 Robert Underly
Robert Underly Mee-Sook Song
Mee-Sook Song Gary L. Dunbar
Gary L. Dunbar Charles L. Weaver
Charles L. Weaver- 1Department of Psychology, Saginaw Valley State University, University Center, MI, USA
- 2Field Neurosciences Institute, Saginaw, MI, USA
- 3Department of Psychology, Central Michigan University, Mount Pleasant, MI, USA
- 4Department of Health Sciences, Saginaw Valley State University, University Center, MI, USA
The neurofibrillary tau pathology and amyloid deposits seen in Alzheimer’s disease (AD) also have been seen in bacteria-infected brains. However, few studies have examined the role of these bacteria in the generation of tau pathology. One suggested link between infection and AD is edentulism, the complete loss of teeth. Edentulism can result from chronic periodontal disease due to infection by Enterococcus faecalis. The current study assessed the ability to generate early Alzheimer-like neurofibrillary epitopes in primary rat cortical neurons through bacterial infection by E. faecalis. Seven-day old cultured neurons were infected with E. faecalis for 24 and 48 h. An upward molecular weight shift in tau by Western blotting (WB) and increased appearance of tau reactivity in cell bodies and degenerating neurites was found in the 48 h infection group for the antibody CP13 (phospho-Serine 202). A substantial increase in reactivity of Alz-50 was seen at 24 and 48 h after infection. Furthermore, extensive microtubule-associated protein 2 (MAP2) reactivity also was seen at 24 and 48 h post-infection. Our preliminary findings suggest a potential link between E. faecalis infection and intracellular changes that may help facilitate early AD-like neurofibrillary pathology.
Highlights
• Enterococcus faecalis used in the generation of AD neurofibrillary epitopes in rat.
• Infection increases Alz-50, phospho-Serine 202 tau, and MAP2 expression.
• Infection by Enterococcus may play a role in early Alzheimer neurofibrillary changes.
Introduction
Neurofibrillary tau pathology and beta amyloid deposition are the classic defining biological markers of Alzheimer’s disease (AD). The appearance and spread of neurofibrillary tangles and amyloid plaques have been shown to correlate with the onset and severity of Alzheimer-type cognitive decline (Braak and Braak, 1995; Sperling et al., 2011). Although these pathological hallmarks have been studied extensively, a mechanism for their onset in AD has yet to be elucidated. During the past three decades, studies have re-examined the presence of bacterial infection in AD brains. Various types of spirochetes, including six periodontal pathogen spirochetes and Borrelia burgdorferi (MacDonald, 1986; MacDonald and Miranda, 1987; Miklossy, 1993; Riviere et al., 2002; Miklossy et al., 2004) and an obligate intracellular bacterium Chlamydophila (Chlamydia) pneumoniae (Balin et al., 1998, 2008; Hammond et al., 2010) have been detected and isolated from Alzheimer brain tissue, yet few studies have examined the involvement of these pathogens in the production of AD-like tau/neurofibrillary pathology (Miklossy et al., 2006).
The relationship between chronic periodontitis and AD has been shown with respect to periodontal pathogens (Treponema denticola, Tannerella forsythia, and Porphyromonas gingivalis) and their virulence factors (Poole et al., 2013, 2015; Singhrao et al., 2014). Infection from Enterococcus faecalis, the bacteria most commonly associated with failed endodontic procedures (Sundqvist et al., 1998; Rocas et al., 2004; Zoletti et al., 2006; Al-Ahmad et al., 2010), secondary endodontic infections (Schirrmeister et al., 2009), urinary tract infections (Zoletti et al., 2006) and nosocomial infection (Courvalin, 2006), also has been implicated in the pathogenesis of chronic periodontitis (Sun et al., 2012). These bacteria can form biofilms, making them extremely difficult to remove and allowing for cell microcolony formation which results in chronic infection (Mohamed and Huang, 2007; Arciola et al., 2008). Edentulism, the complete loss of teeth, also has been shown to be present in a subset of individuals with AD or cerebral atherosclerotic pathologies (Gallo et al., 2005); and has chronic periodontitis as its most common factor. Lastly, it has been suggested that E. faecalis can travel from sites of dental infection to the brain (Mylona et al., 2012); thus making it an attractive candidate associated with AD pathology.
To address if early AD-like neurofibrillary epitopes could be generated by infection with E. faecalis, we examined potential posttranslational modifications of tau in primary rat cortical neuron cell cultures after infection with E. faecalis. Antibodies recognizing conformational change (Alz-50) and phosphorylation (CP13) of tau were used to detect these epitopes after the addition of bacteria. This study demonstrates that E. faecalis increases both Alz-50 and CP13 reactivity, as well as that of microtubule-associated protein 2 (MAP2), in cortical cultured neurons; and provides support for the hypothesis that bacterial infection may play a potential role in Alzheimer pathology.
Materials and Methods
Bacterial Preparation and Primary Neuronal Cultures
Enterococcus faecalis (ATCC 29212) was cultured in 7 ml of brain-heart infusion (BHI) broth in 15 ml conical tubes, and was maintained in an incubator, separate from the neuronal cultures. The bacteria were stored at 37°C in a 5% CO2-humidified incubator. Primary cortical neurons from day-18 Fisher 344 rat embryos (Cat. #A10840, Gibco/Invitrogen) were cultured on poly-d-lysine coated 6- and 12-well cell culture plates to a density of 1.7 × 105 and 8.3 × 104 per well, respectively. Neurons were plated using Neurobasal medium with 2% B27, 1% Penicillin-Streptomycin, 0.5% fetal goat serum (FGS), and 0.5 mM GlutaMax (Cat. #35050, Gibco/Invitrogen). Cells were grown at 37°C in a 5% CO2-humidified incubator with media being changed every 3–4 days. Seven-day old cultured neurons were infected with E. faecalis, with the final concentration of bacteria per well being 7 × 106 bacteria/ml. Neurons were harvested for immunohistochemistry (IHC) and Western blotting (WB) at 24 and 48 h post-infection time points. Experiments were run in triplicate.
