 Gyun Jee Song
Gyun Jee Song Kyoungho Suk*
Kyoungho Suk*- BK21 Plus KNU Biomedical Convergence Program, Department of Pharmacology, Brain Science & Engineering Institute, School of Medicine, Kyungpook National University, Daegu, South Korea
Microglia are the resident innate immune cells of the central nervous system that mediate brain homeostasis maintenance. Microglia-mediated neuroinflammation is a hallmark shared by various neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis. Numerous studies have shown microglial activation phenotypes to be heterogeneous; however, these microglial phenotypes can largely be categorized as being either M1 or M2 type. Although the specific classification of M1 and M2 functionally polarized microglia remains a topic for debate, the use of functional modulators of microglial phenotypes as potential therapeutic approaches for the treatment of neurodegenerative diseases has garnered considerable attention. This review discusses M1 and M2 microglial phenotypes and their relevance in neurodegenerative disease models, as described in recent literature. The modulation of microglial polarization toward the M2 phenotype may lead to development of future therapeutic and preventive strategies for neuroinflammatory and neurodegenerative diseases. Thus, we focus on recent studies of microglial polarization modulators, with a particular emphasis on the small-molecule compounds and their intracellular target proteins.
Introduction: Diversity of Microglial Phenotypes and Disease Relevance
Microglia, the resident immune cells of the central nervous system (CNS), are highly specialized macrophages that play a fundamental role in neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS) (Mandrekar-Colucci and Landreth, 2010; Colonna and Butovsky, 2017). Microglia have been traditionally classified as either of the following: (1) resting with branched morphology and present in healthy brains or (2) activated with amoeboid morphology and present in diseased brains. Recent microglia classifications are more complex. Activated microglia are now recognized as being heterogeneous and plastic, and exist in various phenotypes in the CNS. Microglia can be divided into at least two types (neurotoxic or neuroprotective) based on their function (Kettenmann et al., 2013). Microglia can promote neurotoxicity via the release of several pro-inflammatory mediators, such as nitric oxide, interleukin (IL)-1β, and tumor necrosis factor-alpha (TNF-α) (Hanisch, 2002; Block et al., 2007). Conversely, they can be neuroprotective and neurosupportive, via several mechanisms under certain conditions. For example, neuroprotective roles of microglia include glutamate uptake (Byrnes et al., 2009), removal of dead cell debris and abnormally accumulated proteins (Diaz-Aparicio et al., 2016), and production of neurotrophic factors such as insulin-like growth factor-1 (IGF-1) (Thored et al., 2009), glial cell-derived neurotrophic factor (GDNF) (Lu et al., 2005), and brain-derived neurotrophic factor (BDNF) (Batchelor et al., 1999).
The dual nature of microglial functional polarization is consistent with the general classification of macrophages as being either the M1 (classic pro-inflammatory) or M2 (anti-inflammatory) phenotype (Michelucci et al., 2009). Specific environmental cues induce macrophages to adopt a given functionality. For example, stimulation with either lipopolysaccharide (LPS) or interferon (IFN)-γ induces activation of the classical M1 phenotype, whereas stimulation with either IL-4 or IL-13 induces the M2 activation (Cherry et al., 2014; Loane and Kumar, 2016). Microglia are critical to immune response in the CNS, and unsurprisingly, microglial functional polarization has been implicated in almost all CNS disorders, and in the progression of neurodegenerative diseases (Tang and Le, 2016). Microglia also play key functional roles in recovery from brain injury and in the maintenance of homeostasis in the brain.
Microglia in Neurodegenerative Disease
Neurodegenerative diseases such as AD, PD, amyotrophic lateral sclerosis (ALS), and MS, are characterized by neuronal degeneration in specific regions of the CNS, sharing common pathophysiological mechanisms including inflammation and abnormal protein deposition (Heppner et al., 2015; Chen et al., 2016). AD is the most prevalent neurodegenerative disorder that is characterized by progressive focal cortical atrophy, cognitive dysfunction, β-amyloid deposition, and neuronal degeneration that initiates with neurons of the hippocampus and cortex. PD is the second-most prevalent disorder, and is characterized by a progressive loss of the dopaminergic neurons in the substantia nigra and accumulation of α-synuclein and other abnormal proteins in Lewy bodies. ALS is a chronic degenerative disease characterized by the progressive degeneration of motor neurons. The accumulation of abnormal proteins such as superoxide dismutase (SOD)1 and transactive response (TAR) DNA binding protein 43 kDa (TDP-43) in motor neurons is the major pathological feature. MS is an inflammatory and degenerative disease of the CNS characterized by autoimmunity, monocyte infiltration, microglial activation, demyelination, and neuronal death. Inflammatory mediators, such as IL-1β, IL-6 TNF-α, chemokines, matrix metalloproteinase 2 (MMP2), nitric oxide, and nuclear factor kappa-B (NF-κB), contribute to neurodegeneration and myelin damage in these neurodegenerative diseases. Activated microglia and macrophages produce these inflammatory mediators, orchestrating neuroinflammation, and neurodegeneration. Neuroinflammation is initially a protective response in the brain, however, uncontrolled inflammatory responses are detrimental as they induce the release of high levels of neurotoxic factors such as ROS and TNF-α, and block neuronal regeneration (Chen et al., 2016).
Therefore, demonstrating a microglial functional polarization would potentially uncover new prospects for understanding the way in which neurodegeneration can be promoted by local CNS inflammation. In AD, inflammation is considered to be a pathology that occurs in response to β-amyloid accumulation. Classical (M1) activation of microglia is considered the initial defense mechanism in AD, and is characterized by the release of proinflammatory cytokines. Conversely, alternative (M2) activation is associated with angiogenesis, neurogenesis, anti-inflammatory effects, and the degradation of β-amyloid deposits. Thus, modulation of microglial activation toward the M2 phenotype has been proposed as a novel therapeutic strategy for patients with AD. Indeed, Latta et al. (2015) showed that an enhanced M2 phenotype, induced either by IL-4 overexpression, or treatment with exogenous IL-4, decreased β-amyloid deposition both in vitro and in an animal model. Similarly, M1 polarization of microglia induced by expression of IFN-γ has been shown to increase the amyloid burden in an amyloid precursor protein/presenilin1 (APP/PS1) AD mouse model (Weekman et al., 2014).
Microglia-mediated neuroinflammation is also a hallmark of PD. In the brains of PD patients, microglia exert both neurotoxic and neuroprotective effects depending on the surrounding microenvironment. Persistent microglial activation by damaged neurons and α-synuclein deposition is generally detrimental. Reactive microglia release a range of reactive oxygen species, such as nitric oxide and superoxide-anion and pro-inflammatory cytokines, which exacerbate motor deficits in PD. Therefore, many anti-inflammatory agents have been proposed as promising PD therapeutic agents. Indeed, non-steroidal anti-inflammatory drugs and minocycline have been used in clinical studies for PD patients (Gao and Hong, 2008).
Accumulation of the misfolded ALS-linked mutant SOD1 or TDP-43 is tightly associated with the neurotoxic M1 inflammatory microglial activation (Boillee et al., 2006; Swarup et al., 2011; Huang et al., 2012). Primary microglia isolated from SODG93A transgenic mice are more neurotoxic compared to wild-type microglia, due to an increased production of superoxide and nitric oxide as well as the decreased expression of IGF-I (Xiao et al., 2007). Furthermore, IL-4-induced M2 microglia reduced LPS-induced microglia-mediated motor neuron injury (Zhao et al., 2006) and disease stage-dependent microglial switch from neuroprotective to neurotoxic phenotype has been observed in an ALS mouse model.
