 Camilla Lauridsen1
Camilla Lauridsen1 Sigrid B. Sando1,2
Sigrid B. Sando1,2 Ina Møller2
Ina Møller2 Guro Berge1
Guro Berge1 Precious K. Pomary1
Precious K. Pomary1 Gøril R. Grøntvedt1,2
Gøril R. Grøntvedt1,2 Øyvind Salvesen3
Øyvind Salvesen3 Geir Bråthen1,2
Geir Bråthen1,2 Linda R. White1,2*
Linda R. White1,2*- 1Department of Neuromedicine and Movement Science, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway
- 2Department of Neurology, Trondheim University Hospital, Trondheim, Norway
- 3Unit for Applied Clinical Research, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway
Background: Amyloid beta 1–43 (Aβ43) may be a useful additional biomarker for diagnosing Alzheimer’s disease (AD). We have investigated cerebrospinal fluid (CSF) levels of Aβ43 in patients with early-onset AD in contrast to levels in late-onset AD. For comparison, in addition to the ‘core’ biomarkers, several other analytes were also determined [YKL-40, neurofilament light (NF-L), glial fibrillary acidic protein (GFAP), and progranulin].
Material and Methods: Cerebrospinal fluid samples were obtained from patients with early-onset AD (age ≤ 62, n = 66), late-onset AD (age ≥ 68, n = 25), and groups of cognitively intact individuals (age ≤ 62, n = 41, age ≥ 68, n = 39). Core CSF AD biomarkers [amyloid beta 1–42 (Aβ42), total tau, phosphorylated tau] were analyzed, as well as levels of Aβ43 and other analytes, using commercially available enzyme-linked immunosorbent assays.
Results: Cerebrospinal fluid Aβ43 was significantly reduced in early-onset AD compared to late-onset AD (14.8 ± 7.3 vs. 21.8 ± 9.4 pg/ml, respectively), whereas the levels of Aβ42 in the two AD groups were not significantly different (474.9 ± 142.0 vs. 539.6 ± 159.9 pg/ml, respectively). Aβ43 and all core biomarkers were significantly altered in patients with AD compared to corresponding controls. NF-L was significantly increased in early-onset AD compared to younger controls, an effect not found between the older groups. Relationships between the Aβ peptides and tau proteins, YKL-40, NF-L, GFAP and progranulin were also investigated without finding marked associations. However, age-associated increases in levels of tau proteins, YKL-40, NF-L and GFAP were found with respect to age in healthy controls. Results for these other analytes were similar to previously published data. Aβ43 did not improve diagnostic accuracy in either AD group compared to Aβ42. Discussion: Cerebrospinal fluid Aβ43, but not Aβ42 levels, varied significantly with age in patients with AD. If CSF levels of Aβ peptides reflect amyloid deposition in brain, the possibility arises that there is a difference between Aβ43 and Aβ42 deposition in younger compared to older brain. However, the level of Aβ43 in CSF shows no improvement over Aβ42 regarding diagnostic accuracy.
Introduction
Alzheimer’s disease (AD) is often separated according to age, whereby onset prior to age 65 years is considered to be early-onset AD, while onset from an age of 65 years (which is much more common) is termed late-onset AD. Although the pathological burden of amyloid plaques and neurofibrillary tangles has been shown to be greater in early-onset AD than in patients with late-onset AD (Ho et al., 2002; Marshall et al., 2007), imaging studies have indicated the global burden to be similar between the two groups (Rabinovici et al., 2010), though sometimes with variation in regional anatomical distribution of amyloid (Ossenkoppele et al., 2012; Cho et al., 2013). Such putative differences in the distribution of amyloid pathology have not been found to alter levels of the core cerebrospinal fluid (CSF) biomarkers for AD; amyloid beta 1–42 (Aβ42), total tau (t-tau) and phosphorylated tau (p-tau) protein, in early- compared to late-onset AD (Bouwman et al., 2009; Chiaravalloti et al., 2016). Additionally, several studies have shown no correlation between the core biomarkers and age in AD patients (Bouwman et al., 2009; Mattsson et al., 2009; Popp et al., 2010).
