated with mutations in progranulin. Neurobiol. Aging 30, 739–751. doi:10.1016/j.neurobiolaging.2007.08.022
Kelley, B. J., Haidar, W., Boeve, B. F., Baker, M., Shiung, M., Knopman, D. S., et al. (2010). Alzheimer disease–like phenotype associated with the c.154delA mutation in progranulin. Arch. Neurol. 67, 171–177. doi:10.1001/archneurol.2010.113
Kertesz, A., Ang, L.-C., Jesso, S., MacKinley, J., Baker, M., Brown, P., et al. (2013). Psychosis and hallucinations in frontotemporal dementia with the C9ORF72 mutation: a detailed clinical cohort. Cogn. Behav. Neurol. 26, 146–154. doi:10.1097/WNN.0000000000000008
Kertesz, A., Kawarai, T., Rogaeva, E., St George-Hyslop, P., Poorkaj, P., Bird, T. D., et al. (2000). Familial frontotemporal dementia with ubiquitin-positive, tau-negative inclusions. Neurology 54, 818–827. doi:10.1212/WNL.54.4.818
Kim, E.-J., Park, Y.-E., Kim, D.-S., Ahn, B.-Y., Kim, H.-S., Chang, Y. H., et al. (2011). Inclusion body myopathy with paget disease of bone and frontotemporal dementia linked to VCP p.Arg155Cys in a Korean family. Arch. Neurol. 68, 1–10. doi:10.1001/archneurol.2010.376
Kim, E. J., Rabinovici, G. D., Seeley, W. W., Halabi, C., Shu, H., Weiner, M., et al. (2007). Patterns of MRI atrophy in tau positive and ubiquitin positive frontotemporal lobar degeneration. J. Neurol. Neurosurg. Psychiatry 78, 1375–1378. doi:10.1136/jnnp.2006.114231
Kim, H. J., Kim, N. C., Wang, Y.-D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi:10.1038/nature11922
Kimonis, V. E., Fulchiero, E., Vesa, J., and Watts, G. (2008). VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim. Biophys. Acta 1782, 744–748. doi:10.1016/j.bbadis.2008.09.003
Kovach, M. J., Waggoner, B., Leal, S. M., Gelber, D., Khardori, R., Levenstien, M. A., et al. (2001). Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol. Genet. Metab. 74, 458–475. doi:10.1006/mgme.2001.3256
Kovacs, G. G., Murrell, J. R., Horvath, S., Haraszti, L., Majtenyi, K., Molnar, M. J., et al. (2009). TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov. Disord. 24, 1842–1847. doi:10.1002/mds.22697
Kühnlein, P., Sperfeld, A.-D., Vanmassenhove, B., Van Deerlin, V., Lee, V. M. Y., Trojanowski, J. Q., et al. (2008). Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch. Neurol. 65, 1185–1189. doi:10.1001/archneur.65.9.1185
Kumar, K. R., Needham, M., Mina, K., Davis, M., Brewer, J., Staples, C., et al. (2010). Two Australian families with inclusion-body myopathy, Paget’s disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul. Disord. 20, 330–334. doi:10.1016/j.nmd.2010.03.002
Kwiatkowski, T. J., Bosco, D. A., LeClerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi:10.1126/science.1166066
Lagier-Tourenne, C., Baughn, M., and Rigo, F. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U.S. A. 110, E4530–E4539. doi:10.1073/pnas.1318835110/-/DCSupplemental
Lagier-Tourenne, C., Polymenidou, M., and Cleveland, D. W. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64. doi:10.1093/hmg/ddq137
Lashley, T., Rohrer, J. D., Mahoney, C., Gordon, E., Beck, J., Mead, S., et al. (2014). A pathogenic progranulin mutation and C9orf72 repeat expansion in a family with frontotemporal dementia. Neuropathol. Appl. Neurobiol. 40, 502–513. doi:10.1111/nan.12100
Lattante, S., Le Ber, I., Camuzat, A., Dayan, S., Godard, C., Van Bortel, I., et al. (2013). TREM2 mutations are rare in a French cohort of patients with frontotemporal dementia. Neurobiol. Aging 34, .e1–.e2. doi:10.1016/j.neurobiolaging.2013.04.030
Lavedan, C., Hofmann-Radvanyi, H., Shelbourne, P., Rabes, J. P., Duros, C., Savoy, D., et al. (1993). Myotonic dystrophy: size- and sex-dependent dynamics of CTG meiotic instability, and somatic mosaicism. Am. J. Hum. Genet. 52, 875–883.