Antibodies
Primary antibodies recognizing pathological changes in tau (Alz-50, conformation; and CP13, phospho-Serine 202; generous gift from Dr. Peter Davies, Albert Einstein College of Medicine) were used for IHC and WB. Anti-β-tubulin (Cat. # ab5392, Abcam) and anti-MAP2 (Cat. #ab56676, Abcam) primary antibodies were used as controls in WB and IHC, respectively. The anti-tubulin antibody was used to standardize protein levels; and for lack of availability of a suitable commercial rat-specific pan-tau antibody that provides consistent staining of tau. Horseradish peroxidase (HRP)-labeled secondary antibodies (Southern Biotech, Birmingham, AL, USA) were used for WB, and fluorescence-labeled secondary antibodies (Molecular Probes, Invitrogen) were used for IHC.
Western Blot
Cell lysates were analyzed by SDS–PAGE and Western blot. Briefly, cells were washed with tris-buffered saline (TBS) and solubilized in ice-cold lysis buffer (50 mM Tris, pH 7.4, 5 mM EDTA, 150 mM NaCl, 1 × ProteaseArrest and 1 × PhosphataseArrest [GBiosciences, St. Louis, MO, USA]) containing 1% SDS. Cells immediately were scraped off the plates and transferred to a microcentrifuge tube. After centrifugation, sample buffer was added to the supernatant in an equal volume, and the entire sample was heated at 100°C for 5 min. Proteins were separated in SDS-PAGE (10% polyacrylamide gels; Bio Rad, Hercules, CA, USA) and transferred onto 0.45 μm nitrocellulose membranes. Membranes were blocked in 5% fat-free milk in TBS (milk), and incubated with primary antibodies overnight at 4°C (1:200 for Alz-50, CP13, and tubulin). An enhanced and optimized 4-chloronaphthal (Opti-4CN, Cat. # 170-8235, Bio Rad) detection procedure was then used. Procedures for the non-infection group were always performed first to avoid accidental contamination.
Immunohistochemistry
A general IHC double-labeling technique was performed using the previously mentioned primary antibodies. Cells plated in 12-well culture plates (one control plate and one infected plate) with poly-d-lysine coated coverslips were fixed using 10% buffered formalin. Anti-tau primary antibodies (Alz-50 and CP13; 1:200 dilution) and anti-MAP2 (control; 1:1000 dilution) were added individually to their specified wells and allowed to incubate at 4°C for 24 h. After washes in TBS, primary antibodies were removed and secondary antibodies were added in a dark room to avoid loss of fluorescence. Secondary antibodies (1:500 dilution for all) were removed, and after additional washes in TBS, coverslips were mounted and prepared for confocal fluorescent microscopy analysis (Olympus FluoView FV10i).
Total Volume Quantification of Alz-50
Imaging of antibody-labeled cells was performed using an Olympus confocal microscope and FV-1000 software. Due to the presence of Alz-50-positive cells in young rat brain (Byne et al., 1991), a method for determining total volume of Alz-50 expression, with or without infection, was needed. Volume images from the confocal 3D data sets were processed with IMARIS software (Bitplane AG, Zurich, Switzerland). Briefly, in order to avoid bias in the volume quantification, regions of interest were selected on a similar grid pattern with the same number and location of regions for each slide being analyzed. Images were opened individually and within the z-stack image, a position 1/3 of the way through the stack was selected. This position (field) remained constant for all images being analyzed. After the appropriate fluorescence channel was selected, absolute and lower threshold intensities were set. Volume data were collected through the entire cell by the software, and values given in μm3. Data were exported into Microsoft Excel, and used in an independent samples t-test. Files were coded so that groups were not known to the researcher performing the analysis.
Results
Increased Alz-50, CP13, and MAP2 Reactivity Following Infection
Immunocytochemistry revealed Alz-50 and CP13 reactivity in cell bodies of neurons in control cultures. Alz-50 and CP13 reactivity increased after the introduction of E. faecalis in primary rat cortical neuron cultures. Numerous degenerating (beaded) neurites as well as whole cell bodies were labeled with Alz-50, both 24 and 48 h post-infection (Figure 1). Immunostaining revealed similar changes in the location and abundance of CP13 labeling 48 h post-infection (Figure 2). There was nearly complete absence of CP13 labeling at 24 h. MAP2 immunoreactivity of many neurons was also increased at 48 h post-infection compared to control cells. An independent samples t-test was used to determine the difference in amounts of Alz-50 reactivity between control and infected cells at two time points (Figure 3). Analyses demonstrated a significant increase (p < 0.001; Fields examined: control N = 14, infection N = 21) in Alz-50 reactivity 24 h post-infection when compared to the 24 h control. A significant increase (p = 0.003; Fields examined: control N = 24, infection N = 15) in Alz-50 reactivity was also demonstrated 48 h post-infection, when compared to the 48 h control.
