 Cheng-Wu Zhang
Cheng-Wu Zhang Liting Hang3
Liting Hang3 Kah-Leong Lim
Kah-Leong Lim- 1Neurodegeneration Research Laboratory, National Neuroscience Institute, Singapore, Singapore
- 2Institute of Advanced Materials, Nanjing Tech University, Nanjing, People’s Republic of China
- 3Department of Physiology, National University of Singapore, Singapore, Singapore
- 4Departments of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC, USA
- 5Duke-NUS Graduate Medical School, National University of Singapore, Singapore, Singapore
Parkin is a unique, multifunctional ubiquitin ligase whose various roles in the cell, particularly in neurons, are widely thought to be protective. The pivotal role that Parkin plays in maintaining neuronal survival is underscored by our current recognition that Parkin dysfunction represents not only a predominant cause of familial parkinsonism but also a formal risk factor for the more common, sporadic form of Parkinson’s disease (PD). Accordingly, keen research on Parkin over the past decade has led to an explosion of knowledge regarding its physiological roles and its relevance to PD. However, our understanding of Parkin is far from being complete. Indeed, surprises emerge from time to time that compel us to constantly update the paradigm of Parkin function. For example, we now know that Parkin’s function is not confined to mere housekeeping protein quality control (QC) roles but also includes mitochondrial homeostasis and stress-related signaling. Furthermore, emerging evidence also suggest a role for Parkin in several other major neurodegenerative diseases including Alzheimer’s disease (AD) and Amyotrophic Lateral Sclerosis (ALS). Yet, it remains truly amazing to note that a single enzyme could serve such multitude of functions and cellular roles. Clearly, its activity has to be tightly regulated. In this review, we shall discuss this and how dysregulated Parkin function may precipitate neuronal demise in various neurodegenerative disorders.
Introduction
Parkin is a unique multifunctional ubiquitin ligase whose various roles in the cell, particularly in neurons, are widely thought to be protective. The pivotal role that Parkin plays in maintaining neuronal survival is underscored by our current recognition that Parkin dysfunction represents not only a predominant cause of familial Parkinsonism but also a formal risk factor for the more common, sporadic form of Parkinson’s disease (PD; Dawson and Dawson, 2014). Moreover, emerging evidence also implicates a role for Parkin in Alzheimer’s disease (AD), Amyotrophic Lateral Sclerosis (ALS) and Huntington’s disease (HD; Tsai et al., 2003; Rosen et al., 2010; Hebron et al., 2014). Further, several studies have documented the ability of Parkin to protect neurons against a wide variety of insults, including those mediated by neurotoxins and metallic ions (Kubo et al., 2013). Thus, maintaining an optimal state of Parkin function appears to be an important determinant of neuronal health in general, although dopaminergic (DA) neurons whose degeneration is causative of PD seems particularly sensitive to Parkin deficiency.
Mutations in Parkin were originally identified in Japan more than a decade ago to be a major genetic contributor of Autosomal Recessive Juvenile Parkinsonism (ARJP; Kitada et al., 1998). Following this discovery, several ethnically diverse individuals with early-onset PD (age <45 years) in other parts of the world were also found to carry Parkin mutations, which occur at a frequency of about 10–20 and 50% in sporadic and familial early-onset cases respectively (Lücking et al., 2000; Periquet et al., 2003; Mata et al., 2004). At around the same period, three independent groups demonstrated that Parkin functions as an E2-dependent ubiquitin ligase associated with the ubiquitin-proteasome system (UPS; Imai et al., 2000; Shimura et al., 2000; Zhang et al., 2000), a major intracellular protein degradation machinery, which fuelled intense research on the role of Parkin in neuronal protein homeostasis. The particular excitement in this topic was in part also due to the finding that the first PD-linked gene elucidated before Parkin, i.e., α-synuclein, encodes a presynaptic protein that tends to misfold and that was subsequently found to be a major component of Lewy Bodies (LBs), a histological hallmark of the PD brain (Spillantini et al., 1997). It seemed intuitive therefore to associate UPS dysfunction with PD pathogenesis. Further support for this soon came with the elucidation of another PD-linked gene, i.e., UCH-L1, which encodes yet another component of the UPS. One could therefore readily appreciate that it was then a hugely popular trend to look at the “proteasome theory of PD” (and subsequently, the related “autophagy theory of PD”). Parkin was soon found to be involved in both UPS- and autophagy-associated protein quality control (QC; Lim and Zhang, 2013). However, along the way, several groups (particularly those working with Drosophila Parkin models) also documented an intriguing relationship between Parkin and mitochondrial QC, i.e., mitochondria tends to become abnormal in the absence of functional Parkin (Greene et al., 2003). This phenomenon remains largely unexplained and overlooked until 2008, when a seminal discovery by a team led by Richard Youle demonstrated that Parkin is a key mammalian regulator of mitochondrial autophagy, or “mitophagy” (Narendra et al., 2008). Subsequent studies by his group and several others revealed that Parkin collaborates with another PD-linked gene product known as PINK1 (encoding a mitochondrial-targeted serine/threonine kinase) to mediate mitophagy (Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2010a; Vives-Bauza et al., 2010). Collectively, these reports triggered an explosion of interest among the global mitochondria and PD research community in delineating the pathways involved in Parkin/PINK1-mediated mitophagy, with the excitement ensuing to this date. Meanwhile, the “mitochondrial theory of PD” that was popular in the eighties and nineties but had lay “dormant” subsequently is now enjoying its “renaissance” of support.
Notwithstanding the periodic bias in favor of a particular disease-associated pathway, it is now evident that Parkin is an important regulator of protein and mitochondrial homeostasis that operates against the backdrop of a multitude of intracellular pathways. Accordingly, the ubiquitin ligase itself would need to be exquisitely regulated to fulfil its diverse cellular roles in a timely fashion to maintain optimal neuronal function. In this review, we shall discuss the mechanisms underlying Parkin activity regulation and its involvement in protein/mitochondrial homeostasis and finally, how dysregulated Parkin function may predispose neurons to degeneration.
Structure and Regulation of Parkin Activity
Structurally, the 465 amino acid-containing human Parkin protein is comprised of a ubiquitin-like (Ubl) domain at its N-terminus, a RING1-IBR-RING2 (RBR) domain at its C-terminus and a unique middle segment that links the two domains. A zinc-chelating RING0 domain that is juxtaposed (N-terminally) to the RBR domains resides within the linker segment. Another motif, known as the Repressor Element of Parkin (REP), sits between the IBR and RING2 domains (Figure 1). During Parkin-mediated ubiquitination, E2 enzymes are first recruited to RING1 domain and the charged ubiquitin they carry are then transferred to a catalytic cysteine (C431) in the RING2 domain before being finally transferred to the primary amino group of the substrate via an iso-peptide bond formation. This is an intriguing catalytic mechanism for a RING-containing E3 as the process is reminiscent of HECT domain-containing ubiquitin ligases. We now know that RBR E3 s such as Parkin and HHARI use such a RING-HECT hybrid mechanism for catalysis (Wenzel et al., 2011). Interestingly, structural studies of Parkin revealed that the enzyme exists in a closed, inactivated state under normal conditions. This auto-inhibited state is achieved through an intricate folding of the protein whereby RING0 is inserted between RING1 and RING2 and in so doing occludes the active site on RING2. At the same time, the closed conformation also promotes the REP to adopt a structure that lies across the putative E2-binding site on RING1 thus preventing its recruitment of E2 s (Riley et al., 2013; Trempe et al., 2013; Wauer and Komander, 2013; Figure 1). Consistent with this, both Helen Walden’s group and ours have shown experimentally that the N-terminal region of Parkin represses Parkin-mediated polyubiquitination, although an important difference between the two studies is the repressing element involved, i.e., we have proposed the linker segment (amino acid 152–237 – that contains the RING0 domain) to be the repressing element whereas Walden’s group showed that it is the Ubl domain (Chaugule et al., 2011; Chew et al., 2011). Notwithstanding this, for Parkin to become active, it is obvious that substantial rearrangements of its structure need to take place to allow the dissociation of RING0 from RING2 as well as REP from RING1 among other conformational changes.
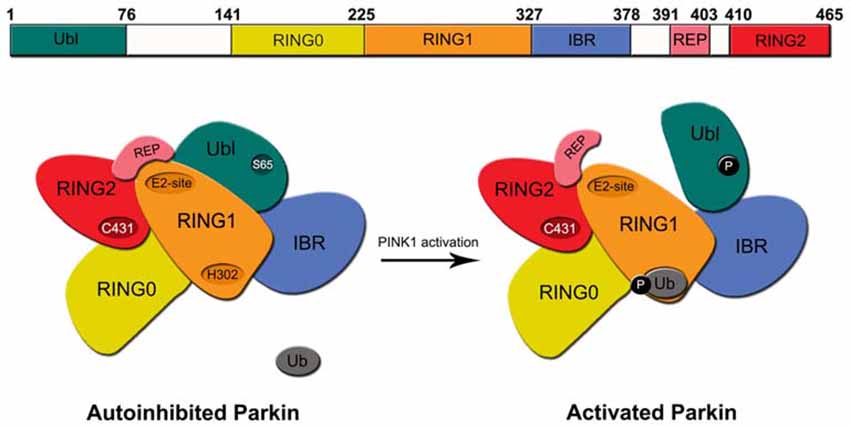
Figure 1. Structure of regulation of Parkin. Top, Schematic depiction of parkin structure. Bottom, A model of Parkin activity regulation—Under normal conditions, Parkin exists in an auto-inhibited state where access to its E2-binding RING1 site is occluded by its Ubl and repressor element of parkin (REP) domains and access to its RING2’s active site is blocked by the RING0 domain. Upon the phosphorylation Parkin’s Ubl domain by PINK1 at Serine 65 (S65) and the concomitant engagement of RING1 with phosphorylated ubiquitin, the Ubl is displaced away from RING1, which led to the structural rearrangement of the various domains of Parkin. The enzyme consequently becomes fully activated.
