 Laura Nadal
Laura Nadal Neus Garcia*†Erica HurtadoAnna SimóMarta TomàsMaria A. Lanuza†Victor CillerosJosep Tomàs*†
Neus Garcia*†Erica HurtadoAnna SimóMarta TomàsMaria A. Lanuza†Victor CillerosJosep Tomàs*†- Unitat d’Histologia i Neurobiologia (UHN), Facultat de Medicina i Ciències de la Salut, Universitat Rovira i Virgili, Reus, Spain
The development of the nervous system involves the overproduction of synapses but connectivity is refined by Hebbian activity-dependent axonal competition. The newborn skeletal muscle fibers are polyinnervated but, at the end of the competition process, some days later, become innervated by a single axon. We used quantitative confocal imaging of the autofluorescent axons from transgenic B6.Cg-Tg (Thy1-YFP)16 Jrs/J mice to investigate the possible cooperation of the muscarinic autoreceptors (mAChR, M1-, M2- and M4-subtypes) and the tyrosine kinase B (TrkB) receptor in the control of axonal elimination after the mice Levator auris longus (LAL) muscle had been exposed to several selective antagonist of the corresponding receptor pathways in vivo. Our previous results show that M1, M2 and TrkB signaling individually increase axonal loss rate around P9. Here we show that although the M1 and TrkB receptors cooperate and add their respective individual effects to increase axonal elimination rate even more, the effect of the M2 receptor is largely independent of both M1 and TrkB receptors. Thus both, cooperative and non-cooperative signaling mechanisms contribute to developmental synapse elimination.
Introduction
During the development of the nervous system, synapses are eliminated on a broad scale (Thompson, 1985; Bourgeois and Rakic, 1993). This allows connectivity to be refined on the basis of Hebbian activity-dependent axonal competition (Jansen and Fladby, 1990; Sanes and Lichtman, 1999). In newborn animals, the skeletal muscle fibers are polyinnervated in the neuromuscular junction area (NMJ; Redfern, 1970; Brown et al., 1976; Ribchester and Barry, 1994), but at the end of the axonal competition, endplates are innervated by a single axon (Benoit and Changeux, 1975; O’Brien et al., 1978; Jansen and Fladby, 1990; Sanes and Lichtman, 1999). This peripheral synapse has been studied extensively as a model for synapse development (Liu et al., 1994; Nguyen and Lichtman, 1996; Chang and Balice-Gordon, 1997; Sanes and Lichtman, 1999; Lanuza et al., 2001; Santafé et al., 2001; Herrera and Zeng, 2003; Wyatt and Balice-Gordon, 2003; Buffelli et al., 2004; Garcia et al., 2010). Several signaling molecules and presynaptic receptors play a role in the axonal competition, which means that the various nerve endings influence one another (Santafé et al., 2009a; Garcia et al., 2010; Nadal et al., 2016). Postsynaptic-derived trophic substances (Nadal et al., 2016) and the participation of glial cells (Lee et al., 2016; Yang et al., 2016) also make a decisive contribution.
In a previous study, we investigated how individual muscarinic acetylcholine receptor (mAChR) subtypes (M1, M2 and M4), adenosine receptors (AR; A1 and A2A) and tropomyosin-related tyrosine kinase B (TrkB) receptors are involved in the control of synapse elimination in the mouse NMJ (Nadal et al., 2016). The data show that mAChR, AR and TrkB signaling lessen the initial chance of axonal elimination (around P5–P7) by extending the period of axonal competition but then increase (around P9) axonal loss rate (Nadal et al., 2016). The three receptor sets promote axonal disconnection at the beginning of the second postnatal week largely independently of the postsynaptic nicotinic acetylcholine receptor (nAChR) cluster maturation. In addition, a real cooperation between some of the mAChR and AR subtypes is observed. More specifically, preliminary results show that both AR subtypes (A1 and A2A) can add their independent effect on axonal loss to the effect of the M1 muscarinic receptor, which leads to greater elimination because of the additive effect of the pathways (Nadal et al., 2016).
In this study, we investigate whether the mAChR subtypes and the TrkB receptor also work together, and whether the respective pathway inhibitors have any additive or occlusive effects and, therefore, whether there is any real cooperation between them in synapse elimination at the NMJ. The main result shows that, like the mAChR and AR relations, the effect of M1 and TrkB receptors can be added to increase axonal loss rate at P9 but that the effect of M2 is largely independent of the TrkB receptors. Thus, cooperative and non-cooperative signaling contribute to synapse elimination, which highlights the importance of axonal competition and loss in the development of neural connectivity.
Materials and Methods
Animals
Transgenic B6.Cg-Tg (Thy1-YFP)16 Jrs/J mice were used (The Jackson Laboratory,Bar Harbor, ME, USA). The mice express spectral variants of GFP (yellow-YFP) at high levels in motor and sensory neurons, and axons are brightly fluorescent all the way to the terminals.
