 Illana Gozes*
Illana Gozes* Yanina Ivashko-Pachima
Yanina Ivashko-Pachima- Elton Laboratory for Molecular Neuroendocrinology, Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Sagol School of Neuroscience & Adams Super Center for Brain Studies, Tel Aviv University, Tel Aviv, Israel
Activity-dependent neuroprotective protein (ADNP) is deregulated in Alzheimer's disease (AD) and in schizophrenia and mutated in autism. In mice, ADNP is essential for brain formation and ADNP haploinsufficiency is associated with cognitive and social deficits and tauopathy. Tauopathy, a major pathology in AD, is also found in ~45% of frontotemporal dementias (FTDs). Tau transcript, a product of a single gene, undergoes alternative splicing. Tau splicing seems to be altered in FTD brain. In transgenic mice overexpressing a mutated tau in the cerebral cortex, significant increases in ADNP transcript expression were observed in the cerebral cortex of young transgenic mice (~disease onset) and a marked decrease with aging as compared to control littermates. ADNP is a member of the SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeling complex also associated with alternative splicing, including tau transcript splicing. Further cellular interactions of ADNP include association with microtubules, with tau being a microtubule—associated protein. NAP (davundetide), a novel drug candidate derived from ADNP, increases ADNP-microtubule association and protects against tauopathy and cognitive deficiencies in mice. Although, NAP did not provide protection in progressive supranuclear palsy (PSP), a pure tauopathy, it increased cognitive scores in amnestic mild cognitively impaired patients and protected functional activity in schizophrenia patients. This mini-review focuses on ADNP in the context of FTD and tau/microtubules and proposes NAP as a novel drug target for future clinical evaluations.
Activity-dependent Neuroprotective Protein (ADNP) Deregulation is Linked to Brain Diseases
Activity-dependent neuroprotective protein (ADNP) that has been discovered in our laboratory (Bassan et al., 1999; Zamostiano et al., 2001) has been associated with various diseases such as Alzheimer's disease (AD)—with ADNP protein levels suggested to be decreased in patient serum samples compared to controls (Yang et al., 2012), schizophrenia—with ADNP mRNA levels increased in lymphocytes of patients compared to controls (Merenlender-Wagner et al., 2015), multiple sclerosis—with ADNP mRNA levels decreased in nucleated blood cells of patients (Braitch et al., 2009), mutated in autism and associated with cognitive deficits (O'Roak et al., 2012a,b; Helsmoortel et al., 2014; Gozes et al., 2015a), and mutated in cancer (Zamostiano et al., 2001). Additional studies identified ADNP single nucleotide polymorphism in the second intron, upstream the protein coding region, as a risk factor to prostate cancer (Al Olama et al., 2014). Our recent bioinformatics showed that ADNP is unique to chordata, a gene shaping the brain of higher organisms (Gozes et al., 2015b). In mice, ADNP is essential for brain formation (Pinhasov et al., 2003; Mandel et al., 2007) and ADNP haploinsufficiency is associated with cognitive and social deficits and tauopathy (Vulih-Shultzman et al., 2007). Here we posit an association of ADNP with frontotemportal degeneration/dementia (FTD).
Pathophysiology of FTD
FTD is a spectrum of neurodegenerative diseases with pathological involvement of the frontal and temporal lobes, which refers to a variety of clinical manifestations of frontotemporal lobar degeneration (FTLD). FTD, the most common subtype of FTLD, is also divided into two main syndromes (Hopkins and Chan, 2015): a behavioral variant (bvFTD), characterized by changes in social and personal conduct with loss of volition and abstract thought, as well as decreased speech output; and primary progressive aphasia (PPA), characterized by a speech and language impairment resulting in mutism and an inability to communicate. PPA is further divided into two syndromes, the agrammatic variant and semantic dementia (Benussi et al., 2015). In certain forms of FTD, parkinsonian symptoms and amyotrophic lateral sclerosis (ALS)-like motor abnormalities may be present. In addition to FTLD subtypes, there are different clinical syndromes [corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and motor neuron diseases] with overlapping clinical/pathological features, leading to confusion in the terminology and to difficulties in diagnosis (Mendez et al., 2007; Rascovsky et al., 2011). Another classification of FTLD subtypes uses neuropathological findings. Mackenzie et al. (2009, 2010), classified two main neuropathological subtypes of FTLD: FTLD with tau-positive inclusions (FTLD-tau), caused by tau mutations or tau deregulation (Gozes, 2010), and FLTD with ubiquitinated inclusions (FTLDU), caused by mutations/deregulation in the TARDBP, GRN, VCP, and CHMP2B genes. A recent review extends these definitions and describes, in depth, the network of RNA and protein interactions in FTD (Fontana et al., 2015), encompassing C9ORF72, a gene mutated in FTD and ALS (DeJesus-Hernandez et al., 2011; Renton et al., 2011) and associated with RNA splicing factors (Cooper-Knock et al., 2015), which in turn may regulate tau. Thus, potential links between C9ORF72 to tau deserve future studies. Interestingly, recent findings identified C9ORF72 repeat expansions in 4 of 334 subjects [1.2% (or 1.8% of 217 tested families)]. All the tested subjects had behavioral phenotypes and also harbored well-known pathogenic mutations in either progranulin (GRN: p.C466LfsX46, p.R493X, p.C31LfsX35) or tau (MAPT: p.P301L), contributing to the pleiotropy of the disease (van Blitterswijk et al., 2013).
