 Jesus Avila1,2*
Jesus Avila1,2* Juan S. Jiménez3
Juan S. Jiménez3 Carmen L. Sayas4
Carmen L. Sayas4 Marta Bolós1,2
Marta Bolós1,2 Juan C. Zabala5
Juan C. Zabala5 Germán Rivas6
Germán Rivas6 Felix Hernández1,2
Felix Hernández1,2- 1Centro de Biología Molecular Severo Ochoa (Consejo Superior de Investigaciones Científicas-UAM), Madrid, Spain
- 2Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas, Madrid, Spain
- 3Departamento de Química Física Aplicada, Universidad Autónoma de Madrid, Madrid, Spain
- 4Centre for Biomedical Research of the Canary Islands, Institute for Biomedical Technologies, University of La Laguna, Tenerife, Spain
- 5Departamento de Biología Molecular, Facultad de Medicina, IDIVAL-Universidad de Cantabria, Santander, Spain
- 6Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas, Madrid, Spain
Tau is a microtubule-associated protein that plays an important role in axonal stabilization, neuronal development, and neuronal polarity. In this review, we focus on the primary, secondary, tertiary, and quaternary tau structures. We describe the structure of tau from its specific residues until its conformation in dimers, oligomers, and larger polymers in physiological and pathological situations.
Tau is a neuronal protein that was found (Weingarten et al., 1975) associated to microtubules, together with other microtubule-associated proteins (see Matus, 1988 for a review). Tau protein plays a role in microtubule assembly and dynamics that may regulate neuron morphology (Drubin and Kirschner, 1986; Caceres and Kosik, 1990; Panda et al., 1999). The roles of tau in axon development and navigation (Dawson et al., 2001; Sayas et al., 2015), dendritic spine function (Ittner et al., 2010), or long-term depression (Takashima, 2012), have been described.
Tau protein(s) is (are) encoded by a single gene, mapt, that is located on chromosome 17 in humans (Neve et al., 1986). This gene is transcribed into a nuclear RNA that yields six different isoforms, lacking or containing exons 2, 3, and 10 by alternative splicing (Avila et al., 2004; Andreadis, 2006). Exon 10 contains a microtubule-binding region similar, but not identical, to other three additional microtubule-binding regions. Thus, isoforms containing exon 10, resulting in tau with four microtubule (tubulin)-binding regions (repeats) are known as tau 4R, whereas alternative spliced isoforms, lacking exon 10, are known as tau 3R.
Tau Primary Structure
As previously indicated, there are mainly six tau isoforms in brain [and central nervous system (CNS)], showing differences in their primary structure due to the presence or absence of some specific exons. In addition, in peripheral nervous system there are additional tau isoforms containing an extra exon, exon 4a (Goedert et al., 1992). However, the longest human CNS tau isoform with 441 residues has usually been chosen as a model to exemplify tau primary structure (Goedert et al., 1989). That isoform contains exons 1, 2, 3, 4, 5, 7, 9, 10, 11, 12, and 13. Within those 441 residues there is a high proportion (80) of phosphorylable residues (serine and threonine) and a low proportion, compared to other proteins, of hydrophobic aminoacids [alanine (34), valine (25), isoleucine (15), leucine (20), methionine (5), phenylalanine (3), tyrosine (5) or tryptophan (0)].
Phylogenetic analyses of proteins with tau-like microtubule-binding regions have indicated the existence of a protein family of microtubule-associated proteins composed by tau, MAP2, and MAP4 proteins. Also, similarities with some proteins from invertebrate organisms and with a histone acetyltransferase subunit (KANSL1), have been recently reported (Sündermann et al., 2016).
Comparison of sequences of tau proteins from different origins has indicated a larger variability at the N-terminal half of tau than at C-terminal tau (Leon-Espinosa et al., 2013). Interestingly, the localization of threonine (or alanine) residues do not present a random distribution and they are present in a higher proportion at the N-terminal region of tau protein (Avila et al., 2012). Regarding the possible function of exons 2 and 3, it has been described that they might play a role in the association of tau protein with different cell membrane proteins like apolipoprotein ApoA1 (preferentially binding to tau with those exons) or synaptophysin (preferentially binding tau without those exons; Liu C. et al., 2016). Curiously, a binding site for NRF2, a transcription factor involved in antioxidant responses, is present in the first intron of the mapt gene and it may facilitate exon 3 inclusion in tau protein, an inclusion that could have a protective role (Wang et al., 2016).
Another important feature related to the primary structure of tau protein is the presence of different types of posttranslational modifications (PTMs) of specific residues. Among these modifications are phosphorylation, acetylation, deamidation, methylation, O-Glycylation, or ubiquitination (Avila et al., 2004; Avila, 2009; Morris et al., 2015; Huang et al., 2016; Iqbal et al., 2016). Furthermore, loss of N-terminal or C-terminal regions by truncation are very well-known (García-Sierra et al., 2008; Avila et al., 2015).
Although tau protein has been considered as an intrinsically disordered protein (IDP), modifications by phosphorylation or other posttranslational modifications could facilitate a “gain” in secondary structure that could facilitate the appearance of α-helix or β-sheet regions (see below).
Focus in Specific Tau Residues
As previously mentioned, primary structure of tau protein can be divided into two regions, N-terminal, and C-terminal regions. The C-terminal part shows less variability among tau proteins from different species. This fact may suggest that some of these conserved residues could be essential for some tau functions. Hence, related to tau functions, we have selected 11 of the conserved residues that could be important for human tau's role. Only three of them are at the N-terminal half of the protein. The chosen residues are: M11, A152, K174, S214, T231, R279, K280, C322, N368, S409, and D421 (Figure 1A shows some of them).
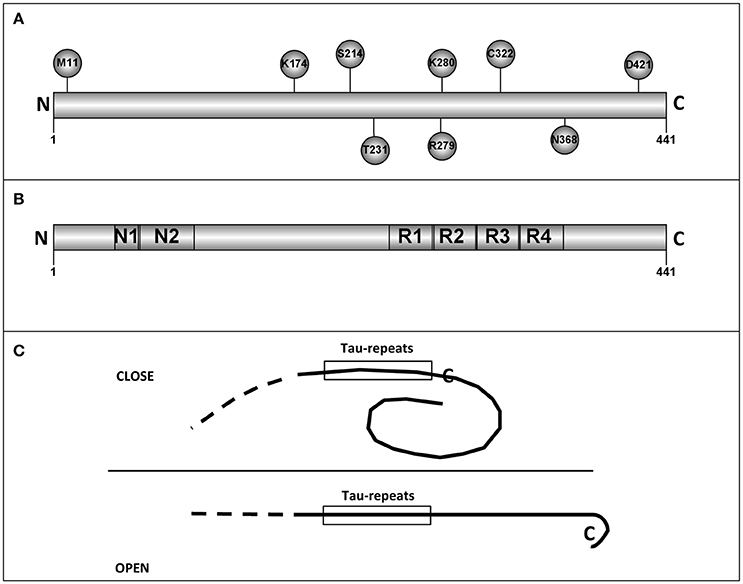
Figure 1. Tau residues and tau regions. (A) The localization in tau molecules of some tau residues is shown. (B) The different tau regions, including the microtubule binding repeats are shown. (C) Intramolecular interactions, like that described for “paper-clip” structure (see text and reference, Jeganathan et al., 2006) could prevent the tau self-interaction by masking the region(s) involved in that interaction.