However, healthy individuals display increased AD pathology with increasing age (Savva et al., 2009). Older control individuals have been found to have decreased CSF Aβ42 levels compared to younger controls (Bouwman et al., 2009), and the level was negatively correlated with age (Popp et al., 2010). Conversely, CSF t-tau and p-tau correlate positively with age in healthy elderly individuals (Blomberg et al., 2001; Glodzik-Sobanska et al., 2009; Jaworski et al., 2009; Alcolea et al., 2015).
Amyloid beta 1–43 (Aβ43), compared to Aβ42, has an additional threonine at the C-terminal through an alternative γ-secretase cleavage of amyloid precursor protein (APP), and is generally considered likely to be even more aggregation-prone than Aβ42 (Jarrett et al., 1993; Saito et al., 2011; Conicella and Fawzi, 2014), though recent kinetic experiments in vitro dispute this (Chemuru et al., 2016). Aβ43 has been hypothesized to play a role in AD pathogenesis despite its low concentration in human brain tissue, and found to be frequent in both neuritic and diffuse extracellular plaques in both familial and sporadic AD (Welander et al., 2009; Keller et al., 2010; Sandebring et al., 2013). In an APP-expressing transgenic mouse model, Aβ43 has been found to be the first amyloid peptide to deposit in brain (Zou et al., 2013), perhaps seeding subsequent Aβ42 deposition (Conicella and Fawzi, 2014). Moreover, knock-in mice bearing the pathogenic presenilin-1 R278I mutation demonstrated overproduction of Aβ43, impaired short-term memory and acceleration of amyloid-β pathology (Saito et al., 2011). Aβ43 correlates positively with age in patients with AD (Bruggink et al., 2013), and correlates closely with CSF Aβ42 both in patients and control individuals (Bruggink et al., 2013; Lauridsen et al., 2016). As far as we know, no study as yet has compared CSF levels of Aβ43 in early-onset AD with late-onset AD.
We have therefore investigated Aβ43 and Aβ42 in CSF from well-characterized cohorts of patients with early- and late-onset AD. The cut-off between these subtypes of AD has been accepted as 65 years of age, but in the present study we excluded patients with age at onset in the 5-year period 63–67 years to highlight potential age differences. Thus only patients with early-onset AD ≤ 62 years of age, or patients with late-onset AD who were aged ≥68 years were included.
In addition to the Aβ species in CSF from these two subgroups of patients with AD and corresponding control groups, levels of t-tau and p-tau were also determined. For further comparison, levels of several other analytes that have been investigated with respect to AD, and to age, were also assessed. These included two other cytoskeletal intermediate filaments; neurofilament light (NF-L) (Petzold et al., 2007; Vagberg et al., 2015; Olsson et al., 2016) and glial fibrillary acidic protein (GFAP) (Vagberg et al., 2015; Wennstrom et al., 2015), found, respectively, in neurons and glia, YKL-40 (also known as chitinase 3-like protein 1), a protease secreted mainly by astrocytes and considered a marker for gliosis and neuroinflammation (Craig-Schapiro et al., 2010), and progranulin, a growth factor believed to have anti-inflammatory and neuroprotective abilities (Jing et al., 2016).
Materials and Methods
Subjects
Study patients were ethnic Norwegians referred to the Department of Neurology, St. Olav’s Hospital (Trondheim University Hospital) by general practitioners, and diagnosed by a neurologist. Some patients were initially diagnosed with amnestic mild cognitive impairment (aMCI, n = 14) according to the International Working Group on Mild Cognitive Impairment criteria (Winblad et al., 2004), but all later developed AD within the next 2 years. Patients with AD were diagnosed according to the NINCDS-ADRDA criteria (McKhann et al., 1984), final total n = 91, whereof 66 were aged ≤62 years at onset (early-onset AD) and 25 were aged ≥68 years at onset of symptoms (late-onset AD).
