 Min Soo Byun1
Min Soo Byun1 Young Min Choe2Bo Kyung Sohn3
Young Min Choe2Bo Kyung Sohn3 Dahyun Yi1Ji Young Han4
Dahyun Yi1Ji Young Han4 Jinsick Park5Hyo Jung Choi4Hyewon Baek6
Jinsick Park5Hyo Jung Choi4Hyewon Baek6 Jun Ho Lee4Hyun Jung Kim7Yu Kyeong Kim8Eun Jin Yoon8Chul-Ho Sohn9Jong Inn Woo10
Jun Ho Lee4Hyun Jung Kim7Yu Kyeong Kim8Eun Jin Yoon8Chul-Ho Sohn9Jong Inn Woo10 Dong Young Lee1,4,10*
Dong Young Lee1,4,10*- 1Institute of Human Behavioral Medicine, Medical Research Center, Seoul National University, Seoul, South Korea
- 2Department of Neuropsychiatry, Ulsan University Hospital, Ulsan, South Korea
- 3Department of Neuropsychiatry, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Seoul, South Korea
- 4Department of Neuropsychiatry, Seoul National University Hospital, Seoul, South Korea
- 5Department of Biomedical Engineering, Hanyang University, Seoul, South Korea
- 6Department of Neuropsychiatry, Kyunggi Provincial Hospital for the Elderly, Yongin, South Korea
- 7Department of Neuropsychiatry, Changsan Convalescent Hospital, Changwon, South Korea
- 8Department of Nuclear Medicine, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Seoul, South Korea
- 9Department of Radiology, Seoul National University Hospital, Seoul, South Korea
- 10Department of Psychiatry, Seoul National University College of Medicine, Seoul, South Korea
Previous literature suggests that Alzheimer's disease (AD) process may contribute to late-life onset depression (LLOD). Therefore, we investigated the association of LLOD with cerebral amyloidosis and neuronal injury, the two key brain changes in AD, along with vascular risks. Twenty nine non-demented individuals who first experienced major depressive disorder (MDD) after age of 60 years were included as LLOD subjects, and 27 non-demented elderly individuals without lifetime experience of MDD were included as normal controls (NC). Comorbid mild cognitive impairment (MCI) was diagnosed in 48% of LLOD subjects and in 0% of NC. LLOD, irrespective of comorbid MCI diagnosis, was associated with prominent prefrontal cortical atrophy. Compared to NC, LLOD subjects with comorbid MCI (LLODMCI) showed increased cerebral 11C-Pittsburg compound B (PiB) retention and plasma beta-amyloid 1–40 and 1–42 peptides, as measures of cerebral amyloidosis; and, such relationship was not observed in overall LLOD or LLOD without MCI (LLODwoMCI). LLOD subjects, particularly the LLODwoMCI, had higher systolic blood pressure (SBP) than NC. When analyzed in the same multiple logistic regression model that included prefrontal gray matter (GM) density, cerebral amyloidosis, and SBP as independent variables, only prefrontal GM density showed a significant independent association with LLOD regardless of MCI comorbidity status. Our findings suggest AD process might be related to LLOD via prefrontal neuronal injury in the MCI stage, whereas vascular processes—SBP elevation, in particular—are associated with LLOD via prefrontal neuronal injury even in cognitively intact or less impaired individuals.
Introduction
Late-life onset depression (LLOD), most commonly defined as depression occurring for the first time at the age of 60 years or older, has distinct clinical characteristics such as high medical comorbidity compared to early-life onset depression (ELOD; Salloway et al., 1996; Potter and Steffens, 2007). In addition, since cerebrovascular changes and vascular risk factors (VRFs) are more commonly observed in LLOD than ELOD subjects, the “vascular depression” hypothesis has been proposed as a possible biological etiology of LLOD (Alexopoulos et al., 1997a,b; Krishnan et al., 1997).
However, previous studies suggested that neurodegenerative processes, Alzheimer's disease (AD) in particular, may contribute to LLOD, which might be an early prodromal symptom of AD (Brommelhoff et al., 2009; Panza et al., 2010). According to prior epidemiological studies, the prevalence rates of depressive symptoms increased within 3 years preceding the diagnosis of AD dementia and were significantly higher in subjects with mild cognitive impairment (MCI) than normal controls (NC; Berger et al., 1999; Lopez et al., 2003). Such findings indicate that AD-related pathological processes, such as beta-amyloid (Aβ) deposition and related neuronal injury, might contribute to the occurrence of LLOD by increasing brain vulnerability (Weisenbach and Kumar, 2014).
In line with epidemiological findings, some studies reported altered plasma levels of Aβ peptides or their ratio, considered potential biomarkers for increased risk of AD or MCI (Song et al., 2011), in elderly individuals with depression (Pomara et al., 2006; Qiu et al., 2007), and proposed the term “amyloid-associated depression” as a prodromal manifestation of AD (Sun et al., 2008). However, since plasma Aβ biomarkers are indirect measures for probing cerebral amyloid burden and direction of change was inconsistent among previous studies, direct quantification of Aβ deposition in in vivo human brain is required to accurately assess the state of cerebral amyloidosis in LLOD subjects. Recently, several studies compared cerebral amyloid burden between subjects with late-life depression and NC using amyloid positron emission tomography (PET) imaging with 11C-Pittsburg compound B (PiB) or 18F-Florbetapir tracer (Butters et al., 2008a; Madsen et al., 2012; Wu et al., 2014). However, all these studies, which showed conflicting results, defined depression as a “late-life depression” (Butters et al., 2008a) or “lifetime history of major depression in the elderly” (Madsen et al., 2012; Wu et al., 2014); these definitions incorporate not only LLOD but also ELOD that recurs or continues in the late-life period (Panza et al., 2010). To date, whether cerebral amyloidosis is associated with pure LLOD and plays a critical role in the development of LLOD remains unclear.
Based on magnetic resonance imaging (MRI) studies, LLOD is closely associated with structural brain changes such as regional cortical atrophy, a marker for neuronal injury (Bobinski et al., 2000; Frisoni et al., 2010; Hampel et al., 2010; Jack et al., 2010, 2011; Vemuri and Jack, 2010; Whitwell and Vemuri, 2011), especially in prefrontal regions (Kumar et al., 1998; Almeida et al., 2003). Although the vascular process has been suggested as their cause (Alexopoulos et al., 1997a), structural brain changes in LLOD may be due to the neuronal injury associated with the pathological process of AD, which is initiated by cerebral Aβ deposition (Jack et al., 2010; Sperling et al., 2011). Thus, to clearly elucidate the integrative amyloid-associated neurobiological process that underlies LLOD, neuronal injury process identified by MRI-measured atrophy, as well as cerebral amyloid burden, should also be examined simultaneously along with the vascular risks. Nevertheless, to date, no studies have investigated cerebral amyloid burden and regional cortical atrophy in LLOD individuals by integrating the perspectives of AD and the vascular process.
