 Suzanne Hood
Suzanne Hood Shimon Amir
Shimon Amir- 1Department of Psychology, Bishop’s University, Sherbrooke, QC, Canada
- 2Department of Psychology, Concordia University, Montreal, QC, Canada
Despite varied etiologies and symptoms, several neurodegenerative diseases—specifically, Alzheimer’s (AD), Parkinson’s (PD), and Huntington’s diseases (HDs)—share the common feature of abnormal circadian rhythms, such as those in behavior (e.g., disrupted sleep/wake cycles), physiological processes (e.g., diminished hormone release) and biochemical activities (e.g., antioxidant production). Circadian disturbances are among the earliest symptoms of these diseases, and the molecular mechanisms of the circadian system are suspected to play a pivotal, and possibly causal, role in their natural histories. Here, we review the common circadian abnormalities observed in ADs, PDs and HDs, and summarize the evidence that the molecular circadian clockwork directly influences the course of these disease states. On the basis of this research, we explore several circadian-oriented interventions proposed as treatments for these neurological disorders.
Circadian Rhythms and Neurodegenerative Diseases
As life expectancy increases globally, the prevalence of neurodegenerative diseases mounts steadily. Worldwide, Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) are among the most prevalent neurodegenerative diseases, and are associated with a significant burden for health care systems (Neurological Health Charities Canada, 2014, September). Despite the varied pathogenesis and diversity of symptoms among them, common to AD, PD and HD are disruptions of circadian rhythms, or the near 24-h cyclic fluctuations in a host of physiological and behavioral processes. A rapidly growing body of research suggests that disturbances in the circadian system precede the emergence of the characteristic cognitive and motor symptoms of these diseases by years (Kondratova and Kondratov, 2012; Hastings and Goedert, 2013; Videnovic et al., 2014a; Abbott and Videnovic, 2016; Mattis and Sehgal, 2016), and may contribute to their onset (Kondratova and Kondratov, 2012; Videnovic and Zee, 2015). Here, we provide a concise overview of the evidence linking the circadian system to these diseases, and examine circadian-oriented approaches to the treatment of AD, PD and HD.
Circadian Rhythms and Cellular Clocks
The circadian system provides an adaptive mechanism for organisms to coordinate cellular processes, physiological functions and behaviors with the predictable 24-h cycle of light and dark on Earth (Bell-Pedersen et al., 2005). In humans, familiar examples of rhythms include daily patterns of sleeping and waking; the rise and fall of core body temperature; heart rate; blood pressure; and release of a wide variety of hormones, such as the nightly surge in melatonin from the pineal gland. The presence of an endogenous timing system in the body is clearly seen in conditions when predictable time-of-day cues are removed, yet near-24 h rhythms in these processes persist nonetheless (Arendt, 2012).
In mammals, the suprachiasmatic nucleus (SCN) houses the master circadian clock, and is found just dorsal to the optic chiasm. Inputs to the SCN from the retinohypothalamic tract provide information about daily light exposure to synchronize the endogenous clockwork to the external environment (Welsh et al., 2010). In turn, the SCN communicates time-of-day information by both synaptic and diffusible signals to a host of peripheral oscillators in a variety of brain regions and organs, such as heart, lungs, liver and adrenal glands. Thus, the SCN serves to coordinate the timing of a distributed network of clocks throughout the body (Mohawk et al., 2012). This coordination is vital for health and well-being: circadian desynchrony is already implicated in a number of disease states, including some cancers, metabolic diseases, and mood disorders such as bipolar disease and major depression (Roybal et al., 2007; McFadden et al., 2014; Stevens et al., 2014; Lucassen et al., 2016; Morris et al., 2016).
As outlined in Figure 1, the circadian timekeeping mechanism is controlled at a cellular level by a group of genes that regulate their own transcription and translation over approximately 24 h via a series of interacting negative feedback loops (for a review see Mohawk et al., 2012). In addition to regulating their own levels of expression, “clock” genes serve as transcription factors for other genes which regulate a variety of functions, including cell division, metabolism, immune responses and oxidative processes (Duffield, 2003; Wilking et al., 2013). Importantly, mutations of the bmal1 and period genes yield an accelerated aging phenotype in Drosophila and mice, with faster rates of tissue decline, impairments in cognitive function and shorter lifespan relative to age-matched wild type controls (Kondratov et al., 2006; Krishnan et al., 2009).
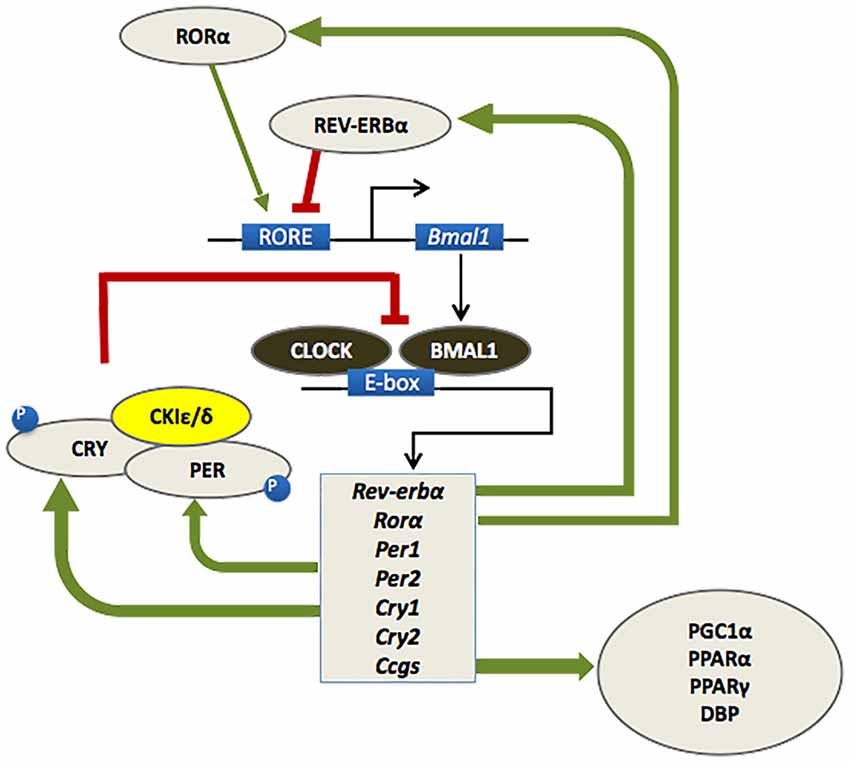
Figure 1. At a molecular level, the mammalian circadian clock is composed of a group of clock genes that regulate their own transcription and translation in a series of interlocking negative feedback loops. Heterodimers of the transcription factors BMAL1 and CLOCK drive the expression of the Period (Per1/Per2) and Cryptochrome (Cry1/Cry2) genes, the nuclear receptors retinoid-related orphan receptor (RORα) and REV-ERBα, and a number of downstream genes referred to as clock-controlled genes (CCGs). The protein products of the Per and Cry genes dimerize and inhibit the transcriptional activity of CLOCK-BMAL1. A number of kinases, such as casein kinase 1ɛ/δ (CK1 ɛ/δ), regulate the activity of PER-CRY dimers at a post-transcriptional level. RORα and REV-ERBα also regulate the transcription of BMAL1, whereby RORα promotes its expression, whereas REV-ERBα inhibits it. This cycle of clock gene expression completes in approximately 24 h (Huang et al., 2011; Mohawk et al., 2012).
