 Naoyuki Sato
Naoyuki Sato Ryuichi Morishita
Ryuichi Morishita- 1Department of Clinical Gene Therapy, Graduate School of Medicine, Osaka University, Suita, Japan
- 2Department of Geriatric Medicine, Graduate School of Medicine, Osaka University, Suita, Japan
Diabetes is a risk factor for Alzheimer disease (AD). Apolipoprotein E (ApoE) and several genes related to AD have recently been identified by genome-wide association studies (GWAS) as being closely linked to lipid metabolism. Lipid metabolism and glucose-energy metabolism are closely related. Here, we review the emerging evidence regarding the roles of lipid and glucose metabolism in the modulation of β-amyloid, tau, and neurodegeneration during the pathogenesis of AD. Disruption of homeostasis of lipid and glucose metabolism affects production and clearance of β-amyloid and tau phosphorylation, and induces neurodegeneration. A more integrated understanding of the interactions among lipid, glucose, and protein metabolism is required to elucidate the pathogenesis of AD and to develop next-generation therapeutic options.
Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disorder that is pathologically characterized by cerebral atrophy (particularly within the hippocampus and temporal and parietal lobes), senile plaques, neurofibrillary tangles (NFT), and neuronal cell death. Familial AD is caused by mutations in the amyloid precursor protein (Goate et al., 1991) and presenilin (Sherrington et al., 1995). These mutations cause overproduction of β-amyloid (Aβ), particularly its longer form, Aβ42, which aggregates in vitro (Jarrett et al., 1993) and forms the initial deposits in the brain (Iwatsubo et al., 1994) to form senile plaques. Apolipoprotein E (ApoE) is an essential regulator of brain cholesterol metabolism and is the strongest genetic risk factor for sporadic AD (Ashford, 2004). In addition to the ApoE gene, recent genome-wide association studies (GWAS) have identified novel risk genes for AD (Hollingworth et al., 2011; Olgiati et al., 2011), and some of these genes are closely associated with lipid metabolism. Moreover, numerous epidemiological studies have demonstrated that patients with diabetes in which glucose-energy metabolism is affected have a significantly higher risk of developing AD (Ott et al., 1999; Kopf and Frölich, 2009; Maher and Schubert, 2009; Matsuzaki et al., 2010). However, the roles of lipid metabolism and glucose-energy metabolism in the pathogenesis of AD are not fully understood (Figure 1). Here, we review the roles of lipid and glucose metabolism in modulating Aβ, tau, and neurodegeneration during the pathogenesis of AD (Table 1) and focus on novel therapy development.
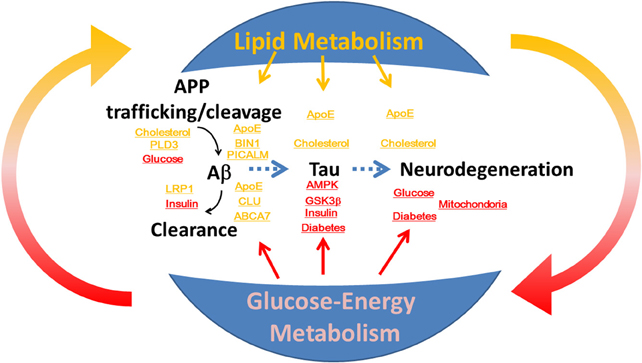
Figure 1. The roles of lipid and glucose metabolism in the modulation of β-amyloid and tau during neurodegeneration and the pathogenesis of Alzheimer disease. Disruption of homeostasis of lipid and glucose metabolism affects production and clearance of β-amyloid and tau phosphorylation, and induces neurodegeneration.
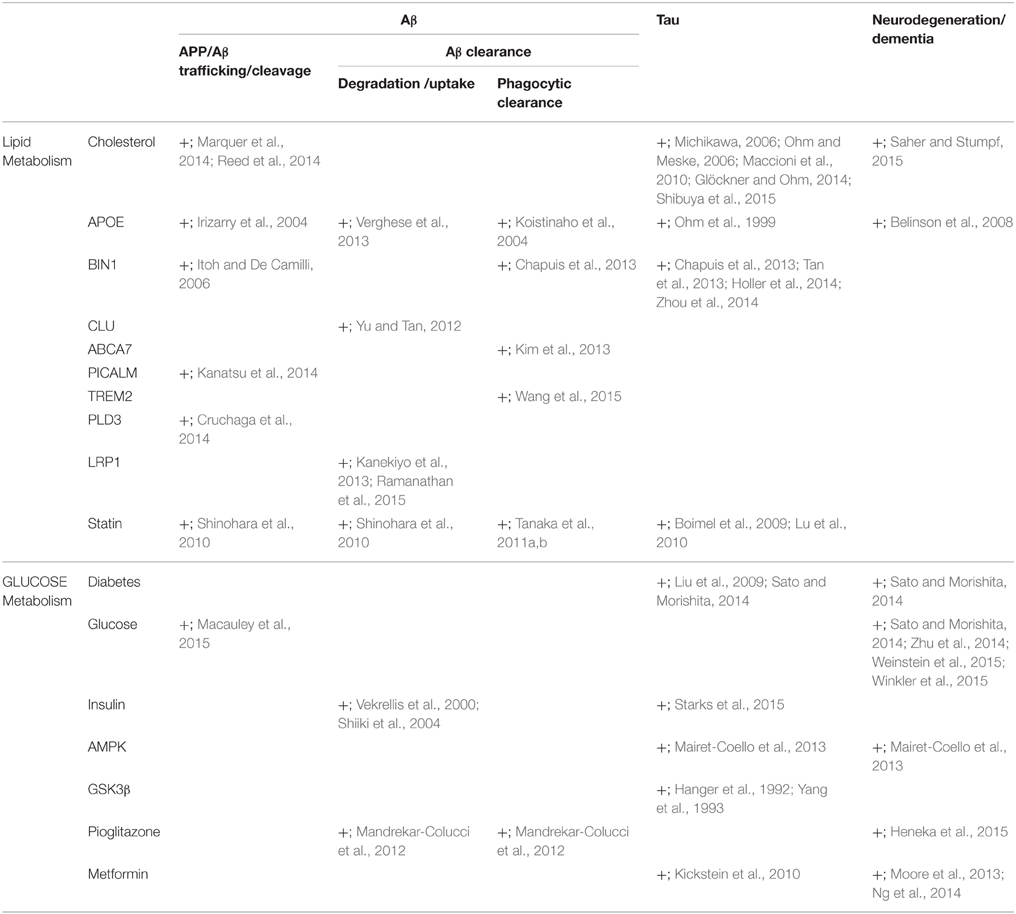
Table 1. Lipid and glucose metabolism are associated with the modulation of β-amyloid and tau and neurodegeneration.