Elucidating the underlying pathophysiology of LLOD, particularly its association with neurodegenerative process such as cerebral amyloidosis and neuronal injury in non-demented elderly individuals will provide us insight on predicting the prognosis of and developing the novel therapeutic approaches for LLOD in clinical practice (Pomara and Sidtis, 2007; Mahgoub and Alexopoulos, 2016). In the present study, we aimed to investigate whether LLOD is associated with cerebral amyloidosis and regional neuronal injury, along with the vascular risks, in elderly individuals with no dementia. We first examined regional cortical atrophy using voxel-by-voxel analysis to identify regional neuronal injury related to LLOD. Next, we investigated the association of LLOD with cerebral amyloidosis, as measured by both cerebral Aβ deposition and plasma Aβ peptides levels, and vascular risks. Finally, we investigated whether regional neuronal injury, cerebral amyloidosis or vascular risks were independently associated with a diagnosis of LLOD. Because a diagnosis of MCI is considered to be associated with increased cerebral amyloidosis (Wolk et al., 2009), the vascular process (Casado Naranjo et al., 2015), and cortical atrophy (Jack et al., 2010), we also explored whether the association of LLOD with cerebral amyloidosis, vascular process, or regional cortical atrophy differs according to the presence of MCI.
Materials and Methods
Participants
Elderly individuals who first experienced a major depressive disorder (MDD), as defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV; First et al., 1996), after the age of 60 years were recruited from the Geriatric Psychiatry Clinic of the Seoul National University Hospital (SNUH) as LLOD subjects. In addition, elderly individuals without lifetime experience of MDD were recruited from the community as NC. To diagnose and characterize onset and course of MDD, as well as to exclude other psychiatric disorders, all participants underwent a standardized interview including the Structured Clinical Interview for DSM-IV (SCID) that is required for the diagnosis of MDD by board-certified psychiatrists at the time of recruitment. Reliable informants were also interviewed and medical records were reviewed for more accurate information. Exclusion criteria for both groups were: (1) dementia according to DSM-IV criteria; (2) comorbid major neuropsychiatric illnesses other than MDD; (3) a history of significant systemic illness/neurologic disorder; (4) major abnormal findings on brain MRI; (5) a history of electroconvulsive therapy; or, (6) contraindications for the MRI scan. The presence of MCI was not included in the exclusion criteria for both groups. Among the initially recruited 59 subjects, 3 LLOD participants were excluded from the final analysis due to significant abnormalities on MRI (i.e., acute infarct, old hemorrhage, and severe brain parenchymal tissue loss). The study was approved by the Institutional Review Board of SNUH and all participants provided written informed consent. This study was conducted in accordance with the recommendations of the current version of the Declaration of Helsinki.
Clinical and Neuropsychological Assessment
Although all LLOD subjects were in MDD state at the time of enrollment, most of them (90%) were remitted from MDD after receiving treatment in the clinic at the time of the detailed examination including neuropsychological tests and MRI/PET scan. Severity of depressive symptoms at the time of the examination was measured using the 17-item Hamilton Rating Scale for Depression (Mulsant et al., 1994), the 30-item Korean version of the Geriatric Depression Scale (GDS; Yesavage et al., 1982; Bae and Cho, 2004) and Montgomery-Åsberg Depression Rating Scale (Montgomery and Asberg, 1979). All subjects received standardized clinical and neuropsychological assessments according to the protocol of the Korean version of the Consortium to Establish a Registry for Alzheimer Disease (CERAD) Assessment Packet (Morris et al., 1989; Lee et al., 2002), the CERAD neuropsychological battery (Lee et al., 2004) and the Stroop test (Seo et al., 2008) by board-certified psychiatrists and neuropsychologists. MCI was diagnosed according to the international consensus criteria (Winblad et al., 2004) and detailed information on the diagnostic criteria for MCI used in this study is described in elsewhere (see Supplementary Text).
The presence or absence of six VRFs, including hypertension, diabetes, dyslipidemia, heart disease, transient ischemic attack, and stroke was systematically assessed for each participant; the participant's history was provided by an informant and the medical records were reviewed. As a composite score of VRFs, the VRF score (VRS) was calculated as the number of the VRFs present and reported as a percentage (DeCarli et al., 2004). The systolic/diastolic blood pressure (SBP/DBP) was measured at supine position by trained nurse using sphygmomanometer and body mass index (BMI) were also assessed.
MRI Acquisition, Pre-processing, and Analyses
MRI scanning was performed using a 3T Siemens TrioTim magnetic resonance scanner (Siemens AG, Erlangen, Germany) to acquire three-dimensional (3-D) T1-weighted magnetization-prepared rapid gradient-echo (MPRAGE) and fluid-attenuated inversion recovery (FLAIR) sequences. Detailed information on MRI sequences and parameters is described in elsewhere (see Supplementary Text).
Voxel-based morphometry (VBM) analysis was performed using Statistical Parametric Mapping 8 (SPM8, Wellcome Department of Cognitive Neurology, London, UK; http://www.fil.ion.ucl.ac.uk/spm/) with the VBM8 toolbox (http://dbm.neuro.uni-jena.de/vbm/) to demonstrate group differences in regional gray matter (GM) density between the NC and LLOD groups. All T1-weighted images of each subject were normalized into standard anatomical space using MNI 152 template with a linear 12-parameter affine transformation. Next, normalized images were segmented into GM, white matter and cerebrospinal fluid. Smoothing at 12-mm full width at half maximum was performed after segmentation and modulation.
For the rating of white matter hyperintensity (WMH), all MRIs with FLAIR sequence were assessed blinded to clinical information by one experienced rater according to the Fazekas scale (Fazekas et al., 1987), where the severity of periventricular and deep WMH was scaled on a 0 (absence) to 3 (severe) separately.
11C-PiB PET Image Acquisition, Pre-processing, and Analyses
Participants also underwent 11C-PiB PET imaging using Biograph PET/CT scanners (Siemens, TN, USA). All participants underwent 11C-PiB PET imaging using Biograph PET/CT scanners (Siemens, TN, USA). For each subject, 550–750 MBq of 11C-PiB was administered by intravenous injection. A 20 min emission scan was obtained in 3D mode starting 50 min after injection and a CT scan was performed for attenuation correction (120 kVp, 40 mAs, pitches of 0.8). PET reconstructions were performed using a point spread function-based iterative algorithm (TrueX; 6 iterations, 21 subsets) with a matrix 256 × 256 in size (74 slices, voxel size: 1.3364 × 1.3364 mm2; slice thickness: 3 mm), and reconstructed images were rearranged onto transaxial, sagittal and coronal images.
Image preprocessing for statistical analyses was performed using SPM8 implemented in Matlab (MathWorks, Natick, MA, USA). The 11C-PiB PET data of each subject were co-registered to individual volumetric magnetic resonance images and then automatically spatially normalized into the standard MNI template in SPM8 using transformation parameters derived from the normalization of individual MRI scans to the template. All normalized images were reformatted with a 2 × 2 × 2 mm voxel. For quantitative normalization of cerebral 11C-PiB uptake values, the cerebellum was used as a reference region (Lopresti et al., 2005) and 11C-PiB retention maps, as region-to-cerebellar ratios, were generated by dividing regional uptake values by the individual mean cerebellar uptake values in the same images.