ADNP Expression is Correlated with the Microtubule Associated Protein Tau that is in Turn Linked to FTD
The neurodegeneration underlying FTD results from cortical and subcortical neuronal loss in the frontal and temporal lobes, whereas tau-positive inclusions may be found in ~45% of FTLDs (Boxer et al., 2013). Tau transcript, a product of a single gene, undergoes alternative splicing. Tau splicing seems to be altered in FTD brain. Alternative splicing around exon 10 of the tau transcript yields tau protein variants including tau protein containing either 3 or 4 microtubule binding repeat domains (tau 3R or 4R), associated with dynamic and stable microtubules, respectively (Goedert et al., 1988, 1989a,b; Goedert and Jakes, 1990). The healthy human brain exhibits a 1/1 ratio of tau 3R/4R and deviation from this ratio are pathological features of FTD taupathies (Goedert et al., 1988, 1989a,b; Hutton et al., 1998; Spillantini et al., 1998; Goedert and Spillantini, 2011).
In FTD mouse model overexpressing a mutated tau (4R) in the cerebral cortex [rTg(tauP301L)4510], significant transient increase in ADNP transcript expression (~three-fold) was observed in young transgenic mice at the disease onset compared to control littermates. This effect has not been seen in the cerebellum that is lacking an expression of a mutated tau. The increase in ADNP paralleled an increase in tau 3R and blocking the mutated tau 4R transgene expression resulted in normalization of ADNP and tau 3R mRNA levels. Furthermore, an aging-augmented decrease in ADNP expression was observed in the in the cerebral cortex of the mutated mice, compared to control littermates (Gozes et al., 2014a; Schirer et al., 2014). These findings suggest an ADNP/tau/FTD interaction.
Mechanistically, we have shown that ADNP is a member of the SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeling complex (Mandel and Gozes, 2007). As such, ADNP binds to heterochromatin protein 1 (Mandel et al., 2007; Mosch et al., 2011). Brahma (Brm), a component of the SWI/SNF complex regulating alternative splicing, showed a similar developmental expression pattern to ADNP in the rTg4510 mice. Immunoprecipitations further suggested a Brm-ADNP interaction. Further protein-protein association was found between ADNP and the polypyrimidine tract-binding protein (PTB)-associated splicing factor (PSF), with PSF being a direct regulator of tau transcript splicing (Schirer et al., 2014). These ADNP protein-protein interactions implicate a potential influence of ADNP expression on tau mRNA splicing. Notably, PSF interacts with peroxisome proliferator-activated receptor gamma (PPARc), a nuclear receptor that plays an essential role in cell proliferation, apoptosis, and inflammation (Esteves et al., 2014), serving as a therapeutic target in AD (Roses et al., 2013). PSF regulation and the regulation of tau splice variant expression have both been associated with learning and memory (Antunes-Martins et al., 2007). Furthermore, PSF (also known as SFPQ, splicing factor, proline, and glutamine-rich) is depleted from the nucleus and accumulates in the cytoplasm in AD and in FTLD, in brain areas affected by tau pathology. This cellular localization is mediated by tau protein over-expression (Ke et al., 2012). In this respect, ADNP haploinsufficiency was associated with cognitive and social deficits and aging-associated tau hyperphosphorylation, neurofibrillary tangle-like accumulation, and neurodegeneration, in mice (Vulih-Shultzman et al., 2007).
ADNP is Linked to Microtubules and Autophagy
Further cellular interactions of ADNP include association with microtubules (Furman et al., 2004; Oz et al., 2014). Specifically, we showed interaction of ADNP with microtubule end binding proteins (EBs) (Oz et al., 2014). Recent findings identified tau as a regulator of the localization and function of EB1 and EB3 in developing neuronal cells (Sayas et al., 2015). Tau is associated with microtubule dynamics and axonal transport, while EB1 is important for axons (Alves-Silva et al., 2012) and EB3 for dendritic spines (Jaworski et al., 2009) and both EB1 and EB3 are important for ADNP-associated neuronal survival (Oz et al., 2014). In this respect, small hairpin RNA ADNP downregulation (~80% robust reduction) paralleled a significant reduction (~50%) in neurite numbers in neuronal-like teratocarcinoma P19 cells (as measured by microtubule associated protein 2 immunoreactivity) (Mandel et al., 2008).
We also could show a direct association of ADNP with microtubule associated protein 1, light chain 3 (in short, LC3), a major component of the autophagosome and a key regulator of autophagy (Merenlender-Wagner et al., 2015). In this respect, autophagy was shown to be reduced in cases of tauopathy (Schaeffer and Goedert, 2012) and microtubule integrity is tightly associated with functional autophagy (Esteves et al., 2014).
Microtubules are also associated with the cellular protein translation process (Ben-Ze'ev et al., 1979), and in this respect, we showed direct ADNP interaction with the eukaryotic translation initiation factor 4E (eIF4E). As this factor is a regulator of autistic phenotype (Gkogkas et al., 2013; Malishkevich et al., 2015), association between ADNP and eIF4E may be underlying some of the behavioral phenotypes observed in FTD.