The Role of Lipid Metabolism in β-Amyloid Modulation
ApoE is an essential regulator of cholesterol metabolism and is the strongest genetic risk factor for AD (Ashford, 2004). The ApoEε4 allele increases the accumulation of senile plaques in patients with AD and in cognitively normal people (Reiman et al., 2009; Morris et al., 2010). Physiologically, ApoE is important for brain function through lipid transport of high-density lipoprotein-like particles (Raber et al., 2000; Ji et al., 2003; Bu, 2009; Filippini et al., 2009; Trachtenberg et al., 2011; Verghese et al., 2011; Wisdom et al., 2011). ApoE isoforms are believed to differentially regulate Aβ aggregation and uptake, degradation, and phagocytic clearance in the brain, and each isoform has distinct functions in regulating brain lipid transport, glucose metabolism, and mitochondrial function (Koistinaho et al., 2004; Verghese et al., 2013).
Moreover, independently of ApoEε4, high levels of low-density lipoprotein cholesterol and low levels of high-density lipoprotein cholesterol are associated with higher amyloid-PET indices (Reed et al., 2014). This observation may be partially explained by an in vitro experiment in which transient membrane cholesterol loading increased Aβ42 secretion (Marquer et al., 2014). Interestingly, lipids also trigger the aggregation of α-synuclein, a major component of Lewy bodies, by stimulating nucleation (Galvagnion et al., 2015). This evidence increases the possibility that lipids might promote protein aggregation.
In addition to the ApoE gene, recent GWAS studies have identified novel risk genes for AD (Hollingworth et al., 2011; Olgiati et al., 2011). These genes include bridging integrator 1 (BIN1), clusterin (CLU, also called apolipoprotein J), ATP-binding cassette transporter A7 (ABCA7), triggering receptor expressed on myeloid cells 2 (TREM2), and phosphatidylinositol-binding clathrin assembly protein (PICALM). CLU is a primary brain cholesterol transport lipoprotein and may behave similarly to ApoE (Calero et al., 1999; Yu and Tan, 2012). ABCA7 is involved in lipid homeostasis (Tanaka et al., 2011b). Deletion of ABCA7 increases Aβ accumulation in amyloid precursor protein transgenic (APP Tg) mice through reduced phagocytic clearance of Aβ (Kim et al., 2013). TREM2 regulates the microglial response through lipid sensing around the senile plaque in an AD mouse model (Wang et al., 2015). BIN1 is involved in endocytosis and membrane trafficking (Itoh and De Camilli, 2006) through phosphatidylinositol binding (Lee et al., 2002; Kojima et al., 2004) and also modulates APP trafficking in neurons (Chapuis et al., 2013). BIN1 expression is also observed in microglia, suggesting a role for BIN1 in Aβ phagocytosis (Chapuis et al., 2013). PICALM also plays important roles in clathrin-mediated endocytosis (Dreyling et al., 1996) through phosphatidylinositol binding (Ford et al., 2001), suggesting involvement in APP trafficking (Xiao et al., 2012). Indeed, γ-secretase is endocytosed in a PICALM-dependent manner (Kanatsu et al., 2014). Interestingly, altering the rate of clathrin-mediated endocytosis of γ-secretase increases Aβ42 production (Kanatsu et al., 2014). ApoE also could modulate γ-secretase cleavage of APP, though all three isoforms of ε2, ε3, and ε4, have similar effects (Irizarry et al., 2004). Recent whole-exome sequencing and functional data indicate that carriers of PLD3 (phospholipase D3) coding variants have a two-fold increased risk for late-onset AD and that PLD3 influences APP processing (Cruchaga et al., 2014).
Although clinical studies have indicated that statins have no beneficial effect on cognitive function (McGuinness et al., 2014), statins administered in midlife might prevent AD in late life by modifying the genetic and non-genetic risk factors for AD (Sato et al., 2012; Shinohara et al., 2014). In experimental settings in vivo and in vitro, statins reduced the Aβ level in the brain (Fassbender et al., 2001; Burns et al., 2006; Ostrowski et al., 2007; Kurinami et al., 2008; Papadopoulos et al., 2014). Aβ reduction by a statin is associated with a reduction in the carboxyl terminal fragment of amyloid precursor protein (APP-CTF; Shinohara et al., 2010). Statins reduce the brain Aβ levels by increasing APP-CTF trafficking through isoprenylation inhibition. Moreover, a statin up-regulated Aβ clearance by up-regulating low-density lipoprotein receptor-related protein 1 (LRP1) expression in the vasculature (Shinohara et al., 2010). LRP1 play roles in the efflux of Aβ from the brain (Ramanathan et al., 2015) and neuronal Aβ uptake and degradation (Kanekiyo et al., 2013). Finally, statins enhance ABCA7-dependent phagocytosis (Tanaka et al., 2011a). Thus, lipid metabolism is involved in modulating Aβ levels.
The Role of Lipid Metabolism in Tau Modulation
Normal tau promotes the assembly and stabilization of microtubules. However, hyperphosphorylated tau sequesters normal tau and disrupts microtubules, forming NFT (Iqbal et al., 1994, 2009). In mice expressing mutant human tau, cellular cholesterol levels were higher in neurons affected by tau pathology (Glöckner and Ohm, 2014). Indeed, impaired cholesterol metabolism is involved in tau hyperphosphorylation (Michikawa, 2006; Ohm and Meske, 2006; Maccioni et al., 2010). Emerging data suggest that BIN1 modulates tau pathology in addition to Aβ (Chapuis et al., 2013; Tan et al., 2013; Holler et al., 2014; Zhou et al., 2014). BIN co-localizes and interacts with tau (Chapuis et al., 2013; Zhou et al., 2014). Therefore, BIN1 levels may correlate with NFTs in AD (Glennon et al., 2013; Holler et al., 2014). In addition to their effects on Aβ metabolism, statins suppress tau hyperphosphorylation induced by excess cholesterol in the brain (Lu et al., 2010) and also reduce NFTs in a tau pathology model (Boimel et al., 2009). Moreover, inhibition of cholesterol metabolism by blocking acyl-coenzyme A:cholesterol acyltransferase 1 activity reduces the amount of mutant human tau in neurons of triple transgenic mice (Shibuya et al., 2015). In comparison with ApoEε3, the presence of the ApoEε4 is reported to be associated with NFT formation (Ohm et al., 1999). These studies demonstrate that the regulation and dysregulation of cholesterol metabolism affect tau pathology in the brain.