As controls, CSF samples were obtained either from non-demented elderly volunteers (n = 35) recruited from societies for retired people or caregivers not genetically related to the patient, or from samples stored in the Neurological Research Biobank at the hospital (n = 45). These latter individuals had been referred to the clinic for suspected neurological conditions, but none was subsequently found. Of the total 80 control individuals, 41 were aged ≤62 years, and 39 were aged ≥68 years. For the control groups, CSF cell count, glucose and protein were within standard physiological limits.
The neurological examination performed on most study participants included the Mini Mental State Examination (MMSE) (Folstein et al., 1975). MMSE was performed on all patients, but for many control individuals there had been no reason to carry out an MMSE during the clinical work-up, and where MMSE was available, the minimum score was 28. For the same reason, APOE genotype was not available for most younger controls. The demographic data are shown in Table 1.
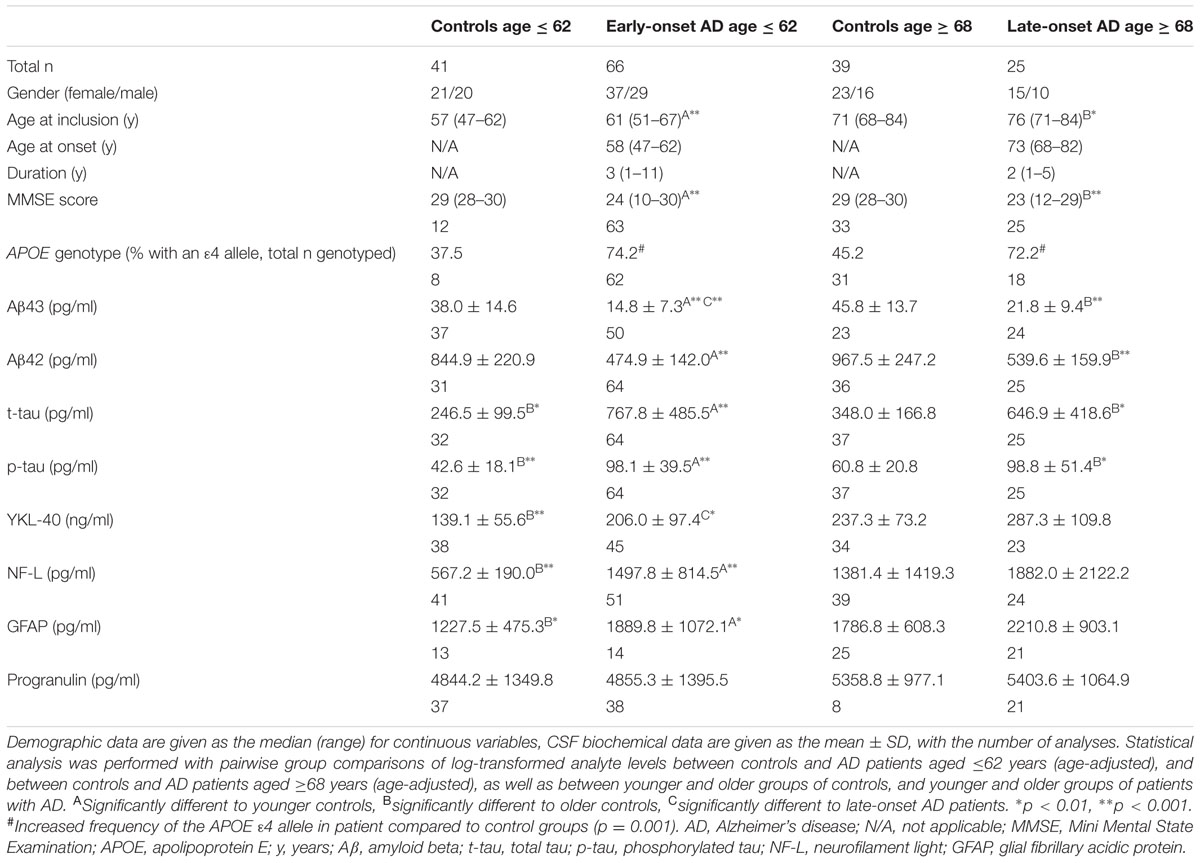
TABLE 1. Demographic and CSF biochemical data.