Microglia isolated from ALS mice at disease onset expressed higher levels of M2 markers and lower levels of the M1 marker, NADPH oxidase (NOX)2, compared with those isolated at the end-stage of ALS, indicating a diminished function of neuroprotective microglia in the late stage of the disease (Liao et al., 2012). Thus, the administration of minocycline delays the pathogenesis of SODG93A mice by selectively attenuating the induction of M1 microglia markers during the progressive phase, without affecting the transient enhancement of M2 microglia markers at the early stage (Kobayashi et al., 2013).
Microglial activation has also been studied extensively in MS patients and in the experimental autoimmune encephalomyelitis (EAE) mouse model. In this pathological condition, microglia release neurotoxic and neurotrophic molecules, pro- and anti-inflammatory cytokines, playing both beneficial and detrimental roles during the demyelination and recovery stages (Correale, 2014). Miron et al. (2013) examined whether M2 phenotypes contribute to regenerative response in the CNS. In their study, the M1 to M2 switch was observed at the initiation of remyelination, 10 days post-injection of lyso-phosphatidylcholine (lecithin). Oligodendrocyte differentiation for regeneration was enhanced by M2 microglia-conditioned medium. M2 polarization of microglia has been proposed to preserve myelin homeostasis after white matter injury in traumatic brain injury (TBI) or cuprizone-induced demyelination models (Chen et al., 2014c; Wang et al., 2015). Furthermore, the protective mechanisms exerted by alternatively activated (M2) microglia have been discussed in recent review articles (Cherry et al., 2014; Du et al., 2016; Tang and Le, 2016). Thus, enhancing the neuroprotective effects of these M2 microglia may be a promising therapeutic approach.
Prion disease is another progressive neurodegenerative disorder, and like many other neurodegenerative diseases, it is characterized by misfolded protein aggregates and neuroinflammation (Burchell and Panegyres, 2016; Stopschinski and Diamond, 2017). In prion disease, misfolded prion protein aggregates propagate by the conversion of normal cellular prion protein (PrPC) to abnormal isoforms, designated pathogenic conformers of the prion protein (PrPSc), which causes rapid neurodegeneration accompanied by spongiform change and neuronal loss in the brain. Furthermore, the crosstalk between misfolded proteins in animal models of Alzheimer’s and prion diseases has been proposed in recent studies (Morales et al., 2010; Fernandez et al., 2017), suggesting that one protein misfolding process may be an important risk factor for the development of other protein aggregation-induced diseases. Importantly, microglial proliferation, activation, and phenotype conversion have been associated with overall prion disease progression (Aguzzi et al., 2013; Grizenkova et al., 2014; Lins et al., 2016).
Specific Markers for the M1 and M2 Phenotypes
M1-type microglia release diverse proinflammatory mediators and free radicals that inhibit brain repair and regeneration. Conversely, microglia of the M2 phenotype improve brain repair and regeneration by enhancing phagocytosis, releasing trophic factors, and reducing brain inflammation. Following stimulation with LPS or IFN-γ, M1 microglia express high levels of inducible nitric oxide synthase (iNOS) and pro-inflammatory cytokines/chemokines such as TNF-α, IL-1β, and CC chemokine ligand (CCL)2. IL-4- or IL-13-stimulated microglia express Arg1, Ym-1, CD200R, IL-10, transforming growth factor (TGF)-β and Fizzl-1, which serve as specific markers for M2 microglia. M1 and M2 microglia represent a spectrum of various activation phenotype rather than single phenotype of each status. Three subtypes of M2 microglia have been proposed, such as M2a (wound-healing and anti-inflammatory phenotype expressing CD206, Fizz-1, Arg1, Ym1), M2b (inflammation modulatory phenotype expressing IL-10, COX2) and M2c (immunosuppressive phenotype expressing CD163) (Franco and Fernandez-Suarez, 2015; Du et al., 2016). More recently, Kumar et al. (2016) proposed a mixed transitional phenotype of microglia called Mtran, co-expressing M1 makers (iNOS and IL-12) and M2 markers (TGF-β and Arg1). In their study, up to 42% of Arg1 (M2 marker)-positive cells co-express the M1 marker iNOS at 7 days post-TBI indicating a significant mixed population of microglia during recovery after injury (Kumar et al., 2016), which was confirmed in the middle cerebral artery occlusion (MCAo) model (Moretti et al., 2016). Furthermore, it is difficult to discriminate between M1 or each M2 subtype in vitro, and even more so in vivo. Recent technical advancements, including the fluorescent analysis of activation markers, cell sorting, and single-cell RNA-seq analysis, have helped to define microglia-specific genes compared to those specific to macrophages and other glia (Butovsky et al., 2014; Crotti and Ransohoff, 2016). However, M1/M2 phenotypes of microglia do not precisely match the microglial classification based on a transcriptomic analysis such as RNA-seq (Yamasaki et al., 2014). Although the binary concept of microglial M1/M2 classification has been recently debated (Ransohoff, 2016), the functional classification of microglia as being either neurotoxic (M1) or neuroprotective (M2) is a useful for illustrating the pathobiology of inflammatory and degenerative CNS disorders. Therefore, we used the M1 and M2 phenotypic classification for activated microglia in this review.
Pharmacological Approaches for Modulating Microglial Phenotypes
Currently available medication is incapable of repairing degenerated neurons, inducing regeneration, and/or preventing further neuronal death in AD, PD, ALS, and/or MS patients. Current drugs are used to reduce the severity of disease-related symptoms by limiting the extent of neuroinflammation in patients (Glass et al., 2010). Nevertheless, although these drugs reduce symptoms, and thus increase the quality of life for these patients, they are not able to repair or regenerate damaged neurons.
Given that the balance of microglial M1/M2 phenotypes has been implicated in the pathogenesis of neurodegenerative diseases, several small-molecule compounds have recently been studied to ascertain their ability to regulate microglial functional polarization, and thereby exert neuroprotection against neurodegenerative diseases in animal models. Elucidating the underlying mechanisms of action and identifying the target proteins of these small-molecule compounds may be essential to design better chemical modulators of microglial polarization and thus effective neuroprotective drugs.
The microglial microenvironment (for example, infection, ischemic injury, β-amyloid depositions, and/or pro-inflammatory mediators such as TNF-α, IL-1β, and nitric oxide), has been shown to play a critical role in determining the microglial polarization state. Several soluble factors released from neurons and astrocytes are suggested to contribute to the determination of microglial phenotypes, as demonstrated by the M2 to M1 microglial phenotypic switch induced by exposure to cell culture medium conditioned by damaged neurons or activated astrocytes. Furthermore, many intracellular molecules are involved in controlling microglial/macrophage polarization. For example, nuclear receptors [peroxisome proliferator-activated receptor (PPAR)γ, PPARδ, retinoid X receptor (RXR)], redox signaling molecules [NOX2, hypoxia-inducible factor (HIF)-1α], NF-κB signaling molecules, and metabolic shift-mediated proteins are all known to control the phenotypic switching of microglia and macrophages (Sica and Mantovani, 2012). Thus, potential exploitation of these molecules may be a promising method for developing novel therapeutic drugs for neurodegenerative diseases.
