 Yiwei Wang
Yiwei Wang Roberta D. Brinton
Roberta D. Brinton- 1Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA, USA
- 2Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, CA, USA
Brain is the most energetically demanding organ of the body, and is thus vulnerable to even modest decline in ATP generation. Multiple neurodegenerative diseases are associated with decline in mitochondrial function, e.g., Alzheimer’s, Parkinson’s, multiple sclerosis and multiple neuropathies. Genetic variances in the mitochondrial genome can modify bioenergetic and respiratory phenotypes, at both the cellular and system biology levels. Mitochondrial haplotype can be a key driver of mitochondrial efficiency. Herein, we focus on the association between mitochondrial haplotype and risk of late onset Alzheimer’s disease (LOAD). Evidence for the association of mitochondrial genetic variances/haplotypes and the risk of developing LOAD are explored and discussed. Further, we provide a conceptual framework that suggests an interaction between mitochondrial haplotypes and two demonstrated risk factors for Alzheimer’s disease (AD), apolipoprotein E (APOE) genotype and chromosomal sex. We posit herein that mitochondrial haplotype, and hence respiratory capacity, plays a key role in determining risk of LOAD and other age-associated neurodegenerative diseases. Further, therapeutic design and targeting that involve mitochondrial haplotype would advance precision medicine for AD and other age related neurodegenerative diseases.
Introduction
A central role of mitochondria in age-related metabolic and neurodegenerative diseases has long been proposed, and an association between mitochondrial dysfunction and Alzheimer’s disease has been long proposed across multiple investigative strategies from analysis of human tissue to cell model systems (Beal, 1996; Gibson et al., 2000; Trimmer et al., 2000; Swerdlow and Khan, 2004; Bubber et al., 2005; Wallace, 2005; Lin and Beal, 2006; Brinton, 2008, 2009; Khusnutdinova et al., 2008; Simpkins et al., 2008; Coskun et al., 2012; Brinton et al., 2015). The “mitochondrial cascade hypothesis”, originally proposed by Swerdlow and Khan (2004) as an explanation for late onset Alzheimer’s disease (LOAD), proposed that inherited mitochondrial genetic variations and mutations accumulated from aging should occupy the apex of the disease cascade. Briefly, this hypothesis maintained that mitochondrial genetic variations and mutations lead to deficient electron transport chain function, resulting in less ATP production, increased free radical production, disrupted calcium homeostasis, beta amyloid production and plaque deposition, and tau phosphorylation and tangle formation. These results in turn lead to further damage of mitochondrial DNA, proteins, and lipids, and the opening of mitochondrial permeability transition pore, which ultimately leads to cell death and neurodegeneration (see Figure 1). Recent studies also indicated disrupted crosstalk between mitochondria and endoplasmic reticulum (ER) via mitochondrial associated membrane (MAM), as well as abnormal mitochondrial dynamics in the etiology of Alzheimer’s disease (AD; Trimmer et al., 2000; Baloyannis et al., 2004; Paillusson et al., 2013; Burté et al., 2015; Zhang et al., 2016; see Figure 1).
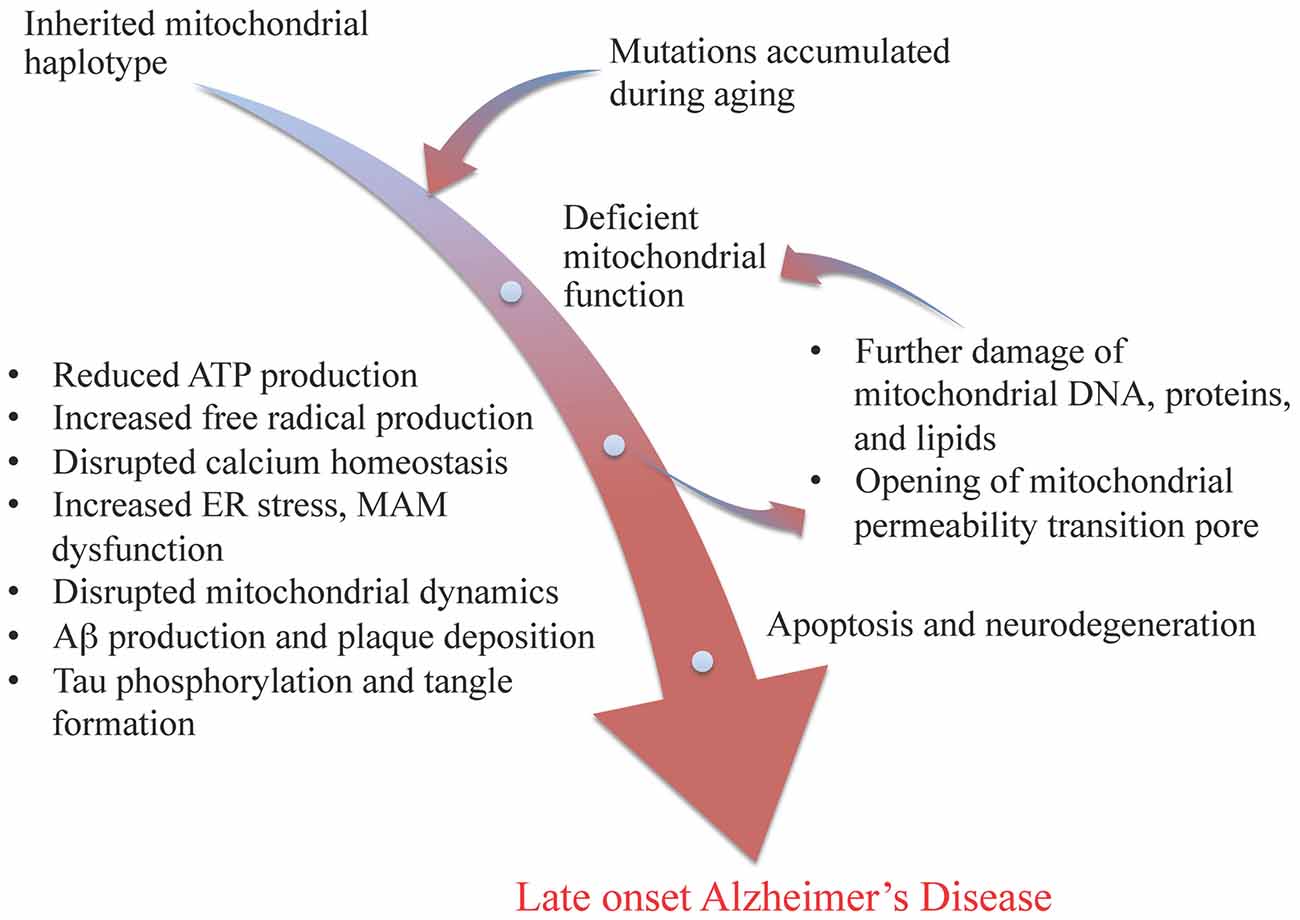
Figure 1. Modified mitochondrial cascade of late-onset Alzheimer’s disease (LOAD). Inherited mitochondrial genetic variations and accumulated mutations during aging lead to deficiency in mitochondrial functions, initiating a cascade of events including reduced ATP production, increased free radical formation and ER stress, disrupted mitochondrial associated membrane (MAM) function and mitochondrial dynamics, as well as amyloid beta plaque and tau tangle formation, which result in further damages of mitochondria and ultimately leads to apoptosis and neurodegeneration, such as LOAD.
Here we review the link between mitochondrial dysfunction and the risk of AD, the contribution of mitochondrial genetic variances to the risk, and how the risk is modulated by other factors, such as apolipoprotein E (APOE) genotype and sex differences.
Mitochondrial Genome and Haplotypes
Unlike many other organelles, mitochondria have their own genome. The human mitochondrial genome is a circular set of 16,569 base pairs encoding 37 genes. Thirteen of these genes encode protein subunits required for four of the five electron transport chain complexes: complex I (NADH ubiquinone oxidoreductase), complex III (cytochrome bc1 complex), complex IV (cytochrome c oxidase), and complex V (ATP synthase); two encode rRNAs for mitochondrial ribosomes (12S and 16S), and 22 encode tRNAs (see Figure 2).