Experiments were performed on the Levator auris longus (LAL) muscle. Neonatal pups of either sex (9 days) were obtained and the date of birth was designated postnatal day 0 (P0). We minimized the variability in our measurements by carefully monitoring the timing of conception. Also, the weights of the individuals were within 5% of the mean for a given day after conception. The mice were cared for in accordance with the guidelines of the European Community’s Council Directive of 24 November 1986 (86/609/EEC) for the humane treatment of laboratory animals. All experiments on animals have been reviewed and approved by the Animal Research Committee of the Universitat Rovira i Virgili (Reference number: 0233).
Injection Procedure
The newborn mice were anesthetized with 2% tribromoethanol (0.15 ml/10 g body weight, i.p.). Mice pups received daily subcutaneous injections in the back of the neck beginning on postnatal day 5 of one or two substances (combinations of two selective mAChR antagonists or of one mAChR antagonist plus the TrkB signaling agent TrkB-Fc). Under aseptic conditions, solutions were administered in 50 μl of sterile physiological saline by subcutaneous injection over the LAL external surface as described elsewhere (Lanuza et al., 2001). The animals received four injections from postnatal day 5, and the LAL muscles were studied on day 9. Control injections were given in exactly the same way as experimental injections, using phosphate buffered saline (PBS) alone. No differences were found between the muscles injected or not with PBS, thus indicating that the injection procedure did not in itself induce changes in the overall morphology of the motor endplate and nerve terminals. The solutions were administered at a concentration in accordance with the previously reported biological action of the substance (Santafé et al., 2004, 2015; Garcia et al., 2010).
Tissue Preparation and Histochemistry
Neonatal pups were given a lethal dose of 2% tribromoethanol. Their heads were removed and fixed in 4% paraformaldehyde for 1.5 h. After washing in PBS, LAL muscles were removed and post-fixed for 45 min. After washing in PBS, Thy1-YFP LAL muscles were incubated in PBS containing a 1/800 dilution of 1 μg/ml tetramethylrhodamine conjugated α-bungarotoxin (α-BTX-TRITC; T1175, Molecular Probes, Eugene, OR, USA) for 1 h at room temperature. Whole muscles were mounted in Mowiol with p-phenylenediamide (Sigma).
Confocal Microscopy and Morphological Analysis
NMJs were analyzed using an inverted Nikon TE-2000 fluorescent microscope (Nikon, Tokyo, Japan) connected to a standard personal computer that was running image analysis software (ACT-1, Nikon). The number of axons per endplate was counted. Because of the difficulty of determining the exact number of axonal inputs for each nAChR cluster, when more than two axons converged at the same synaptic site we classified the NMJs into three groups only: junctions that were monoinnervated, doubly innervated, or innervated by three or more terminal axons. These data enabled us to calculate the “average number of axonal inputs” and the “percentage of polyneuronal innervation” for all fibers receiving two or more axons.
Statistical Analysis
All NMJs visible in their entirety were scored, with a minimum of 100 per muscle. At least six muscles were studied for each age and condition examined. Fisher’s test was applied to compare percentages. The criterion for statistical significance was P < 0.05. The categories were scored and the counting was performed by a person with no knowledge of the age or treatment of the animals. The data are presented as mean ± SD.
Drugs
Selective M1, M2 and M4 mAChR Antagonists
The stock solutions were pirenzepine (PIR) dihydrochloride (1071, Tocris Bioscience) 10 mM; methoctramine (MET; M105, Sigma—Aldrich, St. Louis, MO, USA) 1 mM; muscarinic toxin 3 (MT3; M-140, Alomone Labs) 50 μM. The working solutions used were PIR (10 μM), MET (1 μM) and MT3 (100 nM).
TrkB Receptor-Related Agent
The following stock solutions were used: recombinant human trkB/Fc Chimera (trkB-Fc; 688-TK; R&D Systems), 100 μg/ml. Working solutions were trkB-Fc 5 μg/ml.
Results
mAChR and TrkB Receptors in Axon Loss Control
Figure 1A shows representative confocal immunofluorescence images of the autofluorescent axons in singly- and polyinnervated LAL P9 NMJs from B6.Cg-Tg (Thy1-YFP) mice (hereafter YFP). When the mAChR subtype-selective inhibitors PIR (M1 blocker) and MET (M2 blocker), and the TrkB pathway blocker (a TrkB-Fc chimera) were applied once a day between P5-P8 on the LAL muscle surface, there was a notable delay in the transition to monoinnervation on the NMJ observed at P9 (Nadal et al., 2016). However, the M4 subtype blocker MT3 shows no effect on axonal loss at this time. Figure 1B shows the effect of these selective blockers in increasing order of their relative ability to delay monoinnervation and maintain a high percentage of synapses innervated by three or more axons (PBS-P9 = MT3 < TrkB-Fc < PIR < MET). MET and PIR delayed the three-to-one axon transition whereas TrkB-Fc delayed the two-to-one transition. Therefore, in normal conditions without inhibitors, the two receptor sets (mAChR and TrkB) will contribute to promoting axonal disconnection at the beginning of the second postnatal week (see also Nadal et al., 2016). However, the absolute potency of these receptors in modulating synapse loss cannot be directly compared because the blocking efficacy of the respective selective inhibitors is not the same.