Methionine 11 could be the first residue for a tau truncated protein, in the absence of the first methionine. Tau protein starting at M11, has a decreased interaction with Alz50 antibody suggesting possible changes in the putative tau tertiary structure (Carmel et al., 1996; see below).
Lysine 174 can be acetylated. This modification results in a toxic protein (Min et al., 2015). Serine 214 can be phosphorylated by protein kinase A (Scott et al., 1993) and it has been suggested that this modification might play a positive role in the binding of tau to microtubules (García de Ancos et al., 1993). Threonine 231 is followed by two proline residues. T231 is modified by different proline directed kinases like GSK3, JNK, MAPK or cdk5 (Illenberger et al., 1998; Reynolds et al., 2000). As indicated, T231 can be phosphorylated and the following prolines can have a cis or trans conformation which could regulate the posterior dephosphorylation of P-T231 (Lu et al., 1999). This is an important task since the presence of this phosphoprotein could be toxic (Nakamura et al., 2012). Also, P-T231 is present in peripheral nervous system in submandibular gland and, curiously, the level of phosphorylation decreases with increasing Braak stage of Alzheimer disease (Dugger et al., 2016). Tau is mainly an axonal protein. However, in Alzheimer disease and other tauopathies it is also present at dendritic spines where may play a toxic role. This toxic role could be the consequence of tau modifications like phosphorylation, truncation, or acetylation. The knowledge of the role of acetylated tau in tauopathies is increasing gradually. For example, in the work of Tracy et al. is described, in an elegant way, a possible mechanism explaining the role of acetylated tau (at lysines 274 and 281). In this article, memory impairment is studied by using a mouse model which express human tau with lysine to glutamine mutations to mimic lysine acetylation (Tracy et al., 2016). Acetylated tau reduces the levels of kidney and brain expressed protein (KIBRA) present at synapses and this may produce a decrease in synaptic efficiency (Tracy et al., 2016). More recently, it has been shown that acetylated tau (at lysines 274 and 281) destabilizes the cytoskeleton in the axon initial segment (Sohn et al., 2016).
Arginine 279 can be deaminated (Dan et al., 2013) and deamination results in a reduced ability of modified tau to bind to microtubules (Dan et al., 2013) and perhaps it could facilitate its self-aggregation (Montejo de Garcini et al., 1986).
Lysine 280 loss facilitates tau self-assembly and exacerbates tau toxicity (Gorsky et al., 2016; Wang and Mandelkow, 2016).
Cysteine 322 is modified by oxidation, a modification that regulates tau self-assembly (Kim et al., 2015; Wang and Mandelkow, 2016).
Cleavage of asparagine 368 by an asparaginase results in the appearance of a very toxic fragment (Zhang et al., 2014).
Serine 409 is a good substrate for Rho kinase and other related kinases (Amano et al., 2003). Recently, it has been suggested that ROCK phosphorylates tau protein decreasing its further degradation (mainly for Tau4R). This may result in tau accumulation, as occurs in some tauopathies like corticobasal degeneration or progressive supranuclear palsy. As a results, the use of ROCK inhibitors has been suggested for the treatment of those diseases (Gentry et al., 2016).
Finally, truncation of aspartic 421 facilitates tau aggregation and toxicity (García-Sierra et al., 2008) and phosphorylation at serine 422 could enhance SDS-stable dimer formation (Tiernan et al., 2016).
Other residues could be relevant because they can be modified by phosphorylation (see review by Hanger et al., 2009) or because their mutations could facilitate the development of some tauopathies (for a review see Goedert and Jakes, 2005).
A recent example of aminoacid changes resulting in toxic function is the mutation of alanine 152 to threonine which causes age-dependent neuronal dysfunction, possibly through a mechanism in which NMDA receptors could be involved (Decker et al., 2016; Maeda et al., 2016). The consequences of having A152T mutation are different than those due to the presence of other tau mutations. Commonly, tau mutations could facilitate the assembly of large tau polymers. However, A152T mutation promotes the formation of smaller toxic tau oligomers. In this way, using a C. elegans tauopathy model, expressing A152T tau mutant, it was found a reduced lifespan and severe locomotor deficits. This pathology was due to the presence of the indicated toxic tau oligomers but not to that of insoluble aggregates (Pir et al., 2016).
Tau Regions or Motifs Involved in the Binding of Tau to Other Proteins
Tau, α-Tubulin Binding Protein
The development of the mammalian CNS requires the generation, migration, and differentiation of neurons. Each of these cellular events needs a highly dynamic microtubule cytoskeleton. It is necessary for, among others, the separation of chromatids in mitosis, nuclear translocation during migration, and the outgrowth of neurites. Microtubules are polymers composed of globular subunits of α- and β-tubulin, two members of one of the largest families of genes and proteins (Turk et al., 2015). These two families of tubulins, with an approximate molecular mass of 50 kDa, are GTP-binding proteins and form an αβ-tubulin heterodimer which represents the structural and functional unit of the microtubule. It has been thought for a long time that the GTP-bound to the α subunit (non-exchangeable) is never hydrolyzed being inaccessible for biochemical manipulations, while those bound to the β subunit (exchangeable) are hydrolyzed twice. First, during the formation of the dimer and second, after microtubule incorporation. The process of tubulin folding follows a sophisticated pathway in which several molecular chaperones are involved. α and β subunits are folded independently with the GTP bound to each one. Among all the chaperones involved, including CCT and prefoldin, tubulin folding cofactors are molecular chaperones specifically required for αβ-tubulin dimer formation and the acquisition of its quaternary structure (Serna and Zabala, 2016).
On the other hand, as adult neurons are not dividing cells, they need to stabilize the microtubule cytoskeleton in order to maintain neuronal processes such as the fast axonal transport through long distances. Tau was identified (1–3) as a microtubule associated protein mainly expressed in neurons. The main role of tau is the stabilization of the axonal microtubules contributing to decrease their intrinsic dynamic instability. Furthermore, tau maturation implies a fine regulation of its expression in order to produce appropriate amounts of different isoforms by alternative splicing. This is because the isoforms regulate differentially microtubule dynamics (Amos, 2004). Paclitaxel is considered a mitotic inhibitor for its ability to stabilize microtubules suppressing their dynamic, which is a requirement for a proper cell division. For this reason, it is widely used in cancer treatment. Extensive studies have been directed to the precise understanding of the interaction site in the β-tubulin subunit. Paclitaxel and different microtubule-stabilizing compounds such as epothilones compete at the same general site for binding to microtubules, although paclitaxel does not bind to yeast microtubules, for they share overlapping but not identical binding sites. This difference was found to reside in few amino acid changes in the β-tubulin binding region (Gupta et al., 2003). As described above, tau is an intrinsically disordered and extended polypeptide that contains a microtubule binding domain with three or four binding regions that allow binding to several heterodimers at the microtubule lattice (Figure 1B). These tubulin-binding regions are characterized by the presence of prolines as well as basic residues to counteract the presence of acidic residues at the C-terminus existing on the surface of microtubules. It seems that binding to microtubules in different heterodimers favors the stabilization of the microtubule and such binding occurs in the same site where paclitaxel binds to mammalian microtubules. Paclitaxel and discodermolide, among others, compete with tau for binding the β-tubulin. In fact, in polymerization experiments performed in the presence of such kind of compounds, pelleted microtubules contain less tau polypeptides. In addition, the sequence THVPGGN contained in the tau binding domain is similar to the α-tubulin sequence TVVPGGDL, present in its extended loop (Rowinsky and Donehower, 1995). When for different reasons tau is detached from microtubules, they become destabilized and also due to the fact that tau is a disorganized protein with interacting surfaces exposed, it tends to aggregate forming PHFs. This situation led to the formation of neurofibrillary tangles (see below) and finally to neuron death. In this case, induction of tau degradation might be an important therapeutic approach. Chu et al. (2016) have recently developed peptides derived from α- and β-tubulins that recognized tau proteins connected to peptides recognized by E3 ubiquitin ligases and also to a cell penetrating peptide. They found that some of these peptides induce tau poly-ubiquitination and as a consequence its degradation in an AD mouse model decreasing cytotoxicity (Chu et al., 2016).