Le Ber, I. (2013). Genetics of frontotemporal lobar degeneration: an up-date and diagnosis algorithm. Rev. Neurol. 169, 811–819. doi:10.1016/j.neurol.2013.07.014
Le Ber, I., Camuzat, A., Berger, E., Hannequin, D., Laquerriere, A., Golfier, V., et al. (2009). Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 72, 1669–1676. doi:10.1212/WNL.0b013e3181a55f1c
Le Ber, I., Camuzat, A., Hannequin, D., Pasquier, F., Guedj, E., Rovelet-Lecrux, A., et al. (2008). Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 131, 732–746. doi:10.1093/brain/awn012
Le Ber, I., De Septenville, A., Guerreiro, R., Bras, J., Camuzat, A., Caroppo, P., et al. (2014). Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 35, .e23–.e25. doi:10.1016/j.neurobiolaging.2014.04.010
Lee, M.-J., Chen, T.-F., Cheng, T.-W., and Chiu, M.-J. (2011). rs5848 variant of progranulin gene is a risk of Alzheimer’s disease in the Taiwanese population. Neurodegener. Dis. 8, 216–220. doi:10.1159/000322538
Lee, S. E., Khazenzon, A. M., Trujillo, A. J., Guo, C. C., Yokoyama, J. S., Sha, S. J., et al. (2014). Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain 137, 3047–3060. doi:10.1093/brain/awu248
Lee, S. E., Seeley, W. W., Poorzand, P., Rademakers, R., Karydas, A., Stanley, C. M., et al. (2012). Clinical characterization of bvFTD due to FUS neuropathology. Neurocase 18, 305–317. doi:10.1080/13554794.2011.604637
Liu, E. Y., Russ, J., Wu, K., Neal, D., Suh, E., McNally, A. G., et al. (2014). C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 128, 525–541. doi:10.1007/s00401-014-1286-y
Lomen-Hoerth, C., Anderson, T., and Miller, B. (2002). The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. doi:10.1212/wnl.59.7.1077
Luty, A. A., Kwok, J. B., Thompson, E. M., Blumbergs, P., Brooks, W. S., Loy, C. T., et al. (2008). Pedigree with frontotemporal lobar degeneration – motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 8:32. doi:10.1186/1471-2377-8-32
Mackenzie, I. R., Arzberger, T., Kremmer, E., Troost, D., Lorenzl, S., Mori, K., et al. (2013). Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126, 859–879. doi:10.1007/s00401-013-1181-y
Mackenzie, I. R. A., Neumann, M., Baborie, A., Sampathu, D. M., Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. doi:10.1007/s00401-011-0845-8
Mackenzie, I. R. A., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., et al. (2008). Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 117, 15–18. doi:10.1007/s00401-008-0460-5
Mackenzie, I. R. A., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., et al. (2010). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 119, 1–4. doi:10.1007/s00401-009-0612-2
Mackenzie, I. R. A., Shi, J., Shaw, C. L., Duplessis, D., Neary, D., Snowden, J. S., et al. (2006). Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol. 112, 551–559. doi:10.1007/s00401-006-0123-3
Mahoney, C. J., Beck, J., Rohrer, J. D., Lashley, T., Mok, K., Shakespeare, T., et al. (2012). Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 135, 736–750. doi:10.1093/brain/awr361
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi:10.1016/S1474-4422(12)70043-1
Masellis, M., Momeni, P., Meschino, W., Heffner, R., Elder, J., Sato, C., et al. (2006). Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 129, 3115–3123. doi:10.1093/brain/awl276
Matsuura, T., Fang, P., Lin, X., Khajavi, M., Tsuji, K., Rasmussen, A., et al. (2004). Somatic and germline instability of the ATTCT repeat in spinocerebellar ataxia type 10. Am. J. Hum. Genet. 74, 1216–1224. doi:10.1086/421526
McKhann, G. M., Albert, M. S., Grossman, M., Miller, B., Dickson, D., Trojanowski, J. Q., et al. (2001). Clinical and pathological diagnosis of frontotemporal dementia: report of the work group on frontotemporal dementia and Pick’s disease. Arch. Neurol. 58, 1803–1809. doi:10.1001/archneur.58.11.1803
McMillan, C. T., Irwin, D. J., Avants, B. B., Powers, J., Cook, P. A., Toledo, J. B., et al. (2013). White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J. Neurol. Neurosurg. Psychiatry 84, 949–955. doi:10.1136/jnnp-2012-304418
McMillan, C. T., Russ, J., Wood, E. M., Irwin, D. J., Grossman, M., McCluskey, L., et al. (2015). C9orf72 promoter hypermethylation is neuroprotective: neuroimaging and neuropathologic evidence. Neurology 84, 1622–1630. doi:10.1212/WNL.0000000000001495
Mesulam, M., Johnson, N., Krefft, T. A., Gass, J. M., Cannon, A. D., Adamson, J. L., et al. (2007). Progranulin mutations in primary progressive aphasia. Arch. Neurol. 64, 43. doi:10.1001/archneur.64.1.43
Mesulam, M. M. (1982). Slowly progressive aphasia without generalized dementia. Ann. Neurol. 11, 592–598. doi:10.1002/ana.410110607
Miller, J. W., Smith, B. N., Topp, S. D., Al-Chalabi, A., Shaw, C. E., and Vance, C. (2012). Mutation analysis of VCP in British familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 33, .e1–.e2. doi:10.1016/j.neurobiolaging.2012.06.003