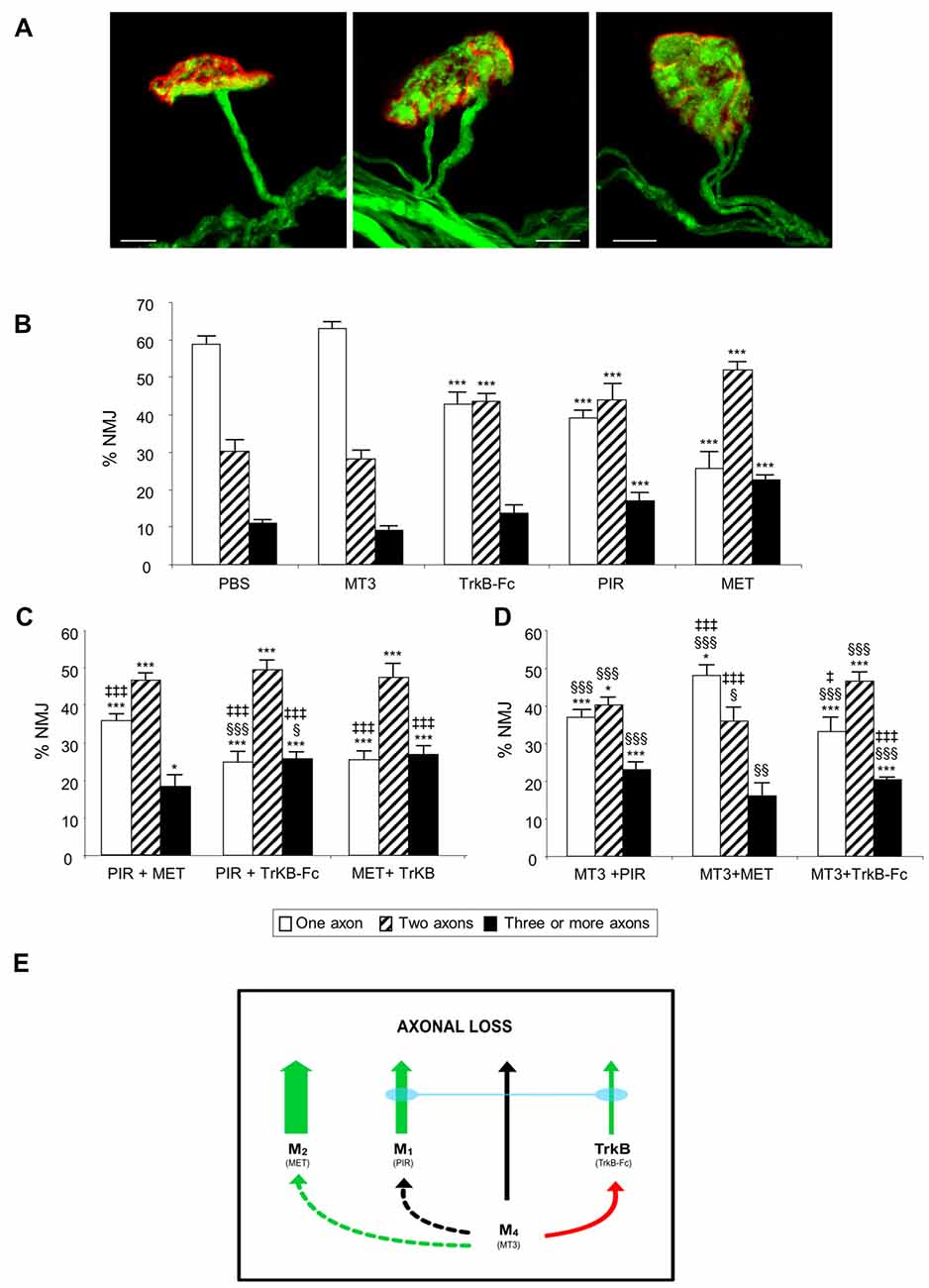
Figure 1. The picture in (A) shows representative confocal immunofluorescence images of singly-, dually- and polyinnervated neuromuscular junction area (NMJ) from P9 YFP mice. Scale bar: 10 μm. The histograms in (B) show the percentage of singly-, doubly- and triply- (or more) innervated NMJs in control (phosphate buffered saline, PBS treated) and levator auris longus (LAL) muscles treated with the inhibitors considered. We show newly reproduced data of previous results to facilitate comparison (Nadal et al., 2016). (C) shows the percentage of NMJs after the simultaneous inhibition of two receptors clearly involved in axonal elimination (those that affect axon loss rate when they are individually blocked (all except M4). The associations of the M4 blocker muscarinic toxin 3 (MT3) with the other inhibitors are represented in (D). An overall representation of the data illustrating the individual role and cooperation of the muscarinic acetylcholine receptor (mAChR) and tyrosine kinase B (TrkB) receptors in developmental axonal loss modulation is shown in the diagram in (E). The green arrows show how effective these receptors are at accelerating axonal elimination (the thicker they are, the greater their effect). The association of the M1 and TrkB blockers results in the addition of their respective effects (blue bond between these receptors). The M4 receptor, which does not by itself affect axonal elimination (black arrow), cooperates positively with M2 (dotted green arrow) and produces some occlusion of the TrkB pathway (red arrow) but does not cooperate with M1 (dotted black arrow). All NMJs visible in their entirety were scored, with a minimum of 100 synapses per muscle. At least six muscles were studied for each age and condition examined. Fisher’s test was applied to compare percentages. When the corresponding antagonist or combinations of two substances were compared with control PBS, significance symbols are: *P < 0.05, ***P < 0.005. §P < 0.05, §§P < 0.01, §§§P < 0.005 when the combination of two substances were compared with the first. ‡P < 0.05, ‡‡‡P < 0.005 when the combination of two substances were compared with the second.