Recent studies have revealed that Parkin activity may be stimulated by a two-step process that involves the phosphorylation of serine 65 (S65) in its Ubl domain (Matsuda et al., 2010; Kondapalli et al., 2012; Shiba-Fukushima et al., 2012) and the phosphorylation of ubiquitin at the same site (Kane et al., 2014; Koyano et al., 2014). Interestingly, whereas phosphorylation of Parkin’s Ubl domain promotes its dissociation from RING1, the phosphorylation of ubiquitin (also at S65) enhances its association with RING1. Thus, despite the fact that Ubl and ubiquitin share marked sequence similarities, their respective interaction with RING1 of Parkin is oppositely regulated by phosphorylation. Precisely how these phosphorylation events could alter the multiple intra-molecular interactions that define the auto-inhibited enzyme to bring about its activation was unclear until very recently. A number of new structural studies on Parkin have now clarified the mechanism of Parkin activation (Kumar et al., 2015; Sauvé et al., 2015; Wauer et al., 2015). In essence, the concerted finding is that the dissociation of Ubl from RING1 (following S65 phosphorylation) exposes a histidine residue (H302) on RING1 that binds to phosphorylated ubiquitin, which leads to the displacement the Ubl domain from its original conformation, loss of structure near the RING1/IBR interface and the overall relieve of Parkin’s auto-inhibited state (Figure 1). RING1 now becomes available for the engagement with E2~ubiquitin conjugate to trigger downstream substrate ubiquitination. Although these structural studies have certainly contributed to a clearer picture of Parkin activation, how the activated enzyme returns to its basal auto-inhibited state remains elusive. Presumably, this will involve the dephosphorylation of S65-phosphorylated Ubl domain and ubiquitin by an unknown phosphatase(s). Moreover, because the above model of Parkin activation is related to its function in mitophagy, it is also unclear whether the same activation mechanism is utilized to promote Parkin-mediated ubiquitination in non-mitophagy events. Parkin’s role in the cell is certainly not restricted to protein turnover during mitophagy. As a matter of fact, the majority of reports before 2008 described its role in protein QC that is not directly related to mitophagy.
Parkin and Protein Homeostasis
As mentioned earlier, Parkin functions as an E3 ligase associated with the UPS, a major proteolytic machinery that normally identifies and degrades unwanted intracellular proteins. In this system, proteins that are destined for proteasome-mediated degradation are added a chain of ubiquitin via a reaction cascade that involves the ubiquitin-activating (E1), -conjugating (E2) and -ligating (E3) enzymes. Through the sequential and repetitive actions of these enzymes, successive isopeptide linkages are formed between the C-terminal glycine carboxyl group (G76) of the ubiquitin moiety being added and the α-amino group of a free lysine (most commonly K48) on the ubiquitin that is attached to the protein. The (G76-K48) polyubiquitinated substrate is then recognized by the 26S proteasome as a target for degradation. It is important to highlight that the ubiquitin sequence contains seven lysine residues (at positions 6, 11, 27, 29, 33, 48 and 63) and that polyubiquitin chain assembly can occur at any of these lysine residues (Peng et al., 2003). In addition, proteins can also be monoubiquitinated. Notably, both K63-linked polyubiquitination and monoubiquitination of proteins are not typically associated with proteasome-mediated ubiquitination.
Along with the original discovery by three independent groups who demonstrated that Parkin functions as a ubiquitin ligase, they also found that disease-associated Parkin mutations compromise its role as an E3 enzyme (Imai et al., 2000; Shimura et al., 2000; Zhang et al., 2000). A logical and popular hypothesis that ensued is that loss of Parkin function would lead to the toxic accumulation of one or several of its substrates, thereby leading to neurodegeneration. This had fuelled intense effort by many laboratories around the world to identify the culprit substrate(s) involved. To date, no less than 25 substrates (or putative substrates) of Parkin have been reported (Table 1). However, none of the Parkin substrates identified thus far is exclusively expressed in DA neurons, which raises the question on why DA neurons in familial Parkinsonism cases linked to Parkin mutations are rather selectively vulnerable to deficient Parkin function. Of course, one could argue that neither is Parkin expression confined to DA neurons and therefore it is perhaps not surprising to note the broad distribution of its substrates as well. Importantly, few from the laundry list of Parkin substrates fulfil an important criterion expected of an “authentic” Parkin substrate, i.e., accumulation in the brains of ARJP patients and Parkin-deficient models. Although the pace of substrate identification has slowed down considerably in recent years, new Parkin substrates continue to emerge periodically, especially those related to the mitochondria. Among the recently isolated Parkin substrates is the zinc finger-containing protein called PARIS (ZNF746; Shin et al., 2011), a major transcriptional repressor of PGC-1α expression, which in turn regulates the transcription of many genes involved in cellular metabolism. Importantly, unlike most previously identified Parkin substrates, PARIS was shown to accumulate in post-mortem brain tissues derived from ARJP and sporadic PD patients, as well as in the ventral midbrain region of mice that is conditionally ablated of Parkin expression (Shin et al., 2011). Based on these findings, the authors proposed that PARIS is an “authentic” Parkin substrate. More recently, and some may think curiously, Parkin was found to interact with the anaphase promoting complex/cyclosome (APC/C) coactivators Cdc20 and Cdh1 to mediate the degradation of several key mitotic regulators independent of APC/C. Accordingly, its deficiency results in the elevated expression of these substrates leading to mitotic defects, genomic instability and tumorigenesis (Lee et al., 2015). Corroborating with these findings, we and others have previously similarly shown that Parkin dysfunction underlies several types of cancers (Cesari et al., 2003; Tay et al., 2010; Yeo et al., 2012). Although not directly related to the current review topic, the several recent reports collectively pointing towards a role for Parkin as a tumor suppressor suggest the interesting possibility that Parkin-related neurodegeneration may arise from aberrant cell cycle re-entry in post-mitotic neurons.
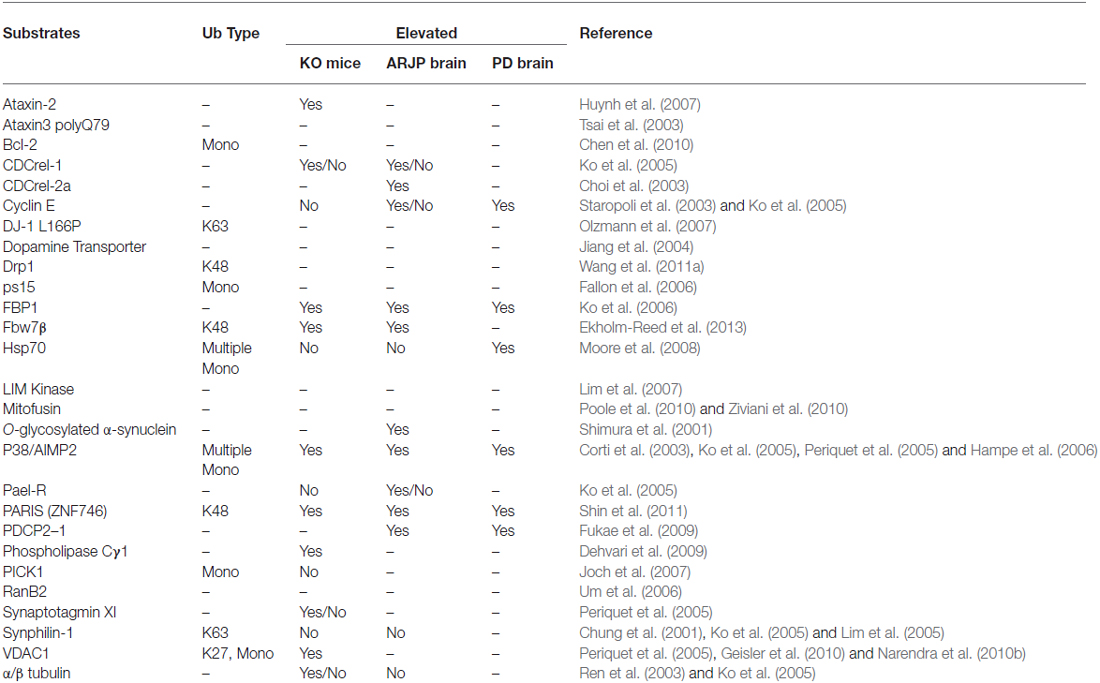
Table 1. A selection of reported Parkin substrates.
Notwithstanding that only a handful of substrates reported to date could fulfil the restricted criterion of a Parkin substrate (as mentioned above), what constitutes an “authentic” substrate for Parkin is really debatable. Although protein ubiquitination is classically associated with proteasome-mediated degradation, the existence of non-classic ubiquitin modifications such as K63-linked polyubiquitination would caution against the fixation on the traditional view that substrates of a ubiquitin ligase must exhibit an accelerated, proteasome-dependent turnover in the presence of the enzyme. This is particularly relevant to Parkin, which we and others have demonstrated to be a multifunctional enzyme capable of mediating alternative ubiquitin topologies such as monoubiquitination and K63-linked polyubiquitination—modifications that are typically uncoupled from the proteasome and often considered as “non-proteolytic” (Doss-Pepe et al., 2005; Lim et al., 2005; Hampe et al., 2006; Matsuda et al., 2006). Our results would suggest that the catalytic function of Parkin is not limited to targeting substrate for degradation by the proteasome. Thus, the lack of accumulation of an identified Parkin substrate in the brains of ARJP patients and Parkin-deficient models does not necessarily mean that it is less than an “authentic” substrate. For example, we have previously reported that Parkin-mediated polyubiquitination of synphilin-1 (an interactor of α-synuclein) normally occurs via K63-linked chains, which does not appear to affect its steady-state turnover (Lim et al., 2005). Not surprisingly, the level of synphilin-1 is neither appreciably altered in ARJP brains nor in brain tissues derived from Parkin null mice (Ko et al., 2005). A corollary to this is that proteasome-independent pathways may also be relevant to Parkin-related neurodegeneration. By virtue of its apparent dissociation from the proteasome, we have originally proposed that Parkin-mediated K63-linked ubiquitination may be involved in cargo diversion during proteasomal stress and accordingly, in the biogenesis of inclusion bodies associated with neurodegenerative diseases (Lim et al., 2006). Our proposal is consistent with the concept of “aggresomes”, which are juxtanuclear inclusion bodies formed in the presence of proteasomal stress and that have been suggested to act as staging grounds for the disposal of protein aggregates via the autophagic route (Kopito, 2000). Supporting our hypothesis, we found that Parkin-mediated K63 polyubiquitination of synphilin-1 promotes its aggregation into aggresome-like inclusion bodies (Lim et al., 2005). Corroborating our findings, Olzmann et al demonstrated that Parkin-mediated K63 polyubiquitination of misfolded DJ-1 couples the protein to the dynein motor complex via the histone deacetylase 6 (HDAC6) adaptor, thereby promoting its sequestration into aggresomes (Olzmann et al., 2007). Importantly, we further identified K63-linked polyubiquitin as a cargo selection signal for macroautophagy-mediated clearance of aggresomes (Tan et al., 2008a,b). Notably, Wang et al. (2012) demonstrated that the formation of aggresomes mediated by mutant superoxide dismutase 1 (SOD1) is dependent on ataxin 3-catalyzed editing of K63-linked polyubiquitin chain on SOD1 (presumably to a correct length) which supports our findings. In a similar development, Hao et al. (2013) demonstrated that the disposal of aggresomes requires the proteasomal deubiquitinating enzyme Poh1, which releases free (K63-linked) ubiquitin chains from aggresomes that activates HDAC6-dependent autophagy clearance of the cargo. In Poh1-deficient cells, aggresome clearance is blocked but may be rescued by the introduction of free K63-linked ubiquitin chains (via microinjection). Finally, by means of ubiquitin linkage-specific antibodies, we recently showed that proteasome inhibition indeed promotes K63-linked ubiquitination of proteins especially in Parkin-expressing cells (Lim et al., 2013). Importantly, we further demonstrated that the recruitment of Ubc13 (an E2 that mediates K63-linked polyubiquitin chain formation exclusively) by Parkin is selectively enhanced under conditions of proteasomal stress, which favors aggresome formation and thereby the clearance of Parkin substrates via autophagy (Lim et al., 2013). Thus, by being capable of mediating both proteasome-associated K48-polyubiquitination and macroautophagy-associated K63-linked polyubiquitination, Parkin may potentially act as an important triage between the two major cellular degradation systems (Figure 2). This multi-functionality of Parkin may in part help explain its apparent broad neuroprotective properties, as the flexibility of ubiquitin linkage usage presumably would allow the enzyme to adapt rapidly to changes in cellular environment.