Circadian Symptoms of Alzheimer’s, Parkinson’s and Huntington’s Diseases
Like other physiological processes, activity of the circadian system changes significantly across the lifespan (for recent reviews see Duffy et al., 2015; Hood and Amir, 2017), with a number of disruptions to rhythms such as the sleep/wake cycle and hormone release emerging in older adulthood. Importantly, some of these age-related disturbances in a number of rhythms resemble the circadian disturbances observed in AD, PD and HD, which are reviewed in the following sections. In noting these similarities, it is vital to recognize that differences exist in the severity and timing of the onset of circadian disturbances in sufferers of AD, PD and HD, compared to their occurrence during otherwise healthy aging. By distinguishing more clearly between changes in rhythms that reflect neurodegenerative processes and those that may not necessarily be pathological, we may be able to identify the development of disease more readily and, potentially, improve prospects for intervention and care.
Sleep/Wake Rhythms
Disturbances in the sleep/wake rhythm are perhaps the most prominent circadian-related symptom in individuals affected by AD, PD, or HD. Nighttime sleep becomes increasingly fragmented as these diseases progress, while nocturnal activity levels and daytime sleepiness increase (Hatfield et al., 2004; Morton et al., 2005; Barone et al., 2009; Merlino et al., 2010; Weissová et al., 2016). In severe cases, minimal differences exist between day and night in bouts of activity and sleep (McCurry et al., 1999). These observations of poorly consolidated rest/activity patterns in humans are paralleled by animal models of each disease state (Morton et al., 2005; Vezoli et al., 2011; Loh et al., 2013; Fifel and Cooper, 2014; Long et al., 2014; Graybeal et al., 2015). In addition, behavioral sleep disorders, such as restless leg syndrome and rapid eye movement behavior disorder (RBD) are highly comorbid with PD (Comella, 2007; Barone et al., 2009). Together, these declines in the normal sleep/wake rhythm and in the quality of sleep are identified as among the most disruptive symptoms of these diseases, and have a profoundly negative impact on quality of life (Barone et al., 2009). Furthermore, they are cited as primary reasons for entering individuals with a neurodegenerative illness into residential care facilities (Pollak and Perlick, 1991; Bianchetti et al., 1995).
Melatonin and Cortisol Rhythms
Disturbances have been documented in the circadian rhythms of melatonin and cortisol release in AD, PD and HD. Common to each is a flattening of the melatonin rhythm, such that the normal nighttime peak is suppressed relative to healthy, age-matched controls (Mishima et al., 1999; Wu et al., 2003; Breen et al., 2014; Kalliolia et al., 2014; Videnovic et al., 2014b; although see Aziz et al., 2009). A decline in the peak of nighttime melatonin release has also been observed in individuals exhibiting pre-clinical cognitive symptoms of dementia (Wu et al., 2003; Waller et al., 2016), and this decline appears to correlate positively with level of daytime sleepiness (Videnovic et al., 2014b).
Changes in the rhythm of cortisol release have also been observed, although these changes are somewhat more varied compared with those in melatonin. The normal cortisol rhythm rises in the early morning, with the peak occurring near waking and the nadir in the late evening (Touitou and Haus, 2000). Minimal change in this rhythm has been observed in individuals with suspected AD or dementia (Hatfield et al., 2004; Waller et al., 2016; although see Giubilei et al., 2001; and Hartmann et al., 1997). In both PD and HD, the diurnal pattern of cortisol release remains rhythmic, yet the total daily amount of cortisol released is elevated (Hartmann et al., 1997; Aziz et al., 2009).
Core Body Temperature Rhythm
The human core body temperature rhythm rises throughout the day to peak in the early evening, then falls throughout the night to reach its nadir in the early morning (Van Someren, 2000). Studies of individuals with AD indicate a delay in the peak of this rhythm and a decrease in its amplitude (Satlin et al., 1995; Harper et al., 2005). In PD, only the amplitude of the rhythm appears to be decreased. This change is attributed to a lowering of peak body temperature relative to healthy age-matched controls (Pierangeli et al., 2001; Zhong et al., 2013). A profound reduction in the amplitude of the temperature rhythm has also been documented in rodent models of HD (Kudo et al., 2011; Fisher et al., 2013).
Mood and Behavior Rhythm
A rhythm in mood and emotional volatility reportedly emerges as neurodegenerative conditions progress. This “sundown syndrome” comprises a daily pattern of increased agitation, emotional volatility, and aggression that peaks in the late afternoon or evening (for review see Bachman and Rabins, 2006). This syndrome is not formally recognized as a clinical condition—indeed, dispute exists as to what behavioral features it includes, and whether increased behavioral disturbances during this time of day truly reflect a clinical phenomenon or a confounding influence, such as reporting bias from caregivers (e.g., Bliwise et al., 1993; Yesavage et al., 2003). However, a number of reports suggest that a small but notable proportion of elderly individuals with dementia do exhibit a predictable diurnal pattern of behavioral and emotional disturbance, particularly among those with severe symptoms (Gallagher-Thompson et al., 1992; Martin et al., 2000). The factors contributing to the expression of agitated behaviors are unknown, but some evidence suggests that this pattern is not a direct consequence of sleep loss (Volicer et al., 2001).
Neurodegenerative Diseases and Clock Gene Expression
Evidence from individuals with AD, PD, or HD and animal models of each disease state indicate abnormalities in the rhythms of bmal1 and per2 expression. In AD, the pattern of change observed in bmal1 mRNA expression is complex. In several brain regions and peripheral tissues, bmal1 mRNA expression remains rhythmic; however, the temporal phase relationships among these tissues differ compared with healthy controls (Cermakian et al., 2011; Weissová et al., 2016). In the pineal gland, the rhythms of bmal1, per1 and cry1 mRNA are lost (Wu et al., 2006). In PD, the bmal1 transcription rhythm in blood cells is blunted in amplitude (Cai et al., 2010; Breen et al., 2014). Furthermore, rodent models of PD exhibit a blunting of rhythmic per2 mRNA and PER2 protein expression in several brain regions downstream of SCN control and in peripheral tissues. For example, loss of dopaminergic innervation to the striatum abolishes the rhythmic expression of PER2 protein in this region (Hood et al., 2010; Gravotta et al., 2011). Similarly, the normal rhythms of per2 mRNA expression in both central and peripheral tissues are disrupted in rodent models of HD (Morton et al., 2005; Maywood et al., 2010).
Does A Faulty Circadian Clock Cause Neurodegenerative Disease?