Gene therapies, including BDNF- or IL4-overexpressing viruses, recombinant proteins such as IL-4, IL-10, IL-13, and/or TGF-β, Etanercept (a TNF-α-antagonist fusion protein), and cell therapies (M2 microglia and macrophages) have been applied as therapeutic tools. However, due to the blood–brain barrier, these therapeutic strategies have limited efficacy, leading to the screening of small synthetic chemical and/or natural compounds as treatments for various brain diseases. In this review, we focus on the effect and the target proteins of these small-molecule compounds that modulate M1/M2 microglial polarization (Table 1).
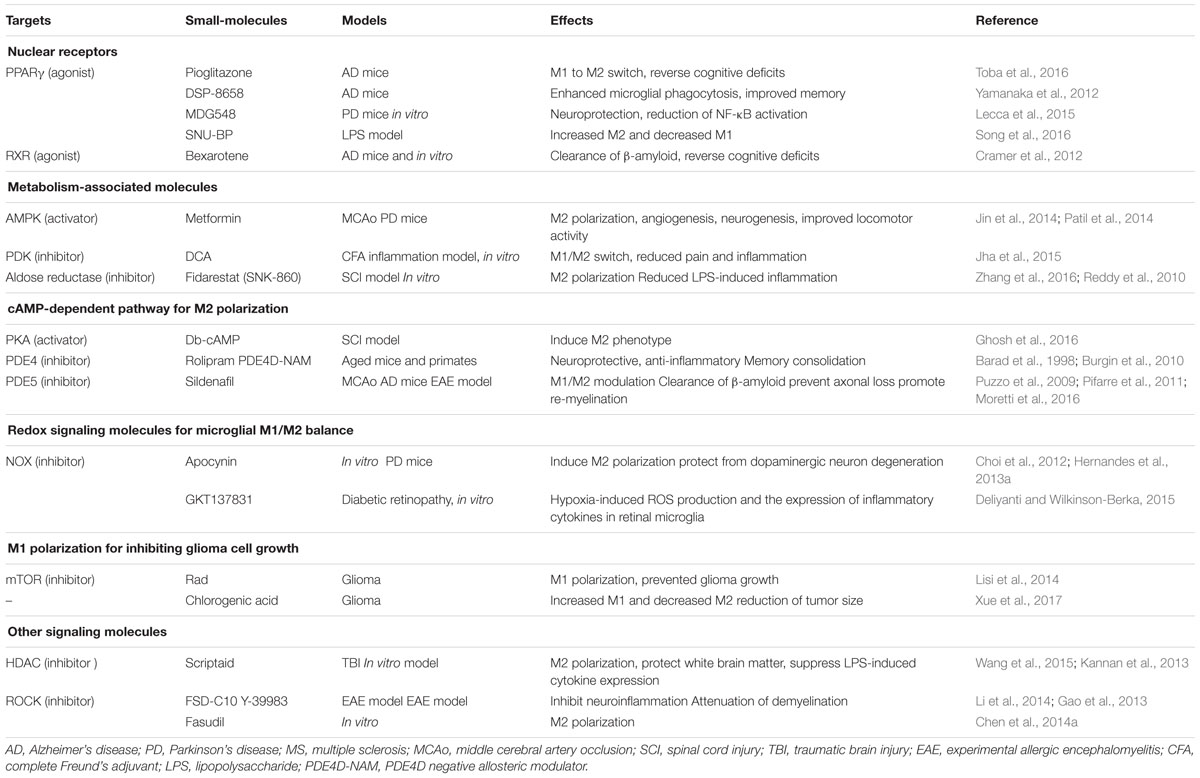
TABLE 1. Pharmacological tools for modulating microglial polarization.
Nuclear Receptors
The nuclear hormone receptor PPAR is a key regulator of the M2 phenotype in macrophages and microglia (Bouhlel et al., 2007; Chawla, 2010). PPAR activation increases phagocytic uptake of amyloid-β plaques, and is neuroprotective in an AD mouse model (Mandrekar-Colucci et al., 2012; Yamanaka et al., 2012). Accordingly, treatment with the PPARγ agonist, pioglitazone, results in the phenotypic switch of proinflammatory M1 to anti-inflammatory M2 microglia. Furthermore, pioglitazone treatment dramatically reduces the levels of soluble and insoluble β-amyloid, and reverses the cognitive deficits in 12-month-old APP/PS1 mice (Toba et al., 2016). A novel selective-PPAR modulator, DSP-8658, was shown to enhance the microglial phagocytosis and improve the spatial memory performance in an AD mouse model (Yamanaka et al., 2012). Similarly, a PPARγ agonist called rosiglitazone has also been shown to induce IL-4 expression in the rat brain as well as age-dependent M1 microglial activation in an IL-4 dependent manner (Loane et al., 2009). A novel compound, MDG548, was found to induce neuroprotection in MPTP-treated mice and decrease LPS-induced NF-κB activation in a dose-dependent manner (Lecca et al., 2015). Finally, an N-carbamolylated urethane compound (SNU-BP) has been identified as a novel PPARγ agonist, and inhibits LPS-induced pro-inflammatory cytokine and nitric oxide production as well as potentiates IL-4-induced M2 marker expression in microglia and astrocytes. SNU-BP was also shown to exhibit an anti-neuroinflammatory effect in an LPS-injected mouse model, probably via the M1/M2 switch (Song et al., 2016).
Alzheimer’s disease (AD) is strongly associated with the impaired clearance of β-amyloid from the brain. The RXR agonist bexarotene (Targretin) has been shown to enhance β-amyloid clearance by activating PPARγ/RXR and liver X receptors (LXRs)/RXR, inducing apoE expression, and promoting microglial phagocytosis (Cramer et al., 2012). It is a highly selective, synthetic retinoid analog with specific affinity for the RXR, which is able to cross the blood–brain barrier and has a favorable FDA-approved safety profile (Skalak et al., 1987; Cramer et al., 2012).
Malibatol A (MA) is a natural resveratrol oligomer extracted from the leaves of the Chinese plant Hopea hainanensis. MA has an anti-inflammatory effect on MCAo mice and LPS-stimulated microglia. MA treatment was found to decrease the expression of M1 (CD16, CD32, and CD86), while increasing that of M2 markers (CD206 and YM-1), in a PPARγ-dependent manner (Pan et al., 2015).
Metabolism-Associated Proteins
Evidence suggests a role for metabolic reprogramming in the modulation of M1/M2 microglial phenotypes (Orihuela et al., 2016). Various studies support the belief that mitochondrial metabolic shifts are associated with microglial polarization. Under normoxic conditions, ATP (energy) production is achieved via oxidative phosphorylation; in contrast, anaerobic glycolysis converts pyruvate into lactate under hypoxic conditions. In the context of the peripheral immune cells, a shift in the cellular metabolism from oxidative phosphorylation to aerobic glycolysis favors the polarization of microglia toward the M1 phenotype. This metabolic switch is promoted by PI3K/AKT and inhibited by AMP-activated protein kinase (AMPK) and IL-10.
AMP-activated protein kinase activation is associated with neuroprotection after stroke (Jin et al., 2014; Venna et al., 2014) via enhanced neurogenesis and lowered blood glucose levels. The AMPK activator metformin promotes both functional recovery and tissue repair following stroke. Mice that administered metformin daily after MCAo exhibited more M2-polarized microglia and macrophages, as well as increased angiogenesis and neurogenesis (Jin et al., 2014). The same study also demonstrated metformin-induced M2 polarization of BV-2 microglial cells dependent upon AMPK activation. Patil et al. (2014) similarly demonstrated the neuroprotective effect of metformin in a PD mouse model, such that impaired locomotor activities and the loss of TH-positive cells were both significantly improved in metformin (500 mg/kg for 21 days)-treated PD mice, compared to controls.