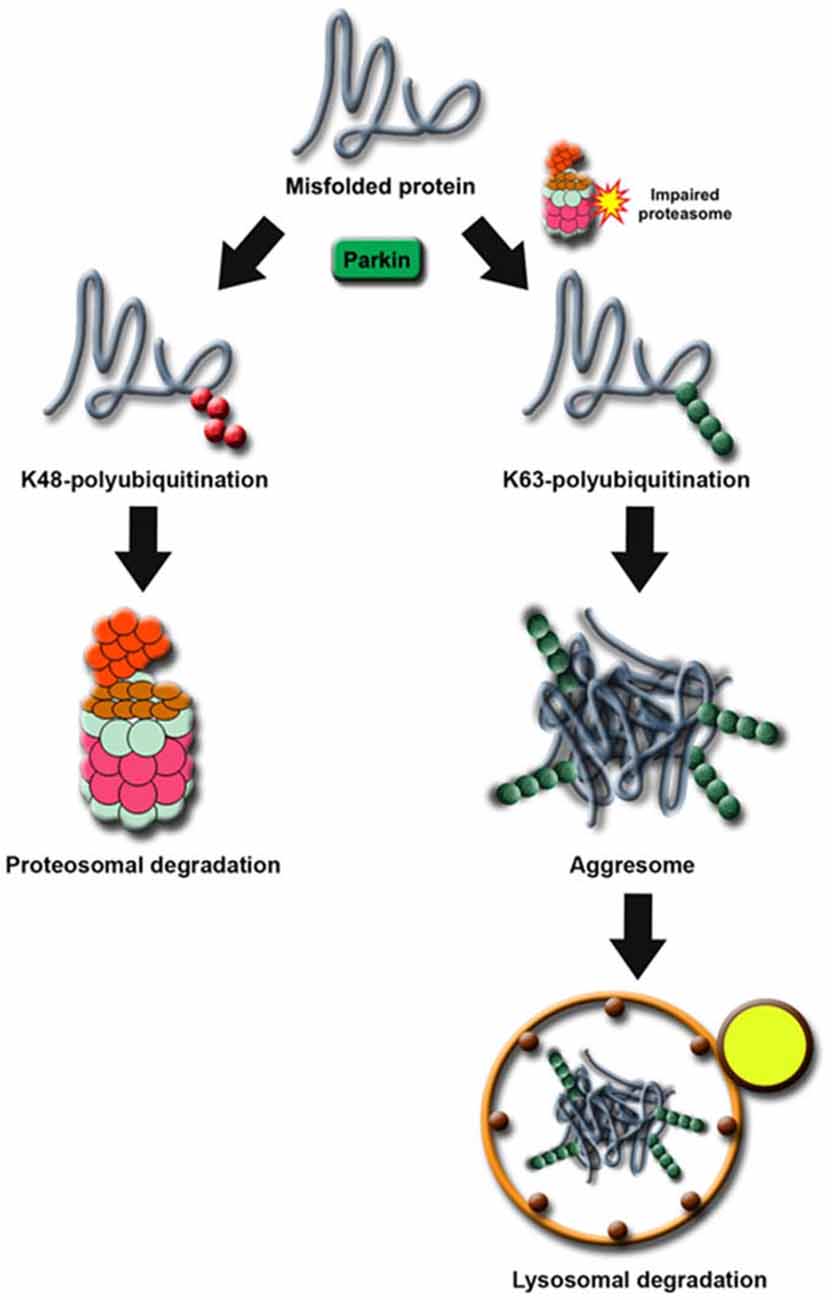
Figure 2. Proposed model of Parkin’s role as a triage between proteasome and autophagy degradation. Under normal cellular conditions, proteins destined for degradation by the proteasome are tagged with a chain of K48-linked ubiquitin. In times of proteolytic stress, the cell switches to K63-linked ubiquitination to divert the protein load originally targeted for proteasomal degradation away from the otherwise overloaded machinery. Parkin facilitates this switch by increasing its affinity for Ubc13 in the presence of proteasome dysfunction.
Parkin and Mitochondrial Homeostasis
One of the first hints that Parkin may play a role in mitochondrial homeostasis aside from its role as a regulator of protein turnover came from a study by Greene et al. (2003) in fruit flies. The authors analyzed adult Drosophila parkin null mutant and observed that the most prominent pathology is not in the brain but in the flight musculature of these mutant flies, which is plagued by muscle degeneration and pronounced mitochondrial lesions. Interestingly, pink1 null flies were subsequently found to phenocopy their parkin-deficient counterparts and importantly, parkin over-expression in pink1−/− flies were shown to be capable of rescuing all the mutant phenotypes tested, although the reverse, does not happen (Clark et al., 2006; Park et al., 2006), suggesting that Parkin acts in the same pathway but downstream of PINK1. Several follow-up studies in flies and other model systems that attempted to explain the mitochondrial phenotype focused initially on the role of Parkin/PINK1 pathway in mitochondrial dynamics, although whether the pathway promotes mitochondrial fission or fusion remains controversial to this date (for a recent review, see Burbulla et al., 2010). Besides potentially participating in mitochondrial dynamics, Parkin, as we have noted earlier, is also involved in mitochondrial biogenesis by virtue of its ability to regulate PGC-1α indirectly through its ability to down-regulate PARIS (Shin et al., 2011), a major transcriptional repressor of PGC-1α (Scarpulla, 2008). The repression of PGC-1α (a key regulator of mitochondrial biogenesis) by PARIS is expected to result in reduced mitochondrial mass, which if unregulated could compromise the ability of the cell to adapt to energy crises.
In recent years, much attention on the relationship between Parkin function and mitochondrial homeostasis has been shifted to its role in mitophagy. As several excellent reviews on Parkin/PINK1-mediated mitophagy have been written on this topic (for example, see Durcan and Fon, 2015; Pickrell and Youle, 2015), we will only briefly describe the model here, as depicted in Figure 3. Until recently, the model describes a linear sequence of events occurring in response to mitochondrial depolarization that culminates in their removal. According to the proposed model (Youle and Narendra, 2011), a key initial event that occurs upon mitochondrial depolarization is the selective accumulation of PINK1 on the outer membrane of the damaged organelle. This accumulation allows PINK1 to recruit Parkin (Okatsu et al., 2012), whose latent ubiquitin ligase activity becomes unmasked along the way in part due to its phosphorylation by PINK1 (Matsuda et al., 2010; Kondapalli et al., 2012). PINK1 also phosphorylates ubiquitin, which binds and activates Parkin (Kane et al., 2014; Koyano et al., 2014). We now know from several recent structural studies discussed above how the dual phosphorylation events mediated by PINK1 lead to the release of Parkin’s auto-inhibited state. Activated Parkin then promotes the ubiquitination and subsequent degradation of many outer mitochondrial membrane (OMM) proteins (Chan et al., 2011; Yoshii et al., 2011). During the process, Parkin-decorated mitochondria progressively cluster towards the peri-nucleus region to form “mito-aggresomes” (Lee et al., 2010), which by virtue of their association with lysosomal components are removed with time in an autophagy-dependent manner. Although this linear model explains the selectivity of the mitophagy process rather elegantly, recent discoveries revealed that a more complicated network of molecular interactions is involved in the disposal of unwanted mitochondria (Heo et al., 2015; Lazarou et al., 2015). In essence, these studies demonstrated that mitochondrial-localized PINK1 is able to recruit the autophagy receptors optineurin (OPTN) and NDP52 in the absence of Parkin and that this phenomenon alone is sufficient to trigger mitophagy, albeit at a low level. The recruitment of OPTN and NDP52 is dependent on PINK1-mediated phosphorylation of ubiquitin, which serves as autophagy signal on the mitochondria. Parkin promotes the process by amplifying the phospho-ubiquitin level generated by PINK1 that otherwise occurs at a low level due to the limited basal ubiquitin availability on mitochondria. In so doing, Parkin mediates a positive feed-forward cycle that generates more ubiquitin substrates on the OMM for PINK1 to phosphorylate that in turn facilitates the recruitment of autophagy receptors thereby enhancing the mitophagy process. Adding to this complexity is the recent elucidation by several groups that deubiquitinating enzymes (DUBs) such as USP8, USP15, USP30 and USP35 can counteract Parkin-mediated mitochondrial ubiquitination and consequently mitophagy, although they generally do not affect the recruitment of Parkin to damaged mitochondria (Bingol et al., 2014; Cornelissen et al., 2014; Durcan et al., 2014; Wang et al., 2015).