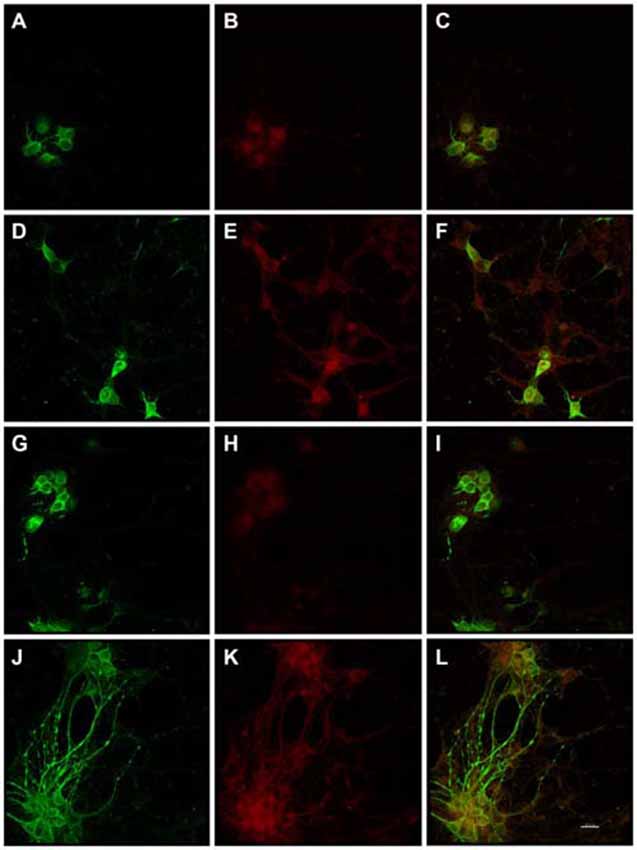
Figure 1. Substantially increased amounts of Alz-50 and MAP2 reactivity were induced in primary rat cortical neurons by exposure to Enterococcus faecalis. MAP2 (control-green) and Alz-50 (red) staining can be seen in both 24 h (A–C) and 48 h (G–I) control cultures (C,I, co-localization). Alz-50 reactivity was apparent in the cell bodies of neurons in control cultures, and confirms that its expression can be found in rat neurons. A striking increase in Alz-50 reactivity was found at both 24 and 48 h post-infection (E,K respectively). Extensive MAP2 reactivity also was seen 48 h post-infection (J), but not at 24 h post-infection (D). Co-localization of MAP2 and Alz-50 staining at 24 and 48 h post-infection is shown in (F,L), respectively. The beaded neurites shown in (J–L) are abundant, and may be indicative of degeneration. Scale bar in L = 20 μm.
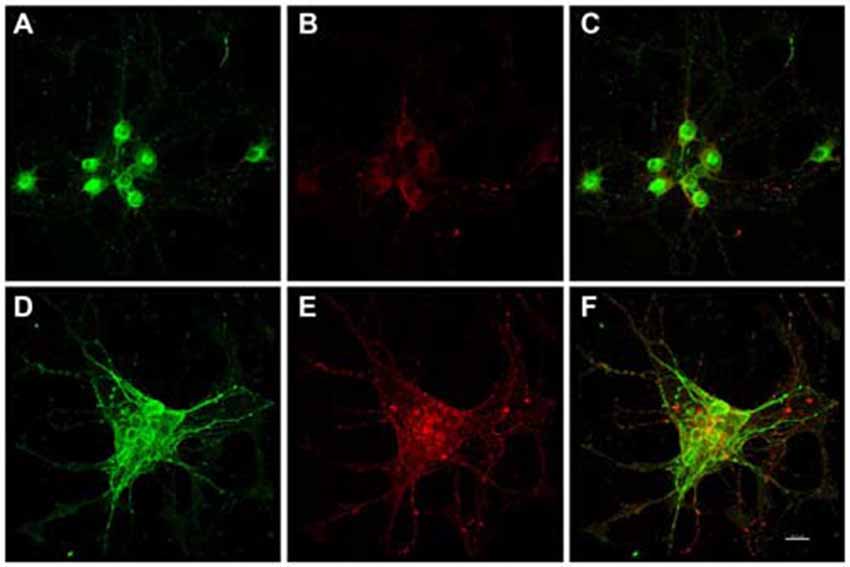
Figure 2. In vitro phospho-Serine 202 expression (CP13 reactivity) was increased in primary rat cortical neurons 48 h after exposure to E. faecalis. MAP2 and CP13 reactivities were seen in the cell body of neurons in control cultures (A,B, respectively), with co-localization demonstrated in (C). Robust CP13 staining was found throughout the cell bodies and degenerating neurites of neurons post-infection (E). Increased immunoreactivity of MAP2 in infected neurons compared to control cells was also revealed (D). MAP2 and CP13 post-infection co-localization is shown in (F). CP13 reactivity was lost 24 h post-infection (data not shown). Scale bar in F = 20 μm.
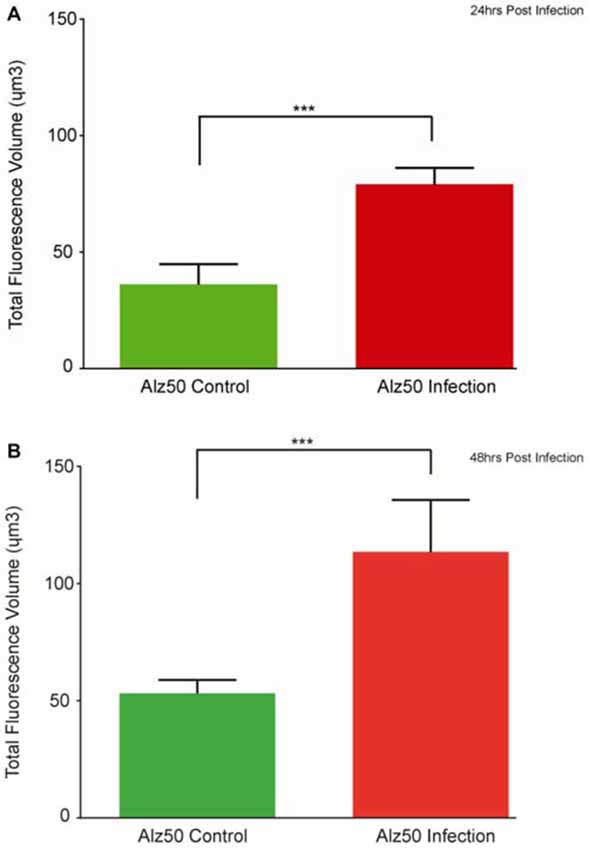
Figure 3. The appearance of Alz-50 labeling of neurons in control cultures necessitated a comparison of intensities found between control and infected plates. Alz-50 staining is found in both control and infected cultures, with significantly more reactivity found at both 24 (A) and 48 h (B) after the introduction of the E. faecalis. ***Represents p < 0.001 (A), p = 0.003 (B).