Pyruvate dehydrogenase kinases (PDKs) are mitochondrial metabolic regulators that modulate pyruvate dehydrogenase (PDH) activity to convert pyruvate either aerobically to acetyl-CoA, or anaerobically to lactate (Jha et al., 2015). In vitro studies support the role of PDK2/4 as promoters of the classical proinflammatory (M1) activation of macrophages. Moreover, the pharmacological PDK inhibitor dichloroacetate (DCA) diminishes complete Freund’s adjuvant (CFA)-induced inflammation and pain via an induced M1-M2 switch (Jha et al., 2015). This suggests that a pathological metabolic shift, and subsequent lactic acid production, each contribute to neuroinflammatory disease progression.
Fidarestat (SNK-860) is an aldose reductase (AR) inhibitor used for the treatment of diabetic neuropathy (Kuzumoto et al., 2006). A recent study showed the incubation of macrophages with LPS to cause a significant increase (6-fold) in the uptake of glucose in a time dependent manner. In contrast, the inhibition of AR via fidarestat treatment decreased LPS-induced glucose uptake by 30% (Reddy et al., 2010). AR expression is enhanced in microglia/macrophages after spinal cord injury (SCI). The study further demonstrated that AR inhibition and/or deficiency in microglia/macrophages favors a phenotype switch toward the M2, rather than the M1 phenotype, and that AR inhibition induces the phosphorylation of cAMP response element-binding protein (CREB) leading to the enhanced expression of the M2 marker, Arg-1 (Zhang et al., 2016).
cAMP-Dependent Pathways and their Regulators
Cyclic adenosine monophosphate (cAMP) is a well-known regulator of microglial function and activation (Ghosh et al., 2012, 2015, 2016). The elevation of cAMP in microglia or macrophages via the application of an adenylyl cyclase activator, synthetic cyclic AMP analoges or phosphodiesterase (PDE) inhibitors, inhibits the production of pro-inflammatory molecules (Gerlo et al., 2011). Interestingly, proinflammatory cytokines such as TNF-α and IL-1β both rapidly reduced cAMP, and increased PDE4 expression in microglia (Ghosh et al., 2012). The protective effect of cAMP on neuronal regeneration has been reported in rat SCI (Neumann et al., 2002) and cerebral ischemia-reperfusion injury models (Niu et al., 2010). Dibutyryl (db)-cAMP is a membrane-permeable derivative of cAMP that exhibits a prolonged response time. Ghosh et al. (2016) investigated the effect of cAMP on the modulation of microglial phenotypes and found that co-treatment of microglial cells with cAMP and IL-4 induced an M2 phenotype (Arg-1+/iNOS-) with concomitant expression of various M2-specific markers including TG2 and FIZZ1. Administration of db-cAMP and IL-4 also promoted M2 phenotypes in the lesions of SCI model mice (Ghosh et al., 2016). Together, these data strongly support that cAMP is a critical determinant of M1-M2 polarization.
PDE4 is an enzyme that negatively regulates cAMP signaling by hydrolyzing cAMP in immune and brain cells. PDE4 inhibitors (including rolipram) are reported to have precognitive, neuroprotective, and anti-inflammatory effects (Barad et al., 1998; Dinter, 2000; Block et al., 2001). PDE4-negative allosteric modulators (NAMs) have been recently developed for specific inhibition of each PDE4 subtype (i.e., PDE4A, B, C, and D). PDE4D-NAM has been shown to improve cognitive performance in healthy rodents (Burgin et al., 2010) and primates (Sutcliffe et al., 2014). PDE4D-NAMs (including D158681, D159153, D159404, and D159687) have also been demonstrated to produce potent cognitive benefits by augmenting signaling via the cAMP/protein kinase A/CREB pathway for memory consolidation (Burgin et al., 2010; Gurney et al., 2015).
PDE4 subtypes are known to modulate the inflammatory response in the brain, such that TNF-α increases PDE4B expression and nuclear translocation in microglia (Ghosh et al., 2012). Of the PDE4 subtypes, PDE4B in particular is highly expressed in activated microglia after TBI and SCI. Importantly, this observation promotes the potential application of therapeutic modulators for the treatment of neuroinflammatory diseases including AD.
PDE5 inhibitor sildenafil has been recently developed as a first-line drug for diabetic patients with erectile dysfunction. Recently PDE5 inhibitors have been proposed as potential therapeutic agents for neuroinflammatory, degenerative, and memory-loss diseases including AD, PD, and MS (Puzzo et al., 2009; Pifarre et al., 2011; Fiorito et al., 2013; Peixoto et al., 2015). Possible underlying mechanisms for the beneficial effects of PDE5 inhibitors include a neuroprotective effect exerted via the cGMP and/or cAMP signaling pathways and an anti-inflammatory-related effect. In an in vitro study of microglia, PDE5 inhibitors were shown to inhibit LPS-induced M1 polarization by decreasing the production of nitric oxide, TNF-α, and IL-1β (Zhao et al., 2016). M2 polarization induced by sildenafil has been shown to provide protection against lesion extension in the late phase of MCAo in neonatal mice (Moretti et al., 2016). Furthermore, the chronic inhibition of PDE-5 has been shown to facilitate the shift from classic (M1) to alternative (M2) macrophage polarization in streptozotocin-induced diabetic mice (Venneri et al., 2015).
Redox Signaling Molecules
Activated microglia-derived oxidative stress is implicated in numerous CNS diseases. The redox status modulates the acquisition of classical microglia activation phenotypes by various mechanisms, including the NOX and NOS-dependent pathways (Bermudez et al., 2016; Haslund-Vinding et al., 2016; Seredenina et al., 2016; Vilhardt et al., 2016). NOX induces M1 polarization of microglia, and thus unsurprisingly, either a NOX inhibitor (apocynin) or genetic depletion has been reported to induce M2 polarization. Choi et al. (2012) showed that pharmacological inhibition of NOX changed microglia from an M1 to an M2 phenotype. Moreover, NOX2 gene-deficient mice (gp91phox-/-) have been found to be completely protected against glial M1 over-activation and dopaminergic neuron degeneration in the 6-hydroxydopamine (6-OHDA)-induced PD mouse model (Hernandes et al., 2013a,b). GKT137831 (NOX1 and NOX4 inhibitor), originally developed for diabetic nephropathy, has been shown to significantly reduce both hypoxia-induced ROS production, and the expression of inflammatory cytokines in retinal microglia (Deliyanti and Wilkinson-Berka, 2015), suggesting that NOX inhibitors may be promising therapeutic agents for diabetic retinopathy and/or neuropathy.