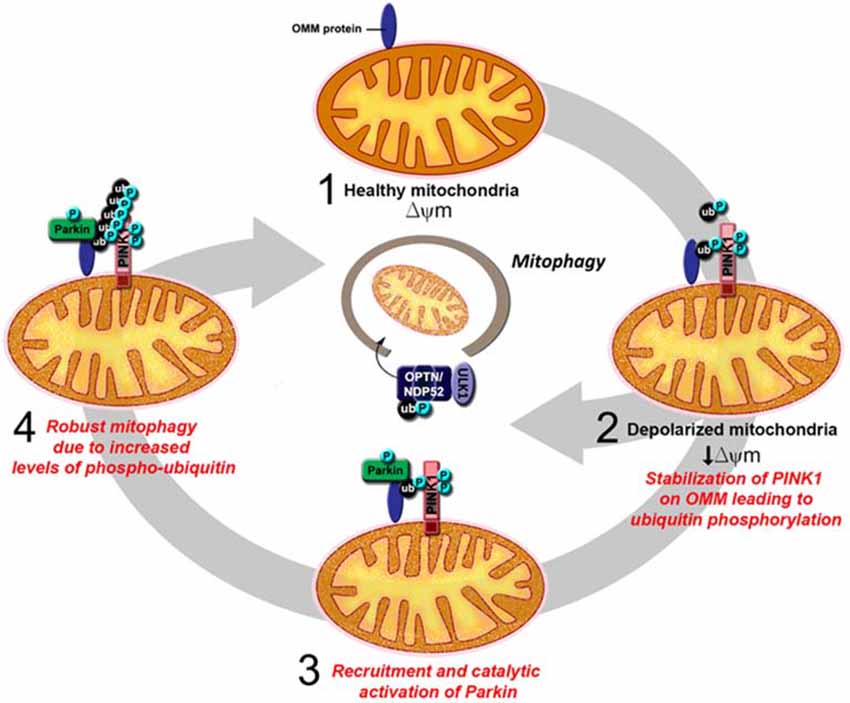
Figure 3. An updated model of PINK1/Parkin-mediated mitophagy. (1) In healthy mitochondria, there is no accumulation of PINK1 on the outer mitochondrial membrane (OMM) as the protein is rapidly imported, processed and degraded. (2) Upon mitochondrial depolarization, full length PINK1 accumulates on the OMM leading to the phosphorylation of ubiquitin on the surface of the mitochondria. This results in the recruitment of the autophagy receptors optineurin (OPTN) and NDP52 and the consequent activation of the mitophagy process, albeit at low level. (3) Parkin is also recruited to the OMM, whose latent activity becomes unmasked due to its interaction with phosphorylated ubiquitin and its phosphorylation by PINK1. (4) Activated Parkin promotes the polyubiqutination of mitochondrial substrates that in turn provides more ubiquitin substrates for PINK1 to phosphorylate. This amplifies the signal for the recruitment of autophagy receptors and results in robust mitophagy.
Notwithstanding the elegance of the proposed mitophagy model, whether the process occurs in post-mitotic neurons that are obligatorily dependent on mitochondrial respiration is contentious. Intuitively, one would logically accept the need for neurons to embark on mitophagy for QC reasons but perhaps in a more limited fashion. An example of this scenario is when damage of mitochondria occurs in at the distal ends of neurons. In this case, the motility of the restricted population of damaged mitochondria has been shown to be arrested. This occurs via the degradation of the mitochondrial motor adaptor protein Miro in a pathway also mediated by PINK1 and Parkin, the effect of which leading to arrested motility suggests that PINK1/Parkin pathway may quarantine damaged mitochondria prior to their clearance (Wang et al., 2011b; Ashrafi et al., 2014). Interestingly, a recent study provided evidence that Parkin-mediated neuronal mitophagy does occur in vivo. The investigators found that the brains of Parkin mutant flies exhibit a significantly decreased rate of mitochondrial protein turnover, which is similar to that produced by general autophagy blockade induced by the genetic ablation of atg7, suggesting that Parkin indeed promotes mitochondrial turnover through autophagy and that the process is physiologically relevant, albeit in the fly (Vincow et al., 2013).
Apart from mitophagy, PINK1/Parkin-dependent mitochondrial QC also occurs via an alternative pathway involving mitochondria-derived vesicles (MDVs). During various forms of mitochondrial stress, MDVs are formed from the budding of membranes from the mitochondria, which mediate the transport of damaged mitochondrial proteins and lipids to other intracellular organelles. The nature of mitochondrial stress is an important determinant of the constituents of its cargo, which in turn determine the fate of MDVs. For example, MDVs containing MAPL, an OMM-anchored protein ligase, exclude other OMM markers such as TOM20 and are selectively targeted to a subpopulation of peroxisomes (Neuspiel et al., 2008). Other MDVs that are not destined to the peroxisome fused instead with late endosomes (or multivesicular bodies), which ultimately target them for lysosomal degradation (Soubannier et al., 2012a). Importantly, the generation of late endosome-targeted mitochondria is dependent on PINK1 and Parkin and occurs under moderate mitochondrial stress as opposed to acute mitochondrial toxin treatment that leads to mitophagy. Furthermore, this phenomenon that is independent of ATG5 and LC3 does not require mitochondrial depolarization, suggesting that this pathway complements mitophagy and contributes to mitochondrial QC (Soubannier et al., 2012a,b; Sugiura et al., 2014). In another scenario, we have recently found that chronic, low dose CCCP treatment promotes Parkin-mediated mitochondrial fusion instead of mitophagy (Norris et al., 2015). In principle, active fusion could protect mitochondrial integrity by complementing damaged mitochondria with their healthy counterparts or by limiting the production of ROS. This adaptive mitochondrial fusion strategy that we have found in the presence of low but chronic mitochondrial stress is likely more physiologically relevant to the disease than high dose mitochondrial toxins commonly used to induce acute neuronal dysfunction. Importantly, we further showed that Parkin, PINK1 and α-synuclein form a regulatory circuit to regulate mitochondrial stress response, thus functionally connecting three key PD-linked genes to the pathogenesis of PD. To clarify, our result does not exclude the need for neuronal mitophagy, especially when mitochondria become chronically damage with time. It does however emphasize the need to pay close attention to the conditions used in mitophagy-related experiments and that mitochondrial QC involves not just its turnover. Whatever it is, it is evident from various studies above that Parkin appears to be involved in the entire spectrum of mitochondrial dynamics, i.e., from biogenesis, fusion/fission, intracellular movements to its exit from the cell.
Parkin Dysregulation and PD
PD is a prevalent neurodegenerative disease that is characterized clinically by a constellation of motoric deficits arising principally from the loss of midbrain DA neurons in the substantia nigra pars compacta (SNpc) that is often accompanied by the presence of LBs in surviving neurons in the SN and other affected brain areas (Braak et al., 2003). Given the pivotal role that Parkin plays in regulating protein and mitochondrial homeostasis, and that DA neurons are usually in a heighten state of stress relative to other neuronal populations, it is perhaps not surprising to note that Parkin dysregulation can result in PD-associated DA neurodegeneration. Moreover, several studies also implicated a more direct role for Parkin in neuroprotective signaling. For example, Parkin-mediated ubiquitination was demonstrated to be important for the activation of major cellular pro-survival pathways such as the NF-κB pathway (Henn et al., 2007), or the suppression of stress-related mitogen-activated protein kinase signaling (e.g., JNK and p38; probably indirectly through its ability to counteract oxidative stress; Cha et al., 2005; Ren et al., 2009). Accordingly, loss of Parkin function would be dire for neuronal survival.
Using phosphorylated ubiquitin as a surrogate marker for mitophagy (or at least the activation of which), Fiesel et al. (2015) recently presented evidence that mitophagy is defective in the brains of PD patients. By means of phospho-specific ubiquitin immunostaining that only recognizes the presence of S65-phosphorylated ubiquitin (pS65-Ub), the authors found that the level of pS65-Ub (that is barely detectable under basal condition) is elevated in cells in response to mitochondrial stress (Fiesel et al., 2015). Importantly, they further found that pS65-Ub accumulates in human brain during aging and in post-mortem PD brain samples. Whether a (hitherto unknown) phosphatase returning pS65-Ub to its basal state is deficient in aged or sporadic PD brains that had resulted in the observed increase in pS65-Ub level is presently unclear. Nonetheless, mitophagy defects seem to be associated with PD pathogenesis. Obviously, Parkin dysfunction can also trigger impairments in neuronal protein homeostasis. Again, using PARIS as an example to illustrate this, its overexpression (via stereotactic injection of viral vector encoding PARIS) in the SN of mice was shown to result in a selective loss of DA neurons. This phenotype can be rescued by either Parkin or PGC-1α co-expression, suggesting that PARIS upregulation may underlie neurodegeneration due to Parkin activation (Shin et al., 2011). Along the same vein, Ekholm-Reed et al. (2013) recently found that the SCF substrate adapter Fbw7β is a target of Parkin during acute oxidative stress and that it accumulates in Parkin-deficient neurons. A consequence of Fbw7β accumulation is that the level of Mcl-1, an anti-apoptotic protein substrate of SCFFbw7β-mediated degradation, is depleted. Loss of Parkin may thus lead to the death of DA neurons through unregulated SCFFbw7β-mediated ubiquitination-dependent proteolysis of Mcl-1, which again illustrates the need to for Parkin to maintain tight homeostatic control over its substrates for optimal neuronal function. However, a more recent study demonstrated that Mcl-1 is also targeted by Parkin for degradation (Carroll et al., 2014) so the verdict regarding the relationship between Parkin and the anti-apoptotic protein Mcl-1 is currently open.
Given the neuroprotective function of Parkin, understanding how Parkin dysregulation occurs in the PD brain is an important endeavor to pursue that has obvious therapeutic implications. From the studies conducted to date, we now appreciate that Parkin dysfunction could happen under a variety of conditions. To begin with, several disease-associated mutations of Parkin directly result in the loss of its enzymatic activity. These include exon deletions that lead to the considerable truncation of the protein and missense mutations that reside on its catalytic RING2 domain. In cases where the missense mutation occurs outside of Parkin’s RING2 domain (i.e., not affecting its catalytic competency), its solubility is often altered that consequently promotes its aggregation and thereby immobilization into inclusion bodies (Ardley et al., 2003; Cookson et al., 2003; Gu et al., 2003; Muqit et al., 2004; Sriram et al., 2005; Wang et al., 2005b; Hampe et al., 2006). Interestingly, among the aggregation-producing Parkin mutations is the R275W substitution, which frequently occurs in the heterozygous state. Although the implied pathogenicity of single heterozygous Parkin mutation contradicts the widely accepted notion that Parkin mutations transmit in a classical autosomal recessive manner, one could envisage that Parkin haploinsufficiency arising from such mutations could increase the risk of heterozygous Parkin carriers for PD especially in view of the importance of optimal Parkin expression to neuroprotection. Indeed, several lines of evidence support the proposal that a single allelic hit on Parkin might be sufficient to cause disease (Hilker et al., 2001; Walter et al., 2004; Buhmann et al., 2005). Alternatively, some mutations of Parkin may compromise its function by destabilizing the protein and accelerating its degradation via the proteasome (Schlehe et al., 2008).