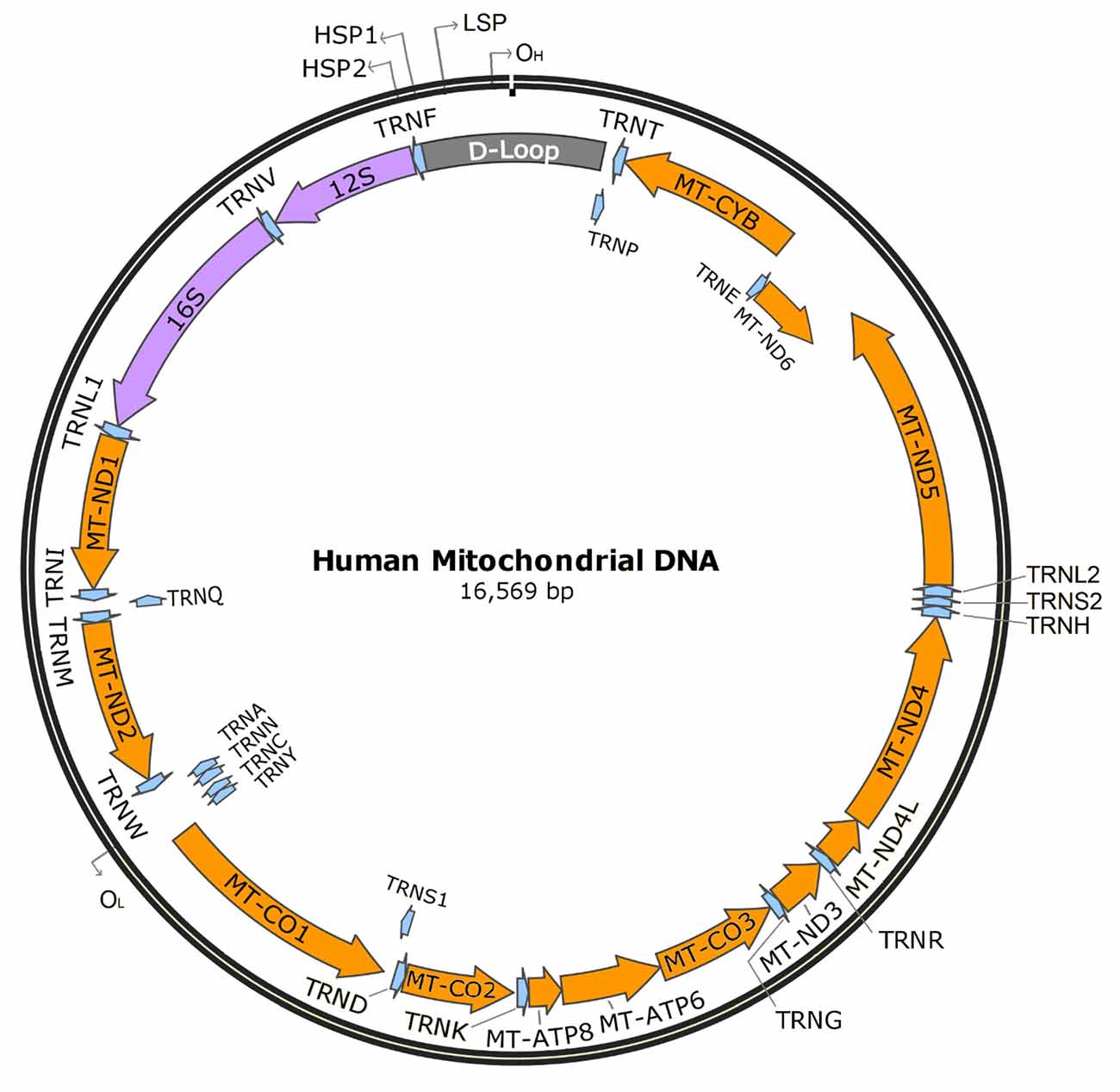
Figure 2. Human mitochondrial DNA. Orange indicates protein-encoding genes (13), purple indicates mitochondrial rRNAs (2), blue indicates tRNAs (22), and gray indicates the D-loop. OH: heavy strand origin, OL: light strand origin, HSP1: major heavy strand promoter, HSP2: minor heavy strand promoter, LSP, light strand promoter.
Mitochondrial retention of their own genome throughout evolution solves two cell biology problems. First, the 13 electron transport subunits coded by mitochondrial DNA solves the problem that if they were generated by nuclear DNA, they would not cross the inner mitochondria membrane due to their high hydrophobicity (Popot and de Vitry, 1990). Second, the eukaryotic mitochondrial genome is transcribed and translated quite differently than the nuclear genome (Mercer et al., 2011). The genetic system of the mitochondria is transcribed as precursor polycistronic transcripts that are subsequently cleaved to generate mRNAs, tRNAs and rRNAs (Mercer et al., 2011). The mRNAs devoted to generating the 13 catalytic subunits required for oxidative phosphorylation are further translated using several non-universal codons unique to the mitochondrial translation machinery (Watanabe, 2010; Watanabe and Yokobori, 2011).
Like nuclear DNA, mitochondrial DNA undergoes mutation, though at a much higher rate (Miyata et al., 1982; Wallace et al., 1987), likely due to higher replication rate, a more mutagenic environment, and less efficient DNA repair (Xu et al., 2012). Unlike the nucleus, the mitochondrial DNA repair mechanism is largely limited to base excision repair. Mismatch as a result of either recombination or repair can lead to single nucleotide polymorphisms (SNPs).
Clusters of specific SNPs in the mitochondrial genome define mitochondrial haplogroups that reflect maternal lineage (Giles et al., 1980; Torroni et al., 1992). For example, the four lineages specific for sub-Saharan Africa are L0, L1, L2 and L3, and haplogroups A, B, C, D, G and F are common in Asia (Stewart and Chinnery, 2015). The major haplogroups within descendants of European ancestry are haplogroups H, I, J, K, M, T, U, V, W and X (Torroni et al., 1996). These haplogroups can be further classified into subhaplogroups or clustered together into superhaplogroups, such as superhaplogroup HV, JT, UK.
Mitochondrial Dysfunction and Increased Risk of Alzheimer’S Disease
AD is a systemic disease with multiple etiologies (Morris et al., 2014). Early evidence linking mitochondrial dysfunction to AD dates back to a 1987 study by Sims et al. (1987), who found a reduced rate of oxygen uptake in the presence and absence of ADP in frontal neocortex of postmortem confirmed AD cases, indicating potential mitochondrial uncoupling in AD patients (Sims et al., 1987).
More direct evidence for the association between mitochondrial dysfunction and AD came later from studies on the activity of electron transport chain enzymes in AD patients and post mortem AD brain tissues. In the early 1990’s, reduced cytochrome c oxidase (complex IV) activity was observed in both platelets and postmortem AD brain (Parker et al., 1990, 1994). The reduction of cytochrome c activity in AD brain was later refined to the temporal cortex and hippocampus (Maurer et al., 2000). Supporting these findings, reduced mRNA levels of cytochrome c oxidase subunit 1 and 3 were observed in AD mid temporal gyrus and cytochrome c oxidase subunit 2 in AD hippocampus (Chandrasekaran et al., 1994; Aksenov et al., 1999). Furthermore, in both AD temporal and parietal cortices, protein levels of cytochrome c oxidase subunits were reduced, especially those encoded by mitochondria DNA (Kish et al., 1999). Although no reduction of cytochrome c oxidase content was observed in platelets of AD patients, cytoplasmic hybrids (cybrids) containing exogenous mitochondria extracted from platelets of AD patients showed less cytochrome c oxidase activity compared to cybrids harboring mitochondria from age-matched controls (Davis et al., 1997; Sheehan et al., 1997; Cardoso et al., 2004). Alternations in gene expression, protein level, and activity of other electron transport chain complexes have also been reported in AD tissues, though the evidence was less compelling and sometimes contradicted (Schagger and Ohm, 1995; Chandrasekaran et al., 1997; Aksenov et al., 1999; Kim et al., 2000; Bosetti et al., 2002; Bubber et al., 2005; Valla et al., 2006).
Upstream to electron transport chain, impairment of the tricarboxylic acid (TCA) cycle enzymes was observed in AD brain. Autopsy-confirmed AD brain had significantly reduced activity in pyruvate dehydrogenase complex, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase complex, whereas the activity of succinate dehydrogenase and malate dehydrogenase were increased (Bubber et al., 2005). The severity of enzyme activity impairment was also correlated with the clinical severity of Alzheimer’s pathology (Bubber et al., 2005). Decreased mitochondrial respiratory capacity was consistent with the higher level of lactate and lower key substrates for TCA cycle in cerebrospinal fluid and blood in AD patients (Redjems-Bennani et al., 1998; Mancuso et al., 2003).
Consistent with deficits in mitochondrial respiratory capacity, AD brain had elevated levels of peroxidation products in the frontal cortex, as well as decreased levels of superoxide dismutase, a radical defensive enzyme, in the frontal cortex, hippocampus, and cerebellum (Richardson, 1993). Increased oxidative stress is consistent with free radical damage of mitochondrial components and loss of mitochondrial membrane potential as observed in cybrids harboring mitochondria with AD origin (Cassarino et al., 1998; Trimmer et al., 2000).
Besides altered bioenergetic capacity, the crosstalk between mitochondria and the ER via MAM, which regulates many key functions of mitochondria, such as calcium uptake, phospholipid exchange, intracellular trafficking, ER stress and mitochondrial biogenesis, was also disrupted in AD (Paillusson et al., 2013; Burté et al., 2015).
Beyond changes in mitochondrial function, distribution and morphology of mitochondria were also different in AD patients. Neurons from AD brain harbored mitochondria of smaller sizes and disrupted mitochondrial cristae morphology, and cybrids created using mitochondria from sporadic AD patients also showed swollen mitochondria and less cristae (Trimmer et al., 2000; Baloyannis et al., 2004). The fission and fusion cycle of mitochondria also seemed disrupted in the hippocampus of AD brain (Zhang et al., 2016). The number of mitochondria was also significantly reduced, likely as a result of increased mitochondria degradation, turnover and autophagy (Hirai et al., 2001).
AD has a pattern of maternal inheritance, where inheritance of AD from mothers are more frequent than from fathers (Duara et al., 1993). The observation of maternal pattern of Alzheimer’s inheritance has been replicated over many decades by multiple groups, and has been linked to deficits in mitochondrial respiration and glucose metabolism apparently early in the aging process (Edland et al., 1996; Mosconi et al., 2011; Liu et al., 2013). In cognitively normal individuals, those with a maternal history of LOAD showed decline in platelet cytochrome c oxidase activity, compared to those with a paternal or no family history of the disease (Edland et al., 1996; Mosconi et al., 2011). In this same cohort, Mosconi et al. (2011) observed significantly lower brain glucose metabolism in persons with a maternal history of Alzheimer’s relative to those with a paternal or no family history of the disease. These data coupled with maternal inheritance of the mitochondrial genome strongly support a role for mitochondrial genetic variances in the etiology of the disease.
Mitochondrial Haplotype is Associated with Different Respiratory, Metabolic and Bioenergetic Phenotypes
Mitochondrial genetic background can also affect metabolism and bioenergetic function. Resting metabolic rate (RMR) and total energy expenditure (TEE) were measured in 294 participants in the health, aging and body composition study (Health ABC; Tranah et al., 2011), including participants of cluster L, which contains common African haplogroups, and cluster N, which contains common European haplogroups (Tranah et al., 2011). Compared to N, cluster L had significantly lower RMR and TEE. Specifically, haplogroups L0, L2 and L3 had significantly lower RMR than haplogroup H and superhaplogroups UK and JT; haplogroup L3 had significantly lower TEE than haplogroup H and superhaplogroups UK and JT; haplogroup L2 had significantly lower TEE than haplogroup H and superhaplogroup JT (Tranah et al., 2011; see Table 1). In a cohort of healthy Spanish males, haplogroup J participants had significantly lower maximum oxygen consumption (VO2max) than non-J participants (Marcuello et al., 2009). This difference was later confirmed in an independent cohort of healthy Spanish males, where haplogroup H was determined to be the driving force for the difference (Martínez-Redondo et al., 2010; see Table 1).