Zinc Accumulation is Associated with Tau Pathology
There is current evidence for a relative increase in intracellular zinc in vulnerable regions of the AD brain (Frederickson et al., 2005a; Berti et al., 2015). Zinc is involved in signal transmission/transduction across synapses and therefore modulates synaptic transmission and plasticity (Frederickson et al., 2005b). Besides its physiological functions, zinc dyshomeostasis can contribute to neuronal and astrocytic cell death (Koh et al., 1996; Bossy-Wetzel et al., 2004). Phosphorylation of tau regulates its binding to microtubules and is also associated with tau aggregation in disease. Aberrant phosphorylation is a key feature of tau isolated from the brains of individuals with AD and many other diseases exhibiting tau pathology (Lee et al., 2001). One likely tau kinase is glycogen synthase kinase 3β (GSK3β) (Medina and Avila, 2014). It has been shown that abnormal high concentration (up to 250 μM) of zinc induces GSK-3β activation and tau release from microtubules (Boom et al., 2009). In terms of FTD, for example, a truncating copper/zinc superoxide dismutase (SOD1) mutation, p.Gly141X, is associated with clinical and pathologic heterogeneity, including FTLD (Nakamura et al., 2015).
Taking into account that increased cellular levels of zink can induce release of tau from microtubules and thereby impair microtubule stability, ADNP replacement therapy that we propose here may be of benefit to prevent microtubule disruption and pathological aggregation of tau (Divinski et al., 2004, 2006; Oz et al., 2012, 2014). This statement leads us to clinical relevance of ADNP and the active ADNP fragment peptide, NAP (NAPVSIPQ, davuentide) (Bassan et al., 1999).
Clinical Prospects Focusing on the ADNP-derived Drug Candidate NAP in the Context of other FTD Drug Developments
To date, there is no disease-modifying treatment for FTD, affecting the underlying disease process. As for symptomatic treatments, FTD patients exhibit serotonin (Franceschi et al., 2005) and dopamine (Rinne et al., 2002) deficits, therefore the main pharmacological treatments used for FTD are based on the neurotransmitter replacement and on medications used to improve the behavioral symptoms (Litvan, 2001).
There are several preclinical studies in search for potential drug candidates for combating FTD. As mentioned above, the repeat expansion in C9ORF72 causes FTD and ALS (c9FTD/ALS). RNA of the expanded repeat [r(GGGGCC)exp] forms nuclear foci or undergoes repeat-associated non- ATG (RAN) translation, producing “c9RAN proteins” (Ash et al., 2013; Mori et al., 2013; Zu et al., 2013). Bioactive small molecules targeting r(GGGGCC)exp were designed and found to significantly inhibit RAN translation and foci formation in cultured cells expressing r(GGGGCC)66 and neurons transdifferentiated from fibroblasts of repeat expansion carriers, presenting a possible–targeted c9FTD/ALS therapeutic (Su et al., 2014).
Searching clinicaltrials.gov and touching on a few selected examples for clinical intervention, reveals that oxytocin affecting emotion is being tested (ClinicalTrials.gov Identifier: NCT01937013). Studies evaluating FRM-0334, a small molecule inhibiting histodeacetylase (HDAC), (NCT02149160) are being carried out for FTD patients with granulin gene mutations. In PSP patients, young plasma transfusion is being tested (NCT02460731). Similarly, recombinant humanized anti-tau antibody, C2N-8E12 (ABBV-8E12), is tested (NCT02494024). Salsalate, a medication that belongs to the salicylate and non-steroidal anti-inflammatory drug (NSAID) classes, is being evaluated as potential drug target as well (NCT02422485). Finally, TPI 287, an abeo-taxane —a synthetic derivative of the taxane diterpenoid drugs used in cancer therapy (which shows blood brain barrier permeability) is being tested for microtubule stabilization (NCT02133846). Together, these potential therapeutics target behavior as well as tau pathology/microtubule stability and inflammation, with potential of symptomatic/disease modifying effects. It is early to estimate potential success of any of the above listed drug candidates, but the broad range of the different approaches should pave the path to future therapeutic interventions. Given the complexity of FTD, it is possible that combination therapies will be required and given the progressive degenerative nature of the disease (s), early detection and early intervention is required.
Here, we focus on ADNP and its potential involvement in FTD. The drug candidate NAP (Bassan et al., 1999) contains the EB1, EB3, ADNP interaction site (SIP), and increases ADNP-microtubule association, i.e., EB3-ADNP association (Oz et al., 2014). NAP enhances dendritic spine formation in an EB3-depedent manner and the NAP protection against zinc intoxication requires EB1 and EB3 (Oz et al., 2014). We have further shown that NAP enhances ADNP-LC3 interaction (with LC3 being a part of the microtubule associated protein 1, and a key component of the autophagy process; Merenlender-Wagner et al., 2015). This finding is complemented by the observations that NAP protects microtubule integrity in parallel with protection of autophagy (Esteves et al., 2014).