Cooperation Between mAChR and TrkB Receptors
Our experiments were designed to investigate the effect on axonal loss of simultaneous incubation with two inhibitors (two antagonists of two different receptors) and reveal the possible occlusive or additive crosstalk effects between the corresponding pathways. Figures 1C,D show the effect of the association of the drugs applied between P5 and P8 and observed at P9. Figure 1C shows the percentage of NMJs—of singly-, doubly- and triply- (or more) innervated endplates—after the simultaneous inhibition of two receptors that, individually, clearly modulate axon loss (see Figure 1B; all except M4). The associations of the M4 blocker MT3 (which does not affect axonal elimination by itself) with the other substances has been represented separately in Figure 1D so that the data is more understandable. The M4 subtype is shown to have a complementary role.
The association of the mAChR blockers PIR and MET shows no additive effect or mutual occlusion in relation to axonal loss (Figure 1C). However, the association of the M1 and TrkB pathway inhibitors (PIR plus TrkB-Fc) results in a clear addition of their respective delaying effects on axonal loss. The percentage of the monoinnervated NMJ after simultaneous exposure to both inhibitors is significantly less (25% of single junctions) than after exposure to only PIR (39%) or only TrkB-Fc (43%). Interestingly, however, the individual effect of the TrkB-Fc does not add to the effect of MET and the result of this dual drug incubation is no different from the effect of MET by itself. The delaying effect of MET on axon loss is the most potent observed in the present experiments and is produced independently of the state of TrkB.
Figure 1B shows that the M4 blocker MT3 by itself has no effect on axonal loss at P9. Figure 1D shows that if MT3 is simultaneously applied with the other blockers it reveal some regulatory or complementary role of M4 on the other receptors. The presence of MT3 does not change the effect of PIR although some occlusion of the potent effect of MET is observed. MET still continues to significantly delay axon loss (the three-to-one transition), however. Interestingly, the presence of MT3 potentiates the delaying effect of TrkB-Fc on axonal loss, which indicates that the respective receptor pathways (M4 and TrkB) are cooperating.
These data are represented in Figure 1E. The green arrows of different thicknesses show how effective these receptors are at accelerating axonal elimination (the thicker they are, the greater their effect; the inhibitor used is in brackets under the name of the receptor). The M4 receptor by itself does not affect axonal elimination at P9 (black arrow). Interestingly, the association of the M1 and TrkB pathway blockers results in the addition of their respective delaying effects on axonal loss, which indicates that the corresponding receptors are cooperating (blue bond between these receptor pathways). However, the potent effect of the M2 cannot be modified with the simultaneous presence of the M1 or TrkB blockers. It seems that the M4 receptor, which does not by itself affect axonal elimination, cooperates positively with M2 (dotted green arrow). Also, M4 produces some occlusion of the TrkB pathway (red arrow) but does not cooperate with M1 (dotted black arrow).
Discussion
The present experiments show evidence of the cooperation between the presynaptic M1, M2 and M4 mAChR subtypes and the TrkB signaling to modulate the conditions of the developmental axonal competition and loss. In a previous study we found that these receptors (as well as presynaptic AR, A1 and A2A subtypes) separately contribute to accelerate synapse elimination around P9 in the mouse NMJ (Nadal et al., 2016). It was thought that the muscarinic autoreceptors of the transmitter acetylcholine (ACh) may allow direct competitive interaction between nerve endings through a differential activity-dependent ACh release. The more active axons may directly punish the less active ones or reward themselves (Santafé et al., 2009a). However, an axon that is eliminated at one NMJ may be successful at another (Tomàs et al., 2011), which suggests that other receptors and local postsynaptic- (and glial cell) derived factors are involved. The involvement of the TrkB signaling described may allow a postsynaptic-derived trophic substance such as Brain-derived neurotrophic factor (BDNF) or neurotrophin-4 (NT-4) to make a contribution (Yoshii and Constantine-Paton, 2010).