Resveratrol (3,5,4′-trihydroxy-trans-stilbene) is a natural phytoalexin found in grape-skin that exerts anti-inflammatory and antioxidant, as well as various other biological effects. Recently, resveratrol has been suggested to have neuroprotective effects against many neurological diseases (Porro et al., 2015; Abdel-Aleem et al., 2016; Ghaiad et al., 2016; Jeong et al., 2016; Shi et al., 2016; Xu et al., 2017), and several underlying mechanisms for this effect have been suggested. For example, resveratrol attenuates hypoxia-induced neurotoxicity by inhibiting microglial activation (Zhang et al., 2015), and similarly constrains amyloid-β-induced microglial activation via NOX (Yao et al., 2015).
mTOR Inhibitors and Chlorogenic Acid: M1 Polarization for Inhibiting Glioma Cell Growth
The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase involved in many cellular processes such as transcription and translation, ribosomal biogenesis, energy metabolism, and autophagy (Laplante and Sabatini, 2012). The inhibition of mTOR has been recently suggested to prevent glioblastoma, the most common and aggressive primary CNS tumor-type, originating from glial cells (Rolle et al., 2012). Rad, an inhibitor of mTOR, was recently found to significantly increase iNOS expression, and to reduce IL-10 expression, suggesting that Rad prevents the acquisition of an M2 phenotype in response to glioma factors promoting classic M1 activation. Thus, mTOR inhibition prevented glioma growth by inhibiting M2 polarization of microglial cells, thereby increasing their anti-tumor cytotoxic potential (Lisi et al., 2014).
Chlorogenic acid, a phenolic compound extracted from natural products, has been shown to exert anti-tumor effects on multiple malignant tumors (Bandyopadhyay et al., 2004; Belkaid et al., 2006). Previous studies on the anti-tumor effects of chlorogenic acid focused on its direct cytotoxic or antioxidant effect (Lewandowska et al., 2016); however, a recent report suggested a novel anti-tumorigenic mechanism for the chlorogenic acid-induced effects, in which it promotes M1 polarization to inhibit tumor growth (Xue et al., 2017). In glioma xenograft mice, chlorogenic acid was shown to increase CD11-positive M1 and decrease CD206-positive M2 cells in tumor tissues, leading to a reduction of the overall tumor size.
Epigenetic Regulators and Other Signaling Molecules
Previous studies have shown that histone deacetylase (HDACs) inhibitors preferentially promote the transcription of neuroprotective genes, and protect against TBI-induced neuronal damage (Lazo-Gomez et al., 2013; Wang et al., 2013) and strongly suppress LPS-induced cytokine expression and release by microglia (Kannan et al., 2013). Recently, Wang et al. (2015) showed that a novel HDAC inhibitor, scriptaid, protected white brain matter from damage by TBI via polarization of microglia/macrophages toward the beneficial M2 phenotype. Scriptaid induced the expression of microglial glycogen synthase kinase 3 beta (GSK3β) and the microglial phenotypic switch from M1 to M2. Media conditioned by Scriptaid treated-microglia conferred a greater protective effect against oxygen and glucose deprivation-induced cell death in oligodendrocytes, as compared to media conditioned by vehicle-treated microglia (Wang et al., 2015).
Rho-associated kinase (ROCK) has been proposed as another potential regulator of the microglial phenotype. ROCK is a serine/threonine kinase and a key regulator in controlling formation of the actin cytoskeleton, as well as cell motility, and cell adhesion. Selective ROCK inhibitors, such as FSD-C10 or Y-39983, have been shown to exhibit therapeutic potential in an EAE model via attenuation of demyelination and neuroinflammation (Gao et al., 2013; Li et al., 2014). More recently, microglial treatment with a ROCK inhibitor Fasudil induced alteration in microglial phenotype polarization and functional plasticity, shifting the M1 to a M2 phenotype (Chen et al., 2014a).
Conclusion
Here, we summarized the current research on pharmacological modulators of microglial phenotypes and their cellular targets. We focused on the effects of these modulators both in vivo, and in in vitro models of neurodegenerative diseases. We divided the cellular targets into groups according to their original functions, these being nuclear receptors, metabolism-associated proteins, proteins regulating the cAMP pathway, redox signaling molecules, and others. These target proteins are potential therapeutic candidates for the effective treatment of neurodegenerative diseases. Furthermore, an improved characterization of target protein functions will enable the design of novel pharmacological compounds to modulate cytotoxic and/or neurotrophic microglial phenotypes at specific stages of neurodegenerative disease. This is owing to the time-dependent microglial activation in the switch from neuroprotective to neurotoxic profiles in chronic diseases. Thus, further studies are essential in cultured cells and animal models, as well as in human patients, to enable the translation of these preliminary findings into clinically applicable interventions for AD, PD, MS, and other neurodegenerative diseases.
Author Contributions
Both authors have made a substantial intellectual contribution to this work, and approved submission of the manuscript. GS and KS formulated the focus of this review focus. GS conducted the literature review and summarized the discussed studies. KS evaluated the manuscript and contributed to the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (Grant Nos. 2015R1A2A1A10051958, 2016M3C7A1904148, 2016R1D1A1B01009186), and by the Korean Health Technology R&D Project, Korean Ministry of Health & Welfare (Grant No. HI14C3331).
References
Abdel-Aleem, G. A., Khaleel, E. F., Mostafa, D. G., and Elberier, L. K. (2016). Neuroprotective effect of resveratrol against brain ischemia reperfusion injury in rats entails reduction of DJ-1 protein expression and activation of PI3K/Akt/GSK3b survival pathway. Arch. Physiol. Biochem. 122, 200–213.
Aguzzi, A., Nuvolone, M., and Zhu, C. (2013). The immunobiology of prion diseases. Nat. Rev. Immunol. 13, 888–902. doi: 10.1038/nri3553
Bandyopadhyay, G., Biswas, T., Roy, K. C., Mandal, S., Mandal, C., Pal, B. C., et al. (2004). Chlorogenic acid inhibits Bcr-Abl tyrosine kinase and triggers p38 mitogen-activated protein kinase-dependent apoptosis in chronic myelogenous leukemic cells. Blood 104, 2514–2522.
Barad, M., Bourtchouladze, R., Winder, D. G., Golan, H., and Kandel, E. (1998). Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc. Natl. Acad. Sci. U.S.A. 95, 15020–15025.
Batchelor, P. E., Liberatore, G. T., Wong, J. Y., Porritt, M. J., Frerichs, F., Donnan, G. A., et al. (1999). Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J. Neurosci. 19, 1708–1716.
Belkaid, A., Currie, J. C., Desgagnes, J., and Annabi, B. (2006). The chemopreventive properties of chlorogenic acid reveal a potential new role for the microsomal glucose-6-phosphate translocase in brain tumor progression. Cancer Cell Int. 6:7.
Bermudez, S., Khayrullina, G., Zhao, Y., and Byrnes, K. R. (2016). NADPH oxidase isoform expression is temporally regulated and may contribute to microglial/macrophage polarization after spinal cord injury. Mol. Cell. Neurosci. 77, 53–64. doi: 10.1016/j.mcn.2016.10.001
Block, F., Schmidt, W., Nolden-Koch, M., and Schwarz, M. (2001). Rolipram reduces excitotoxic neuronal damage. Neuroreport 12, 1507–1511.
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69.
Boillee, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59.
Bouhlel, M. A., Derudas, B., Rigamonti, E., Dievart, R., Brozek, J., Haulon, S., et al. (2007). PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143.