Other Tau Binding Proteins
Actin or protein phosphatase 2A binds to tau, through tau repeats (Correas et al., 1990; Bakota and Brandt, 2016). On the other hand, tau motifs like PXXP may interact with membrane proteins containing SH3 domains (Hwang et al., 1996) or with other proteins, like Fyn tyrosine kinase that can be also localized at the cell membrane (Lau et al., 2016). Tau, N-terminal region, could be involved in the interaction with annexin A2 (Bakota and Brandt, 2016).
The presence of exon 3 facilitates the binding of other proteins such as ApoE, 14-3-3 zeta or synaptotagmin (Liu C. et al., 2016). Also, tau interacts with alpha synuclein, probably through the basic region containing residues 166–226 (Oikawa et al., 2016). As mentioned above, tau interacts with other microtubular proteins, such as the EB1 and EB3 proteins (7), in developing neuronal cells. Characterization of tau binding to EBs is currently under study. Also, tau can interact with other proteins, like the RNA binding protein TIA1, through a RNA intermediate (Vanderweyde et al., 2016). More recently, the interaction of tau with other proteins like ferritin and transferrin has been reported (Jahshan et al., 2016). Also, the residues 139–143 could be involved in the binding to heparin and the residues 336–343 and 347–351 could be involved in chaperone mediated autophagy (Wang et al., 2009). By using a protein-protein mining tool, the laboratory of Dr. Li Weil in China (http://liweilab.genetics.ac.cn/tm/gene.php?st=gn&gn=mapt&gi=4137&ti=9606) have described 172 genes related to tau gene.
Tau Secondary Structure
Tau protein in solution can be considered as an intrinsically disorder protein (IDP). It means that, as probed by different techniques including circular dichroism (CD), Fourier transform infrared spectroscopy (FTIR), X ray diffraction, fluorescence, and some other methods, this protein behaves as a kind of random coil, lacking a well-defined secondary structure (for a full review on intrinsically disordered proteins, IDPs, see reference Skrabana et al., 2006). Tau is a highly soluble and heat-stable protein, in agreement with the lack of a well-defined secondary and indeed tertiary structure. The first report on tau protein structure (Cleveland et al., 1977) appeared 9 years before PHFs were reported to be composed mainly of this protein (Grundke-Iqbal et al., 1986a; Kosik et al., 1986; Nukina and Ihara, 1986). The CD spectrum reported by Cleveland (Cleveland et al., 1977) did not show any relevant secondary structure in a significant extent. This lack of structure was confirmed later by Woody et al. (1983) from proton NMR studies of microtubule- associated proteins. A large number of proteins contain, along their sequence, apparently unordered segments that, on the other hand, possess a clearly defined function once they face their interacting partners. This seems to be the particular case of the so-called conformational diseases. Aggregation of totally or partially disordered proteins has been repeatedly reported to be associated with many human neurodegenerative diseases, including Huntington's, Creutzfeldt-Jakob's, Parkinson's, as well as Alzheimer's disease, and other neurodegenerative disorders as the so-called tauopathies in the case of tau (Avila, 2006). However, in most cases, and the tau protein is one of them, the molecular mechanism of aggregation and even the fine structure of the final aggregate—as it is the case for the PHFs formed by tau protein—remains obscure and controversial.
Research on the secondary structure of tau protein is closely tightened to that concerning the detailed molecular structure of PHFs. Some months after the publication of the reports confirming that tau protein was the main component of PHFs, filaments resembling the PHF structure were obtained “in vitro” from tau protein purified from porcine brain (Montejo de Garcini et al., 1986). Thereafter, the door was open to study the mechanism of tau aggregation leading to PHFs formation and the subsequent search for the inhibition of the aggregation process as a therapeutic target. However, obtaining PHFs from tau protein resulted to be a not so easy task, and it seemed that particular phosphorylation sites on tau had to be phosphorylated (Biernat et al., 1992; Wille et al., 1992). Most intriguing was to reconcile the subtle helical structure found in PHFs with a tau protein devoid of any relevant secondary structure, as deduced from CD and FTIR (Schweers et al., 1994). On the other hand, the detailed molecular structure of PHFs has remained controversial and at the same time no well-defined structure has been reported for tau protein. Already in 1986, before tau was shown to be the main component of PHFs, the characteristic reflection at 4.7 Angstrom, corresponding to a β-pleated sheet structure, was reported to be found after X-ray diffraction on PHFs (Kirschner et al., 1986). However, 8 years later, similarly to tau protein, PHFs were reported to be devoid of any secondary structure, as deduced from X-ray diffraction (Schweers et al., 1994). The presence of β-amyloid in the PHFs preparation was considered as a plausible explanation for the 4.7 Angstrom reflection reported previously. Nevertheless, the same group reported later circular dichroism and FTIR data of a tau derived peptide confirming a pronounced β-structure on assembling. Their data indicated that PHF assembly was dependent of a local sequence in tau protein, at the beginning of the third repeat, forming β-structure (von Bergen et al., 2000). Also, based on X-ray diffraction data, there was a claim that peptides derived from tau protein—but not the full-length protein—formed cross-β structure (Giannetti et al., 2000). On the other hand, evidences for α-helix structure were also reported. Based on CD and FTIR data, Sadqi et al. claimed that PHFs isolated from brain samples of AD patients contained a high proportion of α-helices (Sadqi et al., 2002). A percentage of α-helix higher than β-sheet or unordered structure has also been found for PHFs suspended in Tris buffer (Goux, 2002). A tau fragment corresponding to the microtubule-binding region of the three repeat tau isoforms, as probed by high resolution NMR, has revealed three regions involved in microtubules binding, exhibiting a high preference for α-helix. A β-strand in a stretch between two α-helix segments, alternatively, is involved in the aggregation process to form PHFs (Eliezer et al., 2005). The authors conclude that the cross-β structure might be limited to short stretches at the core of the PHF structure, while α-helices would be found within the core of the microtubule-binding domain, as well as at the N- and C-terminal regions of tau, therefore explaining the high content of α-helices reported (Goux, 2002; Sadqi et al., 2002; Eliezer et al., 2005). This would be in agreement with the observations of Kunjithapatham et al. that proneness of tau to aggregate into α-helical structures could be a consequence of its microtubule-binding capacity (Kunjithapatham et al., 2005).