The automatic anatomic labeling algorithm (Tzourio-Mazoyer et al., 2002) and a region combining method (Reiman et al., 2009) were applied to set regions-of-interest (ROIs) to characterize 11C-PiB retention level in frontal, lateral parietal, posterior cingulate-precuneus (PC-PRC), lateral temporal and basal ganglia (BG) regions. Mean cortical amyloid burden was calculated for a global PiB retention index by averaging the mean ROI value except BG. The image was classified as PiB-positive if the standardized uptake value ratio (SUVR) of 11C-PiB retention was 1.4 or higher in one of the following cortical ROIs: frontal, lateral temporal, lateral parietal, or PC-PRC (Choe et al., 2014).
Plasma Aβ Peptide Level Assessment and Apolipoprotein E (APOE) Genotyping
Fasting blood samples were collected in the morning by venipuncture in tubes containing EDTA as anticoagulant. After centrifugation, plasma samples were aliquoted into polypropylene tubes and stored at −80°C pending biochemical analyses without being thawed and refrozen. Quantification of plasma Aβ isoforms was performed using INNO-BIA plasma Aβ forms assays (Innogenetics, Ghent, Belgium) and the Bio-Plex 200 system with high throughput fluidics (BIO-RAD, Hercules, CA, USA) based on a previous study (Hansson et al., 2010). Genomic DNA was extracted from venous blood and APOE genotyping was performed according to the method described previously (Wenham et al., 1991).
Statistical Analyses
For comparison of demographic and clinical variables between the two groups, independent t-test for continuous variables and chi-square or Fisher's exact test for categorical variables was used (p < 0.05, two-sided). Neuropsychological variables were compared between the two groups using analysis of covariance (ANCOVA) with age, gender, educational level and GDS score as covariates. VBM analysis was performed to identify the region where LLOD subjects showed greater cortical atrophy compared with NC at both uncorrected p < 0.001 (k = 100) and family-wise error (FWE)-corrected p < 0.05 (k = 100), after controlling for age, gender and educational level. Comparison of PiB retention level, as well as plasma Aβ peptide level, between the two groups was performed using ANCOVA after controlling for age, gender and educational level. Multiple logistic regression analysis with diagnostic state (NC vs. LLOD) as a dependent variable was performed to investigate the independent association of regional neuronal injury, cerebral amyloidosis, and vascular risk with occurrence of LLOD after controlling for age, gender, and educational level. All of the abovementioned analyses were performed again after excluding LLOD subjects with MCI (LLODMCI) to characterize the LLOD subjects without MCI (LLODwoMCI) and vice versa.
Results
Demographic, Clinical, and Neuropsychological Characteristics
Differences in terms of age, gender, and educational level, as well as APOE ε4 carrier frequency, were not significant between LLOD subjects and NC (Table 1). Although 26 out of 29 LLOD patients were remitted from MDD at the time of examination, LLOD patients had significantly higher scores on the depression symptom scale than NC. In addition, 48% of LLOD subjects and none of the NC were diagnosed with comorbid MCI.
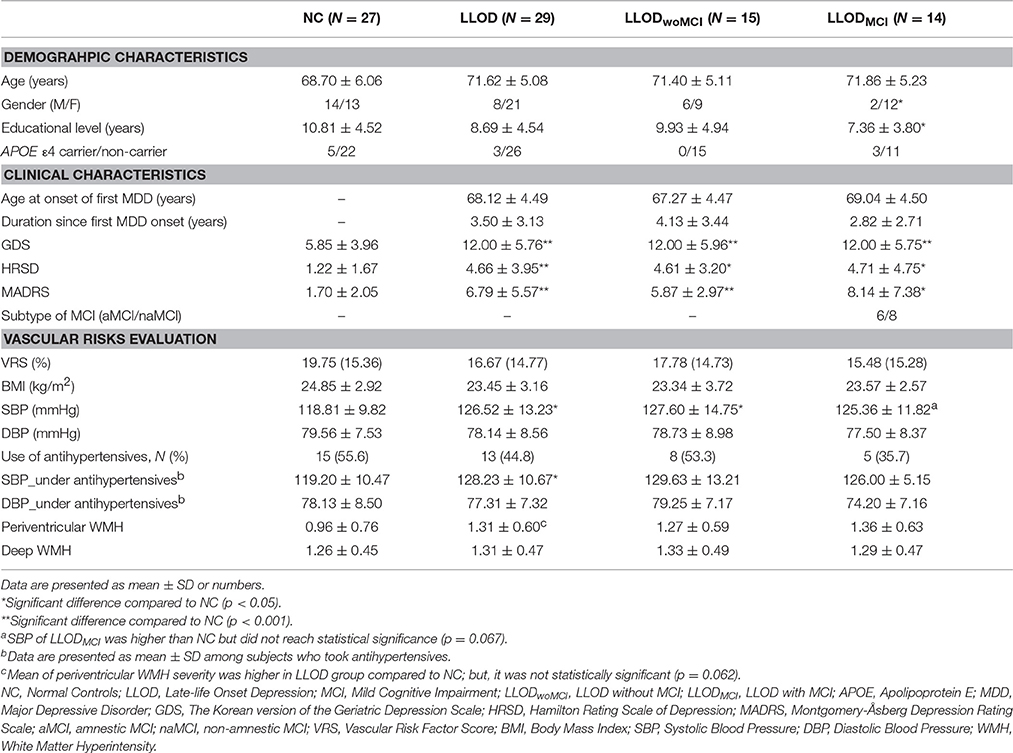
Table 1. Demographic and clinical characteristics of the subjects.
In terms of vascular risk evaluation, no significant differences in VRS, BMI, and DBP were observed between NC and LLOD subjects. However, SBP was significantly higher in LLOD subjects than in NC. In subgroup analysis, LLODwoMCI subjects had significantly higher SBP than NC. Mean of SBP in LLODMCI subjects was higher than that of NC group; but, it was not statistically significant. Even among the subjects treated with antihypertensives, SBP of LLOD group was still significantly higher than NC. Additionally, mean of periventricular WMH severity was higher in LLOD group compared to NC; but, it was not statistically significant.
Compared with NC, LLOD subjects showed significantly greater impairments on multiple cognitive tests including the Mini-mental State Examination (MMSE), Semantic fluency, Word-list memory and Constructional praxis of the CERAD neuropsychological battery, and all Stroop tests (Supplementary Table 1).
Regional Cortical Atrophy in LLOD
On VBM analysis, LLOD individuals demonstrated regional cortical atrophy in the bilateral prefrontal regions including the medial prefrontal areas as well as the bilateral parietal and temporal regions compared with NC at uncorrected p < 0.001, k = 100 (Figure 1A and Supplementary Table 2). After correction for multiple comparisons (FWE-corrected p < 0.05, k = 100), LLOD subjects still showed significant prefrontal atrophy mainly in the right medial prefrontal region (Figure 1B). Prefrontal cortical atrophy with peak voxel located in the right medial prefrontal region (peak MNI: 11, 57, 9) was similarly observed when LLODwoMCI and LLODMCI subjects were compared to NC separately at uncorrected p < 0.001, k = 100 (Figures 1C,D and Supplementary Table 3).