The Role of Lipid Metabolism in Neurodegeneration Modulation
As the largest pool of cholesterol resides in neuronal myelin membranes, disorders that impair sterol synthesis or intracellular trafficking of lipids in neurons cause hypomyelination and neurodegeneration (Saher and Stumpf, 2015). Glial lipid droplets induced by mitochondrial defects also promote neurodegeneration (Liu et al., 2015) suggesting a role for lipid metabolism in glial cells in neurodegeneration. Moreover, the human cortex demonstrates membrane protein oxidation (Granold et al., 2015) and altered phospholipid components during aging (Norris et al., 2015). Other than AD, impaired lipid metabolism has been reported in several neurodegenerative diseases. The huntingtin gene, which is causative for Huntington disease, also seems to play a regulatory role in lipid metabolism (Leoni and Caccia, 2015). Cholesterol metabolism impairment is proportion to the CAG repeat length and to the load of mutant huntingtin leading to neurodegeneration (Leoni and Caccia, 2015). Mucopolysaccharidosis III type C, a progressive neurological pediatric disease, is caused by mutations in the heparan-α-glucosaminide N-acetyltransferase gene and leads to a deficiency in acetyl-CoA: α-glucosaminide N-acetyltransferase (Martins et al., 2015). These results further support the role of lipid metabolism in neurodegeneration. Although reversible, statins might transiently impair cognitive function, especially during the initial administration to patients older than 75 years (Orsi et al., 2001; King et al., 2003; Wagstaff et al., 2003). This effect is probably due in part to cholesterol's modulation of NMDA receptor function (Korinek et al., 2015). Finally, activation of Aβ cascade in ApoEε4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits (Belinson et al., 2008). Taken together, these reports indicate that cholesterol metabolism is tightly linked to neurodegeneration.
The Role of Glucose-Energy Metabolism in β-Amyloid Modulation
Diabetes in midlife is associated with mild cognitive impairment (MCI; Roberts et al., 2014), and impaired glycemia increases the disease progression to dementia in patients with MCI (Morris et al., 2014). However, the mechanisms by which diabetes modifies cognitive function remain unclear (Sato and Morishita, 2013a,b). Diabetes seems to alter brain structure and function through Aβ/tau-dependent and independent mechanisms (Sato and Morishita, 2014). Insulin resistance in midlife is associated with neurodegeneration surrounding senile plaques (Matsuzaki et al., 2010), though retrospective studies suggest that the magnitude of senile plaques is comparable between AD with and without diabetes (Kalaria, 2009). Several groups reported that a high-fat diet causes Aβ accumulation in the brains of wild type rabbits (Sparks et al., 1994) and APP Tg mice (Refolo et al., 2000; Ho et al., 2004). In a murine model of AD, inducing acute hyperglycemia increases Aβ production (Macauley et al., 2015). Moreover, altering insulin and insulin signaling may change Aβ levels in the brain through proteolysis by insulin-degrading enzymes (Vekrellis et al., 2000) and/or Aβ clearance from the brain (Shiiki et al., 2004). Alternative mechanisms might include the accumulation of autophagosomes to enhance amyloidogenic APP processing (Son et al., 2012) or up-regulation of BACE1 (Guglielmotto et al., 2012). APP+-ob/ob mice, generated by crossing diabetic ob/ob mice, display increased Aβ deposition in the cerebral vasculature (Takeda et al., 2010). Whether glycogen synthase kinase-3(GSK3) controls APP processing and Aβ levels in brain is intriguing (Phiel et al., 2003; Sereno et al., 2009; Sofola et al., 2010), but controversial (Jaworski et al., 2011). Anti-diabetic drug, pioglitazone, stimulated Aβ degradation by both microglia and astrocytes in ApoE-dependent manner (Mandrekar-Colucci et al., 2012). Thus, glucose metabolism is also involved in modulating Aβ levels.
The Role of Glucose-Energy Metabolism in Tau Modulation
Several neuropathological studies suggest that the magnitude of NFTs in the brain at autopsy is not different between AD patients with and without diabetes (Kalaria, 2009). However, one report suggests that insulin resistance is associated with higher tau levels in the cerebrospinal fluid (Starks et al., 2015). Moreover, animal studies show that tau phosphorylation is increased in diabetes (Clodfelder-Miller et al., 2006; Jolivalt et al., 2008; Ke et al., 2009; Kim et al., 2009; Qu et al., 2011). For example, tau phosphorylation is increased in db/db mice (Kim et al., 2009), streptozotocin-treated wild type mice (Clodfelder-Miller et al., 2006; Jolivalt et al., 2008; Qu et al., 2011), and mutant human tau mice (Ke et al., 2009). Importantly, in humans, tau phosphorylation sites observed in AD are also increased in the diabetic brain (Liu et al., 2009). Conversely, CSF tau predicts changes in brain glucose metabolism, in turn causing longitudinal cognitive changes (Dowling et al., 2015). An energy-sensor, AMP-activated kinase (AMPK) activation is increased in the AD brain and AMPK phosphorylates Tau (Mairet-Coello et al., 2013). GSK3 also induces tau phosphorylation (Hanger et al., 1992; Yang et al., 1993). Anti-diabetic drug, metformin induces protein phosphatase 2A activity and reduces tau phosphorylation in vitro and in animal models (Kickstein et al., 2010). Therefore, glucose-energy metabolism is closely related to modulation of tau.