One of the main drawbacks to study tau protein structure comes from its flexible and intrinsically disordered nature. This type of proteins, offering a large population of conformations, is not amenable to be studied by crystallographic methods. Therefore, spectroscopy, particularly NMR, as well as CD and Fourier Transform Infrared have been the most relevant sources of structural information. The paper by Mukrasch et al. (2009) is probably the most complete description of the full secondary structure of the longest, 441 amino acid residues, and form of tau protein. The NMR data reveal that 343 out of 441 amino acids comprising tau are devoid of any ordered structure. Only small segments of the sequence present transient elements of secondary structure: six segments display propensity to form β-strands, three segments show poly-Pro helices and, finally two segments composed of ten amino acids, 114–123 and 428–437, within the N-terminal projection domain and the C-terminal domain present transient α-helix structure (Mukrasch et al., 2009; Wang and Mandelkow, 2016). No relevant sequence homology has been found for the 428–437 segment displaying α-helix propensity. On the other hand the α-helix segment at the projection domain 114–123 presents homology with an α-helix segment on cytochrome B5 (Qian et al., 2001). Most interesting is the finding that tau protein, in agreement with previous reports, presents a highly dynamic structure with many compact conformations and a complex network of long range transient contacts. This description is far from the elongated molecule originally described from electron microscopy (Wille et al., 1992), and is closer to the hairpin structure in which both N- and C-terminals contacted with each other (Jeganathan et al., 2006).
Tau protein has a large number of potential sites of phosphorylation (Grundke-Iqbal et al., 1986b; Bancher et al., 1989; Wang and Mandelkow, 2016), 80 Ser or Thr and 5 Tyr. The phosphorylation state of the protein is developmentally regulated and closely related to its function. Phosphorylation is involved in regulating the interaction with different partners as microtubules and nucleic acids (Camero et al., 2014; Multhaup et al., 2015), in the protein location and, indeed it is related to the PHF and tangles formation (Wang and Mandelkow, 2016). In fact one of the initial observations related to the abnormal tau aggregation into PHFs was that tau within this complexes is abnormally hyper-phosphorylated (Grundke-Iqbal et al., 1986b). Tau protein is a dynamic structure displaying a complex network of intramolecular long-range interactions, lacking a well-defined secondary structure -except for a number of short segments displaying some transient proneness to acquire particular structural elements. On the other hand, phosphorylation regulates all tau functions: development, spreading and location, partners interaction and of course PHFs formation and evolution, leading therefore to the question: how does phosphorylation affect the secondary structure of tau? After some mutations or serine and threonine residues in tau into glutamic acid, Bibow et al. have suggested that phosphorylation might induce the opening of the transient folded structure of tau (Bibow et al., 2011). Finally, NMR experiments carried out on a peptide derived from tau protein have shown how phosphorylation stabilizes the α-helix structure on tau (Sibille et al., 2012), therefore suggesting the possibility of a higher content of α-helices in the hyper-phosphorylated tau building PHFs.
Tau Tertiary Structure
Electron microscopy analysis done by Hirokawa's group (Hirokawa et al., 1988) suggested that tau could have an extended fibrillar form. However, by looking at the epitopes recognized by the antibody Alz50, it was found that such antibody reacts with residues present at the N- and C-terminal regions, suggesting a possible tertiary structure in tau protein (Carmel et al., 1996). Although, nuclear magnetic resonance and small-angle X-ray scattering indicate that tau is an intrinsically disordered protein (Qi et al., 2015), a “paper-clip” structure has been suggested for some molecules of tau monomers. This model was obtained by FRET measurements on tryptophan mutants of tau protein (Jeganathan et al., 2006). The presence of a “paper-clip” structure may suggest the presence of intramolecular interactions between, at least, two different regions of tau molecule. If one of those regions is involved in tau self-interaction, the opening of the “paper-clip” structure could facilitate that self-aggregation (see Figure 1C). As indicated, tau is an intrinsically disordered protein and methods to determine its tertiary structure like X-ray crystallography could not be suitable to describe that structure and nuclear magnetic resonance (NMR) studies could be advisable to do it (Lippens et al., 2016).
NMR spectroscopy of isolated tau have shown some conformational changes upon phosphorylation of tau at its threonine 231, suggesting the stabilization of a small α-helix at that region (Sibille et al., 2012); or upon phosphorylation at the Ser 202 and Thr 205, a modification that could be recognized by AT8 antibody. Thus, modification of those sites could result in an unusual turn conformation (Gandhi et al., 2015).
Tau Quaternary Structure
Tau protein could form dimers, oligomers and larger polymers. In the formation of tau dimers or oligomers, cysteine residues may play an important role (Takashima, 2013; Soeda et al., 2015). These cysteine residues are present at the microtubule-binding domain (MTBD) of tau protein that is also involved in tau-tau interaction. Nevertheless, for the formation of tau aggregates or fibers, it is not essential the formation of intramolecular cysteine_cysteine links, and the formation of tau fibers could be facilitated by the presence of polyanions like heparin (Huvent et al., 2014). It is discussed if tau dimers could be toxic (Cowan and Mudher, 2013), but there is some evidence indicating that tau trimers could be the toxic species (Tian et al., 2013). Larger granular tau oligomers, composed by around 36 tau molecules have been also described as possible toxic agents (Maeda et al., 2007). These oligomers may precede the formation of tau filaments found in different tauopathies. In Alzheimer disease, mainly two types of tau filaments have been described: paired helical (PHF) or straight (SF) filaments. By doing “in vitro” analysis, it has been proposed that a difference between PHFs and SFs could be the presence, in PHFs, of sulfated glycosaminoglycans, like heparan sulfate or chondroitin sulfate, associated to tau polymers (Arrasate et al., 1997). On the other hand, highly sulfated, heparan sulfate (heparin) facilitates tau aggregation to form filaments (Goedert et al., 1996; Pérez et al., 1996). In addition, glycation of tau filaments results in the formation of larger aggregates similar to neurofibrillary tangle aggregates (Ledesma et al., 1996).
Tau aggregates and fibrils could have a role in tau spreading. Tau spreading has been a hot topic within recent years (Clavaguera et al., 2009; Sanders et al., 2014; Peeraer et al., 2015). However, since this review is focused on the structure and not in the (toxic) function of tau aggregates, we enclosed reference (Wang and Mandelkow, 2016) for a good review of that topic.
It has been described that tau filaments are assembled by the self-association of tau molecules (Montejo de Garcini et al., 1986), in an anti-parallel arrangement, through the MTBDs. That arrangement could facilitate the interaction through cysteine and other residues present in that domain (Ksiezak-Reding and Yen, 1991; see Figure 2). Tau fragments containing the MTBD could also assemble into fibrillar polymers (Hirokawa et al., 1988; Jeganathan et al., 2006) and even a very small fragment composed by the hexapeptide VQIVYK, present in the MTBD, is able to form filaments (von Bergen et al., 2000).
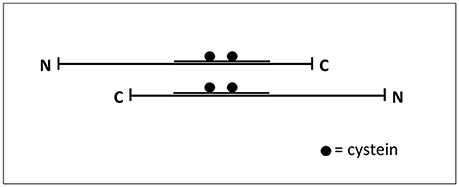
Figure 2. Interaction tau-tau. Tau protein could be found as an extended common long protein that could bind to other proteins, including itself, in this extended fashion (Hirokawa et al., 1988). The Figure shows how two tau molecules could interact, in an antiparallel fashion, through their microtubule-binding domain where cysteine residues are present.
On the other hand, when tau molecules are bound to microtubules, the MTBD is not accessible to facilitate tau-tau association. However, it has been reported that tau, bound to microtubules, could form dimers through its N-terminal region (Feinstein et al., 2016). These regions and also, perhaps, the C-terminal region could play a role in the spatial organization of axonal microtubules, keeping parallel microtubules despite of macromolecular crowding (Méphon-Gaspard et al., 2016).
Although mainly fibrillar polymers could be obtained “in vitro,” using only pure tau proteins, the assembly of the protein could be facilitated upon addition of anionic compounds (see for example Avila et al., 2004, for a review). These fibrillary structures are dynamic polymers, as shown in cell culture models (Guo et al., 2016).