That Parkin mutations transmit in a recessive manner would suggest that the loss of Parkin function predisposes DA neurons to degeneration. An important corollary to this is that any post-translational event that promotes the loss of Parkin function could potentially mimic the effects brought about by overt mutations and be just as detrimental. Further, in view of the suggested contribution of Parkin haploinsufficiency to disease risk, it is reasonable to assume that the down-regulation of normal Parkin function mediated by such events need not result in the total abolition of its enzymatic activity to elicit a pathogenic effect. Notably, we and others have found that a wide variety of PD-linked stressors, including those that produce oxidative and nitrosative stress, induce Parkin solubility alterations and thereby its aggregation in a manner that is analogous to that brought about by several of its missense mutations (Winklhofer et al., 2003; LaVoie et al., 2005; Wang et al., 2005a). Remarkably, dopamine also modifies Parkin in a similar fashion (LaVoie et al., 2005; Wang et al., 2005a). Further, Parkin appears to be uniquely susceptible to dopamine-induced modifications compared to several other related E3 members such as HHARI, Cbl and CHIP (LaVoie et al., 2005, 2007; Wong et al., 2007). Like haploinsufficiency, the biochemical depletion of soluble Parkin levels is expected to increase the vulnerability of susceptible neurons to degeneration. Interestingly, normal Parkin in the brain also becomes progressively more detergent-insoluble with aging (Pawlyk et al., 2003), which may provide an explanation to why age represents a risk factor for PD. Besides stress-induced modifications, Parkin phosphorylation is another post-translational modification that could alter its activity. Unlike PINK1-mediated phosphorylation of Parkin that activates the enzymes, previous studies have demonstrated that Parkin phosphorylation by casein kinase 1 (CK1; at Ser-101, -127, -131, -136, -296, -378) or cyclin-dependent kinase 5 (Cdk5; at Ser-131) down-regulates its activity, and that compound phosphorylation of Parkin by both kinases further leads to its aggregation (Yamamoto et al., 2005; Avraham et al., 2007; Rubio de la Torre et al., 2009), Consistent with this, Parkin phosphorylation was found to be elevated in distinct regions of sporadic PD brains and correlates with increased levels of p25, the activator of CDK5 (Rubio de la Torre et al., 2009). Notwithstanding this, how the various phosphoserines on Parkin modulate its activity remains unclear, especially when the majority of these residues lie outside the regulatory RING0 box of the linker segment of Parkin. Similar to serine phosphorylation, tyrosine phosphorylation of Parkin at residue 143 (Y143) by the src family kinase member, c-Abl, similarly inactivates its enzyme activity and compromises its protective function both in vitro and in vivo. This form of modified Parkin is also found elevated in human post-mortem PD brains (Ko et al., 2010). Besides post-translational modifications, a number of Parkin interactors have also been demonstrated to be capable of inactivating its E3 ligase activity. These include the bcl-2-associated athanogene 5 (BAG5) and 14–3–3η whose association with Parkin reduces its activity and Nrdp1, a RING-finger containing ubiquitin ligase promotes Parkin degradation and depletes the availability of Parkin to the cell (Kalia et al., 2004; Zhong et al., 2005; Sato et al., 2006). These events if unregulated would obviously be detrimental to Parkin’s protective function.
Taken together, it is apparent that loss of Parkin function is not limited to those induced by disease-causing mutations, but also includes several biochemical and protein-protein modifications that can either alter the catalytic function of the E3 ligase directly, or indirectly through promoting its aggregation or degradation. Collectively, these mutation-independent modifications that inactivate Parkin activity provide a mechanism for Parkin dysfunction that is relevant to the pathogenesis of sporadic PD. At the same time, they also suggest therapeutic directions in managing the disease. Interesting, whereas S-nitrosylation of Parkin leads to its inactivation, sulfhydration of Parkin has recently been shown to enhance its activity (Vandiver et al., 2013). Moreover, the levels of sulfhydrated Parkin is markedly depleted in PD brains while nitrosylation increased in tandem, suggesting that hydrogen sulfide donors may counteract S-nitrosylation-mediated inactivation of the enzyme and may accordingly be therapeutic. Similarly, the c-Abl inhibitor, nilotinib, is also protective against DA neuronal loss in a toxin-induced model of PD, although curiously the level of phospho-Y143 Parkin appears to be unchanged (Karuppagounder et al., 2014). These recent studies suitably illustrate the importance of mechanistic insights in guiding therapeutics development.
Parkin Dysregulation and Other Neurodegenerative Diseases
Besides PD, accumulating evidence suggests that Parkin dysfunction may also contribute to the pathogenesis of other neurodegenerative diseases including AD and ALS. Pathologically, the AD brain is characterized by the presence of extracellular amyloid aggregates and intra-neuronal hyperphosphorylated tau aggregates known as neurofibrillary tangles (NFTs). Among the first hints suggesting a relationship between Parkin and AD is the finding by Nemes et al. (2004) that Parkin is a major component cross-linked by gamma-glutamyl-epsilon-lysine bonds in AD-associated NFT. More recently, Corsetti et al. (2015) showed that an N-terminal truncated tau species found in AD individuals can induce deregulated mitophagy by aberrant recruitment of Parkin that result in excessive mitochondrial turnover, synaptic deterioration and neuronal death, thus providing a Parkin-related mechanism in the pathogenesis of AD. Notably, a number of earlier studies have already documented abnormal tau hyperphosphorylation in patients with Parkin mutations (van de Warrenburg et al., 2001; Sánchez et al., 2002), suggesting that Parkin deficiency may precipitate tau pathology. Consistent with this, the accumulation of tau was observed during ageing in Parkin null mice (Rodríguez-Navarro et al., 2007). Interestingly, overexpression of mutant tau in mice against Parkin null background promotes cerebral and systemic amyloidosis (Rodríguez-Navarro et al., 2008) that is accompanied by impairments in memory and exploratory behaviors (Navarro et al., 2008), suggesting that Parkin may also affect amyloidogenesis. Indeed, Rosen et al. (2006) reported that Parkin could lower the level of amyloid burden and protect against its toxicity (albeit in muscle cells). In a series of follow up studies, the same group and others demonstrated the ability of Parkin to facilitate the clearance of the pathogeneic amyloid β42 by reversing its negative effects on the proteasome and also by enhancing beclin-dependent autophagy (Burns et al., 2009; Rosen et al., 2010; Khandelwal et al., 2011). Not surprisingly, Parkin overexpression in an AD mouse model (that is based on altered amyloid precursor protein processing, i.e., APPswe/PSEN1ΔE9 mutant) was found to reduce β-amyloid load and improve hippocampal long term potentiation (Hong et al., 2014). Similar outcomes were also observed when AD animals were treated with nilotinib, which increases parkin solubility and its interaction with Beclin one leading to enhanced amyloid clearance and cognitive performance (Lonskaya et al., 2013).
In a parallel development, several groups working on FUS/TLS and TDP-43 related ALS found an association between these ALS-mutants and Parkin. In an elegant functional genomics study, Yeo’s group found that among the 45 RNA species that were reduced following depletion of TDP-43 or FUS/TLS, one of these encodes Parkin (Lagier-Tourenne et al., 2012). Consistent with this, mice carrying a knock-in copy of mutant TDP-43 exhibit reduced level of Parkin expression (Stribl et al., 2014). Thus, Parkin deficiency may also be interwoven with the pathogenic events in ALS. Indeed, Hebron et al. (2014) recently demonstrated the ability of Parkin to reverse TDP-43-mediated cell death. Apparently, mitochondrial dysfunction may be an underlying denominator responsible for neuronal death in ALS as well. Notably, altered mitochondrial QC was observed in the TDP-43 knock in mutant mice (Stribl et al., 2014). Moreover, mutations in the autophagy/mitophagy receptor OPTN are associated with ALS (Maruyama et al., 2010) and mutant OPTN are apparently incapable of facilitating Parkin/PINK-mediated mitophagy (Wong and Holzbaur, 2014). Finally, despite the anatomical and functional relatedness of HD and PD (i.e., both affect the basal ganglia circuitry and result in movement disorders), only a handful of reports suggest that Parkin deficiency may be a contributor to striatal neurodegeneration in HD. In an earlier study, Parkin was found to protect against the toxicity induced by expanded polyglutamine proteins (Tsai et al., 2003). Subsequent to this, another group found that partial suppression of Parkin aggravates the phenotype in the huntingtin R6/1 mutant mice, albeit only slightly (Rubio et al., 2009). More recently, Parkin/PINK-mediated mitophagy was shown to promote neuroprotection in fly and mouse models of HD (Khalil et al., 2015).
In sum, it is evident from the above discussion that Parkin deficiency is relevant not just to PD but may also be relevant to several other major neurodegenerative diseases. Obviously, this begs the question on why mutations in Parkin are uniquely causative of PD. Although we remain unclear about this, one possible explanation is that human DA neurons are selectively vulnerable to the loss of Parkin function perhaps by virtue of their heighten stress, i.e., Parkin deficiency in this case is the principal driver of disease pathogenesis. In AD and ALS, the depletion of Parkin occurs against the backdrop of other pathogenic factors; the combination of which is likely to aggravate neuronal loss, i.e., Parkin dysfunction is the co-driver. Arguably, some may consider Parkin’s role in these diseases as a passenger, i.e., a consequence of AD or ALS-related pathogenic factors going awry, although several reports demonstrated that Parkin overexpression is beneficial in these cases. Whether Parkin is acting as a driver, co-driver or passenger, we probably could all accept that its optimal expression is important for neuronal homeostasis.