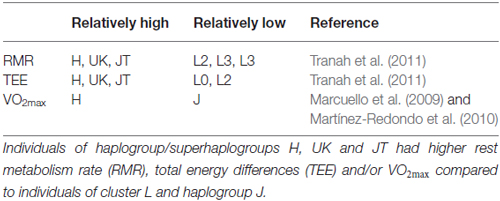
Table 1. Mitochondrial haplogroups/superhaplogroups differentially associated with respiratory phenotypes.
The underlying cellular mechanism of the observed differences across different mitochondrial haplogroups was primarily elucidated using trans-mitochondrial cytoplasmic hybrids, or cybrids, which controlled for the nuclear genetic background to reveal mitochondrial variances. An early cybrids study using cultured A539 human lung carcinoma cells harboring either mitochondria of haplogroup H or T failed to identify any differences in bioenergetics function (Amo et al., 2008). However, differences in bioenergetics and mitochondrial function were identified in multiple later studies using different cell lines and mitochondrial haplogroups. Cybrids constructed from osteosarcoma 143B rho0 cells and platelets from healthy Spanish donors of either haplogroup H or superhaplogroup UK were investigated for mtDNA content (Gómez-Durán et al., 2010). Cybrids harboring UK superhaplogroup were found to have lower mtDNA content, lower mt-rRNA level, reduced protein synthesis, and decreased cytochrome c oxidase amount (Gómez-Durán et al., 2010). UK cybrids also had lower mitochondrial inner membrane potential and higher mitochondrial uncoupling, indicating potentially lower respiratory capacity and reduced ATP production (Gómez-Durán et al., 2010). Similar results were obtained in a later study in middle-age Caucasian males, where OXPHOS capacity normalized to citrate synthase content was found to be reduced by 24% in subjects with haplogroup U background comparing to those with haplogroup H background (Larsen et al., 2014). In 2013, cybrids constructed from human retinal epithelial cell line ARPE-19 and either haplogroup H or J mitochondria showed reduced ATP production and glycolysis in J cybrids (Kenney et al., 2013). In accordance with the observed reduction in mitochondrial respiration, J cybrids also showed lower ROS production (Kenney et al., 2013). Haplogroup J cybrids with chondrocyte nuclear genetic background also demonstrated lower NO levels than non-J cybrids (Fernández-Moreno et al., 2011). Similarly, major Asian mitochondrial haplogroups are also differentially associated with bioenergetic function (Lin et al., 2012). These associations identified in the human are also evident in animal models ranging from Drosophila to mice such that different mitochondrial genetic background is associated with differences in respiratory and metabolic phenotypes, electron transport chain enzyme activities and mitochondrial functions (Pichaud et al., 2012; Scheffler et al., 2012; Latorre-Pellicer et al., 2016).
Although different nuclear genetic background employed in the above studies make it difficult to compare the results across studies, this growing body of evidence supports mitochondrial genetic variance/haplotype as a key factor in the observed differences in bioenergetics and metabolism.
Mitochondrial Genetic Variances/Haplotypes and Haplogroups and the Risk of Alzheimer’S Disease
In an early effort to assess the contribution of mitochondrial DNA variances to pathologies of neurodegenerative diseases, researchers identified a non-synonymous SNP in tRNAGln, mt4336C, that had an increased frequency in a Caucasian cohort of late onset Alzheimer’s and Parkinson disease patients (Shoffner et al., 1993). The contribution of mt4336C was later confirmed in a different North American Caucasian cohort of AD patients (Hutchin and Cortopassi, 1995). Individuals harboring the mt4336C SNP also tended to harbor the mt16304C SNP, and had a more closely related D-loop sequence, which could be traced back to a single phylogenetic node (Shoffner et al., 1993; Hutchin and Cortopassi, 1995). However, two other studies did not confirm either mt4336C or mt16304C as a risk factor for developing AD in similar populations, when blood samples and leukocytes from clinically diagnosed patients were used instead of histopathologically confirmed postmortem AD brain tissues (Wragg et al., 1995; Zsurka et al., 1998). Today, mt16304C is known as a defining SNP for subhaplogroup H5, and mt4336C a defining SNP for subhaplogroup H5a. The above studies constituted the earliest debate over whether mitochondrial genetic variances can modify the risk of developing AD.
In fact, haplogroup H and superhaplogroup HV, which contains haplogroup H and its subhaplogroups, have been the most reported haplogroup in association with increased risk of developing AD (Chinnery et al., 2000; Edland et al., 2002; van der Walt et al., 2004, 2005; Elson et al., 2006; Fesahat et al., 2007; Mancuso et al., 2007; Maruszak et al., 2009, 2011; Santoro et al., 2010; Coto et al., 2011; Ridge et al., 2012; Fachal et al., 2015; see Table 2). A study based on 30 Iranian late onset Alzheimer’s patients and 100 controls found that haplogroup H was significantly more abundant in the disease group (Fesahat et al., 2007; see Table 2). In a Spanish-Caucasian group, haplogroup H and its defining SNP mt7028C were enriched in LOAD patients compared to controls (Coto et al., 2011; see Table 2). In a large Caucasian cohort containing 422 late-onset AD patients and 318 neurologically healthy controls, researchers found that superhaplogroup HV, which contains haplogroup H, had significantly higher presence in LOAD than in control (Maruszak et al., 2011; see Table 2). Finally, a meta-analysis pooling data from five previous studies (some studies including early-onset AD patients) also confirmed the association between haplogroup H and superhaplogroup HV and the risk of developing AD (Maruszak et al., 2011; see Table 2).
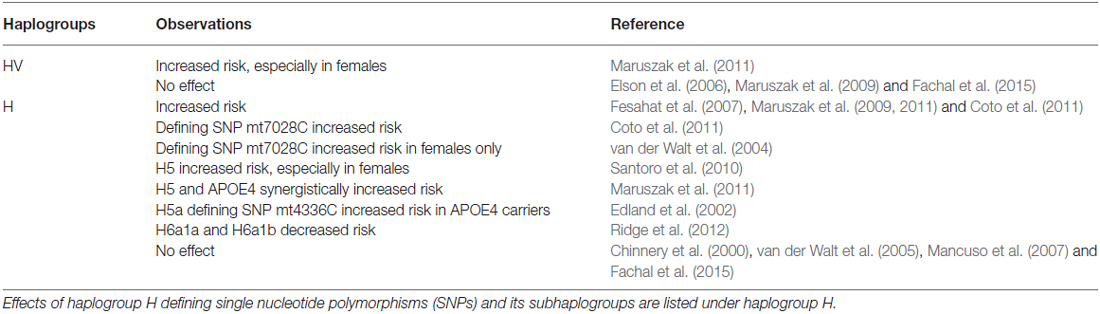
Table 2. Observed effects of mitochondrial superhaplogroup HV and haplogroup H on risk of Alzheimer’s disease (AD).
When mitochondrial DNA was sequenced in greater depth, subhaplogroup H5 was significantly associated with AD compared to haplogroup H of central-northern Italians, (Santoro et al., 2010; see Table 2). However, from the Cache county study on aging and memory in Utah residents, subhaplogroups H6a1a and H6a1b were found to be protective against AD (Ridge et al., 2012; see Table 2). While the protective role of subhaplogroups H6a1a and H6a1b seems contradictory to the overall risk of haplogroup H, the data predict that the observed risks within haplogroup H may be driven by its sub-haplogroups with H5 increases risk of LOAD whereas H6 reduces risk.
The second most studied superhaplogroup is UK, and its member haplogroups U and K, including their subhaplogroups (While haplogroup K is currently recognized as a branch of haplogroup U, early studies classified haplogroups U and K as two parallel haplogroups under superhaplogroup UK. For consistency of referring to previous studies, we will use the earlier classification system throughout this review article; see Table 3). In a Utah based ADNI cohort, superhaplogroup UK was identified as a risk factor for AD (Lakatos et al., 2010; see Table 3). These findings are in contrast to an earlier study conducted in a Poland-based Caucasian population, where no effect of superhaplogroup UK was observed (Maruszak et al., 2009; see Table 3). The disparity may be explained by differences in the distribution of specific subhaplogroups or SNPs in the studied populations. Specifically, while each of the three defining SNPs for haplogroup U (mt11467G, mt12308G and mt12372A) has been identified as a risk factor for AD (Lakatos et al., 2010), subhaplogroup U5a1 and SNP mt16224C, a haplogroup K defining SNP, were shown to be protective (Maruszak et al., 2011; see Table 3).

Table 3. Observed effects of mitochondrial superhaplogroup KU and haplogroups K and U on risk of AD.
Another common European haplogroup studied for its association with AD is haplogroup T, where one study in French-Canadians found that the frequency of SNPs mt709A and mt15928A, both defining SNPs for haplogroup T, were three times higher in controls than in AD patients, suggesting a protective role of haplogroup T (Chagnon et al., 1999; see Table 4). However, the Health, Aging, and Body Composition (Health ABC) study found that haplogroup T had increased risk for dementia when compared to haplogroup H (Tranah et al., 2012). The Health ABC study also found that haplogroup J, also under superhaplogroup JT, was associated with significant decline in cognitive function compared to haplogroup H (Tranah et al., 2012).
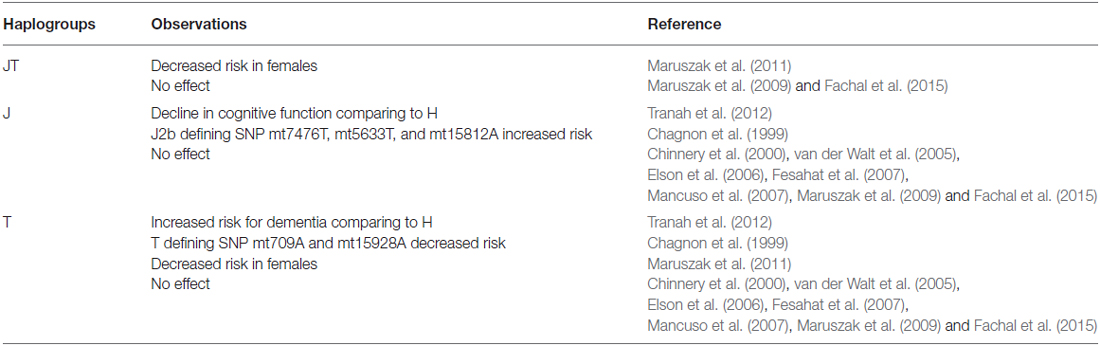
Table 4. Observed effects of mitochondrial superhaplogroup JT and haplogroups J and T on risk of AD.