The Role of Glucose-Energy Metabolism in Neurodegeneration Modulation
Diabetes causes structural deficits in the brain (Sato and Morishita, 2014) indicating that glucose-energy metabolism modulates neurodegeneration. Even in young adults, hyperglycemia is associated with subtle brain injury and impaired attention and memory (Weinstein et al., 2015). Indeed, diabetes reduces the volume of the hippocampus (Moran et al., 2013; Roberts et al., 2014), gray (García-Casares et al., 2014) and white matter (Moran et al., 2013). Gray matter loss occurs in the temporal, anterior cingulate, and frontal lobes (Moran et al., 2013; García-Casares et al., 2014; Roberts et al., 2014; Erus et al., 2015), while white matter loss appears in the frontal and temporal regions (Moran et al., 2013). In patients with AD, gray matter loss occurs in the temporal lobe, hippocampus, entorhinal and parietal lobes (Braak and Braak, 1991; Thompson et al., 2003; Andrade-Moraes et al., 2013), and white matter loss occurs in the temporal region (Mann, 1991). These studies indicate that diabetes causes neurodegeneration in the frontal and temporal lobes and other regions (Sato and Morishita, 2014). The molecular mechanism by which diabetes modulates neurodegeneration has not been fully elucidated, though several possible mechanisms have been proposed. Disturbance of glucose metabolism by GLUT1 deficiency causes neurodegeneration in APP Tg mice (Winkler et al., 2015). Another link between glucose hypometabolism and the progression of AD is the O-GlcNAcylation of proteins (Zhu et al., 2014). Decreased O-GlcNAcylation occurs in AD, which suggests that glucose hypometabolism may impair the protective roles of O-GlcNAc in neurons and lead to neurodegeneration (Zhu et al., 2014). AMPK is an energy-sensor, and AMPK over-activation is sufficient to cause dendritic spine loss (Mairet-Coello et al., 2013). Disturbed mTOR signaling affected by glucose-energy metabolism also causes neurodegeneration through mitochondrial dysfunction and autophagy (Perluigi et al., 2015). An observational study suggests that pioglitazone treatment is associated with a reduced dementia risk in diabetes patients (Heneka et al., 2015). Metformin is also reported to reduce the risk of cognitive decline in diabetes patients (Ng et al., 2014), though other group showed an opposite effect (Moore et al., 2013). These studies indicate that the molecular mechanism by which the dysregulation of glucose-energy metabolism causes neurodegeneration should be targeted to develop novel dementia therapies.
Summary
Recent large, long-term, randomized controlled trials suggest that a multidisciplinary intervention, including exercise and diet, could improve or maintain cognitive function in at-risk elderly people (Ngandu et al., 2015). Exercise and diet alter glucose and lipid metabolism in subjects. As reviewed here, disruption of homeostasis of lipid and glucose metabolism affects production and clearance of β-amyloid and tau phosphorylation, and induces neurodegeneration. Therefore, a more integrated understanding of the interactions among lipid, glucose, and protein metabolism will be required to elucidate the pathogenesis of AD and to develop next-generation therapeutic options.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by grants-in-aid from Japan Promotion of Science; the Japanese Ministry of Education, Culture, Sports, Science and Technology and the Japan Science and Technology Agency (MEXT26293167 and MEXT15K15272 to NS); a Takeda Science Foundation Research Encouragement Grant; a SENSHIN Medical Research Foundation Research Grant; a Novartis Foundation for Gerontological Research Award; and an annual research award grant from the Japanese Society of Anti-aging Medicine (to NS).
References
Andrade-Moraes, C. H., Oliveira-Pinto, A. V., Castro-Fonseca, E., da Silva, C. G., Guimaraes, D. M., Szczupak, D., et al. (2013). Cell number changes in Alzheimer's disease relate to dementia, not to plaques and tangles. Brain 136, 3738–3752. doi: 10.1093/brain/awt273
Ashford, J. W. (2004). APOE genotype effects on Alzheimer's disease onset and epidemiology. J. Mol. Neurosci. 23, 157–165. doi: 10.1385/JMN:23:3:157
Belinson, H., Lev, D., Masliah, E., and Michaelson, D. M. (2008). Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. J. Neurosci. 28, 4690–4701. doi: 10.1523/JNEUROSCI.5633-07.2008
Boimel, M., Grigoriadis, N., Lourbopoulos, A., Touloumi, O., Rosenmann, D., Abramsky, O., et al. (2009). Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J. Neuropathol. Exp. Neurol. 68, 314–325. doi: 10.1097/NEN.0b013e31819ac3cb
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Burns, M. P., Igbavboa, U., Wang, L., Wood, W. G., and Duff, K. (2006). Cholesterol distribution, not total levels, correlate with altered amyloid precursor protein processing in statin-treated mice. Neuromolecular Med. 8, 319–328. doi: 10.1385/NMM:8:3:319
Calero, M., Tokuda, T., Rostagno, A., Kumar, A., Zlokovic, B., Frangione, B., et al. (1999). Functional and structural properties of lipid-associated apolipoprotein J (clusterin). Biochem. J. 344(Pt 2), 375–383. doi: 10.1042/bj3440375
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
Clodfelder-Miller, B. J., Zmijewska, A. A., Johnson, G. V., and Jope, R. S. (2006). Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 55, 3320–3325. doi: 10.2337/db06-0485
Cruchaga, C., Karch, C. M., Jin, S. C., Benitez, B. A., Cai, Y., Guerreiro, R., et al. (2014). Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature 505, 550–554. doi: 10.1038/nature12825
Dowling, N. M., Johnson, S. C., Gleason, C. E., and Jagust, W. J. (2015). The mediational effects of FDG hypometabolism on the association between cerebrospinal fluid biomarkers and neurocognitive function. Neuroimage 105, 357–368. doi: 10.1016/j.neuroimage.2014.10.050