Mobility Shift in Phospho-Tau after Infection
Western blot analysis revealed a loss of CP13 reactivity at 24 h post-infection, and a restoration of reactivity with an upwards molecular weight shift in tau at 48 h post-infection (Figure 4). A similar shift was not seen with the Tubulin control. Immunoblotting failed to yield any reactivity using Alz-50.
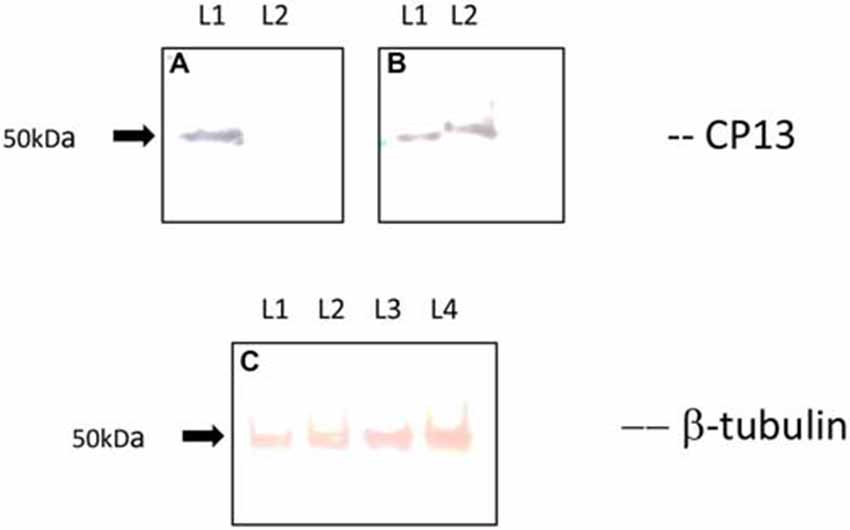
Figure 4. Immunoblot of CP13 and tubulin reactivity in primary rat cortical neurons. CP13 staining of control samples reveals a tau isoform around 50 kDa (A,B; lane 1 [L1]). There is a distinct loss of CP13 reactivity in samples at 24 h post-infection (A; lane 2 [L2]). Blotting of samples at 48 h post-infection demonstrates an upwards shift in mobility of CP13-reactive tau (B; lane 2 [L2]) . The shift in mobility may be the result of increased phosphorylation leading to conformational changes in the protein which causes delayed migration through the gel. β-tubulin was used as a loading control for the amount of protein loaded per lane (C). L1 and L2 reveal β-tubulin staining from control and 24 h post-infection samples, while L3 and L4 reveal β-tubulin staining from control and 48 h post-infection samples.
Discussion
Spirochetes, a phylogenetically distinct group of bacteria, have been detected and isolated in Alzheimer brain tissue. Several types of spirochetes were detected in the AD brain (reviewed by Miklossy, 2011a), including six periodontal pathogen spirochetes: Treponema (T). denticola, T. socranskii, T. pectinovorum, T. amylovorum, T. maltophilum, and T. medium (Riviere et al., 2002) and the Lyme disease spirochete B. burgdorferi (MacDonald and Miranda, 1987), which was shown to induce β-amyloid (Aβ) deposits and increased level of phosphorylated tau successfully in neuronal cell cultures (Miklossy et al., 2006). In vivo studies using intranasal injection of an obligate intracellular bacterium such as C. pneumoniae have been shown to induce beta amyloid deposition in the brains of BALB/c mice (Little et al., 2004). More recently, additional association between periodontal pathogens and AD has been demonstrated (Poole et al., 2013, 2015; Singhrao et al., 2014), along with the association between chronic periodontitis and amyloid load (Kamer et al., 2015). Several extensive reviews of the literature have also reinforced the significant association between AD and detectable evidence of infection (Miklossy, 2011b; Maheshwari and Eslick, 2015; Shoemark and Allen, 2015). Together, these studies suggest a correlation between Alzheimer’s pathology and bacterial infection in brain. Pertinent to the present study, a potential link between oral health and AD has been reviewed by Azaroazhooh et al. (2010). A study by Gatz et al. (2006) demonstrated that substantial tooth loss prior to the age of 35 was a significant risk factor for the development of dementia and AD. Two studies suggested that edentulous patients, not using dentures, were at higher risk for either dementia (Kim et al., 2007) or mortality (Shimazaki et al., 2001). An inverse association was also found between having a low number of teeth and dementia with an absence of the ApoE4 allele (Stein et al., 2007). These studies, along with the personal observation of numerous edentulous AD patients, ultimately led to the examination of whether the powerful dental pathogen, E. faecalis, might induce the biological markers of AD in vitro, which would indicate the potential role of this bacterium in AD pathology. The authors concede, that in the case of positive results, future studies showing more direct evidence of the link between the two is needed. Examination for the remnants of E. faecalis in brains of edentulous and non-edentulous (with severe on-going periodontal disease) AD patients obviously is the first step; and human AD tissue recently has been secured for analysis by the authors. Of particular interest would be those who experienced edentulism due to advanced caries, failed root canal therapy, or untreatable/refractory periodontal disease since enterococci have been demonstrated in these ailments (Kinsel and Lamb, 2001; Sedgley et al., 2004, 2005; Balaei-Gajan et al., 2010; Kouidhi et al., 2011). Nevertheless, the present study further suggests that agents involved in the pathogenesis of chronic periodontal disorders and edentulism may be related to Alzheimer’s pathology; since E. faecalis in vitro can influence the production of pathological epitopes similar to that seen early in AD.