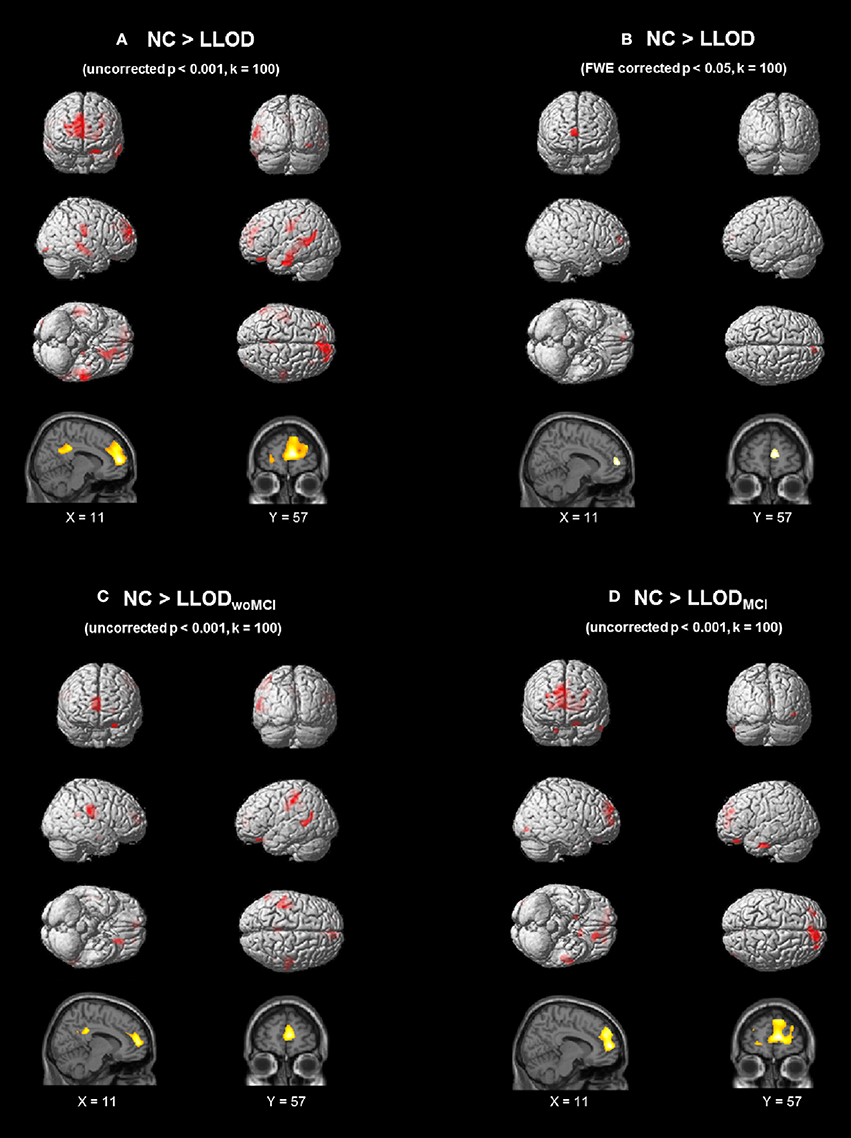
Figure 1. Regional GM atrophy of LLOD subjects compared to NC after controlling age, gender, and educational level (A) at uncorrected p < 0.001 (k = 100) and (B) FWE-corrected p < 0.05 (k = 100). (C) and (D) demonstrated regional GM atrophy in subjects with (C) LLODwoMCI and (D) LLODMCI at uncorrected p < 0.001 (k = 100). NC, Normal Controls; LLOD, Late-life Onset Depression; MCI, Mild Cognitive Impairment; LLODwoMCI, LLOD without MCI; LLODMCI, LLOD with MCI; GM, Gray Matter; FWE, Family-wise Error.
Cerebral PiB Retention and Plasma Aβ Peptide Level in LLOD
Overall, LLOD patients showed no significant differences in both global PiB retention and the PiB-positivity rate compared with NC (Table 2 and Figure 2). Regional PiB retention in the frontal, lateral parietal, PC-PRC, lateral temporal, or BG regions was also not significantly different between NC and LLOD subjects. In terms of plasma Aβ peptides level, no significant differences were observed in all plasma Aβ peptide measurements or their ratios between NC and LLOD subjects (Table 3).
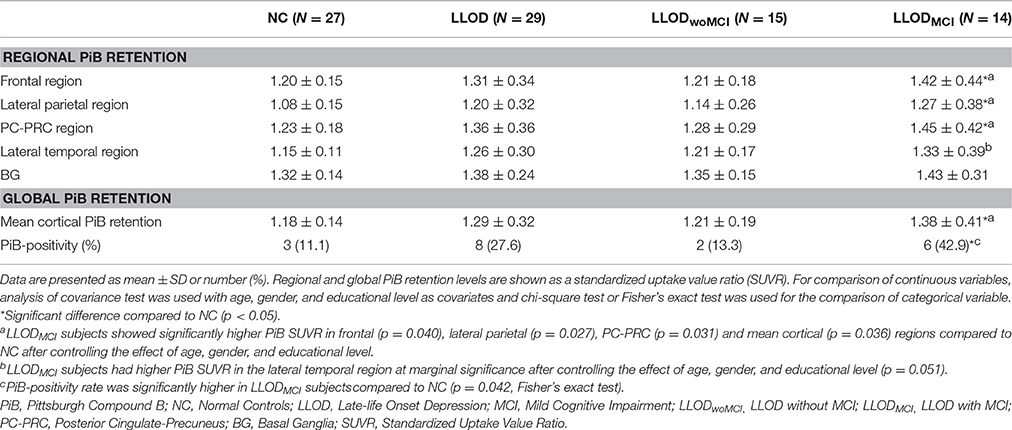
Table 2. Cerebral PiB retention level of subjects.
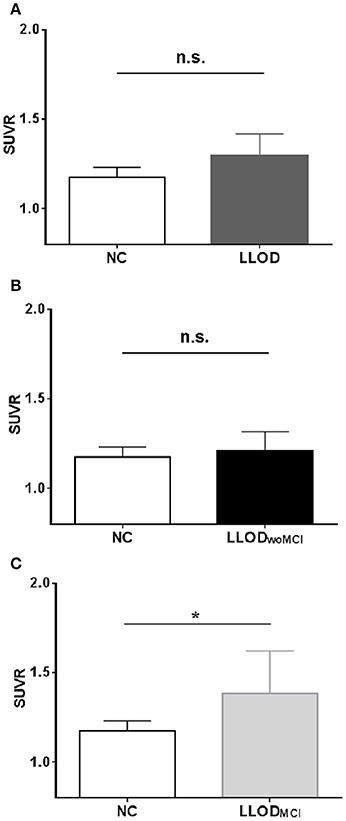
Figure 2. Mean cortical PiB retention level (SUVR) in subjects: (A) NC vs. LLOD, (B) NC vs. LLODwoMCI,(C) NC vs. LLODMCI. *p < 0.05 (after controlling the effect of age, gender, and educational level). PiB, Pittsburgh Compound B; NC, Normal Controls; LLOD, Late-life Onset Depression; MCI, Mild Cognitive Impairment; LLODwoMCI, LLOD without MCI; LLODMCI, LLOD with MCI; SUVR, Standardized Uptake Value Ratio.