Sampling of CSF
Cerebrospinal fluid was collected with patients lying on their side, and lumbar puncture carried out at the level L4/L5 or L5/S1. The first 2.5 mL CSF was used for routine clinical investigation. Aliquots of CSF were collected directly into polypropylene cryovials (Corning) immersed in ice-water. No samples used in this study were contaminated by blood, and so were not centrifuged. All samples were frozen within 30 min of lumbar puncture and stored at -80°C until analysis. Ten samples were thawed and then frozen again before core biomarkers were analyzed. One freeze-thaw cycle has previously been shown to not significantly affect core biomarker results (Le Bastard et al., 2015).
ELISA Assays
Cerebrospinal fluid samples were analyzed using ELISA monoplex kits according to the manufacturers’ instructions [Aβ43 (IBL), Aβ42 (Innogenetics), t-tau (Innogenetics), p-tau (Innogenetics), NF-L (UmanDiagnostics), YKL-40 (Bio-Techne, CSF diluted 1:400), GFAP (BioVendor) and progranulin (Adipogen Life Sciences, CSF diluted 1:15)]. Samples were thawed in ice-water prior to analysis, and all samples were analyzed in duplicate. Cross-reactivity for Aβ42 in the Aβ43 ELISA was given as <1%. Although this would contribute slightly to measurements for Aβ43, it would be a constant for both control and patient groups. Aβ43 was reported to have 50× less affinity than Aβ42 for the antibodies in the Aβ42 kit.
Statistical Analysis
Statistical analyses were carried out using SPSS version 24 (IBM) and Stata version 13.1. Due to multiple testing, p-values < 0.01 were considered statistically significant. Distribution of gender between groups and the distribution of the APOE 𝜀4 allele between groups were assessed with Pearson’s χ2 (chi-square) test. Differences in age at inclusion between patients with early- or late-onset of AD and respective control groups, as well as for MMSE scores and duration of disease, were assessed with the independent samples Mann–Whitney U-test for pairwise comparisons of groups. CSF analyte levels were log-transformed to approximate a normal distribution. Analyte levels were compared for younger and older participants within control and AD patient groups using t-tests for independent samples. However, when comparing analyte levels between controls and AD patients it was necessary to adjust for age because patients were significantly older than controls in both age groups. Analyte levels for the group of younger controls were therefore compared with those of early-onset AD patients, and older controls with those of late-onset AD patients using linear regression and adjusting for age at inclusion. Correlations between analytes, or between analytes and age at inclusion, were calculated with Pearson’s r. Associations are only tentative as both type 1 and type 2 errors can occur even employing a significance level of p < 0.01 as in the present study. Patterns as a whole have been considered more informative than individual correlations. To investigate potential differences in diagnostic accuracy, receiver operating characteristic (ROC) curves were made for Aβ43 and Aβ42, and the area under each ROC curve (AUC) was calculated. Youden’s index was found to determine where the sum of sensitivity and specificity was maximized. AUC was compared between Aβ43 and Aβ42 for controls and AD patients in corresponding groups [DeLong method (DeLong et al., 1988)]. Levels of analyte ratios were compared between groups, but overall did not separate groups more clearly than single analytes, and are therefore not considered further. Ratio data are given in Supplementary Table S1.
Ethics Statement
The study was conducted according to the Helsinki Declaration. Written, informed consent was obtained from all patients or suitable proxies, and from all control individuals. The Neurological Research Biobank has been licensed by the Norwegian Directorate for Health Affairs, and the research was approved by the Regional Committee for Medical Research Ethics (approval 2010/226 REK Midt, 2013/467 REK Midt, 2013/150 REK Sør-Øst).