Momeni, P., Schymick, J., Jain, S., Cookson, M. R., Cairns, N. J., Greggio, E., et al. (2006). Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol. 6:44. doi:10.1186/1471-2377-6-44
Momeni, P., Wickremaratchi, M. M., Bell, J., Arnold, R., Beer, R., Hardy, J., et al. (2010). Familial early onset frontotemporal dementia caused by a novel S356T MAPT mutation, initially diagnosed as schizophrenia. Clin. Neurol. Neurosurg. 112, 917–920. doi:10.1016/j.clineuro.2010.07.015
Montuschi, A., Iazzolino, B., Calvo, A., Moglia, C., Lopiano, L., Restagno, G., et al. (2015). Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J. Neurol. Neurosurg. Psychiatry 86, 168–173. doi:10.1136/jnnp-2013-307223
Moreno, F., Rabinovici, G. D., Karydas, A., Miller, Z., Hsu, S. C., Legati, A., et al. (2015). A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques. Acta Neuropathol. Commun. 3, 542. doi:10.1186/s40478-015-0190-6
Mori, K., Arzberger, T., Grässer, F. A., Gijselinck, I., May, S., Rentzsch, K., et al. (2013a). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893. doi:10.1007/s00401-013-1189-3
Mori, K., Lammich, S., Mackenzie, I. R. A., Forné, I., Zilow, S., Kretzschmar, H., et al. (2013b). hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 125, 413–423. doi:10.1007/s00401-013-1088-7
Mori, K., Weng, S.-M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013c). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi:10.1126/science.1232927
Morita, M., Al-Chalabi, A., Andersen, P. M., Hosler, B., Sapp, P., Englund, E., et al. (2006). A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66, 839–844. doi:10.1212/01.wnl.0000200048.53766.b4
Mosca, L., Lunetta, C., Tarlarini, C., Avemaria, F., Maestri, E., Melazzini, M., et al. (2012). Wide phenotypic spectrum of the TARDBP gene: homozygosity of A382T mutation in a patient presenting with amyotrophic lateral sclerosis, Parkinson’s disease, and frontotemporal lobar degeneration, and in neurologically healthy subject. Neurobiol. Aging 33, .e1–.e4. doi:10.1016/j.neurobiolaging.2012.01.108
Münch, C., Rosenbohm, A., Sperfeld, A.-D., Uttner, I., Reske, S., Krause, B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58, 777–780. doi:10.1002/ana.20631
Munoz, D. G., Neumann, M., Kusaka, H., Yokota, O., Ishihara, K., Terada, S., et al. (2009). FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 118, 617–627. doi:10.1007/s00401-009-0598-9
Munoz, D. G., Ros, R., Fatas, M., Bermejo, F., and de Yebenes, J. G. (2007). Progressive nonfluent aphasia associated with a new mutation V363I in tau gene. Am. J. Alzheimers Dis. Other Demen. 22, 294–299. doi:10.1177/1533317507302320
Murray, M. E., Bieniek, K. F., Banks Greenberg, M., DeJesus-Hernandez, M., Rutherford, N. J., van Blitterswijk, M., et al. (2013). Progressive amnestic dementia, hippocampal sclerosis, and mutation in C9ORF72. Acta Neuropathol. 126, 545–554. doi:10.1007/s00401-013-1161-2
Murray, M. E., DeJesus-Hernandez, M., Rutherford, N. J., Baker, M., Duara, R., Graff-Radford, N. R., et al. (2011). Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 122, 673–690. doi:10.1007/s00401-011-0907-y
Nalbandian, A., Donkervoort, S., Dec, E., Badadani, M., Katheria, V., Rana, P., et al. (2011). The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J. Mol. Neurosci. 45, 522–531. doi:10.1007/s12031-011-9627-y
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi:10.1212/wnl.51.6.1546
Neumann, M., Bentmann, E., Dormann, D., Jawaid, A., DeJesus-Hernandez, M., Ansorge, O., et al. (2011). FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 134, 2595–2609. doi:10.1093/brain/awr201
Neumann, M., Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., and Mackenzie, I. R. A. (2009a). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931. doi:10.1093/brain/awp214
Neumann, M., Roeber, S., Kretzschmar, H. A., Rademakers, R., Baker, M., and Mackenzie, I. R. A. (2009b). Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 118, 605–616. doi:10.1007/s00401-009-0581-5
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi:10.1126/science.1134108
Neve, R. L., Harris, P., Kosik, K. S., Kurnit, D. M., and Donlon, T. A. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387, 271–280. doi:10.1016/0169-328X(86)90033-1
Newsway, V., Fish, M., Rohrer, J. D., Majounie, E., Williams, N., Hack, M., et al. (2010). Perry syndrome due to the DCTN1 G71R mutation: a distinctive levodopa responsive disorder with behavioral syndrome, vertical gaze palsy, and respiratory failure. Mov. Disord. 25, 767–770. doi:10.1002/mds.22950
Nuytemans, K., van der Zee, J., Gijselinck, I., Engelborghs, S., Theuns, J., Kumar-Singh, S., et al. (2007). Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch. Neurol. 64, 1436–1446. doi:10.1001/archneur.64.10.1436