In addition to major European haplogroups, several African and Asian haplogroups have also been reported to be associated with the AD or risk of dementia (see Table 5). For example, in an African American population, haplogroup L1 was found to have increased risk for developing dementia (Tranah et al., 2014). In Asians, subhaplogroups G2a, B4c1 and N9b1 were reported to be associated with AD in Japanese populations, and haplogroup B5 was reported to be associated with AD in Han Chinese (Takasaki, 2008, 2009; Bi et al., 2015; see Table 5).
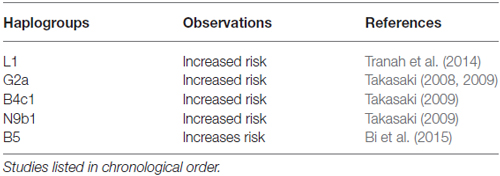
Table 5. Association between some Asian mitochondrial haplogroups and the risk of Alzheimer’s disease.
As reviewed here, the field has not reached consensus on the effect of mitochondrial haplogroups on late onset AD (LOAD). Different sets of haplogroups are identified in different studies and different studies could identify opposite effects of the same mitochondrial haplotype on risk of LOAD. Moreover, multiple studies involving Caucasians of European descendant (European descents in UK and US, Tuscans, Spanish and Old order Amish from Indiana and Ohio) did not detect associations between any haplogroup and AD (Chinnery et al., 2000; van der Walt et al., 2005; Elson et al., 2006; Mancuso et al., 2007; Fachal et al., 2015; see Tables 2–5). While the controversy may be driven by differences in the distribution of subhaplogroups within the studied population or different DNA sources (e.g., postmortem brain tissue vs. peripheral blood), the discrepancies between studies also suggested that as intriguing as the results are, mitochondrial genetic variances are unlikely to be the sole driving force of LOAD. Further, the most consistent risk factor for LOAD is APOE genotype, but it alone is not an absolute determinant. Below, we determine the relationship between mitochondrial haplogroup and APOE genotype on risk of AD.
The Interaction between Mitochondrial Haplogroup and APOE Genotype on Risk of AD
APOEε4 genotype is a widely recognized risk factor for AD, and has been repeatedly confirmed in the studies reviewed herein (Corder et al., 1993; Poirier et al., 1993; Rebeck et al., 1993; Saunders et al., 1993; Carrieri et al., 2001; Edland et al., 2002; Coto et al., 2011; Maruszak et al., 2011). Further, APOEε4 has been associated with mitochondrial dysfunction and glucose hypometabolism in brain (Reiman et al., 2001, 2004, 2005; Mosconi et al., 2004a,b,c, 2005, 2008; Valla et al., 2010; Wolf et al., 2013). Compared to non-carriers, APOEε4 carriers showed reduced cerebral parietal glucose metabolism among cognitive normal elderlies with family history of AD (Small et al., 1995, 2000). In APOEε4 positive MCI patients, reduced regional cerebral metabolic rate of glucose consumption (rCMRglc) was detected in temporoparietal and posterior cingulate cortex (Mosconi et al., 2004b). In AD patients, more severe hypometabolism was detected in the parietal, temporal, and cingulate areas in APOEε4 carriers than non-carriers (Mosconi et al., 2004c; Drzezga et al., 2005). Brain glucose hypometabolism was also more widespread in APOEε4 positive AD patients (Mosconi et al., 2004a). On the therapeutic side, mild-to-moderate AD patients who are APOEε4 carriers were shown to be less responsive to rosiglitazone, which can improve mitochondrial efficiency and glucose metabolism (Risner et al., 2006; Roses et al., 2007).
In longitudinal studies, APOEε4 carriers had significantly greater rCMRglc decline in the vicinity of temporal, posterior cingulate, and prefrontal cortex, basal forebrain, parahippocampal gyrus, and thalamus (Reiman et al., 2001; Mosconi et al., 2008). Decrease of glucose metabolism was also evident in young and middle-aged APOEε4 carriers in posterior cingulate, parietal, temporal, and prefrontal cortex, as well as thalamus (Reiman et al., 2004; Mosconi et al., 2008). The effect of APOEε4 allele has also been shown to be gene dose dependent with APOEε4 homozygote carriers showing greater hypometabolic deficit relative to APOEε3/4 heterozygote carriers (Reiman et al., 2005).
At the cellular level, APOEε4 gene expression in human was associated with down-regulation of genes involved in mitochondrial oxidative phosphorylation and energy metabolism (Xu et al., 2006, 2007). APOEε4 gene expression was also found to be associated with lower mitochondrial cytochrome oxidase activity in posterior cingulate cortex among young adults with family history of AD (Valla et al., 2010). Neurons from humanized APOEε4 knock-in mice had significantly lower amount of all five electron transport chain complexes comparing to those from APOEε3 knock-in mice (Chen et al., 2011). Proteomic analysis revealed decreased expression of proteins involved in the TCA cycle, glucose, lipid and amino acid metabolism in APOEε4 knock-in mice (Shi et al., 2014). Further, cytochrome c levels were significantly lower in ApoEε4 mice compared with ApoEε3 mice (Shi et al., 2014). In vitro studies also suggested that truncated APOEε4 fragment can interact directly with mitochondria and cause mitochondrial dysfunction and neurotoxicity (Chang et al., 2005; Mahley et al., 2007). Given the association between decreased bioenergetic capacity in brain and the risk of AD, an interaction between APOE genotype and mitochondrial haplotypes is possible.
After stratifying by APOEε4 status, three interesting modes of interactions between APOEε4 status and mitochondrial haplogroups in modulating the risk of AD were apparent (see Table 6).

Table 6. Three modes of interactions between APOEε4 status and mitochondrial haplogroups in modulating the risk of AD.
The first mode is a neutralizing effect of mitochondrial haplogroup on the effect of APOEε4 on risk of AD (see Table 6). Early studies identified a non-random association between mitochondrial haplogroup and APOEε4 status in AD patients (Carrieri et al., 2001). Specifically, while APOEε4 carriers had significantly higher odds ratio for AD, those belonging to haplogroups K and U did not, indicating a neutralizing effect of haplogroups K and U on the risk of APOEε4 gene status (Carrieri et al., 2001). The non-random distribution of mitochondrial haplogroups associated with APOEε4 status and the neutralizing effect of mitochondrial haplogroup K on APOEε4 were later confirmed by Maruszak et al. (2011).
The second mode is an enabling effect of APOEε4 on mitochondrial genetic variances as risk factors for AD (see Table 6). In non-APOEε4 carriers, SNP mt4336C (a defining SNP for subhaplogroup H5a) was not an AD risk factor, however, in APOEε4 carriers, the same SNP was a risk factor for AD (Edland et al., 2002). This study indicated that APOE genotype could explain the earlier disparity regarding the association between SNP mt4336C and AD (Shoffner et al., 1993; Hutchin and Cortopassi, 1995; Wragg et al., 1995; Zsurka et al., 1998).
A synergistic effect was also observed between APOEε4 and mitochondrial haplotypes (see Table 6). For example, SNP mt7028C, a defining SNP for haplogroup H, and subhaplogroup H5 were suggested to act synergistically with APOEε4 to increase risk for AD (Coto et al., 2011; Maruszak et al., 2011).
As with the association between mitochondrial haplotype and the risk of late onset AD, the interaction between APOEε4 and mitochondrial genetic variances in modulating the risk of late onset AD remains debatable. Multiple studies failed to identify any correlation between mitochondrial haplogroup and APOEε4 status or failed to identify an interaction between the two on the risk of developing AD (Zsurka et al., 1998; Chinnery et al., 2000; van der Walt et al., 2005; Mancuso et al., 2007; Lakatos et al., 2010; Santoro et al., 2010; Ridge et al., 2013). Collectively, these disparate findings on the association between mitochondrial haplotype, APOE genotype, and risk of AD emphasize the importance of a precision medicine approach that considers mitochondrial genetic variance in combination with nuclear genetics.
Mitochondrial Haplogroup and Sex Differences and Risk of Alzheimer’S Disease
Females are at greater lifetime risk for LOAD, and also have higher prevalence and incidence rate than all age-matched males (Brookmeyer et al., 1998; Mielke et al., 2014; Grimm et al., 2016). The higher risk for female is also evident in faster disease progression and greater burden of AD pathology (Aguero-Torres et al., 1998; Corder et al., 2004; Barnes et al., 2005; Skup et al., 2011; Kelly et al., 2013; Mielke et al., 2014; Grimm et al., 2016). While the underlying mechanism remains to be elucidated, increased mitochondrial oxidative stress may play a role (Schuessel et al., 2004; Mandal et al., 2012). Given the effect of mitochondrial genetic variances on mitochondrial function and risk of AD, it is of interest to identify any interaction between mitochondrial haplotypes and sex difference on risk of LOAD.
While almost all the reviewed studies recognized sex differences on risk of LOAD, only a few studied the effects of mitochondrial genetic variances in each sex (van der Walt et al., 2004; Maruszak et al., 2009, 2011; Santoro et al., 2010; see Table 7). Within these studies, some mitochondrial genetic variances were found to be associated with AD in females only (van der Walt et al., 2004; Maruszak et al., 2009, 2011; Santoro et al., 2010). Superhaplogroup JT, haplogroup T, a haplogroup T defining SNP mt13368A, a haplogroup U defining SNP 12308G and a non-H defining SNP mt7028T were found to exert protective effects only in females (van der Walt et al., 2004; Maruszak et al., 2011; see Table 7). Superhaplogroup HV, haplogroup H, subhaplogroup H5 and a haplogroup H defining SNP mt7028C were identified as risk factors for only in females (Maruszak et al., 2009, 2011; Santoro et al., 2010; see Table 7).