Results
When comparing participant groups, there were no significant differences in the distribution of gender. The median age at inclusion in both younger and older control groups was significantly lower than for corresponding patient age groups. There was no significant difference in the duration of disease between the two patient groups. No significant differences were found in MMSE scores between individuals in the respective control or patient groups. Both patient groups had significantly lower median MMSE scores than their respective control group. There was increased frequency of the APOE 𝜀4 allele in combined patient compared to combined control groups (p = 0.001) (Table 1).
Cerebrospinal fluid levels of the various analytes are shown in Table 1, and scatter plots for amyloid peptides are shown in Figure 1 and in Supplementary Figures S1A–F for the other analytes. Additionally, correlations between CSF levels of Aβ peptides and other analytes, and between Aβ peptide levels and age were calculated. CSF Aβ43 was significantly decreased in patients with early-onset AD compared to late-onset AD, but no significant difference was found between the two patient groups for Aβ42. There were highly significant reductions in the levels of both Aβ43 and Aβ42 in CSF of patients with AD compared to controls. No significant differences in levels of Aβ43 or Aβ42 were found between the two control groups. Both CSF Aβ43 and Aβ42 were excellent at separating corresponding controls from patients in the AD groups, with AUCs of 0.93 or better and no significant difference in AUCs between Aβ43 and Aβ42 (Table 2). Aβ43 and Aβ42 correlated significantly with each other in all four participant groups (r = 0.58–0.85, p ≤ 0.006).
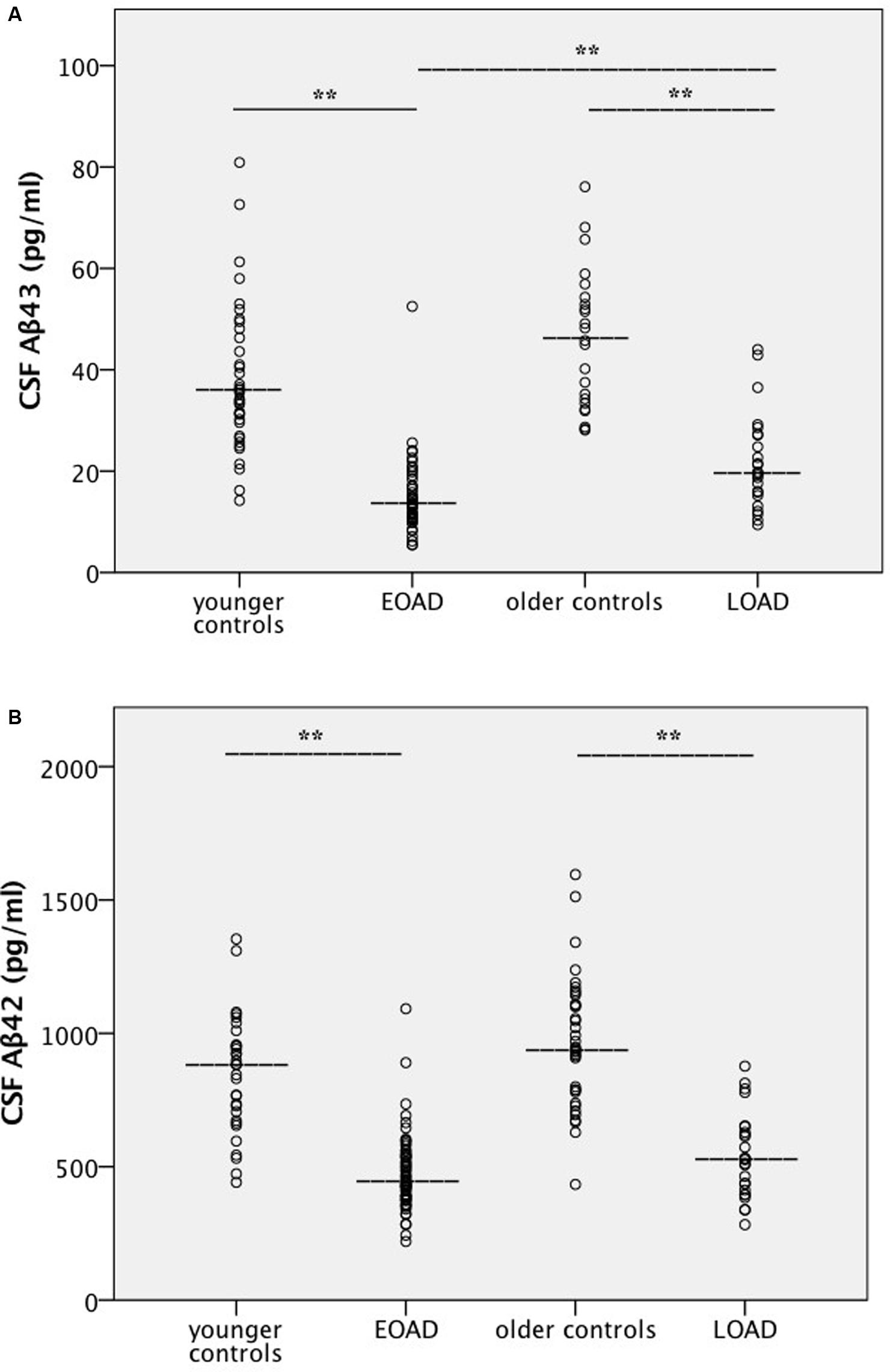
FIGURE 1. (A,B) Amyloid levels in cerebrospinal fluid. Scatter plots for all four participant groups with median lines added for each group. Values for the mean ± 1 SD are given in Table 1. Statistical analysis was performed with pairwise group comparisons of log-transformed analyte levels between controls and AD patients aged ≤62 years (age-adjusted), and between controls and AD patients aged ≥68 years (age-adjusted), as well as between younger and older groups of controls, and younger and older groups of patients with AD. (A) Aβ43, (B) Aβ42. ∗∗Significantly different at the p < 0.001 level. AD, Alzheimer’s disease; Aβ, amyloid beta; EOAD, early-onset AD; LOAD, late-onset AD.

TABLE 2. Diagnostic accuracy of β-amyloids for the separation of controls and patients.