Burchell, J. T., and Panegyres, P. K. (2016). Prion diseases: immunotargets and therapy. Immunotargets Ther. 5, 57–68. doi: 10.2147/ITT.S64795
Burgin, A. B., Magnusson, O. T., Singh, J., Witte, P., Staker, B. L., Bjornsson, J. M., et al. (2010). Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 28, 63–70. doi: 10.1038/nbt.1598
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Byrnes, K. R., Loane, D. J., and Faden, A. I. (2009). Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics 6, 94–107. doi: 10.1016/j.nurt.2008.10.038
Chawla, A. (2010). Control of macrophage activation and function by PPARs. Circ. Res. 106, 1559–1569. doi: 10.1161/CIRCRESAHA.110.216523
Chen, C., Li, Y. H., Zhang, Q., Yu, J. Z., Zhao, Y. F., Ma, C. G., et al. (2014a). Fasudil regulates T cell responses through polarization of BV-2 cells in mice experimental autoimmune encephalomyelitis. Acta Pharmacol. Sin. 35, 1428–1438. doi: 10.1038/aps.2014.68
Chen, S., Zhang, H., Pu, H., Wang, G., Li, W., Leak, R. K., et al. (2014c). n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci. Rep. 4:7458. doi: 10.1038/srep07458
Chen, W. W., Zhang, X., and Huang, W. J. (2016). Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 13, 3391–3396. doi: 10.3892/mmr.2016.4948
Cherry, J. D., Olschowka, J. A., and O’Banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11:98. doi: 10.1186/1742-2094-11-98
Choi, S. H., Aid, S., Kim, H. W., Jackson, S. H., and Bosetti, F. (2012). Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J. Neurochem. 120, 292–301. doi: 10.1111/j.1471-4159.2011.07572.x
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
Correale, J. (2014). The role of microglial activation in disease progression. Mult. Scler. 20, 1288–1295. doi: 10.1177/1352458514533230
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi: 10.1126/science.1217697
Crotti, A., and Ransohoff, R. M. (2016). Microglial physiology and pathophysiology: insights from genome-wide transcriptional profiling. Immunity 44, 505–515. doi: 10.1016/j.immuni.2016.02.013
Deliyanti, D., and Wilkinson-Berka, J. L. (2015). Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina. J. Neuroinflamm. 12:136. doi: 10.1186/s12974-015-0363-z
Diaz-Aparicio, I., Beccari, S., Abiega, O., and Sierra, A. (2016). Clearing the corpses: regulatory mechanisms, novel tools, and therapeutic potential of harnessing microglial phagocytosis in the diseased brain. Neural Regen. Res. 11, 1533–1539.
Dinter, H. (2000). Phosphodiesterase type 4 inhibitors: potential in the treatment of multiple sclerosis? BioDrugs 13, 87–94.
Du, L., Zhang, Y., Chen, Y., Zhu, J., Yang, Y., and Zhang, H. L. (2016). Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. doi: 10.1007/s12035-016-0245-0 [Epub ahead of print].
Fernandez, M. R., Batlle, C., Gil-Garcia, M., and Ventura, S. (2017). Amyloid cores in prion domains: key regulators for prion conformational conversion. Prion 11, 31–39. doi: 10.1080/19336896.2017.1282020
Fiorito, J., Saeed, F., Zhang, H., Staniszewski, A., Feng, Y., Francis, Y. I., et al. (2013). Synthesis of quinoline derivatives: discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 60, 285–294. doi: 10.1016/j.ejmech.2012.12.009
Franco, R., and Fernandez-Suarez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Gao, C., Huang, L., Long, Y., Zheng, J., Yang, J., Pu, S., et al. (2013). Y-39983, a selective Rho-kinase inhibitor, attenuates experimental autoimmune encephalomyelitis via inhibition of demyelination. Neuroimmunomodulation 20, 334–340. doi: 10.1159/000353568
Gao, H. M., and Hong, J. S. (2008). Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365. doi: 10.1016/j.it.2008.05.002
Gerlo, S., Kooijman, R., Beck, I. M., Kolmus, K., Spooren, A., and Haegeman, G. (2011). Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol. Life. Sci. 68, 3823–3841. doi: 10.1007/s00018-011-0757-8
Ghaiad, H. R., Nooh, M. M., El-Sawalhi, M. M., and Shaheen, A. A. (2016). Resveratrol promotes remyelination in cuprizone model of multiple sclerosis: biochemical and histological study. Mol. Neurobiol. doi: 10.1007/s12035-016-9891-5 [Epub ahead of print].
Ghosh, M., Aguirre, V., Wai, K., Felfly, H., Dietrich, W. D., and Pearse, D. D. (2015). The interplay between cyclic AMP, MAPK, and NF-kappaB pathways in response to proinflammatory signals in microglia. Biomed. Res. Int. 2015:308461. doi: 10.1155/2015/308461
Ghosh, M., Garcia-Castillo, D., Aguirre, V., Golshani, R., Atkins, C. M., Bramlett, H. M., et al. (2012). Proinflammatory cytokine regulation of cyclic AMP-phosphodiesterase 4 signaling in microglia in vitro and following CNS injury. Glia 60, 1839–1859. doi: 10.1002/glia.22401
Ghosh, M., Xu, Y., and Pearse, D. D. (2016). Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflammation 13:9. doi: 10.1186/s12974-015-0463-9
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Grizenkova, J., Akhtar, S., Brandner, S., Collinge, J., and Lloyd, S. E. (2014). Microglial Cx3cr1 knockout reduces prion disease incubation time in mice. BMC Neurosci. 15:44. doi: 10.1186/1471-2202-15-44
Gurney, M. E., D’Amato, E. C., and Burgin, A. B. (2015). Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics 12, 49–56. doi: 10.1007/s13311-014-0309-7
Haslund-Vinding, J., McBean, G., Jaquet, V., and Vilhardt, F. (2016). NADPH oxidases in microglia oxidant production: activating receptors, pharmacology, and association with disease. Br. J. Pharmacol. doi: 10.1111/bph.13425 [Epub ahead of print].
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Hernandes, M. S., Cafe-Mendes, C. C., and Britto, L. R. (2013a). NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxid. Med. Cell. Longev. 2013:157857. doi: 10.1155/2013/157857
Hernandes, M. S., Santos, G. D., Cafe-Mendes, C. C., Lima, L. S., Scavone, C., Munhoz, C. D., et al. (2013b). Microglial cells are involved in the susceptibility of NADPH oxidase knockout mice to 6-hydroxy-dopamine-induced neurodegeneration. PLoS ONE 8:e75532. doi: 10.1371/journal.pone.0075532
Huang, C., Tong, J., Bi, F., Zhou, H., and Xia, X. G. (2012). Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J. Clin. Invest. 122, 107–118. doi: 10.1172/JCI59130
Jeong, S. I., Shin, J. A., Cho, S., Kim, H. W., Lee, J. Y., Kang, J. L., et al. (2016). Resveratrol attenuates peripheral and brain inflammation and reduces ischemic brain injury in aged female mice. Neurobiol. Aging 44, 74–84. doi: 10.1016/j.neurobiolaging.2016.04.007
Jha, M. K., Song, G. J., Lee, M. G., Jeoung, N. H., Go, Y., Harris, R. A., et al. (2015). Metabolic connection of inflammatory pain: pivotal role of a pyruvate dehydrogenase kinase-pyruvate dehydrogenase-lactic acid axis. J. Neurosci. 35, 14353–14369. doi: 10.1523/JNEUROSCI.1910-15.2015
Jin, Q., Cheng, J., Liu, Y., Wu, J., Wang, X., Wei, S., et al. (2014). Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav. Immun. 40, 131–142. doi: 10.1016/j.bbi.2014.03.003
Kannan, V., Brouwer, N., Hanisch, U. K., Regen, T., Eggen, B. J., and Boddeke, H. W. (2013). Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J. Neurosci. Res. 91, 1133–1142. doi: 10.1002/jnr.23221
Kettenmann, H., Kirchhoff, F., and Verkhratsky, A. (2013). Microglia: new roles for the synaptic stripper. Neuron 77, 10–18. doi: 10.1016/j.neuron.2012.12.023
Kobayashi, K., Imagama, S., Ohgomori, T., Hirano, K., Uchimura, K., Sakamoto, K., et al. (2013). Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 4:e525. doi: 10.1038/cddis.2013.54
Kumar, A., Alvarez-Croda, D. M., Stoica, B. A., Faden, A. I., and Loane, D. J. (2016). Microglial/macrophage polarization dynamics following traumatic brain injury. J. Neurotrauma 33, 1732–1750.