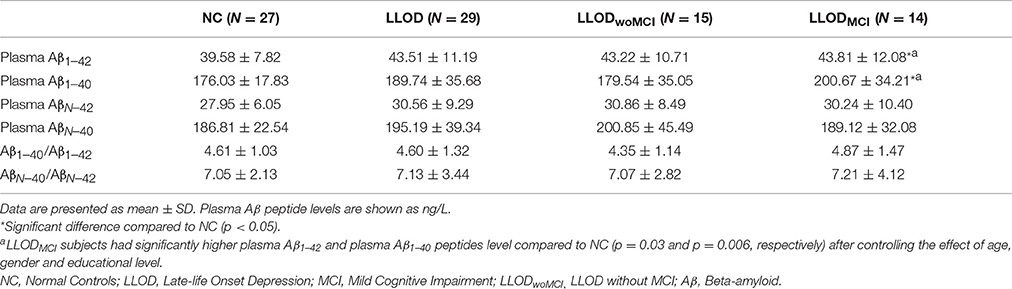
Table 3. Plasma Aβ peptides level of subjects.
In subgroup analysis, LLODwoMCI subjects did not show significant differences in global or regional PiB retention levels, or in the PiB-positivity rate compared with NC. Similarly, plasma Aβ peptides levels were not different between LLODwoMCI subjects and NC. However, LLODMCI subjects had significantly more PiB-positive cases and showed higher global and regional PiB retention than NC (Table 2 and Figure 2). In addition, plasma Aβ1–42 and Aβ1–40 peptide levels were significantly higher in LLODMCI subjects than NC (Table 3).
Independent Association of Regional Neuronal Injury, Cerebral Amyloidosis, and Vascular Process with LLOD Diagnosis
Multiple logistic regression analysis with diagnostic state (NC vs. LLOD) as a dependent variable was performed to investigate the independent relationship of regional neuronal injury, cerebral amyloidosis, and vascular risk with LLOD diagnosis. For this analysis, regional neuronal injury was defined as mean GM density extracted from a prefrontal ROI that was set as a cluster where LLOD subjects showed reduced GM density at FWE-corrected p < 0.05 (peak MNI: 11, 57, 9; T = 5.79; 463 voxels). In addition to prefrontal ROI GM density, global PiB retention, as a measure of cerebral amyloidosis, and SBP, a vascular risk that was significantly different between NC and LLOD subjects, were included in the regression model as independent variables. After adjusting for age, gender and educational level, only prefrontal ROI GM density showed a significant association with LLOD diagnosis [odds ratio (OR) = 0.47 per 0.01; 95% confidence interval (CI): 0.29 – 0.75], whereas global PiB retention (OR = 1.07 per 0.01 SUVR; 95% CI: 0.99 – 1.15) and SBP (OR = 1.06 per 1 mmHg; 95% CI: 0.99 – 1.14) did not in the same model. Moreover, prefrontal ROI GM density was significantly associated with LLODwoMCI diagnosis (OR = 0.25 per 0.01; 95% CI 0.07 – 0.85) or LLODMCI diagnosis (OR = 0.56 per 0.01; 95% CI 0.36 – 0.86) in the regression models, but global PiB retention and SBP were not.
Discussion
We found that nearly half of the subjects who experienced LLOD had comorbid cognitive impairment compatible with MCI, which was not observed in NC. LLOD, irrespective of comorbid MCI diagnosis, was associated with prominent prefrontal cortical atrophy. The LLODMCI subgroup showed increased cerebral PiB retention and plasma Aβ peptide levels as measures of cerebral amyloidosis, compared with NC, while overall LLOD and LLODwoMCI subjects did not. In terms of the vascular process, LLOD individuals had higher SBP than NC, particularly when MCI did not coexist. When simultaneously analyzed in the same model, only prefrontal cortical atrophy showed a significant independent association with LLOD diagnosis, irrespective of MCI status, while cerebral PiB retention and SBP did not. To the best of our knowledge, this study is the first to investigate both in vivo cerebral amyloidosis and regional cortical atrophy simultaneously with vascular risks in the context of LLOD occurrence.
As the accumulation of brain Aβ protein begins 15–20 years before the onset of dementia in AD (Sperling et al., 2011), we aimed to investigate whether LLOD is a secondary result of brain Aβ deposition in the prodromal period of AD dementia. Thus, we strictly recruited only elderly subjects with late-life onset major depression, excluding ELOD cases to increase the homogeneity of study participants. Reportedly, ELOD is a risk factor for developing AD dementia rather than a prodromal phenomenon of AD dementia (Ownby et al., 2006; Byers and Yaffe, 2011). Recent reports on changes in the serum or plasma Aβ1–40/Aβ1–42 ratio even in young-aged major depression patients (Baba et al., 2012) or elderly subjects with ELOD (Pomara et al., 2006; Namekawa et al., 2013) support the link between ELOD and altered Aβ metabolism. Preclinical evidence also suggests that stress responses related to depressive experiences contribute to Aβ accumulation by initiating increased Aβ production (Green et al., 2006; Kang et al., 2007) or interacting with the amyloidogenic process linked to the serotonergic system (Sierksma et al., 2010). Several previous studies have reported elevated brain Aβ deposition or altered plasma Aβ biomarkers in elderly depression (Qiu et al., 2007; Sun et al., 2008; Wu et al., 2014). However, because these studies did not strictly define elderly depression as LLOD, the findings were likely influenced by an etiological contribution of earlier-onset depression to the Aβ-related pathology such that comparison with our results is difficult.
Although most LLOD subjects were in the remitted state, approximately half had comorbid MCI, which was not observed in NC. Considering that structural and functional brain impairments are already prominent in the MCI state (Pihlajamaki et al., 2009; Jack et al., 2010), the higher frequency of comorbid MCI in LLOD subjects indicates that brain damage or pathology is potentially more prevalent in this group, as reported previously (Weisenbach and Kumar, 2014). Our study also revealed prominent prefrontal cortical atrophy in LLOD subjects, regardless of MCI diagnosis. The prefrontal region, especially the medial prefrontal cortex, is important in emotional processing (Elliott and Dolan, 2003). Structural abnormality in the prefrontal region is a robust finding in previous structural imaging studies on late-onset major depression (Kumar et al., 1998) as well as overall MDD (Steele et al., 2007; Lorenzetti et al., 2009). Reductions in neuronal cell size and density in the prefrontal region in MDD were consistently found in previous studies (Kim et al., 2016). Thus, our findings from structural MRI analysis of the cerebral cortex, together with other prior evidences, support that prefrontal neuronal injury is likely a common neural substrate for the emergence of LLOD as well as MDD in general, although underlying etiological factors could be heterogeneous (Butters et al., 2008b; Weisenbach and Kumar, 2014).