In cell cultures, NAP protects neurons and astrocytes against ADNP deficiency (Pascual and Guerri, 2007; Vulih-Shultzman et al., 2007), decreased autophagy (Esteves et al., 2014) microtubule disruption, mitochondrial impairment, and apoptosis [e.g., in the presence of excess zinc (Divinski et al., 2004, 2006) or other toxicities (Zemlyak et al., 2009a,b; Idan-Feldman et al., 2012; Esteves et al., 2014)]. Mechanistically, NAP protects against tauopathy in vitro (Gozes and Divinski, 2004; Shiryaev et al., 2011; Idan-Feldman et al., 2012) and in vivo (Matsuoka et al., 2007, 2008; Vulih-Shultzman et al., 2007; Shiryaev et al., 2009; Jouroukhin et al., 2013; Magen et al., 2014), in part, by enlisting tau back to the microtubules (Shiryaev et al., 2011; Sudo and Baas, 2011; Oz et al., 2012; Quraishe et al., 2013) increasing microtubule dynamics (Oz et al., 2012) and fortifying axonal transport (Jouroukhin et al., 2013; Quraishe et al., 2013). A current working hypothesis for the mechanism by which NAP provides protection is illustrated in Figure 1.
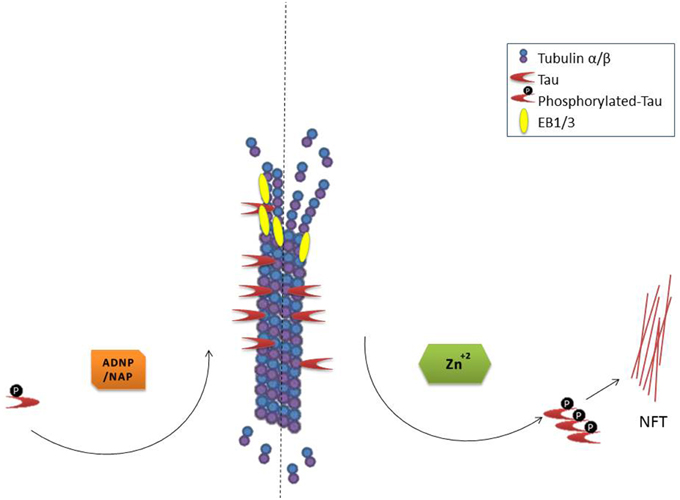
Figure 1. NAP/ADNP protection of microtubules results in protection against tau pathology (NFT, neurofibrillary tangles). Please refer to the text for in depth description.
In mice, NAP protects against tauopathy and cognitive deficiencies in a model of ADNP haploinsufficiency (Vulih-Shultzman et al., 2007) and provides protection in transgenic models of FTD (Shiryaev et al., 2009) and AD (Matsuoka et al., 2007, 2008). However, NAP may also be explored further for pathological conditions other than FTD or AD. As NAP provides protection in the SOD1-G93A transgenic mouse model of ALS (Jouroukhin et al., 2013), this therapeutic approach may be considered in the context of ALS. Furthermore, NAP provides protection in the microtubule associated protein 6 (Map6) deficient mouse model of schizophrenia, protecting autophagy and cognitive functions (Merenlender-Wagner et al., 2014).
We have also derived NAP analogs, which provided cognitive as well as protection against tauopathy in mouse models, e.g., NATLSIHQ (NAT; Gozes et al., 2014a), NAP alpha-aminoisobutyric acid (IsoNAP; Gozes et al., 2014b), and all D-amino acids analog, SALLRSIPA (D-SAL; Shiryaev et al., 2011). SAL/D-SAL may fortify NAP protection (Brenneman et al., 2004) and this is a subject of future investigation.
In men, NAP (davunetide) has a safe clinical profile in relatively large cohorts, altogether exposed to >700 subjects. While, NAP (davunetide) did not provide protection in PSP (Boxer et al., 2014), it increased cognitive scores in amnestic mild cognitively impaired patients (Gozes et al., 2009; Morimoto et al., 2013) and protected functional activity in schizophrenia patients (Javitt et al., 2012; Jarskog et al., 2013). We posit that early intervention with NAP will lead to clinical efficacy.
In conclusion, NAP presents an advantage in being a simple small molecule (a short 8 amino acid peptide, derived from a natural essential protein, ADNP) with brain bioavailability through a non-invasive route. However, in this respect, it is similar to oxytocin. Differing from oxytocin, NAP (davunetide) presents a different, unique mechanism of action (interaction with microtubule end binding proteins and increasing ADNP-associated neuroprotection). Thus, the NAP protection of the microtubule structure relies on the fortification of an endogenous process, unlike taxanes or tau immunotherapy. Together these finding envision NAP as an innovative therapeutic approach in FTD.
Conflict of Interest Statement
NAP (davunetide) and pipeline products are under term sheet agreement for further clinical development (IG).
Acknowledgments
Professor Gozes laboratory is supported by the AMN Foundation, the Israel Ministry of Science, Technology and Space, Israel Science Foundation, CFTAU Montreal Circle of Friends and the Adams family, Adams Super Center for Brain Studies, the Elton Laboratory for Molecular Neuroendocrinology, and the Lily and Avraham Gildor Chair for the Investigation of Growth Factors at Tel Aviv University. IG is a Humboldt Award Recipient, Germany. NAP and related compounds are under patent protection (IG, Inventor). This study is in partial fulfillment for the Ph.D. thesis of YI at the Dr. Miriam and Sheldon G. Adelson Graduate School of Medicine, as the Sackler Faculty of Medicine, Tel Aviv University.