Apart from fibrillary structures there are other tau polymers showing different structures like Pick's bodies or argylophilic grains, among others (see for example Rábano et al., 2013). The assembly of those structures could be facilitated by the presence of other compounds or by differences in the level of tau phosphorylation. Indeed phospho-tau dephosphorylation by protein phosphatase 2A could switch the formation of fibrillary polymers to argylophilic gain-like morphology (Hu et al., 2016). Curiously, injection in mouse brain of brain extracts from patients with tauopathies showing different tau structures could reproduce “in vivo” the appearance, in mouse, of those structures (Clavaguera et al., 2013; Sanders et al., 2014). It suggests that some additional compounds, in addition to tau, could be involved in raising the final morphology of tau structures. Also, those different morphologies could be the consequence of different arrangements of tau molecules, which could be found either in parallel form, mixed of parallel and anti-parallel forms, or anti-parallel form (see Figure 3). For example the hypothetical structure shown in Figure 3 could be the result of assembly of parallel and antiparallel forms. Some of them could aggregate around a cationic (metal) structure, whereas other could form antiparallel aggregates. Also, recently, has been shown that different structures could arise from the mixture of full length tau and tau fragments (Ozcelik et al., 2016). Nevertheless, when the injection in mouse brain was done using only synthetic tau protein, mainly fibrillar polymers were obtained (Iba et al., 2013).
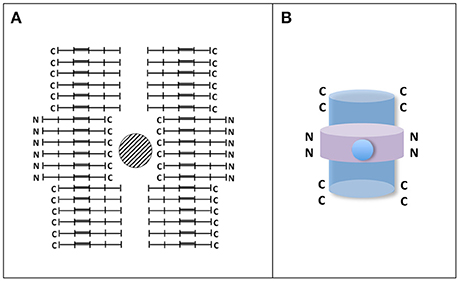
Figure 3. Hypothetical tau circular-like structure. (A) Tau granule (circular-like) structures have been previously reported (Rábano et al., 2013; Takashima, 2013). In the Figure, it is shown an hypothetical closed structure playing with parallel and antiparallel tau molecules. In that structure, a hole in the center surrounded by C-terminal sequences of tau molecule can be found. Since tau C-terminal sequences contain several acidic residues, it is possible the reaction of those residues with metals. It may take place in argylophylic grains. (B) A 3D-like image of the structure is shown. In that image parallel and antiparallel forms, with the C-terminal (blue) or N-terminal (violet), regions of tau molecule at the surface, could be present. It is proposed that those having the C-terminal inside the structure could react with cations.
Although, in a general way, it has been suggested the transition from soluble monomeric tau protein to the formation of large, insoluble, tau aggregates (Takashima, 2013), more studies are needed to test if tau protein follows a protein phase transition in a similar way to that described for the formation of FUS protein aggregates in amyotrophic lateral sclerosis (Patel et al., 2015). Recent findings have established that a variety of intracellular membrane-less organelles and micro-compartments behave as liquid droplets inside the cell that are formed by liquid phase transitions from cytoplasm, suggesting that phase separation play an important role in the functional organization of the cell (Hyman and Simons, 2012). Intrinsic disordered proteins containing low complexity domains (LCD) prone to aggregate have been shown to be more susceptible to phase separate (Hyman and Simons, 2012; Aguzzi and Altmeyer, 2016). In this regard, the assembly of FUS into dynamic non-membrane compartments was found to be dependent on the prion-like domain of the protein, a type of LCD, which is thought to be involved in the formation of toxic aggregates (Patel et al., 2015). In fact, this domain is also present in tau protein and other aggregation-prone proteins associated to aging diseases. Time-dependent FUS aggregation led to the formation of disease-linked solid aggregates and aberrant liquid to solid phase transition was accelerated by ALS patient mutations, suggesting that the pathological aggregates could result from unregulated phase transitions linked to protein modifications that alter its assembly properties. Therefore, factors modulating the relative abundance of the different aggregation states of an amyloid forming protein [as glycosylation (Liu K. et al., 2016) or crowding (Wu et al., 2015) in the case of tau protein] may have a major impact in the formation of dynamic liquid micro-compartments and in the liquid-to-solid phase transition leading to pathological aggregation. The maintenance of the proper balance of physiological phase transitions may constitute a novel therapeutic strategy against neurodegeneration.
Author Contributions
All the authors contributed equally to the outline, drafting, and editing of the present manuscript.
Funding
This work was supported by grants from the Spanish Ministry of Economics and Competitiveness, Plan Estatal de I+D+i.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguzzi, A., and Altmeyer, M. (2016). Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol. 26, 547–558. doi: 10.1016/j.tcb.2016.03.004
Amano, M., Kaneko, T., Maeda, A., Nakayama, M., Ito, M., Yamauchi, T., et al. (2003). Identification of Tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. J. Neurochem. 87, 780–790. doi: 10.1046/j.1471-4159.2003.02054.x
Amos, L. A. (2004). Microtubule structure and its stabilisation. Org. Biomol. Chem. 2, 2153–2160. doi: 10.1039/b403634d
Andreadis, A. (2006). Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog. Mol. Subcell. Biol. 44, 89–107. doi: 10.1007/978-3-540-34449-0_5
Arrasate, M., Perez, M., Valpuesta, J. M., and Avila, J. (1997). Role of glycosaminoglycans in determining the helicity of paired helical filaments. Am. J. Pathol. 151, 1115–1122.
Avila, J. (2006). Tau phosphorylation and aggregation in Alzheimer's disease pathology. FEBS Lett. 580, 2922–2927. doi: 10.1016/j.febslet.2006.02.067
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. doi: 10.1152/physrev.00024.2003
Avila, J., Pallas, N., Bolós, M., Sayas, C. L., and Hernandez, F. (2015). Intracellular and extracellular microtubule associated protein tau as a therapeutic target in Alzheimer disease and other tauopathies. Expert Opin. Ther. Targets 20, 653–661. doi: 10.1517/14728222.2016.1131269
Avila, J., Sayas, C. L., and Villanueva, N. (2012). “Threonine localization in a protein molecule,” in Advances in Medicine and Biology, Vol. 47, ed L. V. Berhardt (New York, NY: Nova Science Publisher), 277–284.
Bakota, L., and Brandt, R. (2016). Tau biology and tau-directed therapies for alzheimer's disease. Drugs 76, 301–313. doi: 10.1007/s40265-015-0529-0
Bancher, C., Brunner, C., Lassmann, H., Budka, H., Jellinger, K., Wiche, G., et al. (1989). Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 477, 90–99. doi: 10.1016/0006-8993(89)91396-6
Bibow, S., Ozenne, V., Biernat, J., Blackledge, M., Mandelkow, E., and Zweckstetter, M. (2011). Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J. Am. Chem. Soc. 133, 15842–15845. doi: 10.1021/ja205836j
Biernat, J., Mandelkow, E. M., Schröter, C., Lichtenberg-Kraag, B., Steiner, B., Berling, B., et al. (1992). The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 11, 1593–1597.