Kuzumoto, Y., Kusunoki, S., Kato, N., Kihara, M., and Low, P. A. (2006). Effect of the aldose reductase inhibitor fidarestat on experimental diabetic neuropathy in the rat. Diabetologia 49, 3085–3093.
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Latta, C. H., Sudduth, T. L., Weekman, E. M., Brothers, H. M., Abner, E. L., Popa, G. J., et al. (2015). Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-beta using BV2 microglial cells and APP/PS1 transgenic mice. J. Neuroinflammation 12:41. doi: 10.1186/s12974-015-0243-6
Lazo-Gomez, R., Ramirez-Jarquin, U. N., Tovar, Y. R. L. B., and Tapia, R. (2013). Histone deacetylases and their role in motor neuron degeneration. Front. Cell. Neurosci. 7:243. doi: 10.3389/fncel.2013.00243
Lecca, D., Nevin, D. K., Mulas, G., Casu, M. A., Diana, A., Rossi, D., et al. (2015). Neuroprotective and anti-inflammatory properties of a novel non-thiazolidinedione PPARgamma agonist in vitro and in MPTP-treated mice. Neuroscience 302, 23–35. doi: 10.1016/j.neuroscience.2015.04.026
Lewandowska, H., Kalinowska, M., Lewandowski, W., Stepkowski, T. M., and Brzoska, K. (2016). The role of natural polyphenols in cell signaling and cytoprotection against cancer development. J. Nutr. Biochem. 32, 1–19. doi: 10.1016/j.jnutbio.2015.11.006
Li, Y. H., Yu, J. Z., Liu, C. Y., Zhang, H., Zhang, H. F., Yang, W. F., et al. (2014). Intranasal delivery of FSD-C0, a novel Rho kinase inhibitor, exhibits therapeutic potential in experimental autoimmune encephalomyelitis. Immunology 143, 219–229. doi: 10.1111/imm.12303
Liao, B., Zhao, W., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152. doi: 10.1016/j.expneurol.2012.06.011
Lins, N., Mourao, L., Trevia, N., Passos, A., Farias, J. A., Assuncao, J., et al. (2016). Virus infections on prion diseased mice exacerbate inflammatory microglial response. Oxid. Med. Cell. Longev. 2016:3974648. doi: 10.1155/2016/3974648
Lisi, L., Laudati, E., Navarra, P., and Dello Russo, C. (2014). The mTOR kinase inhibitors polarize glioma-activated microglia to express a M1 phenotype. J. Neuroinflammation 11:125. doi: 10.1186/1742-2094-11-125
Loane, D. J., Deighan, B. F., Clarke, R. M., Griffin, R. J., Lynch, A. M., and Lynch, M. A. (2009). Interleukin-4 mediates the neuroprotective effects of rosiglitazone in the aged brain. Neurobiol. Aging 30, 920–931.
Loane, D. J., and Kumar, A. (2016). Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 275(Pt 3), 316–327. doi: 10.1016/j.expneurol.2015.08.018
Lu, Y. Z., Lin, C. H., Cheng, F. C., and Hsueh, C. M. (2005). Molecular mechanisms responsible for microglia-derived protection of Sprague-Dawley rat brain cells during in vitro ischemia. Neurosci. Lett. 373, 159–164.
Mandrekar-Colucci, S., Karlo, J. C., and Landreth, G. E. (2012). Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 32, 10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012
Mandrekar-Colucci, S., and Landreth, G. E. (2010). Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 9, 156–167.
Michelucci, A., Heurtaux, T., Grandbarbe, L., Morga, E., and Heuschling, P. (2009). Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 210, 3–12. doi: 10.1016/j.jneuroim.2009.02.003
Miron, V. E., Boyd, A., Zhao, J. W., Yuen, T. J., Ruckh, J. M., Shadrach, J. L., et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218. doi: 10.1038/nn.3469
Morales, R., Estrada, L. D., Diaz-Espinoza, R., Morales-Scheihing, D., Jara, M. C., Castilla, J., et al. (2010). Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J. Neurosci. 30, 4528–4535. doi: 10.1523/JNEUROSCI.5924-09.2010
Moretti, R., Leger, P. L., Besson, V. C., Csaba, Z., Pansiot, J., Di Criscio, L., et al. (2016). Sildenafil, a cyclic GMP phosphodiesterase inhibitor, induces microglial modulation after focal ischemia in the neonatal mouse brain. J. Neuroinflammation 13, 95. doi: 10.1186/s12974-016-0560-4
Neumann, S., Bradke, F., Tessier-Lavigne, M., and Basbaum, A. I. (2002). Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34, 885–893.
Niu, L., Zhou, J., Huang, Y., Chen, Y., and Li, C. (2010). db-Cyclic adenosine monophosphate promotes axon regeneration and motor function recovery in cerebral ischemia-reperfusion rats. Neurol. India 58, 195–200. doi: 10.4103/0028-3886.63786
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Pan, J., Jin, J. L., Ge, H. M., Yin, K. L., Chen, X., Han, L. J., et al. (2015). Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J. Neuroinflamm. 12, 51. doi: 10.1186/s12974-015-0270-3
Patil, S. P., Jain, P. D., Ghumatkar, P. J., Tambe, R., and Sathaye, S. (2014). Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 277, 747–754. doi: 10.1016/j.neuroscience.2014.07.046
Peixoto, C. A., Nunes, A. K., and Garcia-Osta, A. (2015). Phosphodiesterase-5 inhibitors: action on the signaling pathways of neuroinflammation, neurodegeneration, and cognition. Mediat. Inflamm. 2015:940207. doi: 10.1155/2015/940207
Pifarre, P., Prado, J., Baltrons, M. A., Giralt, M., Gabarro, P., Feinstein, D. L., et al. (2011). Sildenafil (Viagra) ameliorates clinical symptoms and neuropathology in a mouse model of multiple sclerosis. Acta Neuropathol. 121, 499–508. doi: 10.1007/s00401-010-0795-6
Porro, C., Cianciulli, A., Calvello, R., and Panaro, M. A. (2015). Reviewing the role of resveratrol as a natural modulator of microglial activities. Curr. Pharm. Des. 21, 5277–5291.