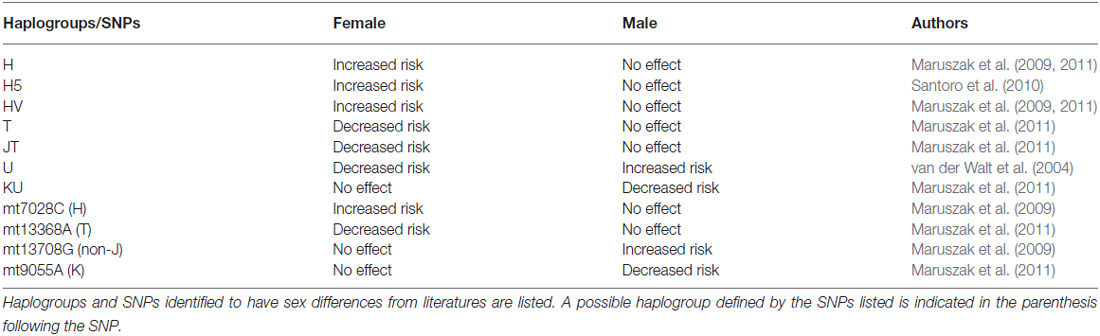
Table 7. Sex differentiates effects of mitochondrial haplogroups on risk of AD.
In contrast, some variances affected only males (see Table 7). Superhaplogroup UK, and SNP mt9055A, a defining SNP for haplogroup K, were found to be associated with reduced risk of AD in males, while SNP mt13708G (for many non-J haplogroups), and SNP mt10398A, a defining SNP for some subhaplogroups of U, were associated with increased risk only in males (van der Walt et al., 2004; Maruszak et al., 2009, 2011; see Table 7). Certain mitochondrial genetic variances also showed opposite effects in each sex. For example, haplogroup U was associated with increased risk in males but decreased risk in females (van der Walt et al., 2004; see Table 7).
These results indicate that previously observed associations between mitochondrial haplogroups and AD could be driven by sex. Conversely, the effects observed in one sex could be diluted in the whole population, or be neutralized by the other sex, resulting in non-significant associations.
Conclusion and Perspectives
Mounting evidence suggested a central role of mitochondrial dysfunction in the etiology of late onset AD. As reviewed above, disruption of mitochondrial bioenergetics, structure and dynamics have all been indicated in AD patients. Given the maternal pattern of inheritance of late onset AD and mitochondrial genome, herein we reviewed the association of mitochondrial genetic variances on bioenergetics, respiratory phenotypes and risk of developing LOAD. While the outcomes remain debatable, a large body of science supports an association between mitochondrial genetic variances and differences in bioenergetics and AD susceptibility. Several factors can help explain the disagreement. First, although most studies were conducted in descendants of European origin, the geographic distribution of participants were drastically different, which could result in diverse nuclear genetic background. Since mitochondria communicate extensively with the nucleus, uncontrolled nuclear background can potentially mask effects of mitochondrial genetic variances. Second, as we have reviewed, observations could be driven by certain subhaplogroups, thus results obtained from different populations may be biased by their dominant subhaplogroup. Third, given the heteroplasmic nature of the mitochondrial DNA, accumulated mutations throughout aging may be haplotype and tissue specific (Wallace, 2005), which could contribute to the discrepancies between studies that had different sources of mtDNA (e.g., brain tissues vs. peripheral blood samples; Wallace, 1994). And last, the criteria for mitochondrial haplogroup assignment evolved during the past two decades. Initial studies used haplotype assignment based on only 10 SNPs in the control region, whereas more recent studies used 138 SNPs across the whole human mitochondrial genome. As a result, some ambiguous assignments were resolved and more subhaplogroups were identified.
The data also indicate that mitochondrial haplotype is one factor among several that impacts risk of AD. This is consistent with the multifactorial nature of aging trajectories and risk for LOAD. A systems biology approach that integrates mitochondrial genetic variances and risk factors such as APOEε4 genotype and sex is a step towards resolving disparities across studies of mitochondrial haplotype and risk of neurodegenerative diseases associated with mitochondrial dysfunction (see Figure 3). More importantly, we propose a precision medicine approach, where nuclear genetic risk factors (especially APOEε4 genotype), mitochondrial haplotypes, and sex differences can be incorporated into future therapeutic designs for LOAD.
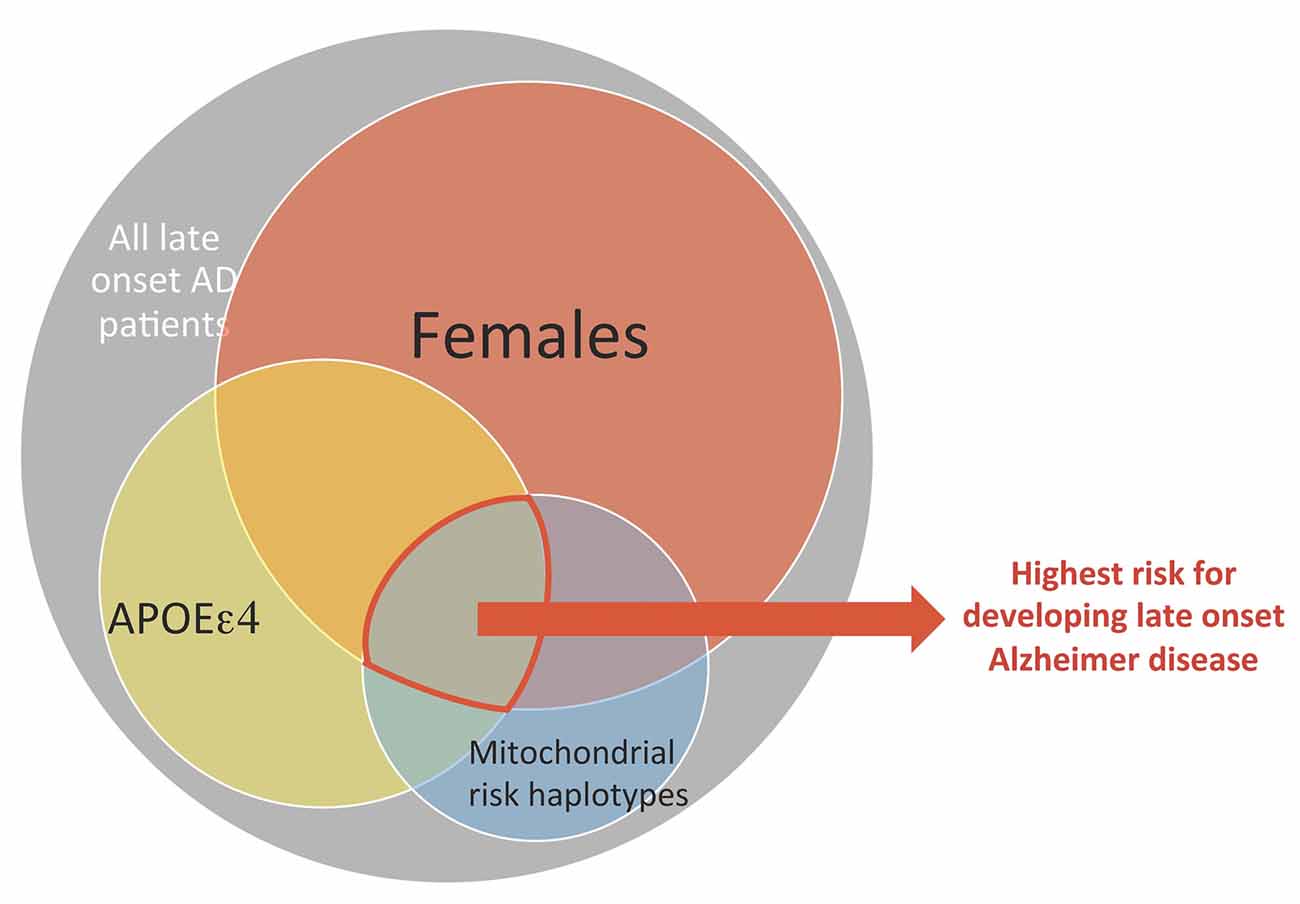
Figure 3. A conceptual framework where the combination of mitochondrial and nuclear genetic risk factors plus sex as a higher risk factor for LOAD. Each of the three factors: sex, APOEε4 genotype and mitochondrial genetic variance has moderate risk for developing late onset AD, but constitutes a much higher risk when combined.
Author Contributions
YW and RDB wrote and reviewed the manuscript together.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by: National Institute on Aging (NIA) grants R01AG032236 and P01AG026572 to RDB, Project 1 to RDB and YW.