Ohshima, S., Tsuboi, Y., Yamamoto, A., Kawakami, M., Farrer, M. J., Kira, J.-I., et al. (2010). Autonomic failures in Perry syndrome with DCTN1 mutation. Parkinsonism Relat. Disord. 16, 612–614. doi:10.1016/j.parkreldis.2010.07.001
Paloneva, J., Kestilä, M., Wu, J., Salminen, A., Bohling, T., Ruotsalainen, V., et al. (2000). Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25, 357–361. doi:10.1038/77153
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662. doi:10.1086/342259
Parkinson, N., Ince, P. G., Smith, M. O., Highley, R., Skibinski, G., Andersen, P. M., et al. (2006). ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077. doi:10.1212/01.wnl.0000231510.89311.8b
Pearson, J. P., Williams, N. M., Majounie, E., Waite, A., Stott, J., Newsway, V., et al. (2011). Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J. Neurol. 258, 647–655. doi:10.1007/s00415-010-5815-x
Pickering-Brown, S. M., Rollinson, S., Plessis Du, D., Morrison, K. E., Varma, A., Richardson, A. M. T., et al. (2008). Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131, 721–731. doi:10.1093/brain/awm331
Pievani, M., Paternicò, D., Benussi, L., Binetti, G., Orlandini, A., Cobelli, M., et al. (2014). Pattern of structural and functional brain abnormalities in asymptomatic granulin mutation carriers. Alzheimer’s & Dementia 10, S354–S363.e1. doi:10.1016/j.jalz.2013.09.009
Pletnikova, O., Sloane, K. L., Renton, A. E., Traynor, B. J., Crain, B. J., Reid, T., et al. (2014). Hippocampal sclerosis dementia with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 35, .e17–.e21. doi:10.1016/j.neurobiolaging.2014.04.009
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature 14, 459–468. doi:10.1038/nn.2779
Poorkaj, P., Bird, T. D., Wijsman, E., Nemens, E., Garruto, R. M., Anderson, L., et al. (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825. doi:10.1002/ana.410430617
Poorkaj, P., Grossman, M., Steinbart, E., Payami, H., Sadovnick, A., Nochlin, D., et al. (2001). Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia. Arch. Neurol. 58, 383–387. doi:10.1001/archneur.58.3.383
Premi, E., Borroni, B., Formenti, A., Gazzina, S., Archetti, S., Gasparotti, R., et al. (2013a). Effect of TMEM106B polymorphism on functional network connectivity in asymptomatic grn mutation carriers. JAMA Neurol. 71, 216–221. doi:10.1001/jamaneurol.2013.4835
Premi, E., Grassi, M., Gazzina, S., Paghera, B., Pepe, D., Archetti, S., et al. (2013b). The neuroimaging signature of frontotemporal lobar degeneration associated with granulin mutations: an effective connectivity study. J Nucl Med 54, 1066–1071. doi:10.2967/jnumed.112.111773
Premi, E., Cauda, F., Gasparotti, R., Diano, M., Archetti, S., Padovani, A., et al. (2014a). Multimodal fMRI resting-state functional connectivity in granulin mutations: the case of fronto-parietal dementia. PLoS ONE 9, e106500. doi:10.1371/journal.pone.0106500
Premi, E., Garibotto, V., Gazzina, S., Formenti, A., Archetti, S., Gasparotti, R., et al. (2014b). Subcortical and deep cortical atrophy in frontotemporal dementia due to granulin mutations. Dement. Geriatr. Cogn. Disord. Extra 4, 95–102. doi:10.1159/000355428
Quadri, M., Cossu, G., Saddi, V., Simons, E. J., Murgia, D., Melis, M., et al. (2011). Broadening the phenotype of TARDBP mutations: the TARDBP Ala382Thr mutation and Parkinson’s disease in Sardinia. Neurogenetics 12, 203–209. doi:10.1007/s10048-011-0288-3
Rabbitts, T. H., Förster, A., Larson, R., and Nathan, P. (1993). Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 4, 175–180. doi:10.1038/ng0693-175
Rademakers, R., Cruts, M., Dermaut, B., Sleegers, K., Rosso, S. M., Van den Broeck, M., et al. (2002). Tau negative frontal lobe dementia at 17q21: significant finemapping of the candidate region to a 4.8 cM interval. Mol. Psychiatry 7, 1064–1074. doi:10.1038/sj.mp.4001198
Rademakers, R., Cruts, M., and Van Broeckhoven, C. (2004). The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 24, 277–295. doi:10.1002/humu.20086
Rademakers, R., Eriksen, J. L., Baker, M., Robinson, T., Ahmed, Z., Lincoln, S. J., et al. (2008). Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum. Mol. Genet. 17, 3631–3642. doi:10.1093/hmg/ddn257
Rademakers, R., Neumann, M., and Mackenzie, I. R. (2012). Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8, 423–434. doi:10.1038/nrneurol.2012.117
Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., and Mackenzie, I. R. A. (2009). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931. doi:10.1093/brain/awp214
Rascovsky, K., Rohrer, J. D., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. doi:10.1093/brain/awr179
Rayaprolu, S., Mullen, B., Baker, M., Lynch, T., Finger, E., Seeley, W. W., et al. (2013). TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 8, 19. doi:10.1186/1750-1326-8-19