References
Al Olama, A. A., Kote-Jarai, Z., Berndt, S. I., Conti, D. V., Schumacher, F., Han, Y., et al. (2014). A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat. Genet. 46, 1103–1109. doi: 10.1038/ng.3094
Alves-Silva, J., Sánchez-Soriano, N., Beaven, R., Klein, M., Parkin, J., Millard, T. H., et al. (2012). Spectraplakins promote microtubule-mediated axonal growth by functioning as structural microtubule-associated proteins and EB1-dependent +TIPs (tip interacting proteins). J. Neurosci. 32, 9143–9158. doi: 10.1523/JNEUROSCI.0416-12.2012
Antunes-Martins, A., Mizuno, K., Irvine, E. E., Lepicard, E. M., and Giese, K. P. (2007). Sex-dependent up-regulation of two splicing factors, Psf and Srp20, during hippocampal memory formation. Learn. Mem. 14, 693–702. doi: 10.1101/lm.640307
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Bassan, M., Zamostiano, R., Davidson, A., Pinhasov, A., Giladi, E., Perl, O., et al. (1999). Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J. Neurochem. 72, 1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x
Benussi, A., Padovani, A., and Borroni, B. (2015). Phenotypic heterogeneity of monogenic frontotemporal dementia. Front. Aging Neurosci. 7:171. doi: 10.3389/fnagi.2015.00171
Ben-Ze'ev, A., Farmer, S. R., and Penman, S. (1979). Mechanisms of regulating tubulin synthesis in cultured mammalian cells. Cell 17, 319–325. doi: 10.1016/0092-8674(79)90157-0
Berti, V., Murray, J., Davies, M., Spector, N., Tsui, W. H., Li, Y., et al. (2015). Nutrient patterns and brain biomarkers of Alzheimer's disease in cognitively normal individuals. J. Nutr. Health Aging 19, 413–423. doi: 10.1007/s12603-014-0534-0
Boom, A., Authelet, M., Dedecker, R., Frederick, C., Van Heurck, R., Daubie, V., et al. (2009). Bimodal modulation of tau protein phosphorylation and conformation by extracellular Zn(2+) in human-tau transfected cells. Biochim. Biophys. Acta 1793, 1058–1067. doi: 10.1016/j.bbamcr.2008.11.011
Bossy-Wetzel, E., Talantova, M. V., Lee, W. D., Schölzke, M. N., Harrop, A., Mathews, E., et al. (2004). Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41, 351–365. doi: 10.1016/S0896-6273(04)00015-7
Boxer, A. L., Gold, M., Huey, E., Gao, F. B., Burton, E. A., Chow, T., et al. (2013). Frontotemporal degeneration, the next therapeutic frontier: molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. 9, 176–188. doi: 10.1016/j.jalz.2012.03.002
Boxer, A. L., Lang, A. E., Grossman, M., Knopman, D. S., Miller, B. L., Schneider, L. S., et al. (2014). Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 13, 676–685. doi: 10.1016/S1474-4422(14)70088-2
Braitch, M., Kawabe, K., Nyirenda, M., Gilles, L. J., Robins, R. A., Gran, B., et al. (2009). Expression of activity-dependent neuroprotective protein in the immune system: possible functions and relevance to multiple sclerosis. Neuroimmunomodulation 17, 120–125. doi: 10.1159/000258695
Brenneman, D. E., Spong, C. Y., Hauser, J. M., Abebe, D., Pinhasov, A., Golian, T., et al. (2004). Protective peptides that are orally active and mechanistically nonchiral. J. Pharmacol. Exp. Ther. 309, 1190–1197. doi: 10.1124/jpet.103.063891
Cooper-Knock, J., Bury, J. J., Heath, P. R., Wyles, M., Higginbottom, A., Gelsthorpe, C., et al. (2015). C9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS ONE 10:e0127376. doi: 10.1371/journal.pone.0127376
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Divinski, I., Holtser-Cochav, M., Vulih-Schultzman, I., Steingart, R. A., and Gozes, I. (2006). Peptide neuroprotection through specific interaction with brain tubulin. J. Neurochem. 98, 973–984. doi: 10.1111/j.1471-4159.2006.03936.x
Divinski, I., Mittelman, L., and Gozes, I. (2004). A femtomolar acting octapeptide interacts with tubulin and protects astrocytes against zinc intoxication. J. Biol. Chem. 279, 28531–28538. doi: 10.1074/jbc.M403197200
Esteves, A. R., Gozes, I., and Cardoso, S. M. (2014). The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson's disease. Biochim. Biophys. Acta 1842, 7–21. doi: 10.1016/j.bbadis.2013.10.003
Fontana, F., Siva, K., and Denti, M. A. (2015). A network of RNA and protein interactions in Fronto Temporal Dementia. Front. Mol. Neurosci. 8:9. doi: 10.3389/fnmol.2015.00009
Franceschi, M., Anchisi, D., Pelati, O., Zuffi, M., Matarrese, M., Moresco, R. M., et al. (2005). Glucose metabolism and serotonin receptors in the frontotemporal lobe degeneration. Ann. Neurol. 57, 216–225. doi: 10.1002/ana.20365
Frederickson, C. J., Cuajungco, M. P., and Frederickson, C. J. (2005b). Is zinc the link between compromises of brain perfusion (excitotoxicity) and Alzheimer's disease? J. Alzheimers Dis. 8, 155–160. discussion 209–215.