Interestingly, we observed that both the presynaptic-derived signal (ACh acting on axonal M1 and M2 mAChRs) and the TrkB-mediated signal (which may be originated by a postsynaptic-derived NT) have the same effect: namely, the acceleration of supernumerary nerve ending elimination. It seems that the outstanding regulatory resources in the NMJ synaptogenesis are committed to achieving monoinnervation. These presynaptic receptors converge in a common intracellular mechanism and a limited repertoire of effector kinases to phosphorylate protein targets and bring about structural and functional changes leading to axon loss. It is well known that in most cells M1 and TrkB operate by stimulating the phospholipase C gamma (PLC gamma) and therefore the protein kinase C (PKC) pathway along with the inositol triphosphate (IP3) pathway, whereas M2–M4 inhibit the adenyl cyclase (AC) and protein kinase A (PKA) pathway (Caulfield, 1993; Felder, 1995; Caulfield and Birdsall, 1998; Nathanson, 2000). In all cases, however, common final changes such as intracellular calcium oscillations can occur (Santafé et al., 2006; Amaral and Pozzo-Miller, 2012). Both PKA and PKC activity changes have been shown to affect pre- and postsynaptic maturation (Lanuza et al., 2001, 2002). Our present data can be related with the intracellular coupling of the receptors to these serine kinases. Though the blocking efficacy of the selective inhibitors of the muscarinic receptors is not assessed here, M2 increases the axonal loss rate most with a slight involvement of the M4 receptor but independently of the M1 and TrkB receptors. This suggests that downregulation of PKA activity through the couple M2–M4 is a key factor in synapse elimination. Concurrently, M1 and TrkB also contribute separately to axonal loss, but their combined action has a potent summed effect similar to the effect of the M2 receptor. This suggest that activation of the PLC gamma-PKC pathway through the couple M1-TrkB may be the other key factor in this process. Thus, a displacement of the PKA/PKC activity ratio to lower values (inhibition of PKA and/or stimulation of PKC) in some nerve endings may have a leading role in synapse elimination. In this context, blockade of PKC in the newborn LAL muscle produces an initial blockade of synapse elimination and a subsequent delay (Lanuza et al., 2002).
In fact, these changes in the kinase activity leading to synapse elimination must occur at least (but not only) in the weakest axons during the competitive interactions. The neurotransmitter release capacity is an important factor in the competing capacity of the various nerve terminals in a NMJ. During development, in the polyinnervated NMJ several nerve endings with different levels of maturation and ACh release capacity get together and compete. The coupling to neurotransmitter release of the considered receptors and kinases is not the same in each of these various endings themselves and in the mature synapses in the adult (Tomàs et al., 2014). So, how does the specific coupling to ACh release of receptors and kinases in the weak and strong axons in competition contribute to axonal loss? As far as serine kinases are concerned, in the adult motor nerve endings both PKA and PKC potentiate ACh release when coupled to neurotransmission (Santafé et al., 2009b). Similarly, the same potentiation is observed in most neuromuscular synapses during development as, for instance, in those formed by the strongest axons (those that evoke the large endplate potential, EPP) in the polyinnervated junctions (Santafé et al., 2004). However, in the weakest endings the inhibition of PKC increases the evoked EPP size indicating that, in normal conditions without any inhibition, this kinase tonically couples to ACh release reduction in these low releasing synapses. Therefore, an M1-TrkB-mediated increase in PKC activity in the weakest endings would debilitate further their ACh release capacity and competitive force and facilitate their elimination. In addition, an M2-mediated PKA downregulation in all nerve endings in competition may differentially affect their ACh release and contribute to elimination. Thus, at this point, there is a significant agreement between the known involvement of these molecules in neurotransmission and axon loss.