Concluding Remarks
It has been 15 years or so since the function of Parkin was originally elucidated. Certainly, we now know much more about the various aspects of the E3 than we did before. Yet, it remains rather amazing at the same time to note that a single ubiquitin ligase could possess such multi-functionality that enables it to perform diverse types of ubiquitination (including mono, K48- and K63-linked ubiquitin modifications) to support a plethora of cellular processes, as well as lend itself as a broad spectrum neuroprotectant. As articulated above, emerging evidence also suggests its relevance to several major neurodegenerative diseases besides PD, and interestingly, to cancer development as well. Thus, Parkin appears to be one of a kind. Accordingly, its activity regulation must be exquisitely controlled in different cell types under different conditions for optimal cellular function. On a related note, given the amazing properties of this RING-containing E3, we thought that it would be appropriate to end this article by suggesting that Parkin be (affectionately) nicknamed as the “Lord of the RING”. Obviously, we are bias here and concede that some readers may consider Parkin as just a “RING-bearer”. Be it the lord or a RING-bearer, we cannot deny the fact that Parkin does have some very enchanting properties.
Author Contributions
CZ and LH drafted the manuscript. CZ generated the table. LH illustrated the figures. TPY provided intellectual inputs and critiques, and contributed to text. KLL conceptualized the flow of the article, revised the draft, polished the text and finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Medical Research Council—Translational Clinical Research Program in Parkinson’s disease, Collaborative Basic Research Grant (KLL) and New Investigator Grant (CZ), and NIH 2R01-NS054022 (TPY). Ms. LH is supported by a graduate scholarship from the National University of Singapore Graduate School for Integrative Sciences and Engineering.
References
Ardley, H. C., Scott, G. B., Rose, S. A., Tan, N. G., Markham, A. F., and Robinson, P. A. (2003). Inhibition of proteasomal activity causes inclusion formation in neuronal and non-neuronal cells overexpressing Parkin. Mol. Biol. Cell 14, 4541–4556. doi: 10.1091/mbc.e03-02-0078
Ashrafi, G., Schlehe, J. S., LaVoie, M. J., and Schwarz, T. L. (2014). Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 206, 655–670. doi: 10.1083/jcb.201401070
Avraham, E., Rott, R., Liani, E., Szargel, R., and Engelender, S. (2007). Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J. Biol. Chem. 282, 12842–12850. doi: 10.1074/jbc.m608243200
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Buhmann, C., Binkofski, F., Klein, C., Büchel, C., Van Eimeren, T., Erdmann, C., et al. (2005). Motor reorganization in asymptomatic carriers of a single mutant Parkin allele: a human model for presymptomatic parkinsonism. Brain 128, 2281–2290. doi: 10.1093/brain/awh572
Burbulla, L. F., Krebiehl, G., and Krüger, R. (2010). Balance is the challenge—the impact of mitochondrial dynamics in Parkinson’s disease. Eur. J. Clin. Invest. 40, 1048–1060. doi: 10.1111/j.1365-2362.2010.02354.x
Burns, M. P., Zhang, L., Rebeck, G. W., Querfurth, H. W., and Moussa, C. E. (2009). Parkin promotes intracellular Abet a1–42 clearance. Hum. Mol. Genet. 18, 3206–3216. doi: 10.1093/hmg/ddp258
Carroll, R. G., Hollville, E., and Martin, S. J. (2014). Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 9, 1538–1553. doi: 10.1016/j.celrep.2014.10.046
Cesari, R., Martin, E. S., Calin, G. A., Pentimalli, F., Bichi, R., McAdams, H., et al. (2003). Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc. Natl. Acad. Sci. U S A 100, 5956–5961. doi: 10.1073/pnas.0931262100
Cha, G. H., Kim, S., Park, J., Lee, E., Kim, M., Lee, S. B., et al. (2005). Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc. Natl. Acad. Sci. U S A 102, 10345–10350. doi: 10.1073/pnas.0500346102
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chaugule, V. K., Burchell, L., Barber, K. R., Sidhu, A., Leslie, S. J., Shaw, G. S., et al. (2011). Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 30, 2853–2867. doi: 10.1038/emboj.2011.204
Chen, D., Gao, F., Li, B., Wang, H., Xu, Y., Zhu, C., et al. (2010). Parkin Mono-ubiquitinates Bcl-2 and regulates autophagy. J. Biol. Chem. 285, 38214–38223. doi: 10.1074/jbc.M110.101469
Chew, K. C., Matsuda, N., Saisho, K., Lim, G. G., Chai, C., Tan, H. M., et al. (2011). Parkin mediates apparent E2-independent monoubiquitination in vitro and contains an intrinsic activity that catalyzes polyubiquitination. PLoS One 6:e19720. doi: 10.1371/journal.pone.0019720
Choi, P., Snyder, H., Petrucelli, L., Theisler, C., Chong, M., Zhang, Y., et al. (2003). SEPT5_v2 is a parkin-binding protein. Brain Res. Mol. Brain Res. 117, 179–189. doi: 10.1016/s0169-328x(03)00318-8
Chung, K. K., Zhang, Y., Lim, K. L., Tanaka, Y., Huang, H., Gao, J., et al. (2001). Parkin ubiquitinates the α-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat. Med. 7, 1144–1150. doi: 10.1038/nm1001-1144
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Cookson, M. R., Lockhart, P. J., McLendon, C., O’Farrell, C., Schlossmacher, M., and Farrer, M. J. (2003). RING finger 1 mutations in Parkin produce altered localization of the protein. Hum. Mol. Genet. 12, 2957–2965. doi: 10.1093/hmg/ddg328
Cornelissen, T., Haddad, D., Wauters, F., Van Humbeeck, C., Mandemakers, W., Koentjoro, B., et al. (2014). The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 23, 5227–5242. doi: 10.1093/hmg/ddu244
Corsetti, V., Florenzano, F., Atlante, A., Bobba, A., Ciotti, M. T., Natale, F., et al. (2015). NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum. Mol. Genet. 24, 3058–3081. doi: 10.1093/hmg/ddv059
Corti, O., Hampe, C., Koutnikova, H., Darios, F., Jacquier, S., Prigent, A., et al. (2003). The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration. Hum. Mol. Genet. 12, 1427–1437. doi: 10.1093/hmg/ddg159
Dawson, T. M., and Dawson, V. L. (2014). Parkin plays a role in sporadic Parkinson’s disease. Neurodegener. Dis. 13, 69–71. doi: 10.1159/000354307
Dehvari, N., Sandebring, A., Flores-Morales, A., Mateos, L., Chuan, Y.-C., Goldberg, M. S., et al. (2009). Parkin-mediated ubiquitination regulates phospholipase C-γ1. J. Cell. Mol. Med. 13, 3061–3068. doi: 10.1111/j.1582-4934.2008.00443.x
Doss-Pepe, E. W., Chen, L., and Madura, K. (2005). Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J. Biol. Chem. 280, 16619–16624. doi: 10.1074/jbc.m413591200
Durcan, T. M., and Fon, E. A. (2015). The three ’P’s of mitophagy: PARKIN, PINK1 and post-translational modifications. Genes Dev. 29, 989–999. doi: 10.1101/gad.262758.115
Durcan, T. M., Tang, M. Y., Pérusse, J. R., Dashti, E. A., Aguileta, M. A., McLelland, G. L., et al. (2014). USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 33, 2473–2491. doi: 10.15252/embj.201489729
Ekholm-Reed, S., Goldberg, M. S., Schlossmacher, M. G., and Reed, S. I. (2013). Parkin-dependent degradation of the F-box protein Fbw7beta promotes neuronal survival in response to oxidative stress by stabilizing Mcl-1. Mol. Cell. Biol. 33, 3627–3643. doi: 10.1128/MCB.00535-13
Fallon, L., Bélanger, C. M. L., Corera, A. T., Kontogiannea, M., Regan-Klapisz, E., Moreau, F., et al. (2006). A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat. Cell Biol. 8, 834–842. doi: 10.1038/ncb1441
Fiesel, F. C., Ando, M., Hudec, R., Hill, A. R., Castanedes-Casey, M., Caulfield, T. R., et al. (2015). (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 16, 1114–1130. doi: 10.15252/embr.201540514
Fukae, J., Sato, S., Shiba, K., Sato, K.-I., Mori, H., Sharp, P. A., et al. (2009). Programmed cell death-2 isoform1 is ubiquitinated by parkin and increased in the substantia nigra of patients with autosomal recessive Parkinson’s disease. FEBS Lett. 583, 521–525. doi: 10.1016/j.febslet.2008.12.055
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B., and Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U S A 100, 4078–4083. doi: 10.1073/pnas.0737556100
Gu, W. J., Corti, O., Araujo, F., Hampe, C., Jacquier, S., Lücking, C. B., et al. (2003). The C289G and C418R missense mutations cause rapid sequestration of human Parkin into insoluble aggregates. Neurobiol. Dis. 14, 357–364. doi: 10.1016/j.nbd.2003.08.011
Hampe, C., Ardila-Osorio, H., Fournier, M., Brice, A., and Corti, O. (2006). Biochemical analysis of Parkinson’s disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum. Mol. Genet. 15, 2059–2075. doi: 10.1093/hmg/ddl131
Hao, R., Nanduri, P., Rao, Y., Panichelli, R. S., Ito, A., Yoshida, M., et al. (2013). Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 51, 819–828. doi: 10.1016/j.molcel.2013.08.016
Hebron, M., Chen, W., Miessau, M. J., Lonskaya, I., and Moussa, C. E. (2014). Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J. Neurochem. 129, 350–361. doi: 10.1111/jnc.12630
Henn, I. H., Bouman, L., Schlehe, J. S., Schlierf, A., Schramm, J. E., Wegener, E., et al. (2007). Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J. Neurosci. 27, 1868–1878. doi: 10.1523/jneurosci.5537-06.2007
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Hilker, R., Klein, C., Ghaemi, M., Kis, B., Strotmann, T., Ozelius, L. J., et al. (2001). Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann. Neurol. 49, 367–376. doi: 10.1002/ana.74.abs
Hong, X., Liu, J., Zhu, G., Zhuang, Y., Suo, H., Wang, P., et al. (2014). Parkin overexpression ameliorates hippocampal long-term potentiation and beta-amyloid load in an Alzheimer’s disease mouse model. Hum. Mol. Genet. 23, 1056–1072. doi: 10.1093/hmg/ddt501
Huynh, D. P., Nguyen, D. T., Pulst-Korenberg, J. B., Brice, A., and Pulst, S. M. (2007). Parkin is an E3 ubiquitin-ligase for normal and mutant ataxin-2 and prevents ataxin-2-induced cell death. Exp. Neurol. 203, 531–541. doi: 10.1016/j.expneurol.2006.09.009
Imai, Y., Soda, M., and Takahashi, R. (2000). Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 275, 35661–35664. doi: 10.1074/jbc.c000447200
Jiang, H., Jiang, Q., and Feng, J. (2004). Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J. Biol. Chem. 279, 54380–54386. doi: 10.1074/jbc.m409282200
Joch, M., Ase, A. R., Chen, C. X. Q., MacDonald, P. A., Kontogiannea, M., Corera, A. T., et al. (2007). Parkin-mediated Monoubiquitination of the PDZ Protein PICK1 regulates the activity of acid-sensing ion channels. Mol. Biol. Cell 18, 3105–3118. doi: 10.1091/mbc.e05-11-1027
Kalia, S. K., Lee, S., Smith, P. D., Liu, L., Crocker, S. J., Thorarinsdottir, T. E., et al. (2004). BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron 44, 931–945. doi: 10.1016/j.neuron.2004.11.026
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Karuppagounder, S. S., Brahmachari, S., Lee, Y., Dawson, V. L., Dawson, T. M., and Ko, H. S. (2014). The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 4:4874. doi: 10.1038/srep04874
Khalil, B., El Fissi, N., Aouane, A., Cabirol-Pol, M. J., Rival, T., and Liévens, J. C. (2015). PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 6:e1617. doi: 10.1038/cddis.2014.581
Khandelwal, P. J., Herman, A. M., Hoe, H. S., Rebeck, G. W., and Moussa, C. E. (2011). Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum. Mol. Genet. 20, 2091–2102. doi: 10.1093/hmg/ddr091
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608.