Given the prevalence of rhythm abnormalities in neurodegenerative diseases, circadian disturbances are increasingly regarded as harbingers of neurodegeneration (e.g., Videnovic and Zee, 2015; Mattis and Sehgal, 2016). Consistent with this idea, several prospective studies have identified excessive daytime sleepiness (Abbott et al., 2005; Bonanni et al., 2005), daytime activity fragmentation (Tranah et al., 2011), and sleep behavior disorders (Iranzo et al., 2013; Schenck et al., 2013; Postuma et al., 2015) as independent predictors of AD, PD and cognitive impairments associated with dementia. In the case of RBD, the vast majority of affected individuals appear to be at risk of developing PD or a related synucleinopathy, particularly if additional non-motor risk factors are also exhibited (Iranzo et al., 2013; Schenck et al., 2013; Postuma et al., 2015).
Are these circadian disruptions a consequence of neurodegeneration affecting clockwork mechanisms in the brain and periphery, or do malfunctioning endogenous clocks directly contribute to disease progression? It is clear that prolonged disruption of normal circadian rhythms yields a variety of negative effects on health via mechanisms including widespread impact on gene transcription and pro-inflammatory processes (Archer and Oster, 2015; Lucassen et al., 2016), which may exacerbate the progress of these pathologies. However, a number of findings indicate that the circadian system may in fact play a more direct role in the etiology of neurodegenerative diseases. For example, single nucleotide polymorphisms of bmal1 and per1 are associated with increased risk of PD (Gu et al., 2015). Furthermore, clock genes regulate the expression of other genes directly implicated in neurocognitive disorders such as AD (Panda et al., 2002; Duffield, 2003; Li et al., 2013). For example, the presenilin-2 gene, which regulates levels of beta amyloid peptide and is linked to familial early onset AD (Levy-Lahad et al., 1995; Giri et al., 2016), is rhythmically expressed in SCN (Esler and Wolfe, 2001; Panda et al., 2002). In peripheral tissues, CLOCK:BMAL dimers regulate the expression of presenilin-2 via transcriptional and post-transcriptional mechanisms (Bélanger et al., 2006). These findings suggest a causal link between clock genes and molecular factors that confer risk of neurodegeneration. To our knowledge, however, no experimental studies have yet demonstrated that manipulation of clock genes affects the expression of presenilin-2 in brain.
Degenerative changes within the SCN itself may play a contributory role in these disease states, although evidence supporting this possibility is not entirely consistent. Some post-mortem studies of brain tissue from sufferers of AD indicate loss of hypothalamic tissue that includes cells in the SCN, a reduction in the expression of the neuropeptides AVP and VIP (Swaab et al., 1985; Stopa et al., 1999), and a decrease in the expression of the melatonin receptor MT1 (Wu et al., 2007; however, see Wang et al., 2015). Rodent models of HD exhibit reduced spontaneous cell firing in the SCN compared with controls (Kudo et al., 2011; although see Pallier et al., 2007), yet no change in SCN cell number (Fahrenkrug et al., 2007). Although coordinated SCN cell firing appears to diminish as a normal part of aging (Farajnia et al., 2012), this reduction occurs at a prematurely young age (3 months) in HD rodent models (Kudo et al., 2011). Given that other hypothalamic structures degenerate in AD, PD and HD (Shan et al., 2015), it is possible that structural changes to the master clock may be a consequence of the progressive course of tissue destruction in each disease state, rather than precede disease onset. Nevertheless, any dysfunction of the master clock is likely to worsen the symptoms of these diseases through downstream effects on peripheral oscillators. Consistent with this idea, levels of beta amyloid peptide in human cerebrospinal fluid have been found to correlate positively with sleep fragmentation (Ju et al., 2013).
Compelling evidence suggests that the circadian system may contribute to neurodegenerative disease states through its involvement in regulating cellular responses to oxidative stress (Kondratova and Kondratov, 2012). Oxidative stress is suspected as a causal factor of neuronal damage, cell death, and mitochondrial dysfunction observed in AD, PD and HD (for a review see Grimm et al., 2011). Clock genes such as bmal1 have been directly implicated in cellular antioxidant responses through downstream regulation of antioxidant response element transcription factors (Lee et al., 2013). Rodents with selective knockouts of bmal1 or the period genes (per1 and per2) exhibit significantly higher rates of oxidative damage in tissues compared with age-matched wild type controls (Kondratov et al., 2006; Jang et al., 2011; Lee et al., 2013). The circadian clock may also regulate oxidative stress via rhythmic release of melatonin, which is an effective free radical scavenger (Reiter et al., 2002). These findings imply that abnormal operation of the molecular clock may create cellular conditions whereby harmful by-products of metabolism and DNA replication accumulate, and mitochondrial damage may develop. In turn, these conditions may promote the pathogenesis of neurodegenerative disease states.
Circadian-Oriented Interventions in Neurodegenerative Disease
If the circadian system is indeed a contributor to neurodegenerative disease, it follows that therapeutic interventions targeting the circadian clock could mitigate symptoms, or perhaps even retard the course of the disease itself. To this end, a number of circadian-oriented therapies have been investigated for AD, PD and HD.
One of the most frequently explored examples of this kind of intervention is the use of bright light therapy. Previous evidence has shown that institutionalized older adults may have very little daily exposure to bright light, particularly those with severe symptoms of dementia (Ancoli-Israel et al., 1997; Shochat et al., 2000). Given the profound effect of light exposure in regulating the timing of the master clock, a number of studies have evaluated whether timed bright light exposure has any beneficial effect on the course of neurodegeneration or its symptoms. To date, results have been mixed (Forbes et al., 2014). Overall, timed light exposure appears to modestly improve the regulation of the circadian system in individuals with neurodegenerative disease. In the case of AD, some positive but short-lived benefits have been reported for timed daily exposure to bright light on the consolidation of activity rhythms in elderly adults with AD (Ancoli-Israel et al., 2003; McCurry et al., 2011) and severe dementia (Ancoli-Israel et al., 2002). In PD, daily light exposure improves sleep/wake rhythms through reducing daytime sleepiness and increasing daytime activity (Videnovic et al., 2017). However, it remains unclear whether timed light exposure lessens cognitive or motor skill decline over time. Although light exposure regimens have yielded some short-term improvements in activities of daily living in individuals with AD or PD, there is not yet sufficient evidence to conclude any long-lasting cognitive or motor benefits of this intervention (Paus et al., 2007; Forbes et al., 2014).