Frederickson, C. J., Koh, J. Y., and Bush, A. I. (2005a). The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 6, 449–462. doi: 10.1038/nrn1671
Furman, S., Steingart, R. A., Mandel, S., Hauser, J. M., Brenneman, D. E., and Gozes, I. (2004). Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 1, 193–199. doi: 10.1017/S1740925X05000013
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi: 10.1038/nature11628
Goedert, M., and Jakes, R. (1990). Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230.
Goedert, M., and Spillantini, M. G. (2011). Pathogenesis of the tauopathies. J. Mol. Neurosci. 45, 425–431. doi: 10.1007/s12031-011-9593-4
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989a). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989b). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399.
Goedert, M., Wischik, C. M., Crowther, R. A., Walker, J. E., and Klug, A. (1988). Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. U.S.A. 85, 4051–4055. doi: 10.1073/pnas.85.11.4051
Gozes, I. (2010). Tau pathology and future therapeutics. Curr. Alzheimer Res. 7, 685–696. doi: 10.2174/156720510793611628
Gozes, I., and Divinski, I. (2004). The femtomolar-acting NAP interacts with microtubules: novel aspects of astrocyte protection. J. Alzheimers. Dis. 6(6 Suppl.), S37–S41.
Gozes, I., Helsmoortel, C., Vandeweyer, G., Van der Aa, N., Kooy, F., and Sermone, S. B. (2015a). The compassionate side of neuroscience: Tony Sermone's undiagnosed genetic journey-ADNP mutation. J. Mol. Neurosci. 56, 751–757. doi: 10.1007/s12031-015-0586-6
Gozes, I., Iram, T., Maryanovsky, E., Arviv, C., Rozenberg, L., Schirer, Y., et al. (2014a). Novel tubulin and tau neuroprotective fragments sharing structural similarities with the drug candidate NAP (Davuentide). J. Alzheimers. Dis. 40(Suppl. 1), S23–S36. doi: 10.3233/JAD-131664
Gozes, I., Schirer, Y., Idan-Feldman, A., David, M., and Furman-Assaf, S. (2014b). NAP alpha-aminoisobutyric acid (IsoNAP). J. Mol. Neurosci. 52, 1–9. doi: 10.1007/s12031-013-0103-8
Gozes, I., Stewart, A., Morimoto, B., Fox, A., Sutherland, K., and Schmechel, D. (2009). Addressing Alzheimer's disease tangles: from NAP to AL-108. Curr. Alzheimer Res. 6, 455–460. doi: 10.2174/156720509789207895
Gozes, I., Yeheskel, A., and Pasmanik-Chor, M. (2015b). Activity-dependent neuroprotective protein (ADNP): a case study for highly conserved chordata-specific genes shaping the brain and mutated in cancer. J. Alzheimers. Dis. 45, 57–73. doi: 10.3233/JAD-142490
Helsmoortel, C., Vulto-van Silfhout, A. T., Coe, B. P., Vandeweyer, G., Rooms, L., van den Ende, J., et al. (2014). A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 46, 380–384. doi: 10.1038/ng.2899
Hopkins, S. A., and Chan, D. (2015). Key emerging issues in frontotemporal dementia. J. Neurol. doi: 10.1007/s00415-015-7880-7. [Epub ahead of print].
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Idan-Feldman, A., Ostritsky, R., and Gozes, I. (2012). Tau and caspase 3 as targets for neuroprotection. Int. J. Alzheimers. Dis. 2012:493670. doi: 10.1155/2012/493670
Jarskog, L. F., Dong, Z., Kangarlu, A., Colibazzi, T., Girgis, R. R., Kegeles, L. S., et al. (2013). Effects of davunetide on N-acetylaspartate and choline in dorsolateral prefrontal cortex in patients with schizophrenia. Neuropsychopharmacology 38, 1245–1252. doi: 10.1038/npp.2013.23
Javitt, D. C., Buchanan, R. W., Keefe, R. S., Kern, R., McMahon, R. P., Green, M. F., et al. (2012). Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr. Res. 136, 25–31. doi: 10.1016/j.schres.2011.11.001
Jaworski, J., Kapitein, L. C., Gouveia, S. M., Dortland, B. R., Wulf, P. S., Grigoriev, I., et al. (2009). Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61, 85–100. doi: 10.1016/j.neuron.2008.11.013
Jouroukhin, Y., Ostritsky, R., Assaf, Y., Pelled, G., Giladi, E., and Gozes, I. (2013). NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: protection against impairments in axonal transport. Neurobiol. Dis. 56, 79–94. doi: 10.1016/j.nbd.2013.04.012
Ke, Y. D., Dramiga, J., Schütz, U., Kril, J. J., Ittner, L. M., Schröder, H., et al. (2012). Tau-mediated nuclear depletion and cytoplasmic accumulation of SFPQ in Alzheimer's and Pick's disease. PLoS ONE 7:e35678. doi: 10.1371/journal.pone.0035678
Koh, J. Y., Suh, S. W., Gwag, B. J., He, Y. Y., Hsu, C. Y., and Choi, D. W. (1996). The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272, 1013–1016. doi: 10.1126/science.272.5264.1013
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Litvan, I. (2001). Therapy and management of frontal lobe dementia patients. Neurology 56(11 Suppl. 4), S41–S45. doi: 10.1212/WNL.56.suppl_4.S41