Ko, H. S., Kim, S. W., Sriram, S. R., Dawson, V. L., and Dawson, T. M. (2006). Identification of far upstream element-binding protein-1 as an authentic parkin substrate. J. Biol. Chem. 281, 16193–16196. doi: 10.1074/jbc.c600041200
Ko, H. S., Lee, Y., Shin, J. H., Karuppagounder, S. S., Gadad, B. S., Koleske, A. J., et al. (2010). Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc. Natl. Acad. Sci. U S A 107, 16691–16696. doi: 10.1073/pnas.1006083107
Ko, H. S., von Coelln, R., Sriram, S. R., Kim, S. W., Chung, K. K., Pletnikova, O., et al. (2005). Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J. Neurosci. 25, 7968–7978. doi: 10.1523/jneurosci.2172-05.2005
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. doi: 10.1016/s0962-8924(00)01852-3
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kubo, S., Hatano, T., Takanashi, M., and Hattori, N. (2013). Can parkin be a target for future treatment of Parkinson’s disease? Expert Opin. Ther Targets 17, 1133–1144. doi: 10.1517/14728222.2013.827173
Kumar, A., Aguirre, J. D., Condos, T. E., Martinez-Torres, R. J., Chaugule, V. K., Toth, R., et al. (2015). Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 34, 2506–2521. doi: 10.15252/embj.201592337
Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R., Vu, A. Q., Baughn, M., Huelga, S. C., et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497. doi: 10.1038/nn.3230
LaVoie, M. J., Cortese, G. P., Ostaszewski, B. L., and Schlossmacher, M. G. (2007). The effects of oxidative stress on parkin and other E3 ligases. J. Neurochem. 103, 2354–2368. doi: 10.1111/j.1471-4159.2007.04911.x
LaVoie, M. J., Ostaszewski, B. L., Weihofen, A., Schlossmacher, M. G., and Selkoe, D. J. (2005). Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 11, 1214–1221. doi: 10.1038/nm1314
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, S. B., Kim, J. J., Nam, H. J., Gao, B., Yin, P., Qin, B., et al. (2015). Parkin regulates mitosis and genomic stability through Cdc20/Cdh1. Mol. Cell 60, 21–34. doi: 10.1016/j.molcel.2015.08.011
Lee, J. Y., Nagano, Y., Taylor, J. P., Lim, K. L., and Yao, T. P. (2010). Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation and HDAC6-dependent mitophagy. J. Cell Biol. 189, 671–679. doi: 10.1083/jcb.201001039
Lim, G. G., Chew, K. C., Ng, X. H., Henry-Basil, A., Sim, R. W., Tan, J. M., et al. (2013). Proteasome inhibition promotes Parkin-Ubc13 interaction and lysine 63-linked ubiquitination. PLoS One 8:e73235. doi: 10.1371/journal.pone.0073235
Lim, K. L., Chew, K. C., Tan, J. M., Wang, C., Chung, K. K., Zhang, Y., et al. (2005). Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25, 2002–2009. doi: 10.1523/jneurosci.4474-04.2005
Lim, K. L., Dawson, V. L., and Dawson, T. M. (2006). Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol. Aging 27, 524–529. doi: 10.1016/j.neurobiolaging.2005.07.023
Lim, M. K., Kawamura, T., Ohsawa, Y., Ohtsubo, M., Asakawa, S., Takayanagi, A., et al. (2007). Parkin interacts with LIM Kinase 1 and reduces its cofilin-phosphorylation activity via ubiquitination. Exp. Cell Res. 313, 2858–2874. doi: 10.1016/j.yexcr.2007.04.016
Lim, K. L., and Zhang, C. W. (2013). Molecular events underlying Parkinson’s disease - an interwoven tapestry. Front. Neurol. 4:33. doi: 10.3389/fneur.2013.00033
Lonskaya, I., Hebron, M. L., Desforges, N. M., Franjie, A., and Moussa, C. E. (2013). Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 5, 1247–1262. doi: 10.1002/emmm.201302771
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). Association between early-onset Parkinson’s disease and mutations in the parkin gene. French Parkinson’s disease genetics study group. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Mata, I. F., Lockhart, P. J., and Farrer, M. J. (2004). Parkin genetics: one model for Parkinson’s disease. Hum. Mol. Genet. 13(Spec No 1), R127–R133. doi: 10.1093/hmg/ddh089
Matsuda, N., Kitami, T., Suzuki, T., Mizuno, Y., Hattori, N., and Tanaka, K. (2006). Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 281, 3204–3209. doi: 10.1074/jbc.m510393200
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Moore, D. J., West, A. B., Dikeman, D. A., Dawson, V. L., and Dawson, T. M. (2008). Parkin mediates the degradation-independent ubiquitination of Hsp70. J. Neurochem. 105, 1806–1819. doi: 10.1111/j.1471-4159.2008.05261.x
Muqit, M. M., Davidson, S. M., Payne Smith, M. D., MacCormac, L. P., Kahns, S., Jensen, P. H., et al. (2004). Parkin is recruited into aggresomes in a stress-specific manner: over-expression of parkin reduces aggresome formation but can be dissociated from parkin’s effect on neuronal survival. Hum. Mol. Genet. 13, 117–135. doi: 10.1093/hmg/ddh012
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010a). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Narendra, D. P., Kane, L. A., Hauser, D. N., Fearnley, I. M., and Youle, R. J. (2010b). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106. doi: 10.4161/auto.6.8.13426
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Navarro, P., Guerrero, R., Gallego, E., Avila, J., Luquin, R., Ruiz, P. J., et al. (2008). Memory and exploratory impairment in mice that lack the Park-2 gene and that over-express the human FTDP-17 mutant Tau. Behav. Brain Res. 189, 350–356. doi: 10.1016/j.bbr.2008.01.017
Nemes, Z., Devreese, B., Steinert, P. M., Van Beeumen, J., and Fésus, L. (2004). Cross-linking of ubiquitin, HSP27, parkin and alpha-synuclein by gamma-glutamyl-epsilon-lysine bonds in Alzheimer’s neurofibrillary tangles. FASEB J. 18, 1135–1137. doi: 10.1096/fj.04-1493fje
Neuspiel, M., Schauss, A. C., Braschi, E., Zunino, R., Rippstein, P., Rachubinski, R. A., et al. (2008). Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108. doi: 10.1016/j.cub.2007.12.038
Norris, K. L., Hao, R., Chen, L. F., Lai, C. H., Kapur, M., Shaughnessy, P. J., et al. (2015). Convergence of Parkin, PINK1 and alpha-Synuclein on stress-induced mitochondrial morphological remodeling. J. Biol. Chem. 290, 13862–13874. doi: 10.1074/jbc.M114.634063
Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3:1016. doi: 10.1038/ncomms2016
Olzmann, J. A., Li, L., Chudaev, M. V., Chen, J., Perez, F. A., Palmiter, R. D., et al. (2007). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038. doi: 10.1083/jcb.200611128
Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. doi: 10.1038/nature04788
Pawlyk, A. C., Giasson, B. I., Sampathu, D. M., Perez, F. A., Lim, K. L., Dawson, V. L., et al. (2003). Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J. Biol. Chem. 278, 48120–48128. doi: 10.1074/jbc.m306889200
Peng, J., Schwartz, D., Elias, J. E., Thoreen, C. C., Cheng, D., Marsischky, G., et al. (2003). A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926. doi: 10.1038/nbt849
Periquet, M., Corti, O., Jacquier, S., and Brice, A. (2005). Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J. Neurochem. 95, 1259–1276. doi: 10.1111/j.1471-4159.2005.03442.x
Periquet, M., Latouche, M., Lohmann, E., Rawal, N., De Michele, G., Ricard, S., et al. (2003). Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 126, 1271–1278. doi: 10.1093/brain/awg136
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Poole, A. C., Thomas, R. E., Yu, S., Vincow, E. S., and Pallanck, L. (2010). The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5:e10054. doi: 10.1371/journal.pone.0010054
Ren, Y., Jiang, H., Yang, F., Nakaso, K., and Feng, J. (2009). Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation. J. Biol. Chem. 284, 4009–4017. doi: 10.1074/jbc.m806245200
Ren, Y., Zhao, J., and Feng, J. (2003). Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J. Neurosci. 23, 3316–3324.