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi:10.1016/j.neuron.2011.09.010
Rizzu, P., van Swieten, J. C., Joosse, M., Hasegawa, M., Stevens, M., Tibben, A., et al. (1999). High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Hum. Genet. 64, 414–421. doi:10.1086/302256
Robinson, A., Davidson, Y., Snowden, J. S., and Mann, D. M. A. (2014). C9ORF72 in dementia with Lewy bodies. J.Neurol. Neurosurg. Psychiatry 85, 1435–1436. doi:10.1136/jnnp-2014-307622
Rohrer, J. D., Crutch, S. J., Warrington, E. K., and Warren, J. D. (2010a). Progranulin-associated primary progressive aphasia: a distinct phenotype? Neuropsychologia 48, 288–297. doi:10.1016/j.neuropsychologia.2009.09.017
Rohrer, J. D., Ridgway, G. R., Modat, M., Ourselin, S., Mead, S., Fox, N. C., et al. (2010b). Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53, 1070–1076. doi:10.1016/j.neuroimage.2009.12.088
Rohrer, J. D., Rossor, M. N., and Warren, J. D. (2010c). Syndromes of nonfluent primary progressive aphasia: a clinical and neurolinguistic analysis. Neurology 75, 603–610. doi:10.1212/wnl.0b013e3181ed9c6b
Rohrer, J. D., Guerreiro, R., Vandrovcova, J., Uphill, J., Reiman, D., Beck, J., et al. (2009). The heritability and genetics of frontotemporal lobar degeneration. Neurology 73, 1451–1456. doi:10.1212/WNL.0b013e3181bf997a
Rohrer, J. D., Isaacs, A. M., Mizielinska, S., Mead, S., Lashley, T., Wray, S., et al. (2015a). C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. 14, 291–301. doi:10.1016/S1474-4422(14)70233-9
Rohrer, J. D., Nicholas, J. M., Cash, D. M., Van Swieten, J., Dopper, E., Jiskoot, L., et al. (2015b). Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the genetic frontotemporal dementia initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 14, 253–262. doi:10.1016/S1474-4422(14)70324-2
Rohrer, J. D., Lashley, T., Schott, J. M., Warren, J. E., Mead, S., Beck, J., et al. (2011a). Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 134, 2565–2581. doi:10.1093/brain/awr198
Rohrer, J. D., Paviour, D., Vandrovcova, J., Hodges, J., de Silva, R., and Rossor, M. N. (2011b). Novel L284R MAPT mutation in a family with an autosomal dominant progressive supranuclear palsy syndrome. Neurodegener. Dis. 8, 149–152. doi:10.1159/000319454
Rohrer, J. D., Rossor, M. N., and Warren, J. D. (2012). Alzheimer’s pathology in primary progressive aphasia. Neurobiol. Aging 33, 744–752. doi:10.1016/j.neurobiolaging.2010.05.020
Rohrer, J. D., and Warren, J. D. (2011). Phenotypic signatures of genetic frontotemporal dementia. Curr. Opin. Neurol. 24, 542–549. doi:10.1097/WCO.0b013e32834cd442
Rohrer, J. D., Warren, J. D., Barnes, J., Mead, S., Beck, J., Pepple, T., et al. (2008). Mapping the progression of progranulin-associated frontotemporal lobar degeneration. Nat. Clin. Pract. Neurol. 4, 455–460. doi:10.1038/ncpneuro0869
Rollinson, S., Snowden, J. S., Neary, D., Morrison, K. E., Mann, D. M. A., and Pickering-Brown, S. M. (2007). TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci. Lett. 419, 1–4. doi:10.1016/j.neulet.2007.03.044
Rosso, S. M., Kamphorst, W., de Graaf, B., Willemsen, R., Ravid, R., Niermeijer, M. F., et al. (2001). Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q21-22. Brain 124, 1948–1957. doi:10.1093/brain/124.10.1948
Rovelet-Lecrux, A., Hannequin, D., Guillin, O., Legallic, S., Jurici, S., Wallon, D., et al. (2010). Frontotemporal dementia phenotype associated with MAPT gene duplication. J. Alzheimers Dis. 21, 897–902. doi:10.3233/JAD-2010-100441
Rubino, E., Rainero, I., Chio, A., Rogaeva, E., Galimberti, D., Fenoglio, P., et al. (2012). SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79, 1556–1562. doi:10.1212/WNL.0b013e31826e25df
Russ, J., Liu, E. Y., Wu, K., Neal, D., Suh, E., Irwin, D. J., et al. (2014). Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 129, 39–52. doi:10.1007/s00401-014-1365-0
Rutherford, N. J., Zhang, Y.-J., Baker, M., Gass, J. M., Finch, N. A., Xu, Y.-F., et al. (2008). Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4:e1000193. doi:10.1371/journal.pgen.1000193
Saint-Aubert, L., Sagot, C., Wallon, D., Hannequin, D., Payoux, P., Nemmi, F., et al. (2014). A case of logopenic primary progressive aphasia with C9ORF72 expansion and cortical florbetapir binding. J. Alzheimers Dis. 42, 413–420. doi:10.3233/JAD-140222
Schofield, E. C., Halliday, G. M., Kwok, J., Loy, C., Double, K. L., and Hodges, J. R. (2010). Low serum progranulin predicts the presence of mutations: a prospective study. J. Alzheimers Dis. 22, 981–984. doi:10.3233/JAD-2010-101032
Schumacher, A., Friedrich, P., Diehl, J., Ibach, B., Schoepfer-Wendels, A., Mueller, J. C., et al. (2009). No association of common VCP variants with sporadic frontotemporal dementia. Neurobiol. Aging 30, 333–335. doi:10.1016/j.neurobiolaging.2007.05.023