Dreyling, M. H., Martinez-Climent, J. A., Zheng, M., Mao, J., Rowley, J. D., and Bohlander, S. K. (1996). The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc. Natl. Acad. Sci. U.S.A. 93, 4804–4809. doi: 10.1073/pnas.93.10.4804
Erus, G., Battapady, H., Zhang, T., Lovato, J., Miller, M. E., Williamson, J. D., et al. (2015). Spatial patterns of structural brain changes in type 2 diabetic patients and their longitudinal progression with intensive control of blood glucose. Diabetes Care 38, 97–104. doi: 10.2337/dc14-1196
Fassbender, K., Simons, M., Bergmann, C., Stroick, M., Lutjohann, D., Keller, P., et al. (2001). Simvastatin strongly reduces levels of Alzheimer's disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 5856–5861. doi: 10.1073/pnas.081620098
Filippini, N., MacIntosh, B. J., Hough, M. G., Goodwin, G. M., Frisoni, G. B., Smith, S. M., et al. (2009). Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl. Acad. Sci. U.S.A. 106, 7209–7214. doi: 10.1073/pnas.0811879106
Ford, M. G., Pearse, B. M., Higgins, M. K., Vallis, Y., Owen, D. J., Gibson, A., et al. (2001). Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291, 1051–1055. doi: 10.1126/science.291.5506.1051
Galvagnion, C., Buell, A. K., Meisl, G., Michaels, T. C., Vendruscolo, M., Knowles, T. P., et al. (2015). Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234. doi: 10.1038/nchembio.1750
García-Casares, N., Berthier, M. L., Jorge, R. E., Gonzalez-Alegre, P., Gutiérrez Cardo, A., Rioja Villodres, J., et al. (2014). Structural and functional brain changes in middle-aged type 2 diabetic patients: a cross-sectional study. J. Alzheimers Dis. 40, 375–386. doi: 10.3233/JAD-131736
Glennon, E. B., Whitehouse, I. J., Miners, J. S., Kehoe, P. G., Love, S., Kellett, K. A., et al. (2013). BIN1 is decreased in sporadic but not familial Alzheimer's disease or in aging. PLoS ONE 8:e78806. doi: 10.1371/journal.pone.0078806
Glöckner, F., and Ohm, T. G. (2014). Tau pathology induces intraneuronal cholesterol accumulation. J. Neuropathol. Exp. Neurol. 73, 846–854. doi: 10.1097/NEN.0000000000000103
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706. doi: 10.1038/349704a0
Granold, M., Moosmann, B., Staib-Lasarzik, I., Arendt, T., Del Rey, A., Engelhard, K., et al. (2015). High membrane protein oxidation in the human cerebral cortex. Redox Biol. 4, 200–207. doi: 10.1016/j.redox.2014.12.013
Guglielmotto, M., Aragno, M., Tamagno, E., Vercellinatto, I., Visentin, S., Medana, C., et al. (2012). AGEs/RAGE complex upregulates BACE1 via NF-κB pathway activation. Neurobiol. Aging 33, 196.e13–196.e27. doi: 10.1016/j.neurobiolaging.2010.05.026
Hanger, D. P., Hughes, K., Woodgett, J. R., Brion, J. P., and Anderton, B. H. (1992). Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62. doi: 10.1016/0304-3940(92)90774-2
Heneka, M. T., Fink, A., and Doblhammer, G. (2015). Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol. 78, 284–294. doi: 10.1002/ana.24439
Ho, L., Qin, W., Pompl, P. N., Xiang, Z., Wang, J., Zhao, Z., et al. (2004). Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. 18, 902–904. doi: 10.1096/fj.03-0978fje
Holler, C. J., Davis, P. R., Beckett, T. L., Platt, T. L., Webb, R. L., Head, E., et al. (2014). Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimers Dis. 42, 1221–1227. doi: 10.3233/JAD-132450
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Iqbal, K., Alonso, A. C., Gong, C. X., Khatoon, S., Singh, T. J., and Grundke-Iqbal, I. (1994). Mechanism of neurofibrillary degeneration in Alzheimer's disease. Mol. Neurobiol. 9, 119–123. doi: 10.1007/BF02816111
Iqbal, K., Liu, F., Gong, C. X., Alonso Adel, C., and Grundke-Iqbal, I. (2009). Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69. doi: 10.1007/s00401-009-0486-3
Irizarry, M. C., Deng, A., Lleo, A., Berezovska, O., Von Arnim, C. A., Martin-Rehrmann, M., et al. (2004). Apolipoprotein E modulates γ-secretase cleavage of the amyloid precursor protein. J. Neurochem. 90, 1132–1143. doi: 10.1111/j.1471-4159.2004.02581.x
Itoh, T., and De Camilli, P. (2006). BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim. Biophys. Acta 1761, 897–912. doi: 10.1016/j.bbalip.2006.06.015
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., and Ihara, Y. (1994). Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 13, 45–53. doi: 10.1016/0896-6273(94)90458-8
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jaworski, T., Dewachter, I., Lechat, B., Gees, M., Kremer, A., Demedts, D., et al. (2011). GSK-3α/β kinases and amyloid production in vivo. Nature 480, E4–E5. discussion: E6. doi: 10.1038/nature10615
Ji, Y., Gong, Y., Gan, W., Beach, T., Holtzman, D. M., and Wisniewski, T. (2003). Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer's disease patients. Neuroscience 122, 305–315. doi: 10.1016/j.neuroscience.2003.08.007
Jolivalt, C. G., Lee, C. A., Beiswenger, K. K., Smith, J. L., Orlov, M., Torrance, M. A., et al. (2008). Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer's disease and correction by insulin. J. Neurosci. Res. 86, 3265–3274. doi: 10.1002/jnr.21787
Kalaria, R. N. (2009). Neurodegenerative disease: diabetes, microvascular pathology and Alzheimer disease. Nat. Rev. Neurol. 5, 305–306. doi: 10.1038/nrneurol.2009.72
Kanatsu, K., Morohashi, Y., Suzuki, M., Kuroda, H., Watanabe, T., Tomita, T., et al. (2014). Decreased CALM expression reduces Aβ42 to total Aβ ratio through clathrin-mediated endocytosis of γ-secretase. Nat. Commun. 5:3386. doi: 10.1038/ncomms4386
Kanekiyo, T., Cirrito, J. R., Liu, C. C., Shinohara, M., Li, J., Schuler, D. R., et al. (2013). Neuronal clearance of amyloid-β by endocytic receptor LRP1. J. Neurosci. 33, 19276–19283. doi: 10.1523/JNEUROSCI.3487-13.2013