Riley, B. E., Lougheed, J. C., Callaway, K., Velasquez, M., Brecht, E., Nguyen, L., et al. (2013). Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 4:1982. doi: 10.1038/ncomms2982
Rodríguez-Navarro, J. A., Casarejos, M. J., Menendez, J., Solano, R. M., Rodal, I., Gomez, A., et al. (2007). Mortality, oxidative stress and tau accumulation during ageing in parkin null mice. J. Neurochem. 103, 98–114. doi: 10.1111/j.1471-4159.2007.04762.x
Rodríguez-Navarro, J. A., Gómez, A., Rodal, I., Perucho, J., Martinez, A., Furió, V., et al. (2008). Parkin deletion causes cerebral and systemic amyloidosis in human mutated tau over-expressing mice. Hum. Mol. Genet. 17, 3128–3143. doi: 10.1093/hmg/ddn210
Rosen, K. M., Moussa, C. E., Lee, H. K., Kumar, P., Kitada, T., Qin, G., et al. (2010). Parkin reverses intracellular beta-amyloid accumulation and its negative effects on proteasome function. J. Neurosci. Res. 88, 167–178. doi: 10.1002/jnr.22178
Rosen, K. M., Veereshwarayya, V., Moussa, C. E., Fu, Q., Goldberg, M. S., Schlossmacher, M. G., et al. (2006). Parkin protects against mitochondrial toxins and beta-amyloid accumulation in skeletal muscle cells. J. Biol. Chem. 281, 12809–12816. doi: 10.1074/jbc.m512649200
Rubio, I., Rodríguez-Navarro, J. A., Tomás-Zapico, C., Ruíz, C., Casarejos, M. J., Perucho, J., et al. (2009). Effects of partial suppression of parkin on huntingtin mutant R6/1 mice. Brain Res. 1281, 91–100. doi: 10.1016/j.brainres.2009.05.039
Rubio de la Torre, E., Luzón-Toro, B., Forte-Lago, I., Minguez-Castellanos, A., Ferrer, I., and Hilfiker, S. (2009). Combined kinase inhibition modulates parkin inactivation. Hum. Mol. Genet. 18, 809–823. doi: 10.1093/hmg/ddn407
Sánchez, M. P., Gonzalo, I., Avila, J., and De Yébenes, J. G. (2002). Progressive supranuclear palsy and tau hyperphosphorylation in a patient with a C212Y parkin mutation. J. Alzheimers Dis. 4, 399–404.
Sato, S., Chiba, T., Sakata, E., Kato, K., Mizuno, Y., Hattori, N., et al. (2006). 14–3-3eta is a novel regulator of parkin ubiquitin ligase. EMBO J. 25, 211–221. doi: 10.1038/sj.emboj.7600774
Sauvé, V., Lilov, A., Seirafi, M., Vranas, M., Rasool, S., Kozlov, G., et al. (2015). A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 34, 2492–2505. doi: 10.15252/embj.201592237
Scarpulla, R. C. (2008). Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N Y Acad. Sci. 1147, 321–334. doi: 10.1196/annals.1427.006
Schlehe, J. S., Lutz, A. K., Pilsl, A., Lämmermann, K., Grgur, K., Henn, I. H., et al. (2008). Aberrant folding of pathogenic Parkin mutants: aggregation versus degradation. J. Biol. Chem. 283, 13771–13779. doi: 10.1074/jbc.M707494200
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2:1002. doi: 10.1038/srep01002
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. doi: 10.1038/77060
Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher, A., Schneider, R., et al. (2001). Ubiquitination of a new form of alpha -synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293, 263–269. doi: 10.1126/science.1060627
Shin, J. H., Ko, H. S., Kang, H., Lee, Y., Lee, Y. I., Pletinkova, O., et al. (2011). PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702. doi: 10.1016/j.cell.2011.02.010
Soubannier, V., McLelland, G. L., Zunino, R., Braschi, E., Rippstein, P., Fon, E. A., et al. (2012a). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. doi: 10.1016/j.cub.2011.11.057
Soubannier, V., Rippstein, P., Kaufman, B. A., Shoubridge, E. A., and McBride, H. M. (2012b). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7:e52830. doi: 10.1371/journal.pone.0052830
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Sriram, S. R., Li, X., Ko, H. S., Chung, K. K., Wong, E., Lim, K. L., et al. (2005). Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum. Mol. Genet. 14, 2571–2586. doi: 10.1093/hmg/ddi292
Staropoli, J. F., McDermott, C., Martinat, C., Schulman, B., Demireva, E., and Abeliovich, A. (2003). Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 37, 735–749. doi: 10.1016/s0896-6273(03)00084-9
Stribl, C., Samara, A., Trümbach, D., Peis, R., Neumann, M., Fuchs, H., et al. (2014). Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 289, 10769–10784. doi: 10.1074/jbc.M113.515940
Sugiura, A., Mclelland, G. L., Fon, E. A., and Mcbride, H. M. (2014). A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 33, 2142–2156. doi: 10.15252/embj.201488104
Tan, J. M., Wong, E. S., Dawson, V. L., Dawson, T. M., and Lim, K. L. (2008a). Lysine 63-linked polyubiquitin potentially partners with p62 to promote the clearance of protein inclusions by autophagy. Autophagy 4, 251–253. doi: 10.4161/auto.5444
Tan, J. M., Wong, E. S., Kirkpatrick, D. S., Pletnikova, O., Ko, H. S., Tay, S. P., et al. (2008b). Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439. doi: 10.1093/hmg/ddm320
Tay, S. P., Yeo, C. W., Chai, C., Chua, P. J., Tan, H. M., Ang, A. X., et al. (2010). Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J. Biol. Chem. 285, 29231–29238. doi: 10.1074/jbc.m110.108241
Trempe, J. F., Sauvé, V., Grenier, K., Seirafi, M., Tang, M. Y., Menade, M., et al. (2013). Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455. doi: 10.1126/science.1237908
Tsai, Y. C., Fishman, P. S., Thakor, N. V., and Oyler, G. A. (2003). Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 278, 22044–22055. doi: 10.1074/jbc.m212235200
Um, J. W., Min, D. S., Rhim, H., Kim, J., Paik, S. R., and Chung, K. C. (2006). Parkin ubiquitinates and promotes the degradation of RanBP2. J. Biol. Chem. 281, 3595–3603. doi: 10.1074/jbc.m504994200
van de Warrenburg, B. P., Lammens, M., Lucking, C. B., Denefle, P., Wesseling, P., Booij, J., et al. (2001). Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 56, 555–557. doi: 10.1212/wnl.56.4.555
Vandiver, M. S., Paul, B. D., Xu, R., Karuppagounder, S., Rao, F., Snowman, A. M., et al. (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 4:1626. doi: 10.1038/ncomms2623
Vincow, E. S., Merrihew, G., Thomas, R. E., Shulman, N. J., Beyer, R. P., Maccoss, M. J., et al. (2013). The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. U S A 110, 6400–6405. doi: 10.1073/pnas.1221132110
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U S A 107, 378–383. doi: 10.1073/pnas.0911187107
Walter, U., Klein, C., Hilker, R., Benecke, R., Pramstaller, P. P., and Dressler, D. (2004). Brain parenchyma sonography detects preclinical parkinsonism. Mov. Disord. 19, 1445–1449. doi: 10.1002/mds.20232
Wang, C., Ko, H. S., Thomas, B., Tsang, F., Chew, K. C., Tay, S. P., et al. (2005a). Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum. Mol. Genet. 14, 3885–3897. doi: 10.1093/hmg/ddi413
Wang, Y., Serricchio, M., Jauregui, M., Shanbhag, R., Stoltz, T., Di Paolo, C. T., et al. (2015). Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11, 595–606. doi: 10.1080/15548627.2015.1034408
Wang, H., Song, P., Du, L., Tian, W., Yue, W., Liu, M., et al. (2011a). Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson’s disease. J. Biol. Chem. 286, 11649–11658. doi: 10.1074/jbc.M110.144238
Wang, C., Tan, J. M., Ho, M. W., Zaiden, N., Wong, S. H., Chew, C. L., et al. (2005b). Alterations in the solubility and intracellular localization of parkin by several familial Parkinson’s disease-linked point mutations. J. Neurochem. 93, 422–431. doi: 10.1111/j.1471-4159.2005.03023.x
Wang, H., Ying, Z., and Wang, G. (2012). Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J. Biol. Chem. 287, 28576–28585. doi: 10.1074/jbc.m111.299990
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011b). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Wauer, T., and Komander, D. (2013). Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 32, 2099–2112. doi: 10.1038/emboj.2013.125
Wauer, T., Simicek, M., Schubert, A., and Komander, D. (2015). Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374. doi: 10.1038/nature14879
Wenzel, D. M., Lissounov, A., Brzovic, P. S., and Klevit, R. E. (2011). UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108. doi: 10.1038/nature09966
Winklhofer, K. F., Henn, I. H., Kay-Jackson, P. C., Heller, U., and Tatzelt, J. (2003). Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J. Biol. Chem. 278, 47199–47208. doi: 10.1074/jbc.m306769200
Wong, Y. C., and Holzbaur, E. L. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U S A 111, E4439–E4448. doi: 10.1073/pnas.1405752111
Wong, E. S., Tan, J. M., Wang, C., Zhang, Z., Tay, S. P., Zaiden, N., et al. (2007). Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J. Biol. Chem. 282, 12310–12318. doi: 10.1074/jbc.m609466200
Yamamoto, A., Friedlein, A., Imai, Y., Takahashi, R., Kahle, P. J., and Haass, C. (2005). Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J. Biol. Chem. 280, 3390–3399. doi: 10.1074/jbc.m407724200
Yeo, C. W., Ng, F. S., Chai, C., Tan, J. M., Koh, G. R., Chong, Y. K., et al. (2012). Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 72, 2543–2553. doi: 10.1158/0008-5472.CAN-11-3060
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028.
Zhang, Y., Gao, J., Chung, K. K., Huang, H., Dawson, V. L., and Dawson, T. M. (2000). Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. U S A 97, 13354–13359. doi: 10.1073/pnas.240347797
Zhong, L., Tan, Y., Zhou, A., Yu, Q., and Zhou, J. (2005). RING finger ubiquitin-protein isopeptide ligase Nrdp1/FLRF regulates parkin stability and activity. J. Biol. Chem. 280, 9425–9430. doi: 10.1074/jbc.m408955200
Keywords: Parkinson’s disease, mitophagy, proteasome, ubiquitin, neurodegeneration, autophagy, protein misfolding, mitochondria
Citation: Zhang C-W, Hang L, Yao T-P and Lim K-L (2016) Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 7:248. doi: 10.3389/fnagi.2015.00248
Received: 21 October 2015; Accepted: 17 December 2015;
Published: 12 January 2016.
Edited by:
Vinood B. Patel, University of Westminster, UKCopyright © 2016 Zhang, Hang, Yao and Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kah-Leong Lim, a2FoX2xlb25nX2xpbUBubmkuY29tLnNn