Mackenzie, I. R., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., et al. (2009). Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 117, 15–18. doi: 10.1007/s00401-008-0460-5
Mackenzie, I. R., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., et al. (2010). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 119, 1–4. doi: 10.1007/s00401-009-0612-2
Magen, I., Ostritsky, R., Richter, F., Zhu, C., Fleming, S. M., Lemesre, V., et al. (2014). Intranasal NAP (davunetide) decreases tau hyperphosphorylation and moderately improves behavioral deficits in mice overexpressing alpha-synuclein. Pharmacol. Res. Perspect. 2:e00065. doi: 10.1002/prp2.65
Malishkevich, A., Amram, N., Hacohen-Kleiman, G., Magen, I., Giladi, E., and Gozes, I. (2015). Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer's pathologies. Transl. Psychiatry 5:e501. doi: 10.1038/tp.2014.138
Mandel, S., and Gozes, I. (2007). Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J. Biol. Chem. 282, 34448–34456. doi: 10.1074/jbc.M704756200
Mandel, S., Rechavi, G., and Gozes, I. (2007). Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev. Biol. 303, 814–824. doi: 10.1016/j.ydbio.2006.11.039
Mandel, S., Spivak-Pohis, I., and Gozes, I. (2008). ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J. Mol. Neurosci. 35, 127–141. doi: 10.1007/s12031-007-9013-y
Matsuoka, Y., Gray, A. J., Hirata-Fukae, C., Minami, S. S., Waterhouse, E. G., Mattson, M. P., et al. (2007). Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer's disease at early pathological stage. J. Mol. Neurosci. 31, 165–170. doi: 10.1385/JMN/31:02:165
Matsuoka, Y., Jouroukhin, Y., Gray, A. J., Ma, L., Hirata-Fukae, C., Li, H. F., et al. (2008). A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer's disease. J. Pharmacol. Exp. Ther. 325, 146–153. doi: 10.1124/jpet.107.130526
Medina, M., and Avila, J. (2014). New insights into the role of glycogen synthase kinase-3 in Alzheimer's disease. Expert Opin. Ther. Targets 18, 69–77. doi: 10.1517/14728222.2013.843670
Mendez, M. F., Shapira, J. S., McMurtray, A., Licht, E., and Miller, B. L. (2007). Accuracy of the clinical evaluation for frontotemporal dementia. Arch. Neurol. 64, 830–835. doi: 10.1001/archneur.64.6.830
Merenlender-Wagner, A., Malishkevich, A., Shemer, Z., Udawela, M., Gibbons, A., Scarr, E., et al. (2015). Autophagy has a key role in the pathophysiology of schizophrenia. Mol. Psychiatry 20, 126–132. doi: 10.1038/mp.2013.174
Merenlender-Wagner, A., Shemer, Z., Touloumi, O., Lagoudaki, R., Giladi, E., Andrieux, A., et al. (2014). New horizons in schizophrenia treatment: autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy 10, 2324–2332. doi: 10.4161/15548627.2014.984274
Mori, K., Arzberger, T., Grässer, F. A., Gijselinck, I., May, S., Rentzsch, K., et al. (2013). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893. doi: 10.1007/s00401-013-1189-3
Morimoto, B. H., Schmechel, D., Hirman, J., Blackwell, A., Keith, J., and Gold, M. (2013). A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 35, 325–336. doi: 10.1159/000348347
Mosch, K., Franz, H., Soeroes, S., Singh, P. B., and Fischle, W. (2011). HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLoS ONE 6:e15894. doi: 10.1371/journal.pone.0015894
Nakamura, M., Bieniek, K. F., Lin, W. L., Graff-Radford, N. R., Murray, M. E., Castanedes-Casey, M., et al. (2015). A truncating SOD1 mutation, p.Gly141X, is associated with clinical and pathologic heterogeneity, including frontotemporal lobar degeneration. Acta Neuropathol. 130, 145–157. doi: 10.1007/s00401-015-1431-2
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Oz, S., Ivashko-Pachima, Y., and Gozes, I. (2012). The ADNP derived peptide, NAP modulates the tubulin pool: implication for neurotrophic and neuroprotective activities. PLoS ONE 7:e51458. doi: 10.1371/journal.pone.0051458
Oz, S., Kapitansky, O., Ivashco-Pachima, Y., Malishkevich, A., Giladi, E., Skalka, N., et al. (2014). The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry 19, 1115–1124. doi: 10.1038/mp.2014.97
Pascual, M., and Guerri, C. (2007). The peptide NAP promotes neuronal growth and differentiation through extracellular signal-regulated protein kinase and Akt pathways, and protects neurons co-cultured with astrocytes damaged by ethanol. J. Neurochem. 103, 557–568. doi: 10.1111/j.1471-4159.2007.04761.x
Pinhasov, A., Mandel, S., Torchinsky, A., Giladi, E., Pittel, Z., Goldsweig, A. M., et al. (2003). Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res. Dev. Brain Res. 144, 83–90. doi: 10.1016/S0165-3806(03)00162-7