However, when considering the real postnatal coupling to ACh release of the mAChR and the TrkB receptor in the different nerve endings (the strongest and the weakest) on developing synapses (Santafé et al., 2004; Garcia et al., 2010), additional interpretative keys are needed. In the mature NMJ, M1 and M2 subtypes modulate evoked transmitter release by positive and negative feedbacks, respectively (Santafé et al., 2003, 2006). However, during NMJ synaptogenesis, the functional significance of the subtypes is different from in the adult. M2 receptors promote release in all nerve endings independently of their ACh release level or maturation state whereas an M1- and M4-mediated reduction in release is observed in the weakest endings on dual junctions (Santafé et al., 2001, 2002, 2003, 2004, 2007, 2009a). Similarly, the BDNF-TrkB pathway contributes to potentiate ACh release in different neuromuscular adult models but the potentiation is not observed in the weakest nerve endings during development and even some ACh release inhibition was observed in the strongest endings (Garcia et al., 2010). Therefore, interpreting the links and molecular relations between transmitter release and elimination of nerve terminals seems more complex than it seemed at first. The involvement of other signaling such as AR can contribute to this complexity (Todd and Robitaille, 2006; Nadal et al., 2016). However, some conclusions can be drawn on the basis of all the above data. First, it should be pointed out that, contrary to what happens in the adult, M1 (and M4) and PKC activity reduces ACh release in the weakest endings and promotes axonal loss. In fact, blocking mAChRs (M1- and/or M4-subtypes) or PKC or voltage-dependent calcium channels (VDCCs; P/Q-, N- or L-type or Ca2+ influx) can lead to similar percentage increases in the size of the synaptic potentials evoked by weak axons (Santafé et al., 2003, 2004, 2007, 2009a,b; Tomàs et al., 2011). Therefore, the M1-PKC pathway may debilitate the ACh release capacity and competitive force of these synaptic contacts and facilitate their elimination. The final target molecules involved may be the VDCC, specially the L-type which is exclusively coupled to ACh release in these weak endings (Santafé et al., 2001) and may contribute to carry high calcium near the molecular mechanism directly involved in axon loss. Second, like M1 signaling, BDNF-TrkB signaling accelerates axon loss. However, it is not so clear whether it is involved in the modulation of ACh release in the nerve endings that are in competition because it does not affect release in the weak axons. Because PKC effectively reduces release in these endings, the TrkB pathway may operate through the IP3 pathway to increase intracellular calcium and modulate the loss of axons. Third, M2 promotes axonal elimination the most. However, unlike the adult, this muscarinic subtype promotes ACh release in all the endings that are in competition, including the weakest endings and the solitary ending that finally wins the competition. Therefore, there is a shift in the M2 coupling during development but how this affects its relation with PKA and how this relates with axonal loss is not known.
Author Contributions
LN: data collection, quantitative analysis, literature search and data interpretation; EH, AS and MT: data collection, NG: statistics; JT, NG and MAL: conception and design, literature search, data interpretation, confocal microscopy and manuscript preparation. JT, NG and MAL contributed equally to this work. All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a grant from the Catalan Government (2014SGR344) and a grant from Ministerio de Economía y Competitividad (MINECO; SAF2015-67143-P).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AC, adenyl cyclase; ACh, acetylcholine; AR, adenosine receptors; BDNF, Brain-derived neurotrophic factor; EPP, evoke endplate potentials; IP3, inositol triphosphate; LAL, Levator auris longus muscle; mAChR, muscarinic acetylcholine receptor; M1, M1-type muscarinic acetylcholine receptor; M2, M2-type muscarinic acetylcholine receptor; M4, M4-type muscarinic acetylcholine receptor; MET, methoctramine; MT-3, muscarinic toxin 3; nAChR, nicotinic acetylcholine receptor; NMJ, neuromuscular junction; NT-4, neurotrophin-4; PBS, phosphate buffered saline; PIR, pirenzepine; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; TrkB, tropomyosin-related kinase B receptor; VDCC, voltage-dependent calcium channels.
References
Amaral, M. D., and Pozzo-Miller, L. (2012). Intracellular Ca2+ stores and Ca2+ influx are both required for BDNF to rapidly increase quantal vesicular transmitter release. Neural Plast. 2012, 2012–203536. doi: 10.1155/2012/203536
Benoit, P., and Changeux, J. P. (1975). Consequences of tenotomy on the evolution of multiinnervation in developing rat soleus muscle. Brain Res. 99, 354–358. doi: 10.1016/0006-8993(75)90036-0
Bourgeois, J. P., and Rakic, P. (1993). Changes of synaptic density in the primary visual cortex of the macaque monkey from fetal to adult stage. J. Neurosci. 13, 2801–2820.
Brown, M. C., Jansen, J. K., and Van Essen, D. (1976). Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. J. Physiol. 261, 387–422. doi: 10.1113/jphysiol.1976.sp011565
Buffelli, M., Busetto, G., Bidoia, C., Favero, M., and Cangiano, A. (2004). Activity-dependent synaptic competition at mammalian neuromuscular junctions. News Physiol. Sci. 19, 85–91. doi: 10.1152/nips.01464.2003
Caulfield, M. P. (1993). Muscarinic receptors–characterization, coupling and function. Pharmacol. Ther. 58, 319–379. doi: 10.1016/0163-7258(93)90027-b
Caulfield, M. P., and Birdsall, N. J. (1998). International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 50, 279–290.
Chang, Q., and Balice-Gordon, R. J. (1997). Nip and tuck at the neuromuscular junction: a role for proteases in developmental synapse elimination. Bioessays 19, 271–275. doi: 10.1002/bies.950190402
Felder, C. C. (1995). Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. 9, 619–625.