Schymick, J. C., Yang, Y., Andersen, P. M., Vonsattel, J. P., Greenway, M., Momeni, P., et al. (2006). Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J. Neurol. Neurosurg. Psychiatry 78, 754–756. doi:10.1136/jnnp.2006.109553
Seelaar, H., Kamphorst, W., Rosso, S. M., Azmani, A., Masdjedi, R., de Koning, I., et al. (2008). Distinct genetic forms of frontotemporal dementia. Neurology 71, 1220–1226. doi:10.1212/01.wnl.0000319702.37497.72
Seelaar, H., Rohrer, J. D., Pijnenburg, Y. A. L., Fox, N. C., and Van Swieten, J. C. (2011). Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J. Neurol. Neurosurg. Psychiatry 82, 476–486. doi:10.1136/jnnp.2010.212225
Sha, S. J., Takada, L. T., Rankin, K. P., Yokoyama, J. S., Rutherford, N. J., Fong, J. C., et al. (2012). Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 79, 1002–1011. doi:10.1212/WNL.0b013e318268452e
Sheng, J., Su, L., Xu, Z., and Chen, G. (2014). Progranulin polymorphism rs5848 is associated with increased risk of Alzheimer’s disease. Gene 542, 141–145. doi:10.1016/j.gene.2014.03.041
Simon-Sanchez, J., Dopper, E. G. P., Cohn-Hokke, P. E., Hukema, R. K., Nicolaou, N., Seelaar, H., et al. (2012). The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135, 723–735. doi:10.1093/brain/awr353
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. doi:10.1038/ng1609
Sleegers, K., Van Damme, P., Engelborghs, S., Gijselinck, I., van der Zee, J., Peeters, K., et al. (2009). Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann. Neurol. 65, 603–609. doi:10.1002/ana.21621
Slowinski, J. L., Schweitzer, K. J., Imamura, A., Uitti, R. J., Strongosky, A. J., Dickson, D. W., et al. (2009). Brainstem atrophy on routine MR study in pallidopontonigral degeneration. J. Neurol. 256, 827–829. doi:10.1007/s00415-009-5013-x
Smith, K. R., Damiano, J., Franceschetti, S., Carpenter, S., Canafoglia, L., Morbin, M., et al. (2012). Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 90, 1102–1107. doi:10.1016/j.ajhg.2012.04.021
Snowden, J. S., Pickering-Brown, S. M., Mackenzie, I. R., Richardson, A. M. T., Varma, A., Neary, D., et al. (2006). Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 129, 3091–3102. doi:10.1093/brain/awl267
Snowden, J. S., Rollinson, S., Thompson, J. C., Harris, J. M., Stopford, C. L., Richardson, A. M. T., et al. (2012). Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 135, 693–708. doi:10.1093/brain/awr355
Soliveri, P., Rossi, G., Monza, D., Tagliavini, F., Piacentini, S., Albanese, A., et al. (2003). A case of dementia parkinsonism resembling progressive supranuclear palsy due to mutation in the tau protein gene. Arch. Neurol. 60, 1454. doi:10.1001/archneur.60.10.1454
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi:10.1016/S1474-4422(13)70090-5
Spillantini, M. G., Murrell, J. R., Goedert, M., Farlow, M. R., Klug, A., and Ghetti, B. (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci.U.S.A. 95, 7737–7741. doi:10.1073/pnas.95.13.7737
Spina, S., Farlow, M. R., Unverzagt, F. W., Kareken, D. A., Murrell, J. R., Fraser, G., et al. (2008). The tauopathy associated with mutation +3 in intron 10 of tau: characterization of the MSTD family. Brain 131, 72–89. doi:10.1093/brain/awm280
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi:10.1126/science.1154584
Stewart, H., Rutherford, N. J., Briemberg, H., Krieger, C., Cashman, N., Fabros, M., et al. (2012). Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 123, 409–417. doi:10.1007/s00401-011-0937-5
Stojkovic, T., Hammouda el, H., Richard, P., de Munain, A. L., Ruiz-Martínez, J., Gonzalez, P. C., et al. (2009). Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul. Disord. 19, 316–323. doi:10.1016/j.nmd.2009.02.012
Suárez-Calvet, M., Camacho, V., Gómez-Ansón, B., Antón, S., Vives-Gilabert, Y., Dols-Icardo, O., et al. (2014). Early cerebellar hypometabolism in patients with frontotemporal dementia carrying the C9orf72 expansion. Alzheimer Dis. Assoc. Disord. doi:10.1097/WAD.0000000000000056
Surampalli, A., Gold, B. T., Smith, C., Castellani, R. J., Khare, M., Yu, H., et al. (2015). A case report comparing clinical, imaging and neuropsychological assessment findings in twins discordant for the VCP p.R155C mutation. Neuromuscul. Disord. 25, 177–183. doi:10.1016/j.nmd.2014.10.003
Synofzik, M., Born, C., Rominger, A., Lummel, N., Schöls, L., Biskup, S., et al. (2014). Targeted high-throughput sequencing identifies a TARDBP mutation as a cause of early-onset FTD without motor neuron disease. Neurobiol. Aging 35, .e1–.e5. doi:10.1016/j.neurobiolaging.2013.10.092
Tan, A. Y., and Manley, J. L. (2009). The TET family of proteins: functions and roles in disease. J. Mol. Cell Biol. 1, 82–92. doi:10.1093/jmcb/mjp025