Puzzo, D., Staniszewski, A., Deng, S. X., Privitera, L., Leznik, E., Liu, S., et al. (2009). Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J. Neurosci. 29, 8075–8086. doi: 10.1523/JNEUROSCI.0864-09.2009
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Reddy, A. B., Srivastava, S. K., and Ramana, K. V. (2010). Aldose reductase inhibition prevents lipopolysaccharide-induced glucose uptake and glucose transporter 3 expression in RAW264.7 macrophages. Int. J. Biochem. Cell Biol. 42, 1039–1045. doi: 10.1016/j.biocel.2010.03.014
Rolle, C. E., Sengupta, S., and Lesniak, M. S. (2012). Mechanisms of immune evasion by gliomas. Adv. Exp. Med. Biol. 746, 53–76. doi: 10.1007/978-1-4614-3146-6_5
Seredenina, T., Nayernia, Z., Sorce, S., Maghzal, G. J., Filippova, A., Ling, S. C., et al. (2016). Evaluation of NADPH oxidases as drug targets in a mouse model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 97, 95–108. doi: 10.1016/j.freeradbiomed.2016.05.016
Shi, N., Zhu, C., and Li, L. (2016). Rehabilitation Training and Resveratrol Improve the Recovery of Neurological and Motor Function in Rats after Cerebral Ischemic Injury through the Sirt1 Signaling Pathway. Biomed Res. Int. 2016:1732163. doi: 10.1155/2016/1732163
Sica, A., and Mantovani, A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795. doi: 10.1172/JCI59643
Skalak, R., Soslowsky, L., Schmalzer, E., Impelluso, T., and Chien, S. (1987). Theory of filtration of mixed blood suspensions. Biorheology 24, 35–52.
Song, G. J., Nam, Y., Jo, M., Jung, M., Koo, J. Y., Cho, W., et al. (2016). A novel small-molecule agonist of PPAR-gamma potentiates an anti-inflammatory M2 glial phenotype. Neuropharmacology 109, 159–169. doi: 10.1016/j.neuropharm.2016.06.009
Stopschinski, B. E., and Diamond, M. I. (2017). The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol. 16, 323–332. doi: 10.1016/S1474-4422(17)30037-6
Sutcliffe, J. S., Beaumont, V., Watson, J. M., Chew, C. S., Beconi, M., Hutcheson, D. M., et al. (2014). Efficacy of selective PDE4D negative allosteric modulators in the object retrieval task in female cynomolgus monkeys (Macaca fascicularis). PLoS ONE 9:e102449. doi: 10.1371/journal.pone.0102449
Swarup, V., Phaneuf, D., Dupre, N., Petri, S., Strong, M., Kriz, J., et al. (2011). Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J. Exp. Med. 208, 2429–2447. doi: 10.1084/jem.20111313
Tang, Y., and Le, W. (2016). Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Thored, P., Heldmann, U., Gomes-Leal, W., Gisler, R., Darsalia, V., Taneera, J., et al. (2009). Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57, 835–849. doi: 10.1002/glia.20810
Toba, J., Nikkuni, M., Ishizeki, M., Yoshii, A., Watamura, N., Inoue, T., et al. (2016). PPARgamma agonist pioglitazone improves cerebellar dysfunction at pre-Abeta deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem. Biophys. Res. Commun. 473, 1039–1044. doi: 10.1016/j.bbrc.2016.04.012
Venna, V. R., Li, J., Hammond, M. D., Mancini, N. S., and McCullough, L. D. (2014). Chronic metformin treatment improves post-stroke angiogenesis and recovery after experimental stroke. Eur. J. Neurosci. 39, 2129–2138. doi: 10.1111/ejn.12556
Venneri, M. A., Giannetta, E., Panio, G., De Gaetano, R., Gianfrilli, D., Pofi, R., et al. (2015). Chronic Inhibition of PDE5 Limits Pro-Inflammatory Monocyte-Macrophage Polarization in Streptozotocin-Induced Diabetic Mice. PLoS ONE 10:e0126580. doi: 10.1371/journal.pone.0126580
Vilhardt, F., Haslund-Vinding, J., Jaquet, V., and McBean, G. (2016). Microglia antioxidant systems and redox signaling. Br. J. Pharmacol. doi: 10.1111/bph.13426 [Epub ahead of print].
Wang, G., Jiang, X., Pu, H., Zhang, W., An, C., Hu, X., et al. (2013). Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway : scriptaid protects against TBI via AKT. Neurotherapeutics 10, 124–142. doi: 10.1007/s13311-012-0157-2
Wang, G., Shi, Y., Jiang, X., Leak, R. K., Hu, X., Wu, Y., et al. (2015). HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc. Natl. Acad. Sci. U.S.A. 112, 2853–2858. doi: 10.1073/pnas.1501441112
Weekman, E. M., Sudduth, T. L., Abner, E. L., Popa, G. J., Mendenhall, M. D., Brothers, H. M., et al. (2014). Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J. Neuroinflammation 11, 127. doi: 10.1186/1742-2094-11-127
Xiao, Q., Zhao, W., Beers, D. R., Yen, A. A., Xie, W., Henkel, J. S., et al. (2007). Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J. Neurochem. 102, 2008–2019.
Xu, P., Zhang, M., Sheng, R., and Ma, Y. (2017). Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Abeta1-42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur. J. Med. Chem. 127, 174–186. doi: 10.1016/j.ejmech.2016.12.045
Xue, N., Zhou, Q., Ji, M., Jin, J., Lai, F., Chen, J., et al. (2017). Chlorogenic acid inhibits glioblastoma growth through repolarizating macrophage from M2 to M1 phenotype. Sci. Rep. 7:39011. doi: 10.1038/srep39011
Yamanaka, M., Ishikawa, T., Griep, A., Axt, D., Kummer, M. P., and Heneka, M. T. (2012). PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 32, 17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012
Yamasaki, R., Lu, H., Butovsky, O., Ohno, N., Rietsch, A. M., Cialic, R., et al. (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533–1549. doi: 10.1084/jem.20132477
Yao, Y., Li, J., Niu, Y., Yu, J. Q., Yan, L., Miao, Z. H., et al. (2015). Resveratrol inhibits oligomeric Abeta-induced microglial activation via NADPH oxidase. Mol. Med. Rep. 12, 6133–6139. doi: 10.3892/mmr.2015.4199
Zhang, Q., Bian, G., Chen, P., Liu, L., Yu, C., Liu, F., et al. (2016). Aldose Reductase Regulates Microglia/Macrophages Polarization Through the cAMP Response Element-Binding Protein After Spinal Cord Injury in Mice. Mol. Neurobiol. 53, 662–676. doi: 10.1007/s12035-014-9035-8
Zhang, Q., Yuan, L., Zhang, Q., Gao, Y., Liu, G., Xiu, M., et al. (2015). Resveratrol attenuates hypoxia-induced neurotoxicity through inhibiting microglial activation. Int. Immunopharmacol. 28, 578–587. doi: 10.1016/j.intimp.2015.07.027
Zhao, S., Yang, J., Wang, L., Peng, S., Yin, J., Jia, L., et al. (2016). NF-kappaB Upregulates Type 5 Phosphodiesterase in N9 Microglial Cells: Inhibition by Sildenafil and Yonkenafil. Mol. Neurobiol. 53, 2647–2658. doi: 10.1007/s12035-015-9293-0
Keywords: pharmacological modulator, microglial polarization, neurodegenerative diseases, neuroinflammation, neuroprotective
Citation: Song GJ and Suk K (2017) Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front. Aging Neurosci. 9:139. doi: 10.3389/fnagi.2017.00139
Received: 07 March 2017; Accepted: 25 April 2017;
Published: 15 May 2017.
Edited by:
Isidre Ferrer, University of Barcelona, SpainReviewed by:
Christiane Charriaut-Marlangue, Institut National de la Santé et de la Recherche Médicale, FranceFranc Llorens, German Center for Neurodegenerative Diseases, Germany
Copyright © 2017 Song and Suk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyoungho Suk, a3N1a0BrbnUuYWMua3I=