Timed administration of melatonin has been investigated for its therapeutic potential in AD, PD and HD. As shown in vitro and in animal models, melatonin has antioxidant and apoptotic properties (Reiter et al., 2002; Wang et al., 2011), and appears to prevent the formation of alpha-synuclein protein aggregations (the primary protein component of Lewy bodies; Ono et al., 2012). However, in randomized controlled clinical trials in humans, the effects of melatonin supplements on sleep quality and activity rhythms have been inconsistent. In individuals with PD, daily doses of melatonin did not improve sleep quality, but were associated with improved self-report measures of sleeping (Medeiros et al., 2007). In trials involving individuals with suspected AD, modest improvements in sleep quality (reduced sleep latency, improved sleep efficiency) and increased total sleep time were observed in some cases (Asayama et al., 2003; Riemersma-van der Lek et al., 2008), particularly when melatonin treatments were combined with daily bright light therapy (Riemersma-van der Lek et al., 2008). However, other trials failed to identify any effects on circadian rhythms of activity, sleep, or cognitive symptoms (Singer et al., 2003; Gehrman et al., 2009; reviewed in Urrestarazu and Iriarte, 2016). A beneficial effect of melatonin supplements has been reported for behaviors associated with sundown syndrome, but this effect has not been found consistently in randomized controlled trials (Riemersma-van der Lek et al., 2008; de Jonghe et al., 2010).
The lack of evidence that timed light exposure and melatonin administration improve the non-circadian symptoms of AD, PD and HD would seem to undermine the idea that the circadian system contributes to the etiology of these neurodegenerative diseases. It is likely, however, that some methodological inconsistencies across trials have contributed to these inconclusive findings. For example, studies evaluating light exposure have varied markedly in the intensity of light used; the timing of light exposure; and clinical characteristics of the participants (Forbes et al., 2014). Similarly, variability in the dosage, timing of administration, and characteristics of the sample under study may have contributed to the inconsistency of findings regarding the impact of melatonin (Urrestarazu and Iriarte, 2016). Furthermore, to our knowledge, no longitudinal studies have yet evaluated whether circadian-oriented interventions moderate the long-term progression of neurodegenerative disease symptoms. Careful consideration of these methodological details and the incorporation of long-term follow-up intervals would be of benefit in the design of future research. As the circadian features of neurodegenerative diseases may reflect a desynchronization of tissue oscillators downstream of SCN control from the master clock, an additional avenue of therapeutic intervention concerns the powerful influence of timed food delivery in the entrainment of circadian rhythms (Maywood et al., 2010; Cermakian et al., 2011). The circadian system retains sensitivity to food as a time cue, or zeitgeber, over the course of healthy aging (Walcott and Tate, 1996). Given that timed food delivery is a highly potent zeitgeber and does not exert its entrainment effects via the SCN (Boulos et al., 1980), it is possible that timed feeding or metabolic cues could serve to re-synchronize disrupted circadian timing (for a review see Kent, 2014). Indeed, some evidence suggests that food intake patterns in individuals with suspected AD or associated dementias vary with the progression of illness. For example, institutionalized elderly with symptoms of AD tend to consume less food in the afternoon and evening compared with those without symptoms of AD, and breakfast becomes the primary meal providing the greatest amount of energy intake for the day (Young and Greenwood, 2001).
Recent studies suggest that imposing restricted meal times could mitigate some of the circadian symptoms of neurodegeneration. In the R6/2 rodent model of HD, the restriction of food access to a 6-h window in the light phase restored a rhythm of locomotor activity and altered clock gene expression patterns in liver, compared with wild type controls (Maywood et al., 2010). The use of a dark-phase restricted feeding schedule also appears to delay the developmental onset of the HD phenotype in R6/2 mice, and increases core body temperature compared with wild type controls (Skillings et al., 2014). Further research into the effects of timed food restriction using animal models of AD and PD would be valuable to pursue.
Conclusions
Taken together, a growing body of evidence strongly implicates the circadian system in the onset and expression of AD, PD and HD. Disruptions to normal rhythmic processes are increasingly recognized as characteristic features of these disease states, and these disruptions may serve as early indicators of developing pathology. At the molecular level, clock genes regulate a number of genes and biochemical processes that contribute directly to neurodegeneration. Although it is currently unclear whether the circadian system plays a causal role in pathogenesis, further research may clarify this relationship. The advancement of knowledge on this subject may foster the development of screening tools to identify individuals at early stages of neurodegeneration, and may perhaps open a new realm of therapeutic interventions. Given the projected increase in the prevalence of neurodegenerative diseases in the coming years (Sosa-Ortiz et al., 2012), these advancements would be both timely and welcome.
Author Contributions
SH and SA wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada (37358.2013), les Fonds de la Recherché en Santé Québec, and the Canadian Institutes for Health Research (MOP142458).
References
Abbott, R. D., Ross, G. W., White, L. R., Tanner, C. M., Masaki, K. H., Nelson, J. S., et al. (2005). Excessive daytime sleepiness and subsequent development of Parkinson disease. Neurology 65, 1442–1446. doi: 10.1212/01.wnl.0000183056.89590.0d
Abbott, S. M., and Videnovic, A. (2016). Chronic sleep disturbance and neural injury: links to neurodegenerative disease. Nat. Sci. Sleep 8, 55–61. doi: 10.2147/NSS.S78947