The Lund and Manchester Groups. (1994). Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester groups. J. Neurol. Neurosurg. Psychiatry 57, 416–418. doi:10.1136/jnnp.57.4.416
Ticozzi, N., Silani, V., LeClerc, A. L., Keagle, P., Gellera, C., Ratti, A., et al. (2009). Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73, 1180–1185. doi:10.1212/WNL.0b013e3181bbff05
Tolboom, N., Koedam, E. L. G. E., Schott, J. M., Yaqub, M., Blankenstein, M. A., Barkhof, F., et al. (2010). Dementia mimicking Alzheimer’s disease owing to a tau mutation: CSF and PET findings. Alzheimer Dis. Assoc. Disord. 24, 303–307. doi:10.1097/WAD.0b013e3181cf35ec
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nature 14, 452–458. doi:10.1038/nn.2778
Troakes, C., Maekawa, S., Wijesekera, L., Rogelj, B., Siklós, L., Bell, C., et al. (2012). An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology 32, 505–514. doi:10.1111/j.1440-1789.2011.01286.x
Tsuboi, Y., Baker, M., Hutton, M. L., Uitti, R. J., Rascol, O., Delisle, M. B., et al. (2002). Clinical and genetic studies of families with the tau N279K mutation (FTDP-17). Neurology 59, 1791–1793. doi:10.1212/01.WNL.0000038909.49164.4B
Urwin, H., Josephs, K. A., Rohrer, J. D., Mackenzie, I. R., Neumann, M., Authier, A., et al. (2010). FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 120, 33–41. doi:10.1007/s00401-010-0698-6
Valdmanis, P. N., Dupre, N., Bouchard, J.-P., Camu, W., Salachas, F., Meininger, V., et al. (2007). Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch. Neurol. 64, 240. doi:10.1001/archneur.64.2.240
van Blitterswijk, M., DeJesus-Hernandez, M., Niemantsverdriet, E., Murray, M. E., Heckman, M. G., Diehl, N. N., et al. (2013). Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988. doi:10.1016/S1474-4422(13)70210-2
Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B., et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416. doi:10.1016/S1474-4422(08)70071-1
Van Deerlin, V. M., Sleiman, P. M. A., Martinez-Lage, M., Chen-Plotkin, A., Wang, L.-S., Graff-Radford, N. R., et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239. doi:10.1038/ng.536
van der Zee, J., Borroni, B., Gijselinck, I., Dillen, L., Van Langenhove, T., Theuns, J., et al. (2013). A Pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 34, 363–373. doi:10.1002/humu.22244
van der Zee, J., Pirici, D., Van Langenhove, T., Engelborghs, S., Vandenberghe, R., Hoffmann, M., et al. (2009). Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 73, 626–632. doi:10.1212/WNL.0b013e3181b389d9
van der Zee, J., and Van Broeckhoven, C. (2014). Year in review. Nat. Rev. Neurol. 10, 1–2. doi:10.1038/nrneurol.2013.270
van der Zee, J., Van Langenhove, T., Kleinberger, G., Sleegers, K., Engelborghs, S., Vandenberghe, R., et al. (2011). TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134, 808–815. doi:10.1093/brain/awr007
van der Zee, J., Van Langenhove, T., Kovacs, G. G., Dillen, L., Deschamps, W., Engelborghs, S., et al. (2014). Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 128, 397–410. doi:10.1007/s00401-014-1298-7
van Herpen, E., Rosso, S. M., Serverijnen, L.-A., Yoshida, H., Breedveld, G., van de Graaf, R., et al. (2003). Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann. Neurol. 54, 573–581. doi:10.1002/ana.10721
Van Langenhove, T., van der Zee, J., Gijselinck, I., Engelborghs, S., Vandenberghe, R., Vandenbulcke, M., et al. (2013). Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol. 70, 365. doi:10.1001/2013.jamaneurol.181
Van Langenhove, T., van der Zee, J., Sleegers, K., Engelborghs, S., Vandenberghe, R., Gijselinck, I., et al. (2010). Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74, 366–371. doi:10.1212/WNL.0b013e3181ccc732
Van Swieten, J., and Spillantini, M. G. (2007). Hereditary frontotemporal dementia caused by tau gene mutations. Brain Pathol. 17, 63–73. doi:10.1111/j.1750-3639.2007.00052.x
van Swieten, J. C., and Heutink, P. (2008). Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 7, 965–974. doi:10.1016/S1474-4422(08)70194-7
Van Swieten, J. C., Stevens, M., Rosso, S. M., Rizzu, P., Joosse, M., de Koning, I., et al. (1999). Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann. Neurol. 46, 617–626. doi:10.1002/1531-8249(199910)46:4<617::AID-ANA10>3.0.CO;2-I
Vance, C., Al-Chalabi, A., Ruddy, D., Smith, B. N., Hu, X., Sreedharan, J., et al. (2006). Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain 129, 868–876. doi:10.1093/brain/awl030
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi:10.1126/science.1165942
Vilariño-Güell, C., Wider, C., Soto-Ortolaza, A. I., Cobb, S. A., Kachergus, J. M., Keeling, B. H., et al. (2009). Characterization of DCTN1 genetic variability in neurodegeneration. Neurology 72, 2024–2028. doi:10.1212/WNL.0b013e3181a92c4c