Antibodies recognizing conformational change (Alz-50) and specific phosphorylations of tau protein (CP13; phospho-Serine 202) have been shown to label pre-tangle neurons; and therefore, are suitable markers of early signs of Alzheimer’s degeneration (Jicha et al., 1997; Weaver et al., 2001; Andorfer et al., 2003, 2005). IHC, performed post-infection, revealed morphological changes in the form of neuritic beading, as well as the increased presence of Alz-50 and CP13 reactivity throughout the entire cell. Because of E. faecalis’ strong virulence, infected neurons tend not to survive in culture much longer than 60 h; so these neurons were harvested at 24 and 48 h post-infection. No changes in reactivity were seen in control samples, which were maintained in culture for 4 weeks.
The Nature of Alz-50 Reactivity
Alz-50 has been shown to label limited cell populations in healthy young rat tissue (Byne et al., 1991) which led to the expectation of finding some Alz-50 reactivity in control primary rat cortical neurons. Using quantitative total volume analysis, the amount of reactivity was deciphered in both the infection and control groups. These data reveal a significant post-infection increase in Alz-50 reactivity at both 24 and 48 h time points. This study provides no evidence that bacterial infection leads to greater amounts of conformational changes of tau. It does, however, confirm the utility of Alz-50 as a marker of epitopes normally found in early AD-like degeneration. Al-Ghoul and Miller (1989) demonstrated Alz-50 labeling in rat neonatal subplate and cortical neurons prior to their elimination from the cortex. A subsequent paper from the same group showed Alz-50 expression in the principal sensory nucleus following trigeminal nerve lesion (Miller et al., 1994). These studies suggest that Alz-50 reactivity may be a marker for neuronal death. A possible substrate reacting with Alz-50 is the fetal Alz-50 clone 1 (FAC1) protein. Previous studies have demonstrated that FAC1 is in abundance in the cell bodies and dendrites of human undifferentiated neurons (Bowser et al., 1995), and have shown an upregulation in FAC1 expression after lesioning of intact rodent tissue (Styren et al., 1997). In the present study, we did not observe Alz-50 reactivity by immunoblotting; and in fact, achieving Alz-50 reactivity when using unpurified and unconcentrated primary cortical rat cell lysate has proven difficult (unpublished raw data). The authors are only familiar with one study demonstrating significant Alz-50 reactivity from rat neurons by immunoblotting; and it was achieved after isolation of the antigen by immunoaffinity chromatography and subsequent concentration (Al-Ghoul and Miller, 1989). Regardless of whether the protein involved is tau or FAC1, it is clear that increased Alz-50 reactivity with the protein is associated with degeneration of the neurons.
Phosphorylation Changes after Infection
The CP13 reactivity seen by immunoblotting in control cell cultures suggests that Serine 202 is normally phosphorylated in a portion of primary rat cortical neurons. The loss of reactivity at 24 h and the recovery of signal at 48 h post-infection together were surprising. However, studies have shown that oxidative stress, in response to ischemia, can induce rapid dephosphorylation of tau (Shackelford and Yeh, 1998), with recovery of the epitope equally rapid for Serine 202/Threonine 205 (Mailliot et al., 2000). Recent studies, also, have demonstrated the ability of E. faecalis to produce oxidative stress through the generation of reactive oxygen species (Golińska et al., 2013; Strickertsson et al., 2013, 2014). Therefore, it may be possible that the bacteria caused local ischemic conditions in the culture dish. Since CP13 is a phosphorylation- and sequence-dependent antibody, dephosphorylation at Serine 202 will result in loss of reactivity. [Note: C.Weaver is the co-generator of the CP13 antibody with P. Davies] The upward molecular weight shift of the tau band in the infection cultures suggests an increase in phosphorylation of tau that may lead to conformational changes that effectively reduces its migration through the acrylamide gel (Lindwall and Cole, 1984; Bretteville et al., 2009). Phosphorylation at one site (Ser 202) may not be enough to generate conformational change in tau. However, it is likely that multiple sites are being phosphorylated since glycogen synthase kinase 3 Alpha and Beta (GSK3α and 3β), microtubule-associated protein kinase 13 (MAPK13), cyclin-dependent kinase 5/p25 (CDK-5/p25), and a host of others can simultaneously phosphorylate Ser 202, Thr 231, Ser 235, and Ser 396/404 (Cavallini et al., 2013). The authors only demonstrated one tau band at 48 h post-infection. It is possible that there would be two bands of tau after re-phosphorylation and subsequent increased phosphorylation. The primary author’s research facilities did not house a film developer, so enhanced chemilluminescence (ECL) was not utilized. The Opti-4CN used in the present study may not have been sensitive enough to display a second tau band similar to that found from control samples. Along with the increase in immunostaining throughout the neurites, it is clear that CP13 reactivity increased as a result of infection. To optimize material for immunoblotting, volume analyses were not performed for CP13 reactivity.
MAP2 and Neurodegeneration
The use of MAP2 as a control marker for primary rat neurons is well documented. Increases in MAP2 staining post-infection were not expected, but are not unreasonable. In AD tissue, MAP2 staining can be found in dystrophic neurites (Neve et al., 1986; Trojanowski et al., 1991; Ashford et al., 1998). While protein levels of MAP2 do not change significantly in AD, its immunoreactivity in rat neuronal cell cultures has been shown to increase in response to serum from AD patients, particularly at 48 h post-application (Brewer and Ashford, 1992). The results of increased MAP2 reactivity and morphological changes to neurites, in this study, provide support for neuritic reorganization and degeneration in this cell culture model.