While the regional neuronal injury was closely associated with LLOD and did not differ according to the comorbid MCI state, the relationship between cerebral amyloidosis and LLOD depended on the presence of MCI. LLODMCI was closely related with increased cerebral PiB retention and plasma Aβ1–42 and Aβ1–40 peptide levels, whereas overall LLOD and LLODwoMCI were not. These findings suggest that cerebral amyloidosis is not the major determinant of LLOD in elderly individuals without cognitive impairment compatible with MCI, but probably exerts an influence on the occurrence of LLOD in subjects who are in the MCI state. Our findings on the differential association of cerebral amyloidosis and LLOD according to MCI comorbidity might also explain inconsistencies among previous findings: elderly subjects with current or history of depression showed increased brain Aβ level when subjects had comorbid MCI diagnosis, whereas those without MCI did not compared to NC (Butters et al., 2008a; Wu et al., 2016). In addition, the proportion of MCI comorbidity in study subjects as well as methodological differences in plasma Aβ measurements might contribute to the mixed results of previous studies on plasma Aβ level in elderly individuals with depression (Pomara et al., 2006; Qiu et al., 2007; Sun et al., 2008; Song et al., 2011; Baba et al., 2012; Namekawa et al., 2013; Osorio et al., 2014).
We also found that SBP in LLOD patients was significantly higher than in NC, particularly in LLODwoMCI subjects. LLOD also tended to have higher WMH than NC. These results support the previous vascular depression hypothesis (Alexopoulos et al., 1997a) and indicate that the vascular process may have a greater impact on the occurrence of LLOD in subjects who are not in the MCI state and have a low probability of underlying AD processes such as cerebral amyloidosis. Progressive cortical thinning in the prefrontal region has also been reported in hypertensive individuals (Gonzalez et al., 2015), suggesting that the prefrontal cortical region is selectively susceptible to blood pressure elevation.
When analyzed in the same multiple logistic regression model that included prefrontal GM density, cerebral amyloidosis and SBP as independent variables, only prefrontal GM density showed a significant independent association with LLOD diagnosis, regardless of MCI comorbidity state, which was not observed for global PiB retention or SBP. These results indicate that cerebral amyloidosis and elevated SBP only indirectly contribute to LLOD occurrence via prefrontal neuronal injury.
The present study had several limitations. First, it used a cross-sectional design and was limited in terms of providing information on the causal contribution of cerebral amyloidosis or blood pressure to LLOD. Second, the sample size for each LLOD subgroup analysis, although conducted for exploratory purposes, was relatively small, which might have reduced the statistical power. Further studies with larger sample sizes and longitudinal follow-ups are needed to address these issues.
In conclusion, our findings suggest that prefrontal neuronal injury is probably a common brain alteration underlying the occurrence of LLOD. Additionally, cerebral amyloidosis might be associated with LLOD via prefrontal neuronal injury in the MCI stage, in which structural and functional brain impairment caused by the AD process begins to become prominent. Meanwhile, the vascular process, SBP elevation in particular, is associated with the occurrence of LLOD through prefrontal neuronal injury even in cognitively intact or less impaired individuals. From a clinical standpoint, an LLOD patient likely has underlying AD pathologies in his/her brain, if the patient is still in the MCI state even after the remission of depression. However, this possibility is low if the patient is cognitively less impaired and not in the MCI state after the remission of depression. Instead, we should consider that vascular process is a main contributor to LLOD in such cases.
Author Contributions
MB and DL contributed to the design of the study, data collection, data analyses, data interpretation, and prepared the report. YC, BS, JH, HC, HB, JL, HK, JW were involved in the collection and analyses of clinical data as well as drafting of the report. DY, JP, YK, EY, and CS participated in image data preprocessing, analyses, and were involved in preparation of the report.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by a grant from Ministry of Science, ICT, and Future Planning (Grant No: NRF-2014M3C7A1046042) and a research grant from Eisai Korea, Inc.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnagi.2016.00236
References
Alexopoulos, G. S., Meyers, B. S., Young, R. C., Campbell, S., Silbersweig, D., and Charlson, M. (1997a). ‘Vascular depression’ hypothesis. Arch. Gen. Psychiatry 54, 915–922.
Alexopoulos, G. S., Meyers, B. S., Young, R. C., Kakuma, T., Silbersweig, D., and Charlson, M. (1997b). Clinically defined vascular depression. Am. J. Psychiatry 154, 562–565.
Almeida, O. P., Burton, E. J., Ferrier, N., McKeith, I. G., and O'Brien, J. T. (2003). Depression with late onset is associated with right frontal lobe atrophy. Psychol. Med. 33, 675–681. doi: 10.1017/S003329170300758X
Baba, H., Nakano, Y., Maeshima, H., Satomura, E., Kita, Y., Suzuki, T., et al. (2012). Metabolism of amyloid-beta protein may be affected in depression. J. Clin. Psychiatry 73, 115–120. doi: 10.4088/JCP.10m06766
Bae, J. N., and Cho, M. J. (2004). Development of the Korean version of the Geriatric Depression Scale and its short form among elderly psychiatric patients. J. Psychosom. Res. 57, 297–305. doi: 10.1016/j.jpsychores.2004.01.004
Berger, A. K., Fratiglioni, L., Forsell, Y., Winblad, B., and Bäckman, L. (1999). The occurrence of depressive symptoms in the preclinical phase of AD: a population-based study. Neurology 53, 1998–2002. doi: 10.1212/WNL.53.9.1998
Bobinski, M., de Leon, M. J., Wegiel, J., Desanti, S., Convit, A., Saint Louis, L. A., et al. (2000). The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer's disease. Neuroscience 95, 721–725. doi: 10.1016/S0306-4522(99)00476-5
Brommelhoff, J. A., Gatz, M., Johansson, B., McArdle, J. J., Fratiglioni, L., and Pedersen, N. L. (2009). Depression as a risk factor or prodromal feature for dementia? Findings in a population-based sample of Swedish twins. Psychol. Aging 24, 373–384. doi: 10.1037/a0015713
Butters, M. A., Klunk, W. E., Mathis, C. A., Price, J. C., Ziolko, S. K., Hoge, J. A., et al. (2008a). Imaging Alzheimer pathology in late-life depression with PET and Pittsburgh Compound-B. Alzheimer Dis. Assoc. Disord. 22, 261–268. doi: 10.1097/WAD.0b013e31816c92bf
Butters, M. A., Young, J. B., Lopez, O., Aizenstein, H. J., Mulsant, B. H., Reynolds, C. F. III, et al. (2008b). Pathways linking late-life depression to persistent cognitive impairment and dementia. Dialogues Clin. Neurosci. 10, 345–357. Available online at: http://www.dialogues-cns.com/publication/pathways-linking-late-life-depression-to-persistent-cognitive-impairment-and-dementia/
Byers, A. L., and Yaffe, K. (2011). Depression and risk of developing dementia. Nat. Rev. Neurol. 7, 323–331. doi: 10.1038/nrneurol.2011.60
Casado Naranjo, I., Portilla Cuenca, J. C., Duque de San Juan, B., García, A. F., Sevilla, R. R., Serrano Cabrera, A., et al. (2015). Association of vascular factors and amnestic mild cognitive impairment: a comprehensive approach. J. Alzheimers. Dis. 44, 695–704. doi: 10.3233/JAD-141770
Choe, Y. M., Sohn, B. K., Choi, H. J., Byun, M. S., Seo, E. H., Han, J. Y., et al. (2014). Association of homocysteine with hippocampal volume independent of cerebral amyloid and vascular burden. Neurobiol. Aging 35, 1519–1525. doi: 10.1016/j.neurobiolaging.2014.01.013
DeCarli, C., Mungas, D., Harvey, D., Reed, B., Weiner, M., Chui, H., et al. (2004). Memory impairment, but not cerebrovascular disease, predicts progression of MCI to dementia. Neurology 63, 220–227. doi: 10.1212/01.WNL.0000130531.90205.EF
Elliott, R., and Dolan, R. J. (2003). “Chapter 7: Functional neuroimaging of depression: a role for medial prefrontal cortex,” in Handbook of Affective Science, eds R. J. Davidson, K. R. Scherer, and H. H. Goldsmith (Oxford; New York, NY: Oxford University Press), 117–128.