Caceres, A., and Kosik, K. S. (1990). Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 343, 461–463. doi: 10.1038/343461a0
Camero, S., Benítez, M. J., Cuadros, R., Hernández, F., Avila, J., and Jiménez, J. S. (2014). Thermodynamics of the interaction between Alzheimer's disease related tau protein and DNA. PLoS ONE 9:e104690. doi: 10.1371/journal.pone.0104690
Carmel, G., Mager, E. M., Binder, L. I., and Kuret, J. (1996). The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J. Biol. Chem. 271, 32789–32795. doi: 10.1074/jbc.271.51.32789
Chu, T. T., Gao, N., Li, Q. Q., Chen, P. G., Yang, X. F., Chen, Y. X., et al. (2016). Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chem. Biol. 23, 453–461. doi: 10.1016/j.chembiol.2016.02.016
Clavaguera, F., Akatsu, H., Fraser, G., Crowther, R. A., Frank, S., Hench, J., et al. (2013). Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 9535–9540. doi: 10.1073/pnas.1301175110
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
Cleveland, D. W., Hwo, S. Y., and Kirschner, M. W. (1977). Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 116, 227–247. doi: 10.1016/0022-2836(77)90214-5
Correas, I., Padilla, R., and Avila, J. (1990). The tubulin-binding sequence of brain microtubule-associated proteins, tau and MAP-2, is also involved in actin binding. Biochem. J. 269, 61–64. doi: 10.1042/bj2690061
Cowan, C. M., and Mudher, A. (2013). Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 4:114. doi: 10.3389/fneur.2013.00114
Dan, A., Takahashi, M., Masuda-Suzukake, M., Kametani, F., Nonaka, T., Kondo, H., et al. (2013). Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer's disease brain. Acta Neuropathol. Commun. 1:54. doi: 10.1186/2051-5960-1-54
Dawson, H. N., Ferreira, A., Eyster, M. V., Ghoshal, N., Binder, L. I., and Vitek, M. P. (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187.
Decker, J. M., Kruger, L., Sydow, A., Dennissen, F., Siskova, Z., Mandelkow, E., et al. (2016). The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 17, 552–569. doi: 10.15252/embr.201541439
Drubin, D. G., and Kirschner, M. W. (1986). Tau protein function in living cells. J. Cell Biol. 103, 2739–2746. doi: 10.1083/jcb.103.6.2739
Dugger, B. N., Whiteside, C. M., Maarouf, C. L., Walker, D. G., Beach, T. G., Sue, L. I., et al. (2016). The presence of select tau species in human peripheral tissues and their relation to Alzheimer's disease. J. Alzheimers Dis. 51, 345–356. doi: 10.3233/JAD-150859
Eliezer, D., Barré, P., Kobaslija, M., Chan, D., Li, X., and Heend, L. (2005). Residual structure in the repeat domain of tau: echoes of microtubule binding and paired helical filament formation. Biochemistry 44, 1026–1036. doi: 10.1021/bi048953n
Feinstein, H. E., Benbow, S. J., LaPointe, N. E., Patel, N., Ramachandran, S., Do, T. D., et al. (2016). Oligomerization of the microtubule associated protein tau is mediated by its N-terminal sequences: implications for normal and pathological tau action. J. Neurochem. 137, 939–954. doi: 10.1111/jnc.13604
Gandhi, N. S., Landrieu, I., Byrne, C., Kukic, P., Amniai, L., Cantrelle, F. X., et al. (2015). A phosphorylation-induced turn defines the alzheimer's disease AT8 antibody epitope on the tau protein. Angew. Chem. Int. Ed Engl. 54, 6819–6823. doi: 10.1002/anie.201501898
García de Ancos, J., Correas, I., and Avila, J. (1993). Differences in microtubule binding and self-association abilities of bovine brain tau isoforms. J. Biol. Chem. 268, 7976–7982.
García-Sierra, F., Mondragón-Rodríguez, S., and Basurto-Islas, G. (2008). Truncation of tau protein and its pathological significance in Alzheimer's disease. J. Alzheimers. Dis. 14, 401–409.
Gentry, E. G., Henderson, B. W., Arrant, A. E., Gearing, M., Feng, Y., Riddle, N. C., et al. (2016). Rho kinase inhibition as a therapeutic for progressive supranuclear palsy and corticobasal degeneration. J. Neurosci. 36, 1316–1323. doi: 10.1523/JNEUROSCI.2336-15.2016
Giannetti, A. M., Lindwall, G., Chau, M. F., Radeke, M. J., Feinstein, S. C., and Kohlstaedt, L. A. (2000). Fibers of tau fragments, but not full length tau, exhibit a cross beta-structure: implications for the formation of paired helical filaments. Protein Sci. 9, 2427–2435. doi: 10.1110/ps.9.12.2427
Goedert, M., and Jakes, R. (2005). Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250. doi: 10.1016/j.bbadis.2004.08.007
Goedert, M., Jakes, R., Spillantini, M. G., Hasegawa, M., Smith, M. J., and Crowther, R. A. (1996). Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553. doi: 10.1038/383550a0
Goedert, M., Spillantini, M. G., and Crowther, R. A. (1992). Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. U.S.A. 89, 1983–1987. doi: 10.1073/pnas.89.5.1983
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Gorsky, M. K., Burnouf, S., Dols, J., Mandelkow, E., and Partridge, L. (2016). Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Sci. Rep. 6:22685. doi: 10.1038/srep22685
Goux, W. J. (2002). The conformations of filamentous and soluble tau associated with Alzheimer paired helical filaments. Biochemistry 41, 13798–13806. doi: 10.1021/bi016079h
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986a). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986b). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Guo, J. L., Buist, A., Soares, A., Callaerts, K., Calafate, S., Stevenaert, F., et al. (2016). The dynamics and turnover of tau aggregates in cultured cells: insights into therapies for tauopathies. J. Biol. Chem. 291, 13175–13193. doi: 10.1074/jbc.M115.712083
Gupta, M. L. Jr., Bode, C. J., Georg, G. I., and Himes, R. H. (2003). Understanding tubulin-Taxol interactions: mutations that impart Taxol binding to yeast tubulin. Proc. Natl. Acad. Sci. U.S.A. 100, 6394–6397. doi: 10.1073/pnas.1131967100
Hanger, D. P., Anderton, B. H., and Noble, W. (2009). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119. doi: 10.1016/j.molmed.2009.01.003
Hirokawa, N., Shiomura, Y., and Okabe, S. (1988). Tau proteins: the molecular structure and mode of binding on microtubules. J. Cell Biol. 107, 1449–1459. doi: 10.1083/jcb.107.4.1449
Hu, W., Zhang, X., Tung, Y. C., Xie, S., Liu, F., and Iqbal, K. (2016). Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement. 12, 1066–1077. doi: 10.1016/j.jalz.2016.01.014
Huang, Y., Wu, Z., and Zhou, B. (2016). Behind the curtain of tauopathy: a show of multiple players orchestrating tau toxicity. Cell. Mol. Life Sci. 73, 1–21. doi: 10.1007/s00018-015-2042-8
Huvent, I., Kamah, A., Cantrelle, F. X., Barois, N., Slomianny, C., Smet-Nocca, C., et al. (2014). A functional fragment of Tau forms fibers without the need for an intermolecular cysteine bridge. Biochem. Biophys. Res. Commun. 445, 299–303. doi: 10.1016/j.bbrc.2014.01.161