References
Aguero-Torres, H., Fratiglioni, L., Guo, Z., Viitanen, M., and Winblad, B. (1998). Prognostic factors in very old demented adults: a seven-year follow-up from a population-based survey in Stockholm. J. Am. Geriatr. Soc. 46, 444–452. doi: 10.1111/j.1532-5415.1998.tb02464.x
Aksenov, M. Y., Tucker, H. M., Nair, P., Aksenova, M. V., Butterfield, D. A., Estus, S., et al. (1999). The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase and NADH dehydrogenase, in different brain regions in Alzheimer’s disease. Neurochem. Res. 24, 767–774. doi: 10.1023/A:1020783614031
Amo, T., Yadava, N., Oh, R., Nicholls, D. G., and Brand, M. D. (2008). Experimental assessment of bioenergetic differences caused by the common European mitochondrial DNA haplogroups H and T. Gene 411, 69–76. doi: 10.1016/j.gene.2008.01.007
Baloyannis, S. J., Costa, V., and Michmizos, D. (2004). Mitochondrial alterations in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 19, 89–93. doi: 10.1177/153331750401900205
Barnes, L. L., Wilson, R. S., Bienias, J. L., Schneider, J. A., Evans, D. A., and Bennett, D. A. (2005). SEx differences in the clinical manifestations of Alzheimer disease pathology. Arch. Gen. Psychiatry 62, 685–691. doi: 10.1001/archpsyc.62.6.685
Beal, M. F. (1996). Mitochondria, free radicals and neurodegeneration. Curr. Opin. Neurobiol. 6, 661–666. doi: 10.1016/s0959-4388(96)80100-0
Bi, R., Zhang, W., Yu, D., Li, X., Wang, H. Z., Hu, Q. X., et al. (2015). Mitochondrial DNA haplogroup B5 confers genetic susceptibility to Alzheimer’s disease in Han Chinese. Neurobiol. Aging 36, 1604.e7–1604.e16. doi: 10.1016/j.neurobiolaging.2014.10.009
Bosetti, F., Brizzi, F., Barogi, S., Mancuso, M., Siciliano, G., Tendi, E. A., et al. (2002). Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 23, 371–376. doi: 10.1016/s0197-4580(01)00314-1
Brinton, R. D. (2008). The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 31, 529–537. doi: 10.1016/j.tins.2008.07.003
Brinton, R. D. (2009). Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci. 30, 212–222. doi: 10.1016/j.tips.2008.12.006
Brinton, R. D., Yao, J., Yin, F., Mack, W. J., and Cadenas, E. (2015). Perimenopause as a neurological transition state. Nat. Rev. Endocrinol. 11, 393–405. doi: 10.1038/nrendo.2015.82
Brookmeyer, R., Gray, S., and Kawas, C. (1998). Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health 88, 1337–1342. doi: 10.2105/ajph.88.9.1337
Bubber, P., Haroutunian, V., Fisch, G., Blass, J. P., and Gibson, G. E. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 57, 695–703. doi: 10.1002/ana.20474
Burté, F., Carelli, V., Chinnery, P. F., and Yu-Wai-Man, P. (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. doi: 10.1038/nrneurol.2014.228
Cardoso, S. M., Proença, M. T., Santos, S., Santana, I., and Oliveira, C. R. (2004). Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 25, 105–110. doi: 10.1016/s0197-4580(03)00033-2
Carrieri, G., Bonafè, M., De Luca, M., Rose, G., Varcasia, O., Bruni, A., et al. (2001). Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Hum. Genet. 108, 194–198. doi: 10.1007/s004390100463
Cassarino, D. S., Swerdlow, R. H., Parks, J. K., Parker, W. D. Jr., and Bennett, J. P. Jr,. (1998). Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer’s disease cybrids. Biochem. Biophys. Res. Commun. 248, 168–173. doi: 10.1006/bbrc.1998.8866
Chagnon, P., Gee, M., Filion, M., Robitaille, Y., Belouchi, M., and Gauvreau, D. (1999). Phylogenetic analysis of the mitochondrial genome indicates significant differences between patients with Alzheimer disease and controls in a French-Canadian founder population. Am. J. Med. Genet. 85, 20–30. doi: 10.1002/(SICI)1096-8628(19990702)85:1<20::AID-AJMG6>3.0.CO;2-K
Chandrasekaran, K., Giordano, T., Brady, D. R., Stoll, J., Martin, L. J., and Rapoport, S. I. (1994). Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res. Mol. Brain Res. 24, 336–340. doi: 10.1016/0169-328x(94)90147-3
Chandrasekaran, K., Hatanpää, K., Rapoport, S. I., and Brady, D. R. (1997). Decreased expression of nuclear and mitochondrial DNA-encoded genes of oxidative phosphorylation in association neocortex in Alzheimer disease. Brain Res. Mol. Brain Res. 44, 99–104. doi: 10.1016/s0169-328x(96)00191-x
Chang, S., Ran Ma, T., Miranda, R. D., Balestra, M. E., Mahley, R. W., and Huang, Y. (2005). Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. U S A 102, 18694–18699. doi: 10.1073/pnas.0508254102
Chen, H. K., Ji, Z. S., Dodson, S. E., Miranda, R. D., Rosenblum, C. I., Reynolds, I. J., et al. (2011). Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 286, 5215–5221. doi: 10.1074/jbc.M110.151084
Chinnery, P. F., Taylor, G. A., Howell, N., Andrews, R. M., Morris, C. M., Taylor, R. W., et al. (2000). Mitochondrial DNA haplogroups and susceptibility to AD and dementia with Lewy bodies. Neurology 55, 302–304. doi: 10.1212/wnl.55.2.302
Corder, E., Saunders, A., Strittmatter, W., Schmechel, D., Gaskell, P., Small, G., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Corder, E. H., Ghebremedhin, E., Taylor, M. G., Thal, D. R., Ohm, T. G., and Braak, H. (2004). The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex and APOE polymorphism. Ann. N Y Acad. Sci. 1019, 24–28. doi: 10.1196/annals.1297.005
Coskun, P., Wyrembak, J., Schriner, S. E., Chen, H. W., Marciniack, C., Laferla, F., et al. (2012). A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 1820, 553–564. doi: 10.1016/j.bbagen.2011.08.008
Coto, E., Gómez, J., Alonso, B., Corao, A. I., Díaz, M., Menéndez, M., et al. (2011). Late-onset Alzheimer’s disease is associated with mitochondrial DNA 7028C/haplogroup H and D310 poly-C tract heteroplasmy. Neurogenetics 12, 345–346. doi: 10.1007/s10048-011-0295-4
Davis, R. E., Miller, S., Herrnstadt, C., Ghosh, S. S., Fahy, E., Shinobu, L. A., et al. (1997). Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc. Natl. Acad. Sci. U S A 94, 4526–4531. doi: 10.1073/pnas.94.9.4526
Drzezga, A., Riemenschneider, M., Strassner, B., Grimmer, T., Peller, M., Knoll, A., et al. (2005). Cerebral glucose metabolism in patients with AD and different APOE genotypes. Neurology 64, 102–107. doi: 10.1212/01.wnl.0000148478.39691.d3
Duara, R., Lopez-Alberola, R. F., Barker, W. W., Loewenstein, D. A., Zatinsky, M., Eisdorfer, C. E., et al. (1993). A comparison of familial and sporadic Alzheimer’s disease. Neurology 43, 1377–1384. doi: 10.1212/wnl.43.7.1377
Edland, S. D., Silverman, J. M., Peskind, E. R., Tsuang, D., Wijsman, E., and Morris, J. C. (1996). Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology 47, 254–256. doi: 10.1212/wnl.47.1.254
Edland, S. D., Tobe, V. O., Rieder, M. J., Bowen, J. D., McCormick, W., Teri, L., et al. (2002). Mitochondrial genetic variants and Alzheimer disease: a case-control study of the T4336C and G5460A variants. Alzheimer Dis. Assoc. Disord. 16, 1–7. doi: 10.1097/00002093-200201000-00001
Elson, J., Herrnstadt, C., Preston, G., Thal, L., Morris, C., Edwardson, J. A., et al. (2006). Does the mitochondrial genome play a role in the etiology of Alzheimer’s disease? Hum. Genet. 119, 241–254. doi: 10.1007/s00439-005-0123-8
Fachal, L., Mosquera-Miguel, A., Pastor, P., Ortega-Cubero, S., Lorenzo, E., Oterino-Durán, A., et al. (2015). No evidence of association between common European mitochondrial DNA variants in Alzheimer, Parkinson and migraine in the Spanish population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 168B, 54–65. doi: 10.1002/ajmg.b.32276
Fernández-Moreno, M., Tamayo, M., Soto-Hermida, A., Mosquera, A., Oreiro, N., Fernández-López, C., et al. (2011). mtDNA haplogroup J modulates telomere length and nitric oxide production. BMC Musculoskelet. Disord. 12:283. doi: 10.1186/1471-2474-12-283
Fesahat, F., Houshmand, M., Panahi, M. S., Gharagozli, K., and Mirzajani, F. (2007). Do haplogroups H and U act to increase the penetrance of Alzheimer’s disease? Cell. Mol. Neurobiol. 27, 329–334. doi: 10.1007/s10571-006-9126-9
Gibson, G. E., Haroutunian, V., Zhang, H., Park, L. C., Shi, Q., Lesser, M., et al. (2000). Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann. Neurol. 48, 297–303. doi: 10.1002/1531-8249(200009)48:3<297::AID-ANA3 >3.3.CO;2-Q
Giles, R. E., Blanc, H., Cann, H. M., and Wallace, D. C. (1980). Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. U S A 77, 6715–6719. doi: 10.1073/pnas.77.11.6715
Gómez-Durán, A., Pacheu-Grau, D., López-Gallardo, E., Díez-Sánchez, C., Montoya, J., López-Pérez, M. J., et al. (2010). Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum. Mol. Genet. 19, 3343–3353. doi: 10.1093/hmg/ddq246
Grimm, A., Mensah-Nyagan, A. G., and Eckert, A. (2016). Alzheimer, mitochondria and gender. Neurosci. Biobehav. Rev. 67, 89–101. doi: 10.1016/j.neubiorev.2016.04.012
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023.