Ke, Y. D., Delerue, F., Gladbach, A., Götz, J., and Ittner, L. M. (2009). Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer's disease. PLoS ONE 4:e7917. doi: 10.1371/journal.pone.0007917
Kickstein, E., Krauss, S., Thornhill, P., Rutschow, D., Zeller, R., Sharkey, J., et al. (2010). Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 21830–21835. doi: 10.1073/pnas.0912793107
Kim, B., Backus, C., Oh, S., Hayes, J. M., and Feldman, E. L. (2009). Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 150, 5294–5301. doi: 10.1210/en.2009-0695
Kim, W. S., Li, H., Ruberu, K., Chan, S., Elliott, D. A., Low, J. K., et al. (2013). Deletion of Abca7 increases cerebral amyloid-β accumulation in the J20 mouse model of Alzheimer's disease. J. Neurosci. 33, 4387–4394. doi: 10.1523/JNEUROSCI.4165-12.2013
King, D. S., Wilburn, A. J., Wofford, M. R., Harrell, T. K., Lindley, B. J., and Jones, D. W. (2003). Cognitive impairment associated with atorvastatin and simvastatin. Pharmacotherapy 23, 1663–1667. doi: 10.1592/phco.23.15.1663.31953
Koistinaho, M., Lin, S., Wu, X., Koger, D., Hanson, J., Higgs, R., et al. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726. doi: 10.1038/nm1058
Kojima, C., Hashimoto, A., Yabuta, I., Hirose, M., Hashimoto, S., Kanaho, Y., et al. (2004). Regulation of Bin1 SH3 domain binding by phosphoinositides. EMBO J. 23, 4413–4422. doi: 10.1038/sj.emboj.7600442
Kopf, D., and Frölich, L. (2009). Risk of incident Alzheimer's disease in diabetic patients: a systematic review of prospective trials. J. Alzheimers Dis. 16, 677–685. doi: 10.3233/JAD-2009-1011
Korinek, M., Vyklicky, V., Borovska, J., Lichnerova, K., Kaniakova, M., Krausova, B., et al. (2015). Cholesterol modulates open probability and desensitization of NMDA receptors. J. Physiol. 593, 2279–2293. doi: 10.1113/jphysiol.2014.288209
Kurinami, H., Sato, N., Shinohara, M., Takeuchi, D., Takeda, S., Shimamura, M., et al. (2008). Prevention of amyloid β-induced memory impairment by fluvastatin, associated with the decrease in amyloid β accumulation and oxidative stress in amyloid β injection mouse model. Int. J. Mol. Med. 21, 531–537. doi: 10.3892/ijmm.21.5.531
Lee, E., Marcucci, M., Daniell, L., Weisz, O. A., Ochoa, G. C., Farsad, K., et al. (2002). Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196. doi: 10.1126/science.1071362
Leoni, V., and Caccia, C. (2015). The impairment of cholesterol metabolism in Huntington disease. Biochim. Biophys. Acta 1851, 1095–1105. doi: 10.1016/j.bbalip.2014.12.018
Liu, L., Zhang, K., Sandoval, H., Yamamoto, S., Jaiswal, M., Sanz, E., et al. (2015). Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. doi: 10.1016/j.cell.2014.12.019
Liu, Y., Liu, F., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2009). Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer's disease. J. Neurochem. 111, 242–249. doi: 10.1111/j.1471-4159.2009.06320.x
Lu, F., Li, X., Suo, A. Q., and Zhang, J. W. (2010). Inhibition of tau hyperphosphorylation and β amyloid production in rat brain by oral administration of atorvastatin. Chin. Med. J. 123, 1864–1870.
Macauley, S. L., Stanley, M., Caesar, E. E., Yamada, S. A., Raichle, M. E., Perez, R., et al. (2015). Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Invest. 125, 2463–2467. doi: 10.1172/JCI79742
Maccioni, R. B., Farías, G., Morales, I., and Navarrete, L. (2010). The revitalized tau hypothesis on Alzheimer's disease. Arch. Med. Res. 41, 226–231. doi: 10.1016/j.arcmed.2010.03.007
Maher, P. A., and Schubert, D. R. (2009). Metabolic links between diabetes and Alzheimer's disease. Expert Rev. Neurother. 9, 617–630. doi: 10.1586/ern.09.18
Mairet-Coello, G., Courchet, J., Pieraut, S., Courchet, V., Maximov, A., and Polleux, F. (2013). The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron 78, 94–108. doi: 10.1016/j.neuron.2013.02.003
Mandrekar-Colucci, S., Karlo, J. C., and Landreth, G. E. (2012). Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J. Neurosci. 32, 10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012
Mann, D. M. (1991). The topographic distribution of brain atrophy in Alzheimer's disease. Acta Neuropathol. 83, 81–86. doi: 10.1007/BF00294434
Marquer, C., Laine, J., Dauphinot, L., Hanbouch, L., Lemercier-Neuillet, C., Pierrot, N., et al. (2014). Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer's disease early phenotypes. Mol. Neurodegener. 9:60. doi: 10.1186/1750-1326-9-60
Martins, C., Hulková, H., Dridi, L., Dormoy-Raclet, V., Grigoryeva, L., Choi, Y., et al. (2015). Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 138, 336–355. doi: 10.1093/brain/awu355
Matsuzaki, T., Sasaki, K., Tanizaki, Y., Hata, J., Fujimi, K., Matsui, Y., et al. (2010). Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 75, 764–770. doi: 10.1212/WNL.0b013e3181eee25f
McGuinness, B., Craig, D., Bullock, R., Malouf, R., and Passmore, P. (2014). Statins for the treatment of dementia. Cochrane Database Syst. Rev. 7:CD007514. doi: 10.1002/14651858.cd007514.pub3
Michikawa, M. (2006). Role of cholesterol in amyloid cascade: cholesterol-dependent modulation of tau phosphorylation and mitochondrial function. Acta Neurol. Scand. Suppl. 185, 21–26. doi: 10.1111/j.1600-0404.2006.00681.x
Moore, E. M., Mander, A. G., Ames, D., Kotowicz, M. A., Carne, R. P., Brodaty, H., et al. (2013). Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 36, 2981–2987. doi: 10.2337/dc13-0229
Moran, C., Phan, T. G., Chen, J., Blizzard, L., Beare, R., Venn, A., et al. (2013). Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care 36, 4036–4042. doi: 10.2337/dc13-0143
Morris, J. C., Roe, C. M., Xiong, C., Fagan, A. M., Goate, A. M., Holtzman, D. M., et al. (2010). APOE predicts amyloid-β but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 67, 122–131. doi: 10.1002/ana.21843