Villa, C., Ghezzi, L., Pietroboni, A. M., Fenoglio, C., Cortini, F., Serpente, M., et al. (2011). A novel MAPT mutation associated with the clinical phenotype of progressive nonfluent aphasia. J. Alzheimers Dis. 26, 19–26. doi:10.3233/JAD-2011-102124
Villemagne, V. L., Fodero-Tavoletti, M. T., Masters, C. L., and Rowe, C. C. (2015). Tau imaging: early progress and future directions. Lancet Neurol. 14, 114–124. doi:10.1016/S1474-4422(14)70252-2
Wang, W.-X., Wilfred, B. R., Madathil, S. K., Tang, G., Hu, Y., Dimayuga, J., et al. (2010). miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 177, 334–345. doi:10.2353/ajpath.2010.091202
Watts, G. D. J., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi:10.1038/ng1332
Whitwell, J. L., Boeve, B. F., Weigand, S. D., Senjem, M. L., Gunter, J. L., Baker, M. C., et al. (2015). Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur. J. Neurol. 22, 745–752. doi:10.1111/ene.12675
Whitwell, J. L., Jack, C. R., Baker, M., Rademakers, R., Adamson, J., Boeve, B. F., et al. (2007). Voxel-Based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch. Neurol. 64, 371. doi:10.1001/archneur.64.3.371
Whitwell, J. L., Jack, C. R., Boeve, B. F., Senjem, M. L., Baker, M., Ivnik, R. J., et al. (2009a). Atrophy patterns in IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology 73, 1058–1065. doi:10.1212/WNL.0b013e3181b9c8b9
Whitwell, J. L., Jack, C. R., Boeve, B. F., Senjem, M. L., Baker, M., Rademakers, R., et al. (2009b). Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 72, 813–820. doi:10.1212/01.wnl.0000343851.46573.67
Whitwell, J. L., Jack, C. R. Jr., Senjem, M. L., Parisi, J. E., Boeve, B. F., Knopman, D. S., et al. (2009c). MRI correlates of protein deposition and disease severity in postmortem frontotemporal lobar degeneration. Neurodegener. Dis. 6, 106–117. doi:10.1159/000209507
Whitwell, J. L., Josephs, K. A., Avula, R., Tosakulwong, N., Weigand, S. D., Senjem, M. L., et al. (2011a). Altered functional connectivity in asymptomatic MAPT subjects: a comparison to bvFTD. Neurology 77, 866–874. doi:10.1212/wnl.0b013e31822c61f2
Whitwell, J. L., Weigand, S. D., Gunter, J. L., Boeve, B. F., Rademakers, R., Baker, M., et al. (2011b). Trajectories of brain and hippocampal atrophy in FTD with mutations in MAPT or GRN. Neurology 77, 393–398. doi:10.1212/WNL.0b013e318227047f
Whitwell, J. L., Weigand, S. D., Boeve, B. F., Senjem, M. L., Gunter, J. L., DeJesus-Hernandez, M., et al. (2012). Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 135, 794–806. doi:10.1093/brain/aws001
Wojtas, A., Heggeli, K. A., Finch, N., Baker, M., DeJesus-Hernandez, M., Younkin, S. G., et al. (2012). C9ORF72 repeat expansions and other FTD gene mutations in a clinical AD patient series from Mayo clinic. Am. J. Neurodegener. Dis. 1, 107–118. doi:10.1016/s1474-4422(12)70046-7
Xi, Z., Zhang, M., Bruni, A. C., Maletta, R. G., Colao, R., Fratta, P., et al. (2015). The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 129, 715–727. doi:10.1007/s00401-015-1401-8
Xi, Z., Zinman, L., Moreno, D., Schymick, J., Liang, Y., Sato, C., et al. (2013). Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 92, 981–989. doi:10.1016/j.ajhg.2013.04.017
Xia, C.-F., Arteaga, J., Chen, G., Gangadharmath, U., Gomez, L. F., Kasi, D., et al. (2013). [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer Dement. 9, 666–676. doi:10.1016/j.jalz.2012.11.008
Yamanaka, K., Sasagawa, Y., and Ogura, T. (2012). Recent advances in p97/VCP/Cdc48 cellular functions. Biochim. Biophys. Acta 1823, 130–137. doi:10.1016/j.bbamcr.2011.07.001
Yan, J., Deng, H. X., Siddique, N., Fecto, F., Chen, W., Yang, Y., et al. (2010). Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75, 807–814. doi:10.1212/WNL.0b013e3181f07e0c
Yokoseki, A., Shiga, A., Tan, C.-F., Tagawa, A., Kaneko, H., Koyama, A., et al. (2008). TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 63, 538–542. doi:10.1002/ana.21392
Keywords: frontotemporal dementia, frontotemporal lobar degeneration, mutation, genetics, neuroimaging
Citation: Benussi A, Padovani A and Borroni B (2015) Phenotypic heterogeneity of monogenic frontotemporal dementia. Front. Aging Neurosci. 7:171. doi: 10.3389/fnagi.2015.00171
Received: 24 June 2015; Accepted: 19 August 2015;
Published: 01 September 2015
Edited by:
Diego Albani, IRCCS Mario Negri Institute for Pharmacological Research, ItalyReviewed by:
Federica Agosta, Vita-Salute San Raffaele University, ItalySabrina Dusi, IRCCS Foundation of the Carlo Besta Neurological Institute, Italy
Sara Batelli, Gene Center of the University of Munich, Germany
Copyright: © 2015 Benussi, Padovani and Borroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Borroni, Clinica Neurologica, Università degli Studi di Brescia, Piazzale Spedali Civili 1, Brescia 25123, Italy,YmJvcnJvbmlAaW53aW5kLml0