Hutchin, T., and Cortopassi, G. (1995). A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proc. Natl. Acad. Sci. U S A 92, 6892–6895. doi: 10.1073/pnas.92.15.6892
Kelly, R. D., Rodda, A. E., Dickinson, A., Mahmud, A., Nefzger, C. M., Lee, W., et al. (2013). Mitochondrial DNA haplotypes define gene expression patterns in pluripotent and differentiating embryonic stem cells. Stem Cells 31, 703–716. doi: 10.1002/stem.1313
Kenney, M. C., Chwa, M., Atilano, S. R., Pavlis, J. M., Falatoonzadeh, P., Ramirez, C., et al. (2013). Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: implications for age-related macular degeneration. PLoS One 8:e54339. doi: 10.1371/journal.pone.0054339
Khusnutdinova, E., Gilyazova, I., Ruiz-Pesini, E., Derbeneva, O., Khusainova, R., Khidiyatova, I., et al. (2008). A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson’s disease. Ann. N Y Acad. Sci. 1147, 1–20. doi: 10.1196/annals.1427.001
Kim, S. H., Vlkolinsky, R., Cairns, N., and Lubec, G. (2000). Decreased levels of complex III core protein 1 and complex V β chain in brains from patients with Alzheimer’s disease and Down syndrome. Cell. Mol. Life Sci. 57, 1810–1816. doi: 10.1007/pl00000661
Kish, S. J., Mastrogiacomo, F., Guttman, M., Furukawa, Y., Taanman, J. W., Dozic, S., et al. (1999). Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer’s disease and in hereditary spinocerebellar ataxia disorders: a nonspecific change? J. Neurochem. 72, 700–707. doi: 10.1046/j.1471-4159.1999.0720700.x
Lakatos, A., Derbeneva, O., Younes, D., Keator, D., Bakken, T., Lvova, M., et al. (2010). Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol. Aging 31, 1355–1363. doi: 10.1016/j.neurobiolaging.2010.04.031
Larsen, S., Díez-Sánchez, C., Rabøl, R., Ara, I., Dela, F., and Helge, J. W. (2014). Increased intrinsic mitochondrial function in humans with mitochondrial haplogroup H. Biochim. Biophys. Acta 1837, 226–231. doi: 10.1016/j.bbabio.2013.10.009
Latorre-Pellicer, A., Moreno-Loshuertos, R., Lechuga-Vieco, A. V., Sánchez-Cabo, F., Torroja, C., Acín-Pérez, R., et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. doi: 10.1038/nature18618
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Lin, T. K., Lin, H. Y., Chen, S. D., Chuang, Y. C., Chuang, J. H., Wang, P. W., et al. (2012). The creation of cybrids harboring mitochondrial haplogroups in the Taiwanese population of ethnic Chinese background: an extensive in vitro tool for the study of mitochondrial genomic variations. Oxid. Med. Cell. Longev. 2012:824275. doi: 10.1155/2012/824275
Liu, Z., Chen, H. H., Li, T. L., Xu, L., and Du, H. Q. (2013). A cross-sectional study on cerebrospinal fluid biomarker levels in cognitively normal elderly subjects with or without a family history of Alzheimer’s disease. CNS Neurosci. Ther. 19, 38–42. doi: 10.1111/cns.12028
Mahley, R. W., Huang, Y., Fau-Weisgraber, K. H., and Weisgraber, K. H. (2007). Detrimental effects of apolipoprotein E4: potential therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 4, 537–540. doi: 10.2174/156720507783018334
Mancuso, M., Filosto, M., Bosetti, F., Ceravolo, R., Rocchi, A., Tognoni, G., et al. (2003). Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Exp. Neurol. 182, 421–426. doi: 10.1016/s0014-4886(03)00092-x
Mancuso, M., Nardini, M., Micheli, D., Rocchi, A., Nesti, C., Giglioli, N. J., et al. (2007). Lack of association between mtDNA haplogroups and Alzheimer’s disease in Tuscany. Neurol. Sci. 28, 142–147. doi: 10.1007/s10072-007-0807-z
Mandal, P. K., Tripathi, M., and Sugunan, S. (2012). Brain oxidative stress: detection and mapping of anti-oxidant marker ‘Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 417, 43–48. doi: 10.1016/j.bbrc.2011.11.047
Marcuello, A., Martínez-Redondo, D., Dahmani, Y., Casajús, J. A., Ruiz-Pesini, E., Montoya, J., et al. (2009). Human mitochondrial variants influence on oxygen consumption. Mitochondrion 9, 27–30. doi: 10.1016/j.mito.2008.10.002
Martínez-Redondo, D., Marcuello, A., Casajús, J. A., Ara, I., Dahmani, Y., Montoya, J., et al. (2010). Human mitochondrial haplogroup H: the highest VO2max consumer–is it a paradox? Mitochondrion 10, 102–107. doi: 10.1016/j.mito.2009.11.005
Maruszak, A., Canter, J. A., Styczynska, M., Zekanowski, C., and Barcikowska, M. (2009). Mitochondrial haplogroup H and Alzheimer’s disease–is there a connection? Neurobiol. Aging 30, 1749–1755. doi: 10.1016/j.neurobiolaging.2008.01.004
Maruszak, A., Safranow, K., Branicki, W., Gaw-da-Walerych, K., Pospiech, E., Gabryelewicz, T., et al. (2011). The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. J. Alzheimers Dis. 27, 197–210. doi: 10.3233/JAD-2011-110710
Maurer, I., Zierz, S., and Möller, H. J. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21, 455–462. doi: 10.1016/s0197-4580(00)00112-3
Mercer, T. R., Neph, S., Dinger, M. E., Crawford, J., Smith, M. A., Shearwood, A. M., et al. (2011). The human mitochondrial transcriptome. Cell 146, 645–658. doi: 10.1016/j.cell.2011.06.051
Mielke, M. M., Vemuri, P., and Rocca, W. A. (2014). Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin. Epidemiol. 6, 37–48. doi: 10.2147/CLEP.s37929
Miyata, T., Hayashida, H., Kikuno, R., Hasegawa, M., Kobayashi, M., and Koike, K. (1982). Molecular clock of silent substitution: at least six-fold preponderance of silent changes in mitochondrial genes over those in nuclear genes. J. Mol. Evol. 19, 28–35. doi: 10.1007/bf02100221
Morris, J. K., Honea, R. A., Vidoni, E. D., Swerdlow, R. H., and Burns, J. M. (2014). Is Alzheimer’s disease a systemic disease? Biochim. Biophys. Acta 1842, 1340–1349. doi: 10.1016/j.bbadis.2014.04.012
Mosconi, L., de Leon, M., Murray, J., Lezi, E, Lu, J., Javier, E., et al. (2011). Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. J. Alzheimers Dis. 27, 483–490. doi: 10.3233/JAD-2011-110866
Mosconi, L., De Santi, S., Brys, M., Tsui, W. H., Pirraglia, E., Glodzik-Sobanska, L., et al. (2008). Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biol. Psychiatry 63, 609–618. doi: 10.1016/j.biopsych.2007.05.030
Mosconi, L., Herholz, K., Prohovnik, I., Nacmias, B., De Cristofaro, M. T., Fayyaz, M., et al. (2005). Metabolic interaction between ApoE genotype and onset age in Alzheimer’s disease: implications for brain reserve. J. Neurol. Neurosurg. Psychiatry 76, 15–23. doi: 10.1136/jnnp.2003.030882
Mosconi, L., Nacmias, B., Sorbi, S., De Cristofaro, M. T., Fayazz, M., Tedde, A., et al. (2004a). Brain metabolic decreases related to the dose of the ApoE e4 allele in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 75, 370–376. doi: 10.1136/jnnp.2003.014993
Mosconi, L., Perani, D., Sorbi, S., Herholz, K., Nacmias, B., Holthoff, V., et al. (2004b). MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology 63, 2332–2340. doi: 10.1212/01.WNL.0000147469.18313.3B
Mosconi, L., Sorbi, S., Nacmias, B., De Cristofaro, M. T., Fayyaz, M., Bracco, L., et al. (2004c). Age and ApoE genotype interaction in Alzheimer’s disease: an FDG-PET study. Psychiatry Res. 130, 141–151. doi: 10.1016/j.pscychresns.2003.12.005
Paillusson, S., Stoica, R., Gomez-Suaga, P., Lau, D. H. W., Mueller, S., Miller, T., et al. (2013). There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 39, 146–157. doi: 10.1016/j.tins.2016.01.008
Parker, W. D. Jr., Filley, C. M., and Parks, J. K. (1990). Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 40, 1302–1303. doi: 10.1212/WNL.40.8.1302
Parker, W. D. Jr., Parks, J., Filley, C. M., and Kleinschmidt-Demasters, B. K. (1994). Electron transport chain defects in Alzheimer’s disease brain. Neurology 44, 1090–1096. doi: 10.1212/WNL.44.6.1090
Pichaud, N., Ballard, J. W., Tanguay, R. M., and Blier, P. U. (2012). Naturally occurring mitochondrial DNA haplotypes exhibit metabolic differences: insight into functional properties of mitochondria. Evolution 66, 3189–3197. doi: 10.1111/j.1558-5646.2012.01683.x
Poirier, J., Bertrand, P., Kogan, S., Gauthier, S., Davignon, J., and Bouthillier, D. (1993). Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 342, 697–699. doi: 10.1016/0140-6736(93)91705-Q
Popot, J. L., and de Vitry, C. (1990). On the microassembly of integral membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 19, 369–403. doi: 10.1146/annurev.biophys.19.1.369
Rebeck, G. W., Reiter, J. S., Strickland, D. K., and Hyman, B. T. (1993). Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 11, 575–580. doi: 10.1016/0896-6273(93)90070-8
Redjems-Bennani, N., Jeandel, C., Lefebvre, E., Blain, H., Vidailhet, M., and Guéant, J. L. (1998). Abnormal substrate levels that depend upon mitochondrial function in cerebrospinal fluid from Alzheimer patients. Gerontology 44, 300–304. doi: 10.1159/000022031
Reiman, E. M., Caselli, R. J., Chen, K., Alexander, G. E., Bandy, D., and Frost, J. (2001). Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: a foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 98, 3334–3339. doi: 10.1073/pnas.061509598
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. U S A 101, 284–289. doi: 10.1073/pnas.2635903100
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2005). Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc. Natl. Acad. Sci. U S A 102, 8299–8302. doi: 10.1073/pnas.0500579102
Richardson, J. S. (1993). Free radicals in the genesis of Alzheimer’s disease. Ann. N Y Acad. Sci. 695, 73–76. doi: 10.1111/j.1749-6632.1993.tb23031.x
Ridge, P. G., Koop, A., Maxwell, T. J., Bailey, M. H., Swerdlow, R. H., Kauwe, J. S., et al. (2013). Mitochondrial haplotypes associated with biomarkers for Alzheimer’s disease. PLoS One 8:e74158. doi: 10.1371/journal.pone.0074158
Ridge, P. G., Maxwell, T. J., Corcoran, C. D., Norton, M. C., Tschanz, J. T., O’Brien, E., et al. (2012). Mitochondrial genomic analysis of late onset Alzheimer’s disease reveals protective haplogroups H6A1A/H6A1B: the cache county study on memory in aging. PLoS One 7:e45134. doi: 10.1371/journal.pone.0045134
Risner, M. E., Saunders, A. M., Altman, J. F., Ormandy, G. C., Craft, S., Foley, I. M., et al. (2006). Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254. doi: 10.1038/sj.tpj.6500369
Roses, A. D., Saunders, A. M., Huang, Y., Strum, J., Weisgraber, K. H., and Mahley, R. W. (2007). Complex disease-associated pharmacogenetics: drug efficacy, drug safety and confirmation of a pathogenetic hypothesis (Alzheimer’s disease). Pharmacogenomics J. 7, 10–28. doi: 10.1038/sj.tpj.6500397
Santoro, A., Balbi, V., Balducci, E., Pirazzini, C., Rosini, F., Tavano, F., et al. (2010). Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS One 5:e12037. doi: 10.1371/journal.pone.0012037
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. S., Pericak-Vance, M. A., Joo, S., et al. (1993). Association of apolipoprotein E allele ɛ4 with late–onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1467. doi: 10.1212/wnl.43.8.1467
Schagger, H., and Ohm, T. G. (1995). Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur. J. Biochem. 227, 916–921. doi: 10.1111/j.1432-1033.1995.0916p.x
Scheffler, K., Krohn, M., Dunkelmann, T., Stenzel, J., Miroux, B., Ibrahim, S., et al. (2012). Mitochondrial DNA polymorphisms specifically modify cerebral beta-amyloid proteostasis. Acta Neuropathol. 124, 199–208. doi: 10.1007/s00401-012-0980-x
Schuessel, K., Leutner, S., Cairns, N. J., Müller, W. E., and Eckert, A. (2004). Impact of gender on upregulation of antioxidant defence mechanisms in Alzheimer’s disease brain. J. Neural Transm. (Vienna) 111, 1167–1182. doi: 10.1007/s00702-004-0156-5
Sheehan, J. P., Swerdlow, R. H., Miller, S. W., Davis, R. E., Parks, J. K., Parker, W. D., et al. (1997). Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J. Neurosci. 17, 4612–4622.