Ancoli-Israel, S., Gehrman, P., Martin, J. L., Shochat, T., Marler, M., Corey-Bloom, J., et al. (2003). Increased light exposure consolidates sleep and strengthens circadian rhythms in severe Alzheimer’s disease patients. Behav. Sleep Med. 1, 22–36. doi: 10.1207/S15402010BSM0101_4
Ancoli-Israel, S., Klauber, M. R., Jones, D. W., Kripke, D. F., Martin, J., Mason, W., et al. (1997). Variations in circadian rhythms of activity, sleep and light exposure related to dementia in nursing-home patients. Sleep 20, 18–23. doi: 10.1093/sleep/20.1.18
Ancoli-Israel, S., Martin, J. L., Kripke, D. F., Marler, M., and Klauber, M. R. (2002). Effect of light treatment on sleep and circadian rhythms in demented nursing home patients. J. Am. Geriatr. Soc. 50, 282–289. doi: 10.1046/j.1532-5415.2002.50060.x
Archer, S. N., and Oster, H. (2015). How sleep and wakefulness influence circadian rhythmicity: effects of insufficient and mistimed sleep on the animal and human transcriptome. J. Sleep Res. 24, 476–493. doi: 10.1111/jsr.12307
Arendt, J. (2012). Biological rhythms during residence in polar regions. Chronobiol. Int. 29, 379–394. doi: 10.3109/07420528.2012.668997
Asayama, K., Yamadera, H., Ito, T., Suzuki, H., Kudo, Y., and Endo, S. (2003). Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. J. Nippon Med. Sch. 70, 334–341. doi: 10.1272/jnms.70.334
Aziz, N. A., Pijl, H., Frölich, M., Schröder-van der Elst, J. P., van der Bent, C., Roelfsema, F., et al. (2009). Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease. J. Neurol. 256, 1961–1965. doi: 10.1007/s00415-009-5196-1
Bachman, D., and Rabins, P. (2006). “Sundowning” and other temporally associated agitation states in dementia patients. Annu. Rev. Med. 57, 499–511. doi: 10.1146/annurev.med.57.071604.141451
Barone, P., Antonini, A., Colosimo, C., Marconi, R., Morgante, L., and Avarello, T. P. (2009). The PRIAMO study: a multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov. Disord. 24, 1641–1649. doi: 10.1002/mds.22643
Bélanger, V., Picard, N., and Cermakian, N. (2006). The circadian regulation of presenilin-2 gene expression. Chronobiol. Int. 23, 747–766. doi: 10.1080/07420520600827087
Bell-Pedersen, D., Cassone, V. M., Earnest, D. J., Golden, S. S., Hardin, P. E., Thomas, T. L., et al. (2005). Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 6, 544–556. doi: 10.1038/nrg1633
Bianchetti, A., Scuratti, A., Zanetti, O., Binetti, G., Frisoni, G. B., Magni, E., et al. (1995). Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia 6, 108–112. doi: 10.1159/000106930
Bliwise, D. L., Carroll, J. S., Lee, K. A., Nekich, J. C., and Dement, W. C. (1993). Sleep and “sundowning” in nursing home patients with dementia. Psychiatry Res. 48, 277–292. doi: 10.1016/0165-1781(93)90078-U
Bonanni, E., Maestri, M., Tognoni, G., Fabbrini, M., Nucciarone, B., Manca, M. L., et al. (2005). Daytime sleepiness in mild and moderate Alzheimer’s disease and its relationship with cognitive impairment. J. Sleep Res. 14, 311–317. doi: 10.1111/j.1365-2869.2005.00462.x
Boulos, Z., Rosenwasser, A. M., and Terman, M. (1980). Feeding schedules and the circadian organization of behavior in the rat. Behav. Brain Res. 1, 39–65. doi: 10.1016/0166-4328(80)90045-5
Breen, D. P., Vuono, R., Nawarathna, U., Fisher, K., Shneerson, J. M., Reddy, A. B., et al. (2014). Sleep and circadian rhythm regulation in early Parkinson’s disease. JAMA Neurol. 71, 589–595. doi: 10.1001/jamaneurol.2014.65
Cai, Y., Liu, S., Sothern, R. B., Xu, S., and Chan, P. (2010). Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease. Eur. J. Neurol. 17, 550–554. doi: 10.1111/j.1468-1331.2009.02848.x
Cermakian, N., Lamont, E. W., Boudreau, P., and Boivin, D. B. (2011). Circadian clock gene expression in brain regions of Alzheimer’s disease patients and control subjects. J. Biol. Rhythms 26, 160–170. doi: 10.1177/0748730410395732
Comella, C. L. (2007). Sleep disorders in Parkinson’s disease: an overview. Mov. Disord. 22, S367–S373. doi: 10.1002/mds.21682
de Jonghe, A., Korevaar, J. C., van Munster, B. C., and de Rooij, S. E. (2010). Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. are there implications for delirium? A systematic review. Int. J. Geriatr. Psychiatry 25, 1201–1208. doi: 10.1002/gps.2454
Duffield, G. E. (2003). DNA microarray analyses of circadian timing: the genomic basis of biological time. J. Neuroendocrinol. 15, 991–1002. doi: 10.1046/j.1365-2826.2003.01082.x
Duffy, J. F., Zitting, K. M., and Chinoy, E. D. (2015). Aging and circadian rhythms. Sleep Med. Clin. 10, 423–434. doi: 10.1016/j.jsmc.2015.08.002
Esler, W. P., and Wolfe, M. S. (2001). A portrait of Alzheimer secretases—new features and familiar faces. Science 293, 1449–1454. doi: 10.1126/science.1064638
Fahrenkrug, J., Popovic, N., Georg, B., Brundin, P., and Hannibal, J. (2007). Decreased VIP and VPAC2 receptor expression in the biological clock of the R6/2 Huntington’s disease mouse. J. Mol. Neurosci. 31, 139–148. doi: 10.1385/JMN/31:02:139
Farajnia, S., Michel, S., Deboer, T., vanderLeest, H. T., Houben, T., and Rohling, J. H. (2012). Evidence for neuronal desynchrony in the aged suprachiasmatic nucleus clock. J. Neurosci. 32, 5891–5899. doi: 10.1523/JNEUROSCI.0469-12.2012
Fifel, K., and Cooper, H. M. (2014). Loss of dopamine disrupts circadian rhythms in a mouse model of Parkinson’s disease. Neurobiol. Dis. 71, 359–369. doi: 10.1016/j.nbd.2014.08.024
Fisher, S. P., Black, S. W., Schwartz, M. D., Wilk, A. J., Chen, T. M., Lincoln, W. U., et al. (2013). Longitudinal analysis of the electroencephalogram and sleep phenotype in the R6/2 mouse model of Huntington’s disease. Brain 136, 2159–2172. doi: 10.1093/brain/awt132
Forbes, D., Blake, C. M., Thiessen, E. J., Peacock, S., and Hawranik, P. (2014). Light therapy for improving cognition, activities of daily living, sleep, challenging behaviour and psychiatric disturbances in dementia. Cochrane Database Syst. Rev. CD003946. doi: 10.1002/14651858.CD003946.pub4
Gallagher-Thompson, D., Brooks, J. O., Bliwise, D., Leader, J., and Yesavage, J. A. (1992). The relations among caregiver stress, “sundowning” symptoms and cognitive decline in Alzheimer’s disease. J. Am. Geriat. Soc. 40, 807–810. doi: 10.1111/j.1532-5415.1992.tb01853.x