Hwang, S. C., Jhon, D. Y., Bae, Y. S., Kim, J. H., and Rhee, S. G. (1996). Activation of phospholipase C-gamma by the concerted action of tau proteins and arachidonic acid. J. Biol. Chem. 271, 18342–18349. doi: 10.1074/jbc.271.31.18342
Hyman, A. A., and Simons, K. (2012). Cell biology. Beyond oil and water–phase transitions in cells. Science 337, 1047–1049. doi: 10.1126/science.1223728
Iba, M., Guo, J. L., McBride, J. D., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J. Neurosci. 33, 1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013
Illenberger, S., Zheng-Fischhöfer, Q., Preuss, U., Stamer, K., Baumann, K., Trinczek, B., et al. (1998). The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer's disease. Mol. Biol. Cell 9, 1495–1512. doi: 10.1091/mbc.9.6.1495
Iqbal, K., Liu, F., and Gong, C. X. (2016). Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 12, 15–27. doi: 10.1038/nrneurol.2015.225
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jahshan, A., Esteves-Villanueva, J. O., and Martic-Milne, S. (2016). Evaluation of ferritin and transferrin binding to tau protein. J. Inorg. Biochem. doi: 10.1016/j.jinorgbio.2016.06.022
Jeganathan, S., von Bergen, M., Brutlach, H., Steinhoff, H. J., and Mandelkow, E. (2006). Global hairpin folding of tau in solution. Biochemistry 45, 2283–2293. doi: 10.1021/bi0521543
Kim, D., Lim, S., Haque, M. M., Ryoo, N., Hong, H. S., Rhim, H., et al. (2015). Identification of disulfide cross-linked tau dimer responsible for tau propagation. Sci. Rep. 5:15231. doi: 10.1038/srep15231
Kirschner, D. A., Abraham, C., and Selkoe, D. J. (1986). X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation. Proc. Natl. Acad. Sci. U.S.A. 83, 503–507. doi: 10.1073/pnas.83.2.503
Kosik, K. S., Joachim, C. L., and Selkoe, D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 83, 4044–4048. doi: 10.1073/pnas.83.11.4044
Ksiezak-Reding, H., and Yen, S. H. (1991). Structural stability of paired helical filaments requires microtubule-binding domains of tau: a model for self-association. Neuron 6, 717–728. doi: 10.1016/0896-6273(91)90169-Z
Kunjithapatham, R., Oliva, F. Y., Doshi, U., Pérez, M., Avila, J., and Muñoz, V. (2005). Role for the alpha-helix in aberrant protein aggregation. Biochemistry 44, 149–156. doi: 10.1021/bi048564t
Lau, D. H., Hogseth, M., Phillips, E. C., O'Neill, M. J., Pooler, A. M., Noble, W., et al. (2016). Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer's disease. Acta Neuropathol. Commun. 4, 49. doi: 10.1186/s40478-016-0317-4
Ledesma, M. D., Medina, M., and Avila, J. (1996). The in vitro formation of recombinant tau polymers: effect of phosphorylation and glycation. Mol. Chem. Neuropathol. 27, 249–258. doi: 10.1007/BF02815107
León-Espinosa, G., Garcia, E., García-Escudero, V., Hernández, F., Defelipe, J., and Avila, J. (2013). Changes in tau phosphorylation in hibernating rodents. J. Neurosci. Res. 91, 954–962. doi: 10.1002/jnr.23220
Lippens, G., Landrieu, I., Smet, C., Huvent, I., Gandhi, N. S., Gigant, B., et al. (2016). NMR meets tau: insights into its function and pathology. Biomolecules 6:E28. doi: 10.3390/biom6020028
Liu, C., Song, X., Nisbet, R., and Götz, J. (2016). Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J. Biol. Chem. 291, 8173–8188. doi: 10.1074/jbc.M115.641902
Liu, K., Liu, Y., Li, L., Qin, P., Iqbal, J., Deng, Y., et al. (2016). Glycation alter the process of tau phosphorylation to change tau isoforms aggregation property. Biochim. Biophys. Acta 1862, 192–201. doi: 10.1016/j.bbadis.2015.12.002
Lu, P. J., Wulf, G., Zhou, X. Z., Davies, P., and Lu, K. P. (1999). The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399, 784–788. doi: 10.1038/21650
Maeda, S., Djukic, B., Taneja, P., Yu, G. Q., Lo, I., Davis, A., et al. (2016). Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 17, 530–551. doi: 10.15252/embr.201541438
Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., et al. (2007). Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. doi: 10.1021/bi061359o
Matus, A. (1988). Microtubule-associated proteins: their potential role in determining neuronal morphology. Annu. Rev. Neurosci. 11, 29–44. doi: 10.1146/annurev.ne.11.030188.000333
Méphon-Gaspard, A., Boca, M., Pioche-Durieu, C., Desforges, B., Burgo, A., Hamon, L., et al. (2016). Role of tau in the spatial organization of axonal microtubules: keeping parallel microtubules evenly distributed despite macromolecular crowding. Cell Mol. Life Sci. 73, 3745–3760. doi: 10.1007/s00018-016-2216-z
Min, S. W., Chen, X., Tracy, T. E., Li, Y., Zhou, Y., Wang, C., et al. (2015). Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 21, 1154–1162. doi: 10.1038/nm.3951
Montejo de Garcini, E., Serrano, L., and Avila, J. (1986). Self assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem. Biophys. Res. Commun. 141, 790–796. doi: 10.1016/S0006-291X(86)80242-X
Morris, M., Knudsen, G. M., Maeda, S., Trinidad, J. C., Ioanoviciu, A., Burlingame, A. L., et al. (2015). Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18, 1183–1189. doi: 10.1038/nn.4067
Mukrasch, M. D., Bibow, S., Korukottu, J., Jeganathan, S., Biernat, J., Griesinger, C., et al. (2009). Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 7:e34. doi: 10.1371/journal.pbio.1000034
Multhaup, G., Huber, O., Buée, L., and Galas, M. C. (2015). Amyloid Precursor Protein (APP) metabolites APP intracellular fragment (AICD), Aβ42, and tau in nuclear roles. J. Biol. Chem. 290, 23515–23522. doi: 10.1074/jbc.R115.677211
Nakamura, K., Greenwood, A., Binder, L., Bigio, E. H., Denial, S., Nicholson, L., et al. (2012). Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell 149, 232–244. doi: 10.1016/j.cell.2012.02.016
Neve, R. L., Harris, P., Kosik, K. S., Kurnit, D. M., and Donlon, T. A. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387, 271–280. doi: 10.1016/0169-328X(86)90033-1
Nukina, N., and Ihara, Y. (1986). One of the antigenic determinants of paired helical filaments is related to tau protein. J. Biochem. 99, 1541–1544.
Oikawa, T., Nonaka, T., Terada, M., Tamaoka, A., Hisanaga, S. I., and Hasegawa, M. (2016). α-Synuclein fibrils exhibit gain-of-toxic-function, promoting tau aggregation and inhibiting microtubule assembly. J. Biol. Chem. 291, 15046–15056. doi: 10.1074/jbc.M116.736355
Ozcelik, S., Sprenger, F., Skachokova, Z., Fraser, G., Abramowski, D., Clavaguera, F., et al. (2016). Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol. Psychiatry. doi: 10.1038/mp.2015.228. [Epub ahead of print].