Shi, L., Du, X., Zhou, H., Tao, C., Liu, Y., Meng, F., et al. (2014). Cumulative effects of the ApoE genotype and gender on the synaptic proteome and oxidative stress in the mouse brain. Int. J. Neuropsychopharmacol. 17, 1863–1879. doi: 10.1017/s1461145714000601
Shoffner, J. M., Brown, M. D., Torroni, A., Lott, M. T., Cabell, M. F., Mirra, S. S., et al. (1993). Mitochondrial DNA variants observed in alzheimer disease and parkinson disease patients. Genomics 17, 171–184. doi: 10.1006/geno.1993.1299
Simpkins, J. W., Yang, S.-H., Sarkar, S. N., and Pearce, V. (2008). Estrogen actions on mitochondria–Physiological and pathological implications. Mol. Cell. Endocrinol. 290, 51–59. doi: 10.1016/j.mce.2008.04.013
Sims, N. R., Finegan, J. M., Blass, J. P., Bowen, D. M., and Neary, D. (1987). Mitochondrial function in brain tissue in primary degenerative dementia. Brain Res. 436, 30–38. doi: 10.1016/0006-8993(87)91553-8
Skup, M., Zhu, H., Wang, Y., Giovanello, K. S., Lin, J. A., Shen, D., et al. (2011). Sex differences in grey matter atrophy patterns among AD and aMCI patients: results from ADNI. Neuroimage 56, 890–906. doi: 10.1016/j.neuroimage.2011.02.060
Small, G. W., Ercoli, L. M., Silverman, D. H., Huang, S. C., Komo, S., Bookheimer, S. Y., et al. (2000). Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 97, 6037–6042. doi: 10.1073/pnas.090106797
Small, G. W., Mazziotta, J. C., Collins, M. T., Baxter, L. R., Phelps, M. E., Mandelkern, M. A., et al. (1995). APolipoprotein e type 4 allele and cerebral glucose metabolism in relatives at risk for familial alzheimer disease. JAMA 273, 942–947. doi: 10.1001/jama.273.12.942
Stewart, J. B., and Chinnery, P. F. (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542. doi: 10.1038/nrg3966
Swerdlow, R. H., and Khan, S. M. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 63, 8–20. doi: 10.1016/j.mehy.2003.12.045
Takasaki, S. (2008). Mitochondrial SNPs associated with Japanese centenarians, Alzheimer’s patients and Parkinson’s patients. Comput. Biol. Chem. 32, 332–337. doi: 10.1016/j.compbiolchem.2008.03.014
Takasaki, S. (2009). Mitochondrial haplogroups associated with Japanese centenarians, Alzheimer’s patients, Parkinson’s patients, type 2 diabetic patients and healthy non-obese young males. J Genet Genomics 36, 425–434. doi: 10.1016/s1673-8527(08)60132-0
Torroni, A., Huoponen, K., Francalacci, P., Petrozzi, M., Morelli, L., Scozzari, R., et al. (1996). Classification of european mtDNAs from an analysis of three european populations. Genetics 144, 1835–1850.
Torroni, A., Schurr, T. G., Yang, C. C., Szathmary, E. J., Williams, R. C., Schanfield, M. S., et al. (1992). Native American mitochondrial DNA analysis indicates that the Amerind and the Nadene populations were founded by two independent migrations. Genetics 130, 153–162.
Tranah, G. J., Manini, T. M., Lohman, K. K., Nalls, M. A., Kritchevsky, S., Newman, A. B., et al. (2011). Mitochondrial DNA variation in human metabolic rate and energy expenditure. Mitochondrion 11, 855–861. doi: 10.1016/j.mito.2011.04.005
Tranah, G. J., Nalls, M. A., Katzman, S. M., Yokoyama, J. S., Lam, E. T., Zhao, Y., et al. (2012). Mitochondrial DNA sequence variation associated with dementia and cognitive function in the elderly. J. Alzheimers Dis. 32, 357–372.
Tranah, G. J., Yokoyama, J. S., Katzman, S. M., Nalls, M. A., Newman, A. B., Harris, T. B., et al. (2014). Mitochondrial DNA sequence associations with dementia and amyloid-beta in elderly African Americans. Neurobiol. Aging 35, 442.e1–442.e8. doi: 10.1016/j.neurobiolaging.2013.05.023
Trimmer, P. A., Swerdlow, R. H., Parks, J. K., Keeney, P., Bennett, J. P. Jr., Miller, S. W., et al. (2000). Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp. Neurol. 162, 37–50. doi: 10.1006/exnr.2000.7333
Valla, J., Schneider, L., Niedzielko, T., Coon, K. D., Caselli, R., Sabbagh, M. N., et al. (2006). Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion 6, 323–330. doi: 10.1016/j.mito.2006.10.004
Valla, J., Yaari, R., Wolf, A. B., Kusne, Y., Beach, T. G., Roher, A. E., et al. (2010). Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimers Dis. 22, 307–313. doi: 10.3233/JAD-2010-100129
van der Walt, J. M., Dementieva, Y. A., Martin, E. R., Scott, W. K., Nicodemus, K. K., Kroner, C. C., et al. (2004). Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci. Lett. 365, 28–32. doi: 10.1016/j.neulet.2004.04.051
van der Walt, J. M., Scott, W. K., Slifer, S., Gaskell, P. C., Martin, E. R., Welsh-Bohmer, K., et al. (2005). Maternal lineages and alzheimer disease risk in the old order amish. Hum. Genet. 118, 115–122. doi: 10.1007/s00439-005-0032-x
Wallace, D. C. (1994). Mitochondrial DNA sequence variation in human evolution and disease. Proc. Natl. Acad. Sci. U S A 91, 8739–8746. doi: 10.1073/pnas.91.19.8739
Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Wallace, D. C., Ye, J. H., Neckelmann, S. N., Singh, G., Webster, K. A., and Greenberg, B. D. (1987). Sequence analysis of cDNAs for the human and bovine ATP synthase beta subunit: mitochondrial DNA genes sustain seventeen times more mutations. Curr. Genet. 12, 81–90. doi: 10.1007/bf00434661
Watanabe, K. (2010). Unique features of animal mitochondrial translation systems. Proc. Jan. Acad. Ser. B Phys. Biol. Sci. 86, 11–39. doi: 10.2183/pjab.86.11
Watanabe, K., and Yokobori, S. (2011). tRNA modification and genetic code variations in animal mitochondria. J. Nucleic Acids 2011:623095. doi: 10.4061/2011/623095
Wolf, A. B., Caselli, R. J., Reiman, E. M., and Valla, J. (2013). APOE and neuroenergetics: an emerging paradigm in Alzheimer’s disease. Neurobiol. Aging 34, 1007–1017. doi: 10.1016/j.neurobiolaging.2012.10.011
Wragg, M. A., Talbot, C. J., Morris, J. C., Lendon, C. L., and Goate, A. M. (1995). No association found between Alzheimer’s disease and a mitochondrial tRNA glutamine gene variant. Neurosci. Lett. 201, 107–110. doi: 10.1016/0304-3940(95)12146-3
Xu, P. T., Li, Y. J., Qin, X. J., Kroner, C., Green-Odlum, A., Xu, H., et al. (2007). A SAGE study of apolipoprotein E3/3, E3/4 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Mol. Cell. Neurosci. 36, 313–331. doi: 10.1016/j.mcn.2007.06.009
Xu, P. T., Li, Y. J., Qin, X. J., Scherzer, C. R., Xu, H., Schmechel, D. E., et al. (2006). Differences in apolipoprotein E3/3 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Neurobiol. Dis. 21, 256–275. doi: 10.1016/j.nbd.2005.07.004
Xu, S., Schaack, S., Seyfert, A., Choi, E., Lynch, M., and Cristescu, M. E. (2012). High mutation rates in the mitochondrial genomes of daphnia pulex. Mol. Biol. Evol. 29, 763–769. doi: 10.1093/molbev/msr243
Zhang, L., Trushin, S., Christensen, T. A., Bachmeier, B. V., Gateno, B., Schroeder, A., et al. (2016). Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 6:18725. doi: 10.1038/srep18725
Keywords: mitochondria, haplogroup, Alzheimer’s disease, APOE, sex
Citation: Wang Y and Brinton RD (2016) Triad of Risk for Late Onset Alzheimer’s: Mitochondrial Haplotype, APOE Genotype and Chromosomal Sex. Front. Aging Neurosci. 8:232. doi: 10.3389/fnagi.2016.00232
Received: 03 August 2016; Accepted: 20 September 2016;
Published: 04 October 2016.
Edited by:
George E. Barreto, Pontifical Xavierian University, ColombiaReviewed by:
Panteleimon Giannakopoulos, University of Geneva, SwitzerlandRussell H. Swerdlow, University of Kansas, USA
Copyright © 2016 Wang and Brinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta D. Brinton, cmJyaW50b25AdXNjLmVkdQ==