Gehrman, P. R., Connor, D. J., Martin, J. L., Shochat, T., Corey-Bloom, J., and Ancoli-Israel, S. (2009). Melatonin fails to improve sleep or agitation in double-blind randomized placebo-controlled trial of institutionalized patients with Alzheimer disease. Am. J. Geriatr. Psychiatry 17, 166–169. doi: 10.1097/JGP.0b013e318187de18
Giri, M., Zhang, M., and Lü, Y. (2016). Genes associated with Alzheimer’s disease: an overview and current status. Clin. Interv. Aging 11, 665–681. doi: 10.2147/CIA.S105769
Giubilei, F., Patacchioli, F. R., Antonini, G., Sepe Monti, M., Tisei, P., Bastianello, S., et al. (2001). Altered circadian cortisol secretion in Alzheimer’s disease: clinical and neuroradiological aspects. J. Neurosci. Res. 66, 262–265. doi: 10.1002/jnr.1219
Gravotta, L., Gavrila, A. M., Hood, S., and Amir, S. (2011). Global depletion of dopamine using intracerebroventricular 6-hydroxydopamine injection disrupts normal circadian wheel-running patterns and PERIOD2 expression in the rat forebrain. J. Mol. Neurosci. 45, 162–171. doi: 10.1007/s12031-011-9520-8
Graybeal, J. J., Bozzelli, P. L., Graybeal, L. L., Groeber, C. M., McKnight, P. E., Cox, D. N., et al. (2015). Human ApoE ɛ4 alters circadian rhythm activity, IL-1β and GFAP in CRND8 mice. J. Alzheimers Dis. 43, 823–834. doi: 10.3233/JAD-132009
Grimm, S., Hoehn, A., Davies, K. J., and Grune, T. (2011). Protein oxidative modifications in the ageing brain: consequence for the onset of neurodegenerative disease. Free Radic. Res. 45, 73–88. doi: 10.3109/10715762.2010.512040
Gu, Z., Wang, B., Zhang, Y. B., Ding, H., Zhang, Y., Yu, J., et al. (2015). Association of ARNTL and PER1 genes with Parkinson’s disease: a case-control study of han chinese. Sci. Rep. 5:15891. doi: 10.1038/srep15891
Harper, D. G., Volicer, L., Stopa, E. G., McKee, A. C., Nitta, M., and Satlin, A. (2005). Disturbance of endogenous circadian rhythm in aging and Alzheimer disease. Am. J. Geriatr. Psychiatry 13, 359–368. doi: 10.1176/appi.ajgp.13.5.359
Hartmann, A., Veldhuis, J. D., Deuschle, M., Standhardt, H., and Heuser, I. (1997). Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol. Aging 18, 285–289. doi: 10.1016/s0197-4580(97)80309-0
Hastings, M. H., and Goedert, M. (2013). Circadian clocks and neurodegenerative diseases: time to aggregate? Curr. Opin. Neurobiol. 23, 880–887. doi: 10.1016/j.conb.2013.05.004
Hatfield, C. F., Herbert, J., van Someren, E. J., Hodges, J. R., and Hastings, M. H. (2004). Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer’s dementia. Brain 127, 1061–1074. doi: 10.1093/brain/awh129
Hood, S., and Amir, S. (2017). The aging clock: circadian rhythms and later life. J. Clin. Invest. 127, 437–446. doi: 10.1172/JCI90328
Hood, S., Cassidy, P., Cossette, M. P., Weigl, Y., Verwey, M., Robinson, B., et al. (2010). Endogenous dopamine regulates the rhythm of expression of the clock protein PER2 in the rat dorsal striatum via daily activation of D2 dopamine receptors. J. Neurosci. 30, 14046–14058. doi: 10.1523/JNEUROSCI.2128-10.2010
Huang, W., Ramsey, K. M., Marcheva, B., and Bass, J. (2011). Circadian rhythms, sleep and metabolism. J. Clin. Invest. 121, 2133–2141. doi: 10.1172/JCI46043
Iranzo, A., Tolosa, E., Gelpi, E., Molinuevo, J. L., Valldeoriola, F., Serradell, M., et al. (2013). Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. 12, 443–453. doi: 10.1016/S1474-4422(13)70056-5
Jang, Y. S., Lee, M. H., Lee, S. H., and Bae, K. (2011). Cu/zn superoxide dismutase is differentially regulated in period gene-mutant mice. Biochem. Biophys. Res. Commun. 409, 22–27. doi: 10.1016/j.bbrc.2011.04.099
Ju, Y. E., McLeland, J. S., Toedebusch, C. D., Xiong, C., Fagan, A. M., Duntley, S. P., et al. (2013). Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 70, 587–593. doi: 10.1001/jamaneurol.2013.2334
Kalliolia, E., Silajdžić, E., Nambron, R., Hill, N. R., Doshi, A., Frost, C., et al. (2014). Plasma melatonin is reduced in Huntington’s disease. Mov. Disord. 29, 1511–1515. doi: 10.1002/mds.26003
Kent, B. A. (2014). Synchronizing an aging brain: can entraining circadian clocks by food slow Alzheimer’s disease? Front. Aging Neurosci. 6:234. doi: 10.3389/fnagi.2014.00234
Kondratov, R. V., Kondratova, A. A., Gorbacheva, V. Y., Vykhovanets, O. V., and Antoch, M. P. (2006). Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 20, 1868–1873. doi: 10.1101/gad.1432206
Kondratova, A. A., and Kondratov, R. V. (2012). The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 13, 325–335. doi: 10.1038/nrn3208
Krishnan, N., Kretzschmar, D., Rakshit, K., Chow, E., and Giebultowicz, J. M. (2009). The circadian clock gene period extends healthspan in aging Drosophila melanogaster. Aging 1, 937–948. doi: 10.18632/aging.100103
Kudo, T., Schroeder, A., Loh, D. H., Kuljis, D., Jordan, M. C., Roos, K. P., et al. (2011). Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp. Neurol. 228, 80–90. doi: 10.1016/j.expneurol.2010.12.011
Lee, J., Moulik, M., Fang, Z., Saha, P., Zou, F., Xu, Y., et al. (2013). Bmal1 and β-cell clock are required for adaptation to circadian disruption and their loss of function leads to oxidative stress-induced β-cell failure in mice. Mol. Cell. Biol. 33, 2327–2338. doi: 10.1128/MCB.01421-12
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977. doi: 10.1126/science.7638622
Li, J. Z., Bunney, B. G., Meng, F., Hagenauer, M. H., Walsh, D. M., Vawter, M. P., et al. (2013). Circadian patterns of gene expression in the human brain and disruption in major depressive disorder. Proc. Natl. Acad. Sci. U S A 110, 9950–9955. doi: 10.1073/pnas.1305814110
Loh, D. H., Kudo, T., Truong, D., Wu, Y., and Colwell, C. S. (2013). The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS One 8:e69993. doi: 10.1371/journal.pone.0069993
Long, D. M., Blake, M. R., Dutta, S., Holbrook, S. D., Kotwica-Rolinska, J., Kretzschmar, D., et al. (2014). Relationships between the circadian system and Alzheimer’s disease-like symptoms in drosophila. PloS One 9:e106068. doi: 10.1371/journal.pone.0106068
Lucassen, E. A., Coomans, C. P., van Putten, M., de Kreij, S. R., van Genugten, J. H., Sutorius, R. P., et al. (2016). Environmental 24-hr cycles are essential for health. Curr. Biol. 26, 1843–1853. doi: 10.1016/j.cub.2016.05.038
Martin, J., Marler, M., Shochat, T., and Ancoli-Israel, S. (2000). Circadian rhythms of agitation in institutionalized patients with Alzheimer’s disease. Chronobiol. Int. 17, 405–418. doi: 10.1081/cbi-100101054