Panda, D., Miller, H. P., and Wilson, L. (1999). Rapid treadmilling of brain microtubules free of microtubule-associated proteins in vitro and its suppression by tau. Proc. Natl. Acad. Sci. U.S.A. 96, 12459–12464. doi: 10.1073/pnas.96.22.12459
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Peeraer, E., Bottelbergs, A., Van Kolen, K., Stancu, I. C., Vasconcelos, B., Mahieu, M., et al. (2015). Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 73, 83–95. doi: 10.1016/j.nbd.2014.08.032
Pérez, M., Valpuesta, J. M., Medina, M. E., Montejo de Garcini, E., and Avila, J. (1996). Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J. Neurochem. 67, 1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x
Pir, G. J., Choudhary, B., Mandelkow, E., and Mandelkow, E. M. (2016). Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 11, 33. doi: 10.1186/s13024-016-0096-1
Qi, H., Cantrelle, F. X., Benhelli-Mokrani, H., Smet-Nocca, C., Buée, L., Lippens, G., et al. (2015). Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry 54, 1525–1533. doi: 10.1021/bi5014613
Qian, C., Yao, Y., Ye, K., Wang, J., Tang, W., Wang, Y., et al. (2001). Effects of charged amino-acid mutation on the solution structure of cytochrome b5 and binding between cytochrome b5 and cytochrome c. Protein Sci. 10, 2451–2459. doi: 10.1110/ps.ps.12401
Rábano, A., Cuadros, R., Calero, M., Hernàndez, F., and Avila, J. (2013). Specific profile of tau isoforms in argyrophylic grain disease. J. Exp. Neurosci. 7, 51–59. doi: 10.4137/JEN.S12202
Reynolds, C. H., Gibb, G. M., and Lovestone, S. (2000). Tau phosphorylation both in vitro and in cells. Methods Mol. Med. 32, 375–393. doi: 10.1385/1-59259-195-7:375
Rowinsky, E. K., and Donehower, R. C. (1995). Paclitaxel (taxol). N.Engl. J. Med. 332, 1004–1014. doi: 10.1056/NEJM199504133321507
Sadqi, M., Hernández, F., Pan, U., Pérez, M., Schaeberle, M. D., Avila, J., et al. (2002). Alpha-helix structure in Alzheimer's disease aggregates of tau-protein. Biochemistry 41, 7150–7155. doi: 10.1021/bi025777e
Sanders, D. W., Kaufman, S. K., DeVos, S. L., Sharma, A. M., Mirbaha, H., Li, A., et al. (2014). Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. doi: 10.1016/j.neuron.2014.04.047
Sayas, C. L., Tortosa, E., Bollati, F., Ramírez-Ríos, S., Arnal, I., and Avila, J. (2015). Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J. Neurochem. 133, 653–667. doi: 10.1111/jnc.13091
Schweers, O., Schönbrunn-Hanebeck, E., Marx, A., and Mandelkow, E. (1994). Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 269, 24290–24297.
Scott, C. W., Spreen, R. C., Herman, J. L., Chow, F. P., Davison, M. D., Young, J., et al. (1993). Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly. J. Biol. Chem. 268, 1166–1173.
Serna, M., and Zabala, J. C. (2016). Tubulin Folding and Degradation. Chichester: John Wiley & Sons Ltd.
Sibille, N., Huvent, I., Fauquant, C., Verdegem, D., Amniai, L., Leroy, A., et al. (2012). Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered Tau protein. Proteins 80, 454–462. doi: 10.1002/prot.23210
Skrabana, R., Sevcik, J., and Novak, M. (2006). Intrinsically disordered proteins in the neurodegenerative processes: formation of tau protein paired helical filaments and their analysis. Cell. Mol. Neurobiol. 26, 1085–1097. doi: 10.1007/s10571-006-9083-3
Soeda, Y., Yoshikawa, M., Almeida, O. F., Sumioka, A., Maeda, S., Osada, H., et al. (2015). Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat. Commun. 6:10216. doi: 10.1038/ncomms10216
Sohn, P. D., Tracy, T. E., Son, H. I., Zhou, Y., Leite, R. E., Miller, B. L., et al. (2016). Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol. Neurodegener. 11, 47. doi: 10.1186/s13024-016-0109-0
Sündermann, F., Fernandez, M. P., and Morgan, R. O. (2016). An evolutionary roadmap to the microtubule-associated protein MAP Tau. BMC Genomics 17:264. doi: 10.1186/s12864-016-2590-9
Takashima, A. (2012). GSK-3β and memory formation. Front. Mol. Neurosci. 5:47. doi: 10.3389/fnmol.2012.00047
Takashima, A. (2013). Tauopathies and tau oligomers. J. Alzheimers. Dis. 37, 565–568. doi: 10.3233/JAD-130653
Tian, H., Davidowitz, E., Lopez, P., Emadi, S., Moe, J., and Sierks, M. (2013). Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013:260787. doi: 10.1155/2013/260787
Tiernan, C. T., Combs, B., Cox, K., Morfini, G., Brady, S. T., Counts, S. E., et al. (2016). Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport. Exp. Neurol. 283, 318–329. doi: 10.1016/j.expneurol.2016.06.030
Tracy, T. E., Sohn, P. D., Minami, S. S., Wang, C., Min, S. W., Li, Y., et al. (2016). Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron. 90, 245–260. doi: 10.1016/j.neuron.2016.03.005
Turk, E., Wills, A. A., Kwon, T., Sedzinski, J., Wallingford, J. B., and Stearns, T. (2015). Zeta-tubulin is a member of a conserved tubulin module and is a component of the centriolar basal foot in multiciliated cells. Curr. Biol. 25, 2177–2183. doi: 10.1016/j.cub.2015.06.063
Vanderweyde, T., Apicco, D. J., Youmans-Kidder, K., Ash, P. E., Cook, C., Lummertz da Rocha, E., et al. (2016). Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 15, 1455–1466. doi: 10.1016/j.celrep.2016.04.045
von Bergen, M., Friedhoff, P., Biernat, J., Heberle, J., Mandelkow, E. M., and Mandelkow, E. (2000). Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134. doi: 10.1073/pnas.97.10.5129
Wang, X., Campbell, M. R., Lacher, S. E., Cho, H. Y., Wan, M., Crowl, C. L., et al. (2016). A polymorphic antioxidant response element links NRF2/sMAF binding to enhanced MAPT expression and reduced risk of Parkinsonian disorders. Cell Rep. 15, 830–842. doi: 10.1016/j.celrep.2016.03.068
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 22–35. doi: 10.1038/nrn.2015.1
Wang, Y., Martinez-Vicente, M., Krüger, U., Kaushik, S., Wong, E., Mandelkow, E. M., et al. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170. doi: 10.1093/hmg/ddp367
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Wille, H., Drewes, G., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (1992). Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 118, 573–584. doi: 10.1083/jcb.118.3.573
Woody, R. W., Clark, D. C., Roberts, G. C., Martin, S. R., and Bayley, P. M. (1983). Molecular flexibility in microtubule proteins: proton nuclear magnetic resonance characterization. Biochemistry 22, 2186–2192. doi: 10.1021/bi00278a020
Wu, Y., Teng, N., and Li, S. (2015). Effects of macromolecular crowding and osmolyte on human Tau fibrillation. Int. J. Biol. Macromol. 90, 27–36. doi: 10.1016/j.ijbiomac.2015.11.091
Keywords: motifs, tau residues, posttranslational modifications, PHFs, tubuling binding, aggregation, microtubule-associated proteins
Citation: Avila J, Jiménez JS, Sayas CL, Bolós M, Zabala JC, Rivas G and Hernández F (2016) Tau Structures. Front. Aging Neurosci. 8:262. doi: 10.3389/fnagi.2016.00262
Received: 19 August 2016; Accepted: 21 October 2016;
Published: 08 November 2016.
Edited by:
Xiongwei Zhu, Case Western Reserve University, USAReviewed by:
Cheng-Xin Gong, New York State Institute for Basic Research in Developmental Disabilities, USAYingjun Zhao, Sanford Burnham Prebys Medical Discovery Institute, USA
Copyright © 2016 Avila, Jiménez, Sayas, Bolós, Zabala, Rivas and Hernández. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesus Avila, amF2aWxhQGNibS5jc2ljLmVz