A significant positive association between Aβ42 and age at inclusion was found in younger controls (r = 0.55, p = 0.001) and in early-onset AD (r = 0.38, p = 0.002). For older controls a trend was found for a negative correlation (r = -0.42, p = 0.012), but this was lost in late-onset AD. For Aβ43, a positive correlation with age at inclusion was found in the early-onset AD group (r = 0.43, p = 0.002), but the association was not significant in the other three participant groups.
Results for t-tau and p-tau were similar in nature, and both correlated with each other in all groups (r = 0.76–0.93, all p < 0.001). Their levels were significantly increased in patients compared to the corresponding control group, but there was no difference between patients with early- or late-onset AD. However, the older control group had significantly higher levels of the tau species compared to younger controls (Table 1). Associations between tau proteins and Aβ peptides were found only in younger controls (r = 0.43, p = 0.016 to r = 0.52, p = 0.003), not older controls or either AD group.
YKL-40 was not significantly increased in patients compared to the respective control group. However, a significant increase was found between early- and late-onset AD, as well as between younger and older controls. There was a pattern for a relationship between the Aβ peptides and YKL-40 in younger controls and early-onset AD, but correlation coefficients were low (all r = 0.33–0.44, p < 0.05 except for Aβ43 and YKL-40 in early-onset AD, p = 0.003).
A highly significant increase in the level of NF-L was found in early-onset AD compared to younger controls, but this difference was lost between late-onset AD and older controls. There was no difference between the two groups of patients, but older controls had significantly higher levels of NF-L compared to younger controls. Levels of GFAP in patients with early-onset AD were significantly higher than in younger controls. Older controls also had significantly increased levels compared to the younger controls, but no significant differences between the patient groups were found. No significant group differences in progranulin levels were found in this material.
Discussion
The most interesting result in this study is that the reduction in CSF levels of Aβ43 was more marked in early-onset compared to late-onset AD, and therefore seems to be age-related. This difference was not found for Aβ42. As expected, there was a clear and highly significant reduction in the concentration of both Aβ43 and Aβ42 in the CSF of the patient groups compared to corresponding controls. However, the data do not suggest that Aβ43 has better diagnostic accuracy for AD than Aβ42.