Garcia, N., Tomàs, M., Santafé, M. M., Besalduch, N., Lanuza, M. A., and Tomàs, J. (2010). The interaction between tropomyosin-related kinase B receptors and presynaptic muscarinic receptors modulates transmitter release in adult rodent motor nerve terminals. J. Neurosci. 30, 16514–16522. doi: 10.1523/JNEUROSCI.2676-10.2010
Herrera, A. A., and Zeng, Y. (2003). Activity-dependent switch from synapse formation to synapse elimination during development of neuromuscular junctions. J. Neurocytol. 32, 817–833. doi: 10.1023/b:neur.0000020626.29900.fb
Jansen, J. K., and Fladby, T. (1990). The perinatal reorganization of the innervation of skeletal muscle in mammals. Prog. Neurobiol. 34, 39–90. doi: 10.1016/0301-0082(90)90025-c
Lanuza, M. A., Garcia, N., Santafé, M., González, C. M., Alonso, I., Nelson, P. G., et al. (2002). Pre- and postsynaptic maturation of the neuromuscular junction during neonatal synapse elimination depends on protein kinase C. J. Neurosci. Res. 67, 607–617. doi: 10.1002/jnr.10122
Lanuza, M. A., Garcia, N., Santafé, M., Nelson, P. G., Fenoll-Brunet, M. R., and Tomas, J. (2001). Pertussis toxin-sensitive G-protein and protein kinase C activity are involved in normal synapse elimination in the neonatal rat muscle. J. Neurosci. Res. 63, 330–340. doi: 10.1002/1097-4547(20010215)63:4<330::aid-jnr1027>3.0.co;2-w
Lee, Y., Li, Y., Mikesh, M., Smith, I., Nave, K.-A., Schwab, M. H., et al. (2016). Neuregulin1 displayed on motor axons regulates terminal Schwann cell-mediated synapse elimination at developing neuromuscular junctions. Proc. Natl. Acad. Sci. U S A 113, E479–E487. doi: 10.1073/pnas.1519156113
Liu, Y., Fields, R. D., Festoff, B. W., and Nelson, P. G. (1994). Proteolytic action of thrombin is required for electrical activity-dependent synapse reduction. Proc. Natl. Acad. Sci. U S A 91, 10300–10304. doi: 10.1073/pnas.91.22.10300
Nadal, L., Garcia, N., Hurtado, E., Simó, A., Tomàs, M., Lanuza, M. A., et al. (2016). Presynaptic muscarinic acetylcholine autoreceptors (M1, M2 and M4 subtypes), adenosine receptors (A1 and A2A) and tropomyosin-related kinase B receptor (TrkB) modulate the developmental synapse elimination process at the neuromuscular junction. Mol. Brain 9:67. doi: 10.1186/s13041-016-0248-9
Nathanson, N. M. (2000). A multiplicity of muscarinic mechanisms: enough signaling pathways to take your breath away. Proc. Natl. Acad. Sci. U S A 97, 6245–6247. doi: 10.1073/pnas.97.12.6245
Nguyen, Q. T., and Lichtman, J. W. (1996). Mechanism of synapse disassembly at the developing neuromuscular junction. Curr. Opin. Neurobiol. 6, 104–112. doi: 10.1016/s0959-4388(96)80015-8
O’Brien, R. A., Ostberg, A. J., and Vrbová, G. (1978). Observations on the elimination of polyneuronal innervation in developing mammalian skeletal muscle. J. Physiol. 282, 571–582. doi: 10.1113/jphysiol.1978.sp012482
Redfern, P. A. (1970). Neuromuscular transmission in new-born rats. J. Physiol. 209, 701–709. doi: 10.1113/jphysiol.1970.sp009187
Ribchester, R. R., and Barry, J. A. (1994). Spatial versus consumptive competition at polyneuronally innervated neuromuscular junctions. Exp. Physiol. 79, 465–494. doi: 10.1113/expphysiol.1994.sp003781
Sanes, J. R., and Lichtman, J. W. (1999). Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 22, 389–442. doi: 10.1146/annurev.neuro.22.1.389
Santafé, M. M., Garcia, N., Lanuza, M. A., Tomàs, M., Besalduch, N., and Tomàs, J. (2009a). Presynaptic muscarinic receptors, calcium channels, and protein kinase C modulate the functional disconnection of weak inputs at polyinnervated neonatal neuromuscular synapses. J. Neurosci. Res. 87, 1195–1206. doi: 10.1002/jnr.21934
Santafé, M. M., Garcia, N., Lanuza, M. A., Tomàs, M., and Tomàs, J. (2009b). Interaction between protein kinase C and protein kinase A can modulate transmitter release at the rat neuromuscular synapse. J. Neurosci. Res. 87, 683–690. doi: 10.1002/jnr.21885