Aβ42 and Enterococcus faecalis
A recent study has shown that Aβ42 functions as an antimicrobial peptide (AMP) that expresses activity against E. faecalis (Soscia et al., 2010). The group demonstrated that Aβ42 was present at significantly greater levels in the temporal lobe of AD patients compared to age-matched non-AD brains. These findings suggest that increased levels of Aβ42 in AD brain tissue could be due to an immune response of Aβ42 to an infection-inducing microorganism present in the tissue. These data lend support to the idea that the pathology seen in AD could be related to an infectious agent.
Conclusion
These preliminary data suggest that pathologic epitopes generated in early AD may be influenced by infection with E. faecalis. While the nature of the type of protein eliciting Alz-50 reactivity is unknown, it is important to recognize that reactivity is significantly increased after infection. As a result, further research on the implications of bacterial infection and AD is warranted.
Author Contributions
RU, performed all aspects of the experiments, and wrote the manuscript. CLW, organized, designed, and performed all parts of the experiments; and edited the manuscript. M-SS, performed the cell culture work. GLD, helped to oversee the cell culture work; and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Fred Bonds, DDS and David Regiani, DDS for their insight into dental infection by enterococci. The work presented in this manuscript was supported by a faculty research award from the Saginaw Valley State University, a collaboration grant from the Field Neurosciences Institute (Saginaw, MI, USA), and a mentorship award from the Support of Mentors and Students (SOMAS-URM) national program (Davidson College, Davidson, NC, USA) to CLW.
References
Al-Ahmad, A., Maier, J., Follo, M., Spitzmuller, B., Wittmer, A., Hellwig, E., et al. (2010). Food-borne enterococci integrate into oral biofilm: an in vivo study. J. Endod. 36, 1812–1819. doi: 10.1016/j.joen.2010.08.011
Al-Ghoul, W. M., and Miller, M. W. (1989). Transient expression of Alz-50 immunoreactivity in developing rat neocortex: a marker for naturally occurring neuronal death? Brain Res. 481, 361–367. doi: 10.1016/0006-8993(89)90815-9
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle re-entry and cell death intransgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454. doi: 10.1523/jneurosci.4637-04.2005
Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K. L., Barde, Y. A., et al. (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590. doi: 10.1046/j.1471-4159.2003.01879.x
Arciola, C. R., Baldassarri, L., Campoccia, D., Creti, R., Pirini, V., Huebner, J., et al. (2008). Strong biofilm production, antibiotic multi-resistance and high gelE expression in epidemic clones of Enterococcus faecalis from orthopaedic implant infections. Biomaterials 29, 580–586. doi: 10.1016/j.biomaterials.2007.10.008
Ashford, J. W., Soultanian, N. S., Zhang, S. X., and Geddes, J. W. (1998). Neuropil threads are collinear with MAP2 immunostaining in neuronal dendrites of Alzheimer brain. J. Neuropathol. Exp. Neurol. 57, 972–978. doi: 10.1097/00005072-199810000-00009
Azaroazhooh, A., Quinonez, C., Hajizadeh, A., Ko, C., Chow, C., Zanon, C., et al. (2010). Does Poor Oral Health Have Cognitive Impact? University of Toronto Faculty of Dentistry, Toronto.
Balaei-Gajan, E., Shirmohammadi, A., Abashov, R., Agazadeh, M., and Faramarzie, M. (2010). Detection of Enterococcus faecalis in subgingival biofilm of patients with chronic refractory periodontitis. Med. Oral Patol. Oral Cir. Bucal 15, e667–e670. doi: 10.4317/medoral.15.e667
Balin, B. J., Gérard, H. C., Arking, E. J., Appelt, D. M., Branigan, P. J., Abrams, J. T., et al. (1998). Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. 187, 23–42. doi: 10.1007/s004300050071
Balin, B. J., Little, C. S., Hammond, C. J., Appelt, D. M., Whittum-Hudson, J. A., Gérard, H. C., et al. (2008). Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J. Alzheimers Dis. 13, 371–380.
Bowser, R., Giambrone, A., and Davies, P. (1995). FAC1, a novel gene identified with the monoclonal antibody Alz50, is developmentally regulated in human brain. Dev. Neurosci. 17, 20–37. doi: 10.1159/000315743
Braak, H., and Braak, E. (1995). Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–278; discussion 278–284. doi: 10.1016/0197-4580(95)00021-6
Bretteville, A., Ando, K., Ghestem, A., Loyens, A., Bégard, S., Beauvillain, J. C., et al. (2009). Two-dimensional electrophoresis of tau mutants reveals specific phosphorylation pattern likely linked to early tau conformational changes. PLoS One 4:e4843. doi: 10.1371/journal.pone.0004843
Brewer, G. J., and Ashford, J. W. (1992). Human serum stimulates Alzheimer markers in cultured hippocampal neurons. J. Neurosci. Res. 33, 355–369. doi: 10.1002/jnr.490330302
Byne, W., Mattiace, L., Kress, Y., and Davies, P. (1991). Alz-50 immunoreactivity in the hypothalamus of the normal and Alzheimer human and the rat. J. Comp. Neurol. 306, 602–612. doi: 10.1002/cne.903060406
Cavallini, A., Brewerton, S., Bell, A., Sargent, S., Glover, S., Hardy, C., et al. (2013). An unbiased approach to identifying tau kinases that phosphorylate tau sites associated with Alzheimer’s Disease. J. Biol. Chem. 288, 23331–23347. doi: 10.1074/jbc.M113.463984
Courvalin, P. (2006). Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 42, S25–S34. doi: 10.1086/491711
Gallo, C., Magliarditi, A., Beghetto, M., Petena, C., and Borella, L. (2005). Oral health status and physiology in dementia patients. Paper Presented at 8th World Congress on Preventative Dentistry, Liverpool.