Morris, J. K., Vidoni, E. D., Honea, R. A., and Burns, J. M. (2014). Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol. Aging 35, 585–589. doi: 10.1016/j.neurobiolaging.2013.09.033
Ng, T. P., Feng, L., Yap, K. B., Lee, T. S., Tan, C. H., and Winblad, B. (2014). Long-term metformin usage and cognitive function among older adults with diabetes. J. Alzheimers Dis. 41, 61–68. doi: 10.3233/JAD-131901
Ngandu, T., Lehtisalo, J., Solomon, A., Levälahti, E., Ahtiluoto, S., Antikainen, R., et al. (2015). A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet 385, 2255–2263. doi: 10.1016/S0140-6736(15)60461-5
Norris, S. E., Friedrich, M. G., Mitchell, T. W., Truscott, R. J., and Else, P. L. (2015). Human prefrontal cortex phospholipids containing docosahexaenoic acid increase during normal adult aging, whereas those containing arachidonic acid decrease. Neurobiol. Aging 36, 1659–1669. doi: 10.1016/j.neurobiolaging.2015.01.002
Ohm, T. G., and Meske, V. (2006). Cholesterol, statins and tau. Acta Neurol. Scand. Suppl. 185, 93–101. doi: 10.1111/j.1600-0404.2006.00692.x
Ohm, T. G., Scharnagl, H., März, W., and Bohl, J. (1999). Apolipoprotein E isoforms and the development of low and high Braak stages of Alzheimer's disease-related lesions. Acta Neuropathol. 98, 273–280. doi: 10.1007/s004010051080
Olgiati, P., Politis, A. M., Papadimitriou, G. N., De Ronchi, D., and Serretti, A. (2011). Genetics of late-onset Alzheimer's disease: update from the alzgene database and analysis of shared pathways. Int. J. Alzheimers Dis. 2011:832379. doi: 10.4061/2011/832379
Orsi, A., Sherman, O., and Woldeselassie, Z. (2001). Simvastatin-associated memory loss. Pharmacotherapy 21, 767–769. doi: 10.1592/phco.21.7.767.34577
Ostrowski, S. M., Wilkinson, B. L., Golde, T. E., and Landreth, G. (2007). Statins reduce amyloid-β production through inhibition of protein isoprenylation. J. Biol. Chem. 282, 26832–26844. doi: 10.1074/jbc.M7026 40200
Ott, A., Stolk, R. P., van Harskamp, F., Pols, H. A., Hofman, A., and Breteler, M. M. (1999). Diabetes mellitus and the risk of dementia: the rotterdam study. Neurology 53, 1937–1942. doi: 10.1212/WNL.53.9.1937
Papadopoulos, P., Tong, X. K., and Hamel, E. (2014). Selective benefits of simvastatin in bitransgenic APPSwe, Ind/TGF-β1 mice. Neurobiol. Aging 35, 203–212. doi: 10.1016/j.neurobiolaging.2013.07.010
Perluigi, M., Di Domenico, F., and Butterfield, D. A. (2015). mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. doi: 10.1016/j.nbd.2015.03.014. [Epub ahead of print].
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3α regulates production of Alzheimer's disease amyloid-β peptides. Nature 423, 435–439. doi: 10.1038/nature01640
Qu, Z., Jiao, Z., Sun, X., Zhao, Y., Ren, J., and Xu, G. (2011). Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res. 1383, 300–306. doi: 10.1016/j.brainres.2011.01.084
Raber, J., Wong, D., Yu, G. Q., Buttini, M., Mahley, R. W., Pitas, R. E., et al. (2000). Apolipoprotein E and cognitive performance. Nature 404, 352–354. doi: 10.1038/35006165
Ramanathan, A., Nelson, A. R., Sagare, A. P., and Zlokovic, B. V. (2015). Impaired vascular-mediated clearance of brain amyloid β in Alzheimer's disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 7:136. doi: 10.3389/fnagi.2015.00136
Reed, B., Villeneuve, S., Mack, W., DeCarli, C., Chui, H. C., and Jagust, W. (2014). Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 71, 195–200. doi: 10.1001/jamaneurol.2013.5390
Refolo, L. M., Malester, B., LaFrancois, J., Bryant-Thomas, T., Wang, R., Tint, G. S., et al. (2000). Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 7, 321–331. doi: 10.1006/nbdi.2000.0304
Reiman, E. M., Chen, K., Liu, X., Bandy, D., Yu, M., Lee, W., et al. (2009). Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825. doi: 10.1073/pnas.0900345106
Roberts, R. O., Knopman, D. S., Przybelski, S. A., Mielke, M. M., Kantarci, K., Preboske, G. M., et al. (2014). Association of type 2 diabetes with brain atrophy and cognitive impairment. Neurology 82, 1132–1141. doi: 10.1212/WNL.0000000000000269
Saher, G., and Stumpf, S. K. (2015). Cholesterol in myelin biogenesis and hypomyelinating disorders. Biochim. Biophys. Acta 1851, 1083–1094. doi: 10.1016/j.bbalip.2015.02.010
Sato, N., and Morishita, R. (2013a). Plasma aβ: a possible missing link between Alzheimer disease and diabetes. Diabetes 62, 1005–1006. doi: 10.2337/db12-1549
Sato, N., and Morishita, R. (2013b). Roles of vascular and metabolic components in cognitive dysfunction of Alzheimer disease: short- and long-term modification by non-genetic risk factors. Front. Aging Neurosci. 5:64. doi: 10.3389/fnagi.2013.00064
Sato, N., and Morishita, R. (2014). Brain alterations and clinical symptoms of dementia in diabetes: aβ/τ-dependent and independent mechanisms. Front. Endocrinol. (Lausanne) 5:143. doi: 10.3389/fendo.2014.00143
Sato, N., Shinohara, M., Rakugi, H., and Morishita, R. (2012). Dual effects of statins on Aβ metabolism: upregulation of the degradation of APP-CTF and Aβ clearance. Neurodegener. Dis. 10, 305–308. doi: 10.1159/000334534
Serenó, L., Coma, M., Rodríguez, M., Sánchez-Ferrer, P., Sánchez, M. B., Gich, I., et al. (2009). A novel GSK-3β inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367. doi: 10.1016/j.nbd.2009.05.025
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shibuya, Y., Niu, Z., Bryleva, E. Y., Harris, B. T., Murphy, S. R., Kheirollah, A., et al. (2015). Acyl-coenzyme A:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer's disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiol. Aging 36, 2248–2259. doi: 10.1016/j.neurobiolaging.2015.04.002