Mattis, J., and Sehgal, A. (2016). Circadian rhythms, sleep and disorders of aging. Trends Endocrinol. Metab. 27, 192–203. doi: 10.1016/j.tem.2016.02.003
Maywood, E. S., Fraenkel, E., McAllister, C. J., Wood, N., Reddy, A. B., Hastings, M. H., et al. (2010). Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J. Neurosci. 30, 10199–10204. doi: 10.1523/JNEUROSCI.1694-10.2010
McCurry, S. M., Logsdon, R. G., Teri, L., Gibbons, L. E., Kukull, W. A., Bowen, J. D., et al. (1999). Characteristics of sleep disturbance in community-dwelling Alzheimer’s disease patients. J. Geriatr. Psychiatry Neurol. 12, 53–59. doi: 10.1177/089198879901200203
McCurry, S. M., Pike, K. C., Vitiello, M. V., Logsdon, R. G., Larson, E. B., and Teri, L. (2011). Increasing walking and bright light exposure to improve sleep in community-dwelling persons with Alzheimer’s disease: results of a randomized, controlled trial. J. Am. Geriatr. Soc. 59, 1393–1402. doi: 10.1111/j.1532-5415.2011.03519.x
McFadden, E., Jones, M. E., Schoemaker, M. J., Ashworth, A., and Swerdlow, A. J. (2014). The relationship between obesity and exposure to light at night: cross-sectional analyses of over 100,000 women in the breakthrough generations study. Am. J. Epidemiol. 180, 245–250. doi: 10.1093/aje/kwu117
Medeiros, C. A., Carvalhedo de Bruin, P. F., Lopes, L. A., Magalhães, M. C., de Lourdes Seabra, M., and de Bruin, V. M. (2007). Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson’s disease. a randomized, double blind, placebo-controlled study. J. Neurol. 254, 459–464. doi: 10.1007/s00415-006-0390-x
Merlino, G., Piani, A., Gigli, G. L., Cancelli, I., Rinaldi, A., Baroselli, A., et al. (2010). Daytime sleepiness is associated with dementia and cognitive decline in older italian adults: a population-based study. Sleep Med. 11, 372–377. doi: 10.1016/j.sleep.2009.07.018
Mishima, K., Tozawa, T., Satoh, K., Matsumoto, Y., Hishikawa, Y., and Okawa, M. (1999). Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol. Psychiatry 45, 417–421. doi: 10.1016/s0006-3223(97)00510-6
Mohawk, J. A., Green, C. B., and Takahashi, J. S. (2012). Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 35, 445–462. doi: 10.1146/annurev-neuro-060909-153128
Morris, C. J., Purvis, T. E., Hu, K., and Scheer, F. A. (2016). Circadian misalignment increases cardiovascular disease risk factors in humans. Proc. Natl. Acad. Sci. U S A 113, E1402–E1411. doi: 10.1073/pnas.1516953113
Morton, A. J., Wood, N. I., Hastings, M. H., Hurelbrink, C., Barker, R. A., and Maywood, E. S. (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 25, 157–163. doi: 10.1523/JNEUROSCI.3842-04.2005
Neurological Health Charities Canada. (2014). Mapping connections: An understanding of neurological conditions in Canada. Available online at: http://www.phac-aspc.gc.ca/publicat/cd-mc/mc-ec/index-eng.php
Ono, K., Mochizuki, H., Ikeda, T., Nihira, T., Takasaki, J., Teplow, D. B., et al. (2012). Effect of melatonin on α-synuclein self-assembly and cytotoxicity. Neurobiol. Aging 33, 2172–2185. doi: 10.1016/j.neurobiolaging.2011.10.015
Pallier, P. N., Maywood, E. S., Zheng, Z., Chesham, J. E., Inyushkin, A. N., Dyball, R., et al. (2007). Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J. Neurosci. 27, 7869–7878. doi: 10.1523/jneurosci.0649-07.2007
Panda, S., Antoch, M. P., Miller, B. H., Su, A. I., Schook, A. B., Straume, M., et al. (2002). Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109, 307–320. doi: 10.1016/s0092-8674(02)00722-5
Paus, S., Schmitz-Hübsch, T., Wüllner, U., Vogel, A., Klockgether, T., and Abele, M. (2007). Bright light therapy in Parkinson’s disease: a pilot study. Mov. Disord. 22, 1495–1498. doi: 10.1002/mds.21542
Pierangeli, G., Provini, F., Maltoni, P., Barletta, G., Contin, M., Lugaresi, E., et al. (2001). Nocturnal body core temperature falls in Parkinson’s disease but not in multiple-system atrophy. Mov. Disord. 16, 226–232. doi: 10.1002/mds.1039
Pollak, C. P., and Perlick, D. (1991). Sleep problems and institutionalization of the elderly. J. Geriatr. Psychiatry Neurol. 4, 204–210. doi: 10.1177/089198879100400405
Postuma, R. B., Gagnon, J. F., Bertrand, J. A., Génier Marchand, D., and Montplaisir, J. Y. (2015). Parkinson risk in idiopathic REM sleep behavior disorder: preparing for neuroprotective trials. Neurology 84, 1104–1113. doi: 10.1212/WNL.0000000000001364
Reiter, R. J., Tan, D. X., Manchester, L. C., and El-Sawi, M. R. (2002). Melatonin reduces oxidant damage and promotes mitochondrial respiration: implications for aging. Ann. N Y Acad. Sci. 959, 238–250. doi: 10.1111/j.1749-6632.2002.tb02096.x
Riemersma-van der Lek, R. F., Swaab, D. F., Twisk, J., Hol, E. M., Hoogendijk, W. J., and Van Someren, E. J. (2008). Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA 299, 2642–2655. doi: 10.1001/jama.299.22.2642
Roybal, K., Theobold, D., Graham, A., DiNieri, J. A., Russo, S. J., Krishnan, V., et al. (2007). Mania-like behavior induced by disruption of CLOCK. Proc. Natl. Acad. Sci. U S A 104, 6406–6411. doi: 10.1073/pnas.0609625104
Satlin, A., Volicer, L., Stopa, E. G., and Harper, D. (1995). Circadian locomotor activity and core-body temperature rhythms in Alzheimer’s disease. Neurobiol. Aging 16, 765–771. doi: 10.1016/0197-4580(95)00059-N
Schenck, C. H., Boeve, B. F., and Mahowald, M. W. (2013). Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-years update on a previously reported series. Sleep Med. 14, 744–748. doi: 10.1016/j.sleep.2012.10.009
Shan, L., Dauvilliers, Y., and Siegel, J. M. (2015). Interactions of the histamine and hypocretin systems in CNS disorders. Nat. Rev. Neurol. 11, 401–413. doi: 10.1038/nrneurol.2015.99
Shochat, T., Martin, J., Marler, M., and Ancoli-Israel, S. (2000). Illumination levels in nursing home patients: effects on sleep and activity rhythms. J. Sleep Res. 9, 373–379. doi: 10.1046/j.1365-2869.2000.00221.x
Singer, C., Tractenberg, R. E., Kaye, J., Schafer, K., Gamst, A., Grundman, M., et al. (2003). A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep 26, 893–901. doi: 10.1093/sleep/26.7.893
Skillings, E. A., Wood, N. I., and Morton, A. J. (2014). Beneficial effects of environmental enrichment and food entrainment in the R6/2 mouse model of Huntington’s disease. Brain Behav. 4, 675–686. doi: 10.1002/brb3.235