Gatz, M., Mortimer, J. A., Fratiglioni, L., Johansson, B., Berg, S., Reynolds, C. A., et al. (2006). Potentially modifiable risk factors for dementia in identical twins. Alzheimer’s and Dementia. Alzheimers Dement. 2, 110–117. doi: 10.1016/j.jalz.2006.01.002
Golińska, E., Tomusiak, A., Gosiewski, T., Wiecek, G., Machul, A., Mikołajczyk, D., et al. (2013). Virulence factors of Enterococcus strains isolated from patients with inflammatory bowel disease. World J. Gastroenterol. 19, 3562–3572. doi: 10.3748/wjg.v19.i23.3562
Hammond, C. J., Hallock, L. R., Howanksi, R. J., Appelt, D. M., Little, C. S., and Balin, B. J. (2010). Immunohistological detection of Chlamydia pneumonia in the Alzheimer’s disease brain. BMC Neurosci. 11, 121–133. doi: 10.1186/1471-2202-11-121
Jicha, G., Bowser, R., Kazam, I., and Davies, P. (1997). Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J. Neurosci. Res. 48, 128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e
Kamer, A. R., Pirraglia, E., Tsui, W., Rusinek, H., Vallabhajosula, S., Mosconi, L., et al. (2015). Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging 36, 627–633. doi: 10.1016/j.neurobiolaging.2014.10.038
Kim, J. M., Stewart, R., Prince, M., Kim, S. W., Yang, S. J., Shin, I. S., et al. (2007). Dental health, nutritional status and recent-onset dementia in a Korean community population. Int. J. Geriatr. Psychiatry 22, 850–855. doi: 10.1002/gps.1750
Kinsel, R. P., and Lamb, R. E. (2001). Development of gingival esthetics in the terminal dentition patient prior to dental implant placement using a full-arch transitional fixed prosthesis: a case report. Int. J. Oral Maxillofac. Implants 16, 583–589.
Kouidhi, B., Zmantar, T., Mahdouani, K., Hentati, H., and Bakhrouf, A. (2011). Antibiotic resistance and adhesion properties of oral Enterococci associated to dental caries. BMC Microbiol. 11:155. doi: 10.1186/1471-2180-11-155
Lindwall, G., and Cole, R. D. (1984). The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. J. Biol. Chem. 259, 12241–12245.
Little, C. S., Hammond, C. J., MacIntyre, A., Balin, B. J., and Appelt, D. M. (2004). Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol. Aging 25, 419–429. doi: 10.1016/s0197-4580(03)00127-1
MacDonald, A. B. (1986). Borrelia in the brains of patients dying with dementia. JAMA 256, 2195–2196. doi: 10.1001/jama.1986.03380160053011
MacDonald, A. B., and Miranda, J. M. (1987). Concurrent neocortical borreliosis and Alzheimer’s disease. Hum. Pathol. 18, 759–761. doi: 10.1016/s0046-8177(87)80252-6
Maheshwari, P., and Eslick, G. D. (2015). Bacterial infection and Alzheimer’s disease: a meta-analysis. J. Alzheimers Dis. 43, 957–966. doi: 10.3233/JAD-140621
Mailliot, C., Podevin-Dimster, V., Rosenthal, R. E., Sergeant, N., Delacourte, A., Fiskum, G., et al. (2000). Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J. Cereb. Blood Flow Metab. 20, 543–549. doi: 10.1097/00004647-200003000-00013
Miklossy, J. (1993). Alzheimer’s disease—a spirochetosis? Neuroreport 4, 841–848. doi: 10.1097/00001756-199307000-00002
Miklossy, J. (2011a). Alzheimer’s disease—a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflammation 8:90. doi: 10.1186/1742-2094-8-90
Miklossy, J. (2011b). Emerging roles of pathogens in Alzheimer disease. Expert Rev. Mol. Med. 13:e30. doi: 10.1017/s1462399411002006
Miklossy, J., Khalili, K., Gern, L., Ericson, R. L., Darekar, P., Bolle, L., et al. (2004). Borrelia burgdorferi persists in the brain in chronic Lyme neuroborreliosis and may be associated with Alzheimer disease. J. Alzheimers Dis. 6, 639–649; discussion 673–681.
Miklossy, J., Kis, A., Radenovic, A., Miller, L., Forro, L., Martins, R., et al. (2006). Beta-amyloid depositon and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol. Aging 27, 228–236. doi: 10.1016/j.neurobiolaging.2005.01.018
Miller, M. W., Murtaugh, M., and Kuhn, P. E. (1994). Developmental expression of a 56 kDa protein isolated from rat neocortex. Brain Res. Dev. Brain Res. 81, 260–268. doi: 10.1016/0165-3806(94)90312-3
Mohamed, J., and Huang, D. (2007). Biofilm formation by enterococci. J. Med. Microbiol. 56, 1581–1588. doi: 10.1099/jmm.0.47331-0
Mylona, E., Vadala, C., Papastamopoulos, V., and Skoutelis, A. (2012). Brain abscess caused by Enterococcus faecalis following a dental procedure in a patient with hereditary hemorrhagic telangiectasia. J. Clin. Microbiol. 50, 1807–1809. doi: 10.1128/JCM.06658-11
Neve, R. L., Selkoe, D. J., Kurnit, D. M., and Kosik, K. S. (1986). A cDNA for a human microtubule associated protein 2 epitope in the Alzheimer neurofibrillary tangle. Brain Res. 387, 193–196. doi: 10.1016/0169-328x(86)90011-2
Poole, S., Singhrao, S. K., Chukkapalli, S., Rivera, M., Velsko, I., Kesavalu, L., et al. (2015). Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE−/− mice brains. J. Alzheimers Dis. 43, 67–80. doi: 10.3233/JAD-140315
Poole, S., Singhrao, S. K., Kesavalu, L., Curtis, M. A., and Crean, S. (2013). Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimers Dis. 36, 665–677. doi: 10.3233/JAD-121918