Santafé, M. M., Garcia, N., Lanuza, M. A., Uchitel, O. D., Salon, I., and Tomàs, J. (2002). Decreased calcium influx into the neonatal rat motor nerve terminals can recruit additional neuromuscular junctions during the synapse elimination period. Neuroscience 110, 147–154. doi: 10.1016/S0306-4522(01)00543-7
Santafé, M. M., Garcia, N., Lanuza, M. A., Uchitel, O. D., and Tomás, J. (2001). Calcium channels coupled to neurotransmitter release at dually innervated neuromuscular junctions in the newborn rat. Neuroscience 102, 697–708. doi: 10.1016/s0306-4522(00)00507-8
Santafé, M. M., Lanuza, M. A., Garcia, N., and Tomàs, J. (2006). Muscarinic autoreceptors modulate transmitter release through protein kinase C and protein kinase A in the rat motor nerve terminal. Eur. J. Neurosci. 23, 2048–2056. doi: 10.1111/j.1460-9568.2006.04753.x
Santafé, M. M., Lanuza, M. A., Garcia, N., Tomàs, M., and Tomàs, J. (2007). Coupling of presynaptic muscarinic autoreceptors to serine kinases in low and high release conditions on the rat motor nerve terminal. Neuroscience 148, 432–440. doi: 10.1016/j.neuroscience.2007.06.017
Santafé, M. M., Priego, M., Obis, T., Garcia, N., Tomàs, M., Lanuza, M. A., et al. (2015). Adenosine receptors and muscarinic receptors cooperate in acetylcholine release modulation in the neuromuscular synapse. Eur. J. Neurosci. 42, 1775–1787. doi: 10.1111/ejn.12922
Santafé, M. M., Salon, I., Garcia, N., Lanuza, M. A., Uchitel, O. D., and Tomàs, J. (2003). Modulation of ACh release by presynaptic muscarinic autoreceptors in the neuromuscular junction of the newborn and adult rat. Eur. J. Neurosci. 17, 119–127. doi: 10.1046/j.1460-9568.2003.02428.x
Santafé, M. M., Salon, I., Garcia, N., Lanuza, M. A., Uchitel, O. D., and Tomàs, J. (2004). Muscarinic autoreceptors related with calcium channels in the strong and weak inputs at polyinnervated developing rat neuromuscular junctions. Neuroscience 123, 61–73. doi: 10.1016/j.neuroscience.2003.09.012
Thompson, W. J. (1985). Activity and synapse elimination at the neuromuscular junction. Cell. Mol. Neurobiol. 5, 167–182. doi: 10.1007/bf00711091
Todd, K. J., and Robitaille, R. (2006). Purinergic modulation of synaptic signalling at the neuromuscular junction. Pflugers Arch. 452, 608–614. doi: 10.1007/s00424-006-0068-3
Tomàs, J., Santafé, M. M., Garcia, N., Lanuza, M. A., Tomàs, M., Besalduch, N., et al. (2014). Presynaptic membrane receptors in acetylcholine release modulation in the neuromuscular synapse. J. Neurosci. Res. 92, 543–554. doi: 10.1002/jnr.23346
Tomàs, J., Santafé, M. M., Lanuza, M. A., García, N., Besalduch, N., and Tomàs, M. (2011). Silent synapses in neuromuscular junction development. J. Neurosci. Res. 89, 3–12. doi: 10.1002/jnr.22494
Wyatt, R. M., and Balice-Gordon, R. J. (2003). Activity-dependent elimination of neuromuscular synapses. J. Neurocytol. 32, 777–794. doi: 10.1023/b:neur.0000020623.62043.33
Yang, J., Yang, H., Liu, Y., Li, X., Qin, L., Lou, H., et al. (2016). Astrocytes contribute to synapse elimination via type 2 inositol 1,4,5-trisphosphate receptor-dependent release of ATP. ELife 5:e15043. doi: 10.7554/eLife.15043
Keywords: motor nerve terminal, cholinergic synapses, neuromuscular junction, presynaptic muscarinic acetylcholine receptors, TrkB receptor, synapse elimination
Citation: Nadal L, Garcia N, Hurtado E, Simó A, Tomàs M, Lanuza MA, Cilleros V and Tomàs J (2017) Presynaptic Muscarinic Acetylcholine Receptors and TrkB Receptor Cooperate in the Elimination of Redundant Motor Nerve Terminals during Development. Front. Aging Neurosci. 9:24. doi: 10.3389/fnagi.2017.00024
Received: 29 December 2016; Accepted: 27 January 2017;
Published: 08 February 2017.
Edited by:
Antonio Camins, University of Barcelona, SpainReviewed by:
Daniel Ortuño-Sahagún, Centro Universitario de Ciencias de la Salud, MexicoJordi Olloquequi, Universidad Autónoma de Chile, Chile
Copyright © 2017 Nadal, Garcia, Hurtado, Simó, Tomàs, Lanuza, Cilleros and Tomàs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neus Garcia, bWFyaWFkZWxlc25ldXMuZ2FyY2lhQHVydi5jYXQ=
Josep Tomàs, am9zZXBtYXJpYS50b21hc0B1cnYuY2F0
† These authors have contributed equally to this work.