Sosa-Ortiz, A. L., Acosta-Castillo, I., and Prince, M. J. (2012). Epidemiology of dementias and Alzheimer’s disease. Arch. Med. Res. 43, 600–608. doi: 10.1016/j.arcmed.2012.11.003
Stevens, R. G., Brainard, G. C., Blask, D. E., Lockley, S. W., and Motta, M. E. (2014). Breast cancer and circadian disruption from electric lighting in the modern world. CA Cancer J Clin. 64, 207–218. doi: 10.3322/caac.21218
Stopa, E. G., Volicer, L., Kuo-Leblanc, V., Harper, D., Lathi, D., Tate, B., et al. (1999). Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J. Neuropathol. Exp. Neurol. 58, 29–39. doi: 10.1097/00005072-199901000-00004
Swaab, D. F., Fliers, E., and Partiman, T. S. (1985). The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 342, 37–44. doi: 10.1016/0006-8993(85)91350-2
Touitou, Y., and Haus, E. (2000). Alterations with aging of the endocrine and neuroendocrine circadian system in humans. Chronobiol. Int. 17, 369–390. doi: 10.1081/cbi-100101052
Tranah, G. J., Blackwell, T., Stone, K. L., Ancoli-Israel, S., Paudel, M. L., Ensrud, K. E., et al. (2011). Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann. Neurol. 70, 722–732. doi: 10.1002/ana.22468
Urrestarazu, E., and Iriarte, J. (2016). Clinical management of sleep disturbances in Alzheimer’s disease: current and emerging strategies. Nat. Sci. Sleep 8, 21–33. doi: 10.2147/NSS.S76706
Van Someren, E. J. (2000). More than a marker: interaction between the circadian regulation of temperature and sleep, age-related changes and treatment possibilities. Chronobiol. Int. 17, 313–354. doi: 10.1081/cbi-100101050
Vezoli, J., Fifel, K., Leviel, V., Dehay, C., Kennedy, H., Cooper, H. M., et al. (2011). Early presymptomatic and long-term changes of rest activity cycles and cognitive behavior in a MPTP-monkey model of Parkinson’s disease. PLoS One 6:e23952. doi: 10.1371/journal.pone.0023952
Videnovic, A., Klerman, E. B., Wang, W., Marconi, A., Kuhta, T., and Zee, P. C. (2017). Timed light therapy for sleep and daytime sleepiness associated with Parkinson disease: a randomized clinical trial. JAMA Neurol. 74, 411–418. doi: 10.1001/jamaneurol.2016.5192
Videnovic, A., Lazar, A. S., Barker, R. A., and Overeem, S. (2014a). “The clocks that time us”—circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 10, 683–693. doi: 10.1038/nrneurol.2014.206
Videnovic, A., Noble, C., Reid, K. J., Peng, J., Turek, F. W., Marconi, A., et al. (2014b). Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol. 71, 463–469. doi: 10.1001/jamaneurol.2013.6239
Videnovic, A., and Zee, P. C. (2015). Consequences of circadian disruption on neurologic health. Sleep Med. Clin. 10, 469–480. doi: 10.1016/j.jsmc.2015.08.004
Volicer, L., Harper, D. G., Manning, B. C., Goldstein, R., and Satlin, A. (2001). Sundowning and circadian rhythms in Alzheimer’s disease. Am. J. Psychiatry 158, 704–711. doi: 10.1176/appi.ajp.158.5.704
Walcott, E. C., and Tate, B. A. (1996). Entrainment of aged, dysrhythmic rats to a restricted feeding schedule. Physiol. Behav. 60, 1205–1208. doi: 10.1016/s0031-9384(96)00215-6
Waller, K. L., Mortensen, E. L., Avlund, K., Fagerlund, B., Lauritzen, M., Gammeltoft, S., et al. (2016). Melatonin and cortisol profiles in late midlife and their association with age-related changes in cognition. Nat. Sci. Sleep 8, 47–53. doi: 10.2147/NSS.S75946
Wang, J. L., Lim, A. S., Chiang, W. Y., Hsieh, W. H., Lo, M. T., Schneider, J. A., et al. (2015). Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann. Neurol. 78, 317–322. doi: 10.1002/ana.24432
Wang, X., Sirianni, A., Pei, Z., Cormier, K., Smith, K., and Jiang, J. (2011). The melatonin MT1 receptor axis modulates mutant huntingtin-mediated toxicity. J. Neurosci. 31, 14496–14507. doi: 10.1523/JNEUROSCI.3059-11.2011
Weissová, K., Bartoš, A., Sládek, M., Nováková, M., and Sumová, A. (2016). Moderate changes in the circadian system of Alzheimer’s disease patients detected in their home environment. PLoS One 11:e0146200. doi: 10.1371/journal.pone.0146200
Welsh, D. K., Takahashi, J. S., and Kay, S. A. (2010). Suprachiasmatic nucleus: cell autonomy and network properties. Annu. Rev. Physiol. 72, 551–577. doi: 10.1146/annurev-physiol-021909-135919
Wilking, M., Ndiaye, M., Mukhtar, H., and Ahmad, N. (2013). Circadian rhythm connections to oxidative stress: implications for human health. Antioxid. Redox Signal. 19, 192–208. doi: 10.1089/ars.2012.4889
Wu, Y. H., Feenstra, M. G., Zhou, J. N., Liu, R. Y., Toranõ, J. S., Van Kan, H. J., et al. (2003). Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J. Clin. Endocrinol. Metab. 88, 5898–5906. doi: 10.1210/jc.2003-030833
Wu, Y. H., Fischer, D. F., Kalsbeek, A., Garidou-Boof, M. L., van der Vliet, J., van Heijningen, C., et al. (2006). Pineal clock gene oscillation is disturbed in Alzheimer’s disease, due to functional disconnection from the “master clock”. FASEB J. 20, 1874–1876. doi: 10.1096/fj.05-4446fje
Wu, Y. H., Zhou, J. N., Van Heerikhuize, J., Jockers, R., and Swaab, D. F. (2007). Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol. Aging 28, 1239–1247. doi: 10.1016/j.neurobiolaging.2006.06.002
Yesavage, J. A., Friedman, L., Ancoli-Israel, S., Bliwise, D., Singer, C., Vitiello, M. V., et al. (2003). Development of diagnostic criteria for defining sleep disturbance in Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 16, 131–139. doi: 10.1177/0891988703255684
Young, K. W., and Greenwood, C. E. (2001). Shift in diurnal feeding patterns in nursing home residents with Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 56, M700–M706. doi: 10.1093/gerona/56.11.m700
Keywords: circadian rhythms, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, neurodegeneration, sleep, clock genes
Citation: Hood S and Amir S (2017) Neurodegeneration and the Circadian Clock. Front. Aging Neurosci. 9:170. doi: 10.3389/fnagi.2017.00170
Received: 10 January 2017; Accepted: 15 May 2017;
Published: 30 May 2017.
Edited by:
Catarina Oliveira, University of Coimbra, PortugalReviewed by:
Lakshmi Rajagopal, Northwestern University, United StatesUmesh Gangishetti, Emory University, United States
Takayoshi Ubuka, Monash University Malaysia, Malaysia
Copyright © 2017 Hood and Amir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shimon Amir, c2hpbW9uLmFtaXJAY29uY29yZGlhLmNh