Quraishe, S., Cowan, C. M., and Mudher, A. (2013). NAP (davunetide) rescues neuronal dysfunction in a Drosophila model of tauopathy. Mol. Psychiatry 18, 834–842. doi: 10.1038/mp.2013.32
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134(Pt 9), 2456–2477. doi: 10.1093/brain/awr179
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rinne, J. O., Laine, M., Kaasinen, V., Norvasuo-Heilä, M. K., Någren, K., and Helenius, H. (2002). Striatal dopamine transporter and extrapyramidal symptoms in frontotemporal dementia. Neurology 58, 1489–1493. doi: 10.1212/WNL.58.10.1489
Roses, A. D., Lutz, M. W., Crenshaw, D. G., Grossman, I., Saunders, A. M., and Gottschalk, W. K. (2013). TOMM40 and APOE: requirements for replication studies of association with age of disease onset and enrichment of a clinical trial. Alzheimers Dement. 9, 132–136. doi: 10.1016/j.jalz.2012.10.009
Sayas, C. L., Tortosa, E., Bollati, F., Ramírez-Ríos, S., Arnal, I., and Avila, J. (2015). Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J. Neurochem. 133, 653–667. doi: 10.1111/jnc.13091
Schaeffer, V., and Goedert, M. (2012). Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy 8, 1686–1687. doi: 10.4161/auto.21488
Schirer, Y., Malishkevich, A., Ophir, Y., Lewis, J., Giladi, E., and Gozes, I. (2014). Novel marker for the onset of frontotemporal dementia: early increase in activity-dependent neuroprotective protein (ADNP) in the face of Tau mutation. PLoS ONE 9:e87383. doi: 10.1371/journal.pone.0087383
Shiryaev, N., Jouroukhin, Y., Giladi, E., Polyzoidou, E., Grigoriadis, N. C., Rosenmann, H., et al. (2009). NAP protects memory, increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model. Neurobiol. Dis. 34, 381–388. doi: 10.1016/j.nbd.2009.02.011
Shiryaev, N., Pikman, R., Giladi, E., and Gozes, I. (2011). Protection against tauopathy by the drug candidates NAP (davunetide) and D-SAL: biochemical, cellular and behavioral aspects. Curr. Pharm. Des. 17, 2603–2612. doi: 10.2174/138161211797416093
Spillantini, M. G., Murrell, J. R., Goedert, M., Farlow, M. R., Klug, A., and Ghetti, B. (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U.S.A. 95, 7737–7741. doi: 10.1073/pnas.95.13.7737
Su, Z., Zhang, Y., Gendron, T. F., Bauer, P. O., Chew, J., Yang, W. Y., et al. (2014). Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 83, 1043–1050. doi: 10.1016/j.neuron.2014.07.041
Sudo, H., and Baas, P. W. (2011). Strategies for diminishing katanin-based loss of microtubules in tauopathic neurodegenerative diseases. Hum. Mol. Genet. 20, 763–778. doi: 10.1093/hmg/ddq521
van Blitterswijk, M., Baker, M. C., DeJesus-Hernandez, M., Ghidoni, R., Benussi, L., Finger, E., et al. (2013). C9ORF72 repeat expansions in cases with previously identified pathogenic mutations. Neurology 81, 1332–1341. doi: 10.1212/WNL.0b013e3182a8250c
Vulih-Shultzman, I., Pinhasov, A., Mandel, S., Grigoriadis, N., Touloumi, O., Pittel, Z., et al. (2007). Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J. Pharmacol. Exp. Ther. 323, 438–449. doi: 10.1124/jpet.107.129551
Yang, M. H., Yang, Y. H., Lu, C. Y., Jong, S. B., Chen, L. J., Lin, Y. F., et al. (2012). Activity-dependent neuroprotector homeobox protein: a candidate protein identified in serum as diagnostic biomarker for Alzheimer's disease. J. Proteomics 75, 3617–3629. doi: 10.1016/j.jprot.2012.04.017
Zamostiano, R., Pinhasov, A., Gelber, E., Steingart, R. A., Seroussi, E., Giladi, E., et al. (2001). Cloning and characterization of the human activity-dependent neuroprotective protein. J. Biol. Chem. 276, 708–714. doi: 10.1074/jbc.M007416200
Zemlyak, I., Sapolsky, R., and Gozes, I. (2009a). NAP protects against cytochrome c release: inhibition of the initiation of apoptosis. Eur. J. Pharmacol. 618, 9–14. doi: 10.1016/j.ejphar.2009.07.013
Zemlyak, I., Sapolsky, R., and Gozes, I. (2009b). NAP protects against cyanide-related microtubule destruction. J. Neural Transm. 116, 1411–1416. doi: 10.1007/s00702-009-0252-7
Keywords: acitivity-dependent neuroprotective protein (ADNP), NAP (davunetide), microtubules, Alzheimer's disease, neuroprotection
Citation: Gozes I and Ivashko-Pachima Y (2015) ADNP: in search for molecular mechanisms and innovative therapeutic strategies for frontotemporal degeneration. Front. Aging Neurosci. 7:205. doi: 10.3389/fnagi.2015.00205
Received: 06 August 2015; Accepted: 12 October 2015;
Published: 29 October 2015.
Edited by:
Diego Albani, Istituto Di Ricerche Farmacologiche Mario Negri—IRCCS, ItalyReviewed by:
Catarina Oliveira, University of Coimbra, PortugalSabina Tahirovic, DZNE-München, Germany
Copyright © 2015 Gozes and Ivashko-Pachima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Illana Gozes, aWdvemVzQHBvc3QudGF1LmFjLmls