Shiiki, T., Ohtsuki, S., Kurihara, A., Naganuma, H., Nishimura, K., Tachikawa, M., et al. (2004). Brain insulin impairs amyloid-β(1-40) clearance from the brain. J. Neurosci. 24, 9632–9637. doi: 10.1523/JNEUROSCI.2236-04.2004
Shinohara, M., Sato, N., Kurinami, H., Takeuchi, D., Takeda, S., Shimamura, M., et al. (2010). Reduction of brain β-amyloid (Aβ) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Aβ clearance. J. Biol. Chem. 285, 22091–22102. doi: 10.1074/jbc.M110.102277
Shinohara, M., Sato, N., Shimamura, M., Kurinami, H., Hamasaki, T., Chatterjee, A., et al. (2014). Possible modification of Alzheimer's disease by statins in midlife: interactions with genetic and non-genetic risk factors. Front. Aging Neurosci. 6:71. doi: 10.3389/fnagi.2014.00071
Sofola, O., Kerr, F., Rogers, I., Killick, R., Augustin, H., Gandy, C., et al. (2010). Inhibition of GSK-3 ameliorates Aβ pathology in an adult-onset Drosophila model of Alzheimer's disease. PLoS Genet. 6:e1001087. doi: 10.1371/journal.pgen.1001087
Son, S. M., Song, H., Byun, J., Park, K. S., Jang, H. C., Park, Y. J., et al. (2012). Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy 8, 1842–1844. doi: 10.4161/auto.21861
Sparks, D. L., Scheff, S. W., Hunsaker, J. C. III, Liu, H., Landers, T., and Gross, D. R. (1994). Induction of Alzheimer-like β-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126, 88–94. doi: 10.1006/exnr.1994.1044
Starks, E. J., O'Grady, J. P., Hoscheidt, S. M., Racine, A. M., Carlsson, C. M., Zetterberg, H., et al. (2015). Insulin resistance is associated with higher cerebrospinal fluid tau levels in asymptomatic APOEϵ4 carriers. J. Alzheimers Dis. 46, 525–533. doi: 10.3233/JAD-150072
Takeda, S., Sato, N., Uchio-Yamada, K., Sawada, K., Kunieda, T., Takeuchi, D., et al. (2010). Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. U.S.A. 107, 7036–7041. doi: 10.1073/pnas.1000645107
Tan, M. S., Yu, J. T., and Tan, L. (2013). Bridging integrator 1 (BIN1): form, function, and Alzheimer's disease. Trends Mol. Med. 19, 594–603. doi: 10.1016/j.molmed.2013.06.004
Tanaka, N., Abe-Dohmae, S., Iwamoto, N., Fitzgerald, M. L., and Yokoyama, S. (2011a). HMG-CoA reductase inhibitors enhance phagocytosis by upregulating ATP-binding cassette transporter A7. Atherosclerosis 217, 407–414. doi: 10.1016/j.atherosclerosis.2011.06.031
Tanaka, N., Abe-Dohmae, S., Iwamoto, N., and Yokoyama, S. (2011b). Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J. Atheroscler. Thromb. 18, 274–281. doi: 10.5551/jat.6726
Thompson, P. M., Hayashi, K. M., de Zubicaray, G., Janke, A. L., Rose, S. E., Semple, J., et al. (2003). Dynamics of gray matter loss in Alzheimer's disease. J. Neurosci. 23, 994–1005.
Trachtenberg, A. J., Filippini, N., Cheeseman, J., Duff, E. P., Neville, M. J., Ebmeier, K. P., et al. (2011). The effects of APOE on brain activity do not simply reflect the risk of Alzheimer's disease. Neurobiol. Aging 33, 618.e1–618.e13. doi: 10.1016/j.neurobiolaging.2010.11.01
Vekrellis, K., Ye, Z., Qiu, W. Q., Walsh, D., Hartley, D., Chesneau, V., et al. (2000). Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665.
Verghese, P. B., Castellano, J. M., Garai, K., Wang, Y., Jiang, H., Shah, A., et al. (2013). ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 110:E1807–E1816. doi: 10.1073/pnas.1220484110
Verghese, P. B., Castellano, J. M., and Holtzman, D. M. (2011). Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 10, 241–252. doi: 10.1016/S1474-4422(10)70325-2
Wagstaff, L. R., Mitton, M. W., Arvik, B. M., and Doraiswamy, P. M. (2003). Statin-associated memory loss: analysis of 60 case reports and review of the literature. Pharmacotherapy 23, 871–880. doi: 10.1592/phco.23.7.871.32720
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Weinstein, G., Maillard, P., Himali, J. J., Beiser, A. S., Au, R., Wolf, P. A., et al. (2015). Glucose indices are associated with cognitive and structural brain measures in young adults. Neurology 84, 2329–2337. doi: 10.1212/WNL.0000000000001655
Winkler, E. A., Nishida, Y., Sagare, A. P., Rege, S. V., Bell, R. D., Perlmutter, D., et al. (2015). GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 18, 521–530. doi: 10.1038/nn.3966
Wisdom, N. M., Callahan, J. L., and Hawkins, K. A. (2011). The effects of apolipoprotein E on non-impaired cognitive functioning: a meta-analysis. Neurobiol. Aging 32, 63–74. doi: 10.1016/j.neurobiolaging.2009.02.003
Xiao, Q., Gil, S. C., Yan, P., Wang, Y., Han, S., Gonzales, E., et al. (2012). Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J. Biol. Chem. 287, 21279–21289. doi: 10.1074/jbc.M111.338376
Yang, S. D., Song, J. S., Yu, J. S., and Shiah, S. G. (1993). Protein kinase FA/GSK-3 phosphorylates tau on Ser235-Pro and Ser404-Pro that are abnormally phosphorylated in Alzheimer's disease brain. J. Neurochem. 61, 1742–1747. doi: 10.1111/j.1471-4159.1993.tb09811.x
Yu, J. T., and Tan, L. (2012). The role of clusterin in Alzheimer's disease: pathways, pathogenesis, and therapy. Mol. Neurobiol. 45, 314–326. doi: 10.1007/s12035-012-8237-1
Zhou, Y., Hayashi, I., Wong, J., Tugusheva, K., Renger, J. J., and Zerbinatti, C. (2014). Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer's disease. PLoS ONE 9:e103187. doi: 10.1371/journal.pone.0103187
Keywords: cholesterol, diabetes, Aβ, tau, neurodegeneration
Citation: Sato N and Morishita R (2015) The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 7:199. doi: 10.3389/fnagi.2015.00199
Received: 14 June 2015; Accepted: 04 October 2015;
Published: 23 October 2015.
Edited by:
Roxana Octavia Carare, University of Southampton, UKReviewed by:
Emmanuel Planel, Centre Hospitalier de l'Université Laval, CanadaRamesh Kandimalla, Texas Tech University, USA
Copyright © 2015 Sato and Morishita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naoyuki Sato, bnNhdG9AY2d0Lm1lZC5vc2FrYS11LmFjLmpw