Fazekas, F., Chawluk, J. B., Alavi, A., Hurtig, H. I., and Zimmerman, R. A. (1987). MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. AJR Am. J. Roentgenol. 149, 351–356. doi: 10.2214/ajr.149.2.351
First, M. B., Spitzer, R. L., Gibbon, M., and Williams, J. B. W. (1996). Structured Clinical Interview for DSM-IV Axis I Disorders, Clinician Version (SCID-CV). Washington, DC: American Psychiatric Press, Inc.
Frisoni, G. B., Fox, N. C., Jack, C. R. Jr., Scheltens, P., and Thompson, P. M. (2010). The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 6, 67–77. doi: 10.1038/nrneurol.2009.215
Gonzalez, C. E., Pacheco, J., Beason-Held, L. L., and Resnick, S. M. (2015). Longitudinal changes in cortical thinning associated with hypertension. J. Hypertens. 33, 1242–1248. doi: 10.1097/HJH.0000000000000531
Green, K. N., Billings, L. M., Roozendaal, B., McGaugh, J. L., and LaFerla, F. M. (2006). Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J. Neurosci. 26, 9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006
Hampel, H., Frank, R., Broich, K., Teipel, S. J., Katz, R. G., Hardy, J., et al. (2010). Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nat. Rev. Drug Discov. 9, 560–574. doi: 10.1038/nrd3115
Hansson, O., Zetterberg, H., Vanmechelen, E., Vanderstichele, H., Andreasson, U., Londos, E., et al. (2010). Evaluation of plasma Abeta(40) and Abeta(42) as predictors of conversion to Alzheimer's disease in patients with mild cognitive impairment. Neurobiol. Aging 31, 357–367. doi: 10.1016/j.neurobiolaging.2008.03.027
Jack, C. R. Jr., Albert, M., Knopman, D. S., McKhann, G. M., Sperling, R. A., Carrillo, M., et al. (2011). Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers. Dement. 7, 257–262. doi: 10.1016/j.jalz.2011.03.004
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/S1474-4422(09)70299-6
Kang, J. E., Cirrito, J. R., Dong, H., Csernansky, J. G., and Holtzman, D. M. (2007). Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc. Natl. Acad. Sci. U.S.A. 104, 10673–10678. doi: 10.1073/pnas.0700148104
Kim, H. K., Nunes, P. V., Oliveira, K. C., Young, L. T., and Lafer, B. (2016). Neuropathological relationship between major depression and dementia: a hypothetical model and review. Prog. Neuropsychopharmacol. Biol. Psychiatry 67, 51–57. doi: 10.1016/j.pnpbp.2016.01.008
Krishnan, K. R., Hays, J. C., and Blazer, D. G. (1997). MRI-defined vascular depression. Am. J. Psychiatry 154, 497–501. doi: 10.1176/ajp.154.4.497
Kumar, A., Jin, Z., Bilker, W., Udupa, J., and Gottlieb, G. (1998). Late-onset minor and major depression: early evidence for common neuroanatomical substrates detected by using MRI. Proc. Natl. Acad. Sci. U.S.A. 95, 7654–7658. doi: 10.1073/pnas.95.13.7654
Lee, D. Y., Lee, K. U., Lee, J. H., Kim, K. W., Jhoo, J. H., Kim, S. Y., et al. (2004). A normative study of the CERAD neuropsychological assessment battery in the Korean elderly. J. Int. Neuropsychol. Soc. 10, 72–81. doi: 10.1017/S1355617704101094
Lee, J. H., Lee, K. U., Lee, D. Y., Kim, K. W., Jhoo, J. H., Kim, J. H., et al. (2002). Development of the Korean version of the Consortium to Establish a Registry for Alzheimer's Disease Assessment Packet (CERAD-K): clinical and neuropsychological assessment batteries. J. Gerontol. B Psychol. Sci. Soc. Sci. 57, P47–P53. doi: 10.1093/geronb/57.1.P47
Lopez, O. L., Becker, J. T., Sweet, R. A., Klunk, W., Kaufer, D. I., Saxton, J., et al. (2003). Psychiatric symptoms vary with the severity of dementia in probable Alzheimer's disease. J. Neuropsychiatry Clin. Neurosci. 15, 346–353. doi: 10.1176/jnp.15.3.346
Lopresti, B. J., Klunk, W. E., Mathis, C. A., Hoge, J. A., Ziolko, S. K., Lu, X., et al. (2005). Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J. Nucl. Med. 46, 1959–1972. Available online at: http://jnm.snmjournals.org/content/46/12/1959.long
Lorenzetti, V., Allen, N. B., Fornito, A., and Yücel, M. (2009). Structural brain abnormalities in major depressive disorder: a selective review of recent MRI studies. J. Affect. Disord. 117, 1–17. doi: 10.1016/j.jad.2008.11.021
Madsen, K., Hasselbalch, B. J., Frederiksen, K. S., Haahr, M. E., Gade, A., Law, I., et al. (2012). Lack of association between prior depressive episodes and cerebral [11C]PiB binding. Neurobiol. Aging 33, 2334–2342. doi: 10.1016/j.neurobiolaging.2011.11.021
Mahgoub, N., and Alexopoulos, G. S. (2016). Amyloid Hypothesis: is there a role for Antiamyloid Treatment in Late-Life Depression? Am. J. Geriatr. Psychiatry 24, 239–247. doi: 10.1016/j.jagp.2015.12.003
Montgomery, S. A., and Asberg, M. (1979). A new depression scale designed to be sensitive to change. Br. J. Psychiatry 134, 382–389. doi: 10.1192/bjp.134.4.382
Morris, J. C., Heyman, A., Mohs, R. C., Hughes, J. P., van Belle, G., Fillenbaum, G., et al. (1989). The Consortium to establish a registry for alzheimer's disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology 39, 1159–1165.