The increased deposition of parenchymal Aβ species in the AD brain has been suggested as the reason for the reduced amounts of Aβ peptides measured in CSF (usually Aβ42), based on the idea that less may be available for passage over the brain-CSF barrier (Fagan et al., 2006). Recent results from imaging studies showed that although CSF Aβ43 is strongly associated with cerebral amyloid deposits, even at early stages of clinical cognitive impairment (subjective cognitive decline and MCI), there were no relative differences in deposition between Aβ42 and Aβ43. Aβ43 therefore provided no diagnostic improvement over the established marker Aβ42 (Almdahl et al., 2017). Also in the present study comparing early- and late-onset AD versus the corresponding control group, no improvement to diagnostic accuracy was found for Aβ43 compared to Aβ42.
It is not immediately obvious why Aβ43 would be reduced more in early-onset than late-onset AD, other than that there is an age difference between the patient groups. However, two studies comparing amyloid imaging in early- and late-onset AD report regional (though not identical) differences in fibrillar amyloid deposition (Ossenkoppele et al., 2012; Cho et al., 2013). It is therefore possible there are age-related differences in the topographical deposition of Aβ peptides, but whether this would produce differences in CSF concentrations of the peptides in early-onset compared to late-onset AD remains unclear. We did not find a similar reduction for CSF Aβ42 in early-onset AD, and this result is very similar to previously published data (Gronning et al., 2012).
In healthy individuals, several studies have found little or no correlation between age and Aβ42 levels in CSF (Hansson et al., 2006; Bouwman et al., 2009; Mattsson et al., 2009; Popp et al., 2010). Similarly in patients with AD, a number of articles report no correlation between age and Aβ42 levels (Bouwman et al., 2009; Mattsson et al., 2009; Popp et al., 2010). Our data were similar in this respect. Even though significant correlations between age and Aβ42 in controls and patients were found, none were strong, and there was no pattern. The significance of weak correlations is dependent on the number of samples included and the significance level applied. Given that even strong correlations do not guarantee biological relevance, it can be questioned whether these fairly weak correlations are sufficiently reliable to warrant speculation of underlying physiological changes.
Generally speaking, our results for the core biomarkers in controls and in AD, as well as the association with age, agree broadly with previous studies (Blomberg et al., 2001; Bouwman et al., 2009; Glodzik-Sobanska et al., 2009; Popp et al., 2010; Alcolea et al., 2015; Chiaravalloti et al., 2016; Olsson et al., 2016). Age is also important for other substances analyzed in the present study. In recent years several reports have shown that YKL-40 increases throughout middle-age in cognitively healthy individuals, suggesting that a certain level of neuroinflammation is physiological in normal aging (Alcolea et al., 2015; Sutphen et al., 2015), as well as being an aspect of AD (Wennstrom et al., 2015). The present study agrees with the finding of increased YKL-40 levels with increased age. There was a pattern of positive correlations between Aβ species and YKL-40 in the younger groups. Most of the correlations were rather weak so it is uncertain whether this represents a physiological relationship, but tentatively agrees with previous data indicating that markers of inflammation, including YKL-40, and Aβ42 in normal aging and the early AD pathological process, are related (Alcolea et al., 2015). NF-L is well known to be increased in the CSF of patients with AD (Petzold et al., 2007; Olsson et al., 2016), and the present results are in accordance with this, but only in connection with early-onset AD. No significant difference was found for CSF NF-L between late-onset AD and older controls, which probably reflects the increase of CSF NF-L in normal aging (Vagberg et al., 2015). Similarly, we found an increase in CSF GFAP in patients with early-onset AD compared to younger controls, but again perhaps due to the increase in CSF levels of GFAP with age (Vagberg et al., 2015), this difference was lost between late-onset AD and older controls. The results for the older groups agree with one study (Wennstrom et al., 2015), but not with another study that found increased GFAP levels in AD patients compared to controls (Jesse et al., 2009). No changes in the concentration of progranulin in CSF from patients with AD were found compared to controls, as previously demonstrated (Morenas-Rodriguez et al., 2016).