Riviere, G., Riviere, K., and Smith, K. (2002). Molecular and immunological evidence of oral treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 17, 113–118. doi: 10.1046/j.0902-0055.2001.00100.x
Rocas, I. N., Siqueira, J. F., and Santos, K. R. (2004). Association of Enterococcus faecalis with different forms of periradicular diseases. J. Endod. 30, 315–320. doi: 10.1097/00004770-200405000-00004
Schirrmeister, J. F., Liebenow, A. L., Pelz, K., Wittmer, A., Serr, A., Hellwig, E., et al. (2009). New bacterial compositions in root-filled teeth with periradicular lesions. J. Endod. 35, 169–174. doi: 10.1016/j.joen.2008.10.024
Sedgley, C. M., Lennan, S. L., and Clewell, D. B. (2004). Prevalence, phenotype and genotype of oral enterococci. Oral Microbiol. Immunol. 19, 95–101. doi: 10.1111/j.0902-0055.2004.00122.x
Sedgley, C. M., Nagel, A. C., Shelburne, C. E., Clewell, D. B., Appelbe, O., and Molander, A. (2005). Quantitative real-time PCR detection of oral Enterococcus faecalis in humans. Arch. Oral Biol. 50, 575–583. doi: 10.1016/j.archoralbio.2004.10.017
Shackelford, D. A., and Yeh, R. Y. (1998). Dephosphorylation of tau during transient forebrain ischemia in the rat. Mol. Chem. Neuropathol. 34, 103–120. doi: 10.1007/bf02815073
Shimazaki, Y., Soh, I., Saito, T., Yamashita, Y., Koga, T., Miyazaki, H., et al. (2001). Influence of dentition status on physical disability, mental impairment and mortality in institutionalized elderly people. J. Dent. Res. 80, 340–345. doi: 10.1177/00220345010800010801
Shoemark, D. K., and Allen, S. J. (2015). The microbiome and disease: reviewing the links between the oral microbiome, aging and Alzheimer’s disease. J. Alzheimers Dis. 43, 725–738. doi: 10.3233/JAD-141170
Singhrao, S. K., Harding, A., Simmons, T., Robinson, S., Kesavalu, L., and Crean, S. (2014). Oral inflammation, tooth loss, risk factors and association with progression of Alzheimer’s disease. J. Alzheimers Dis. 42, 723–737. doi: 10.3233/JAD-140387
Soscia, S., Kirby, J., Washicosky, K., Tucker, S., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS One 5:e9505. doi: 10.1371/journal.pone.0009505
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Towards defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Stein, P. S., Desrosiers, M., Donegan, S. J., Yepes, J. F., and Kryscio, R. J. (2007). Tooth loss, dementia and neuropathology in the nun study. J. Am. Dent. Assoc. 138, 1314–1322. doi: 10.14219/jada.archive.2007.0046
Strickertsson, J. A., Desler, C., Martin-Bertelsen, T., Machado, A. M., Wadstrom, T., Winther, O., et al. (2013). Enterococcus faecalis infection causes inflammation, intracelular oxphos-independent ROS production, and DNA damage in human gastric cancer cells. PLoS One 8:e63147. doi: 10.1371/journal.pone.0063147
Strickertsson, J. A., Rasmussen, L. J., and Friis-Hansen, L. (2014). Enterococcus faecalis infection and reactive oxygen species down-regulates the miR-17-92 cluster in gastric adenocarcinoma cell culture. Genes (Basel) 5, 726–738. doi: 10.3390/genes5030726
Styren, S. D., Bowser, R., and Dekosky, S. T. (1997). Expression of fetal Alz-50 reactive clone 1 (FAC1) in dentate gyrus following entorhinal cortex lesion. J. Comp. Neurol. 386, 555–561. doi: 10.1002/(sici)1096-9861(19971006)386:4<555::aid-cne3>3.3.co;2-2
Sun, J., Sundsfjord, A., and Song, X. (2012). Enterococcus faecalis from patients with chronic periodontitis: virulence and antimicrobial resistance traits and determinants. Eur. J. Clin. Microbiol. Infect. Dis. 31, 267–272. doi: 10.1007/s10096-011-1305-z
Sundqvist, G., Figdor, D., Persson, S., and Sjögren, U. (1998). Microbiologic analysis of teeth with failed endodontic treatment and the outcome of conservative re-treatment. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 85, 86–93. doi: 10.1016/s1079-2104(98)90404-8
Trojanowski, J. Q., Newman, P. D., Hill, W. D., and Lee, V. M. (1991). Human olfactory epithelium in normal aging, Alzheimer’s disease and other neurodegenerative disorders. J. Comp. Neurol. 310, 365–376. doi: 10.1002/cne.903100307
Weaver, C. L., Espinoza, M., Kress, Y., and Davies, P. (2001). Conformational changes as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol. Aging 21, 719–727. doi: 10.1016/s0197-4580(00)00157-3
Keywords: Alzheimer’s disease, Enterococcus faecalis, edentulism, chronic periodontitis, tau, phosphorylation, Alz-50
Citation: Underly R, Song M-S, Dunbar GL and Weaver CL (2016) Expression of Alzheimer-Type Neurofibrillary Epitopes in Primary Rat Cortical Neurons Following Infection with Enterococcus faecalis. Front. Aging Neurosci. 7:259. doi: 10.3389/fnagi.2015.00259
Received: 11 September 2015; Accepted: 24 December 2015;
Published: 20 January 2016.
Edited by:
Fernanda Laezza, University of Texas Medical Branch, USAReviewed by:
Ramesh Kandimalla, Emory University, USAJudith Miklossy, Prevention Alzheimer International Foundation and International Alzheimer Research Center, Switzerland
Copyright © 2016 Underly, Song, Dunbar and Weaver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles L. Weaver, Y2x3ZWF2ZTFAc3ZzdS5lZHU=