Mulsant, B. H., Sweet, R., Rifai, A. H., Pasternak, R. E., McEachran, A., and Zubenko, G. S. (1994). The use of the hamilton rating scale for depression in elderly patients with cognitive impairment and physical illness. Am. J. Geriatr. Psychiatry 2, 220–229. doi: 10.1097/00019442-199400230-00006
Namekawa, Y., Baba, H., Maeshima, H., Nakano, Y., Satomura, E., Takebayashi, N., et al. (2013). Heterogeneity of elderly depression: increased risk of Alzheimer's disease and Abeta protein metabolism. Prog. Neuropsychopharmacol. Biol. Psychiatry 43, 203–208. doi: 10.1016/j.pnpbp.2012.12.016
Osorio, R. S., Gumb, T., and Pomara, N. (2014). Soluble amyloid-β levels and late-life depression. Curr. Pharm. Des. 20, 2547–2554. doi: 10.2174/13816128113199990502
Ownby, R. L., Crocco, E., Acevedo, A., John, V., and Loewenstein, D. (2006). Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch. Gen. Psychiatry 63, 530–538. doi: 10.1001/archpsyc.63.5.530
Panza, F., Frisardi, V., Capurso, C., D'Introno, A., Colacicco, A. M., Imbimbo, B. P., et al. (2010). Late-life depression, mild cognitive impairment, and dementia: possible continuum? Am. J. Geriatr. Psychiatry 18, 98–116. doi: 10.1097/JGP.0b013e3181b0fa13
Pihlajamaki, M., Jauhiainen, A. M., and Soininen, H. (2009). Structural and functional MRI in mild cognitive impairment. Curr. Alzheimer Res. 6, 179–185. doi: 10.2174/156720509787602898
Pomara, N., Doraiswamy, P. M., Willoughby, L. M., Roth, A. E., Mulsant, B. H., Sidtis, J. J., et al. (2006). Elevation in plasma Abeta42 in geriatric depression: a pilot study. Neurochem. Res. 31, 341–349. doi: 10.1007/s11064-005-9029-z
Pomara, N., and Sidtis, J. (2007). Possible therapeutic implication of Abeta disturbances in depression. Int. J. Geriatr. Psychiatry 22, 931–932. doi: 10.1002/gps.1763
Potter, G. G., and Steffens, D. C. (2007). Contribution of depression to cognitive impairment and dementia in older adults. Neurologist 13, 105–117. doi: 10.1097/01.nrl.0000252947.15389.a9
Qiu, W. Q., Sun, X., Selkoe, D. J., Mwamburi, D. M., Huang, T., Bhadela, R., et al. (2007). Depression is associated with low plasma Abeta42 independently of cardiovascular disease in the homebound elderly. Int. J. Geriatr. Psychiatry 22, 536–542. doi: 10.1002/gps.1710
Reiman, E. M., Chen, K., Liu, X., Bandy, D., Yu, M., Lee, W., et al. (2009). Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825. doi: 10.1073/pnas.0900345106
Salloway, S., Malloy, P., Kohn, R., Gillard, E., Duffy, J., Rogg, J., et al. (1996). MRI and neuropsychological differences in early- and late-life-onset geriatric depression. Neurology 46, 1567–1574. doi: 10.1212/WNL.46.6.1567
Seo, E. H., Lee, D. Y., Choo, I. H., Kim, S. G., Kim, K. W., Youn, J. C., et al. (2008). Normative study of the stroop color and word test in an educationally diverse elderly population. Int. J. Geriatr. Psychiatry 23, 1020–1027. doi: 10.1002/gps.2027
Sierksma, A. S., van den Hove, D. L., Steinbusch, H. W., and Prickaerts, J. (2010). Major depression, cognitive dysfunction and Alzheimer's disease: is there a link? Eur. J. Pharmacol. 626, 72–82. doi: 10.1016/j.ejphar.2009.10.021
Song, F., Poljak, A., Valenzuela, M., Mayeux, R., Smythe, G. A., and Sachdev, P. S. (2011). Meta-analysis of plasma amyloid-beta levels in Alzheimer's disease. J. Alzheimers Dis. 26, 365–375. doi: 10.3233/JAD-2011-101977
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Steele, J. D., Currie, J., Lawrie, S. M., and Reid, I. (2007). Prefrontal cortical functional abnormality in major depressive disorder: a stereotactic meta-analysis. J. Affect. Disord. 101, 1–11. doi: 10.1016/j.jad.2006.11.009
Sun, X., Steffens, D. C., Au, R., Folstein, M., Summergrad, P., Yee, J., et al. (2008). Amyloid-associated depression: a prodromal depression of Alzheimer disease? Arch. Gen. Psychiatry 65, 542–550. doi: 10.1001/archpsyc.65.5.542
Tzourio-Mazoyer, N., Landeau, B., Papathanassiou, D., Crivello, F., Etard, O., Delcroix, N., et al. (2002). Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 15, 273–289. doi: 10.1006/nimg.2001.0978
Vemuri, P., and Jack, C. R. Jr. (2010). Role of structural MRI in Alzheimer's disease. Alzheimers Res. Ther. 2, 23. doi: 10.1186/alzrt47
Weisenbach, S. L., and Kumar, A. (2014). Current understanding of the neurobiology and longitudinal course of geriatric depression. Curr. Psychiatry Rep. 16:463. doi: 10.1007/s11920-014-0463-y
Wenham, P. R., Price, W. H., and Blandell, G. (1991). Apolipoprotein E genotyping by one-stage PCR. Lancet 337, 1158–1159.
Whitwell, J. L., and Vemuri, P. (2011). Assessing subtle structural changes in Alzheimer's disease patients. Methods Mol. Biol. 711, 535–550. doi: 10.1007/978-1-61737-992-5_27
Winblad, B., Palmer, K., Kivipelto, M., Jelic, V., Fratiglioni, L., Wahlund, L. O., et al. (2004). Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 256, 240–246. doi: 10.1111/j.1365-2796.2004.01380.x
Wolk, D. A., Price, J. C., Saxton, J. A., Snitz, B. E., James, J. A., Lopez, O. L., et al. (2009). Amyloid imaging in mild cognitive impairment subtypes. Ann. Neurol. 65, 557–568. doi: 10.1002/ana.21598
Wu, K. Y., Hsiao, I. T., Chen, C. S., Chen, C. H., Hsieh, C. J., Wai, Y. Y., et al. (2014). Increased brain amyloid deposition in patients with a lifetime history of major depression: evidenced on 18F-florbetapir (AV-45/Amyvid) positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 41, 714–722. doi: 10.1007/s00259-013-2627-0
Wu, K.-Y., Liu, C.-Y., Chen, C.-S., Chen, C.-H., Hsiao, I.-T., Hsieh, C.-J., et al. (2016). Beta-amyloid deposition and cognitive function in patients with major depressive disorder with different subtypes of mild cognitive impairment: 18F-florbetapir (AV-45/Amyvid) PET study. Eur. J. Nucl. Med. Mol. Imaging 43, 1067–1076. doi: 10.1007/s00259-015-3291-3
Keywords: late-life onset depression, Alzheimer's disease, cerebral amyloidosis, neuronal injury, blood pressure
Citation: Byun MS, Choe YM, Sohn BK, Yi D, Han JY, Park J, Choi HJ, Baek H, Lee JH, Kim HJ, Kim YK, Yoon EJ, Sohn C-H, Woo JI and Lee DY (2016) Association of Cerebral Amyloidosis, Blood Pressure, and Neuronal Injury with Late-Life Onset Depression. Front. Aging Neurosci. 8:236. doi: 10.3389/fnagi.2016.00236
Received: 29 June 2016; Accepted: 23 September 2016;
Published: 13 October 2016.
Edited by:
Hanting Zhang, West Virginia University, USAReviewed by:
Yu-Min Kuo, National Cheng Kung University, TaiwanRamesh Kandimalla, Texas Tech University, USA
Copyright © 2016 Byun, Choe, Sohn, Yi, Han, Park, Choi, Baek, Lee, Kim, Kim, Yoon, Sohn, Woo and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Young Lee, c2VsZnBzeUBzbnUuYWMua3I=