Taken together, few differences were detected between early- and late-onset AD when analyzing CSF for potential markers of disease, even though the two groups had been clearly defined with respect to a difference in age. When comparing patients and controls, more differences were associated with early-onset rather than late-onset AD, perhaps because both patients and controls tend to suffer more comorbidities with increasing age which can cloud differences between patients with AD and controls. The main strength of the present study was to employ clinically well-defined patient and control cohorts that were large enough to distinguish differences and similarities in Aβ43 and Aβ42. In light of an earlier report (Lauridsen et al., 2016), future studies should probably concentrate on examining CSF Aβ43 and Aβ42 in relation to early stages of the AD process, including amnestic MCI, subjective cognitive decline, and cognitively intact individuals who have a pathological pattern of core biomarkers in CSF, or increased amyloid deposition in brain.
Author Contributions
CL planned and performed the laboratory work together with IM, PP, and GBe. SS was responsible for clinical aspects together with GG and GBr. CL analyzed the data and ØS advised on statistical analysis. LW was responsible for study design and data collation together with CL and SS. LW, SS and GBr supervised the project. CL and LW wrote the manuscript. All authors contributed to critical revision and finalization of the manuscript.
Funding
CL holds a Ph.D. scholarship from the Dementia Disease Initiation (DDI) Consortium, through the Research Council of Norway (NASATS-NevroNor grant 217780/H10).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors are sincerely grateful to all study participants.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnagi.2017.00210/full#supplementary-material
References
Alcolea, D., Martinez-Lage, P., Sanchez-Juan, P., Olazaran, J., Antunez, C., Izagirre, A., et al. (2015). Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology 85, 626–633. doi: 10.1212/wnl.0000000000001859
Almdahl, I. S., Lauridsen, C., Selnes, P., Kalheim, L. F., Coello, C., Gajdzik, B., et al. (2017). Cerebrospinal fluid levels of amyloid beta 1-43 mirror 1-42 in relation to imaging biomarkers of Alzheimer’s disease. Front. Aging Neurosci. 9:9. doi: 10.3389/fnagi.2017.00009
Blomberg, M., Jensen, M., Basun, H., Lannfelt, L., and Wahlund, L. O. (2001). Cerebrospinal fluid tau levels increase with age in healthy individuals. Dement. Geriatr. Cogn. Disord. 12, 127–132. doi: 10.1159/000051246
Bouwman, F. H., Schoonenboom, N. S., Verwey, N. A., van Elk, E. J., Kok, A., Blankenstein, M. A., et al. (2009). CSF biomarker levels in early and late onset Alzheimer’s disease. Neurobiol. Aging 30, 1895–1901. doi: 10.1016/j.neurobiolaging.2008.02.007
Bruggink, K. A., Kuiperij, H. B., Claassen, J. A., and Verbeek, M. M. (2013). The diagnostic value of CSF amyloid-beta(43) in differentiation of dementia syndromes. Curr. Alzheimer Res. 10, 1034–1040. doi: 10.2174/15672050113106660168
Chemuru, S., Kodali, R., and Wetzel, R. (2016). C-terminal threonine reduces Abeta43 amyloidogenicity compared with Abeta42. J. Mol. Biol. 428(2 Pt A), 274–291. doi: 10.1016/j.jmb.2015.06.008
Chiaravalloti, A., Koch, G., Toniolo, S., Belli, L., Lorenzo, F. D., Gaudenzi, S., et al. (2016). Comparison between early-onset and late-onset Alzheimer’s disease patients with amnestic presentation: CSF and (18)F-FDG PET study. Dement. Geriatr. Cogn. Dis. Extra 6, 108–119. doi: 10.1159/000441776
Cho, H., Seo, S. W., Kim, J. H., Suh, M. K., Lee, J. H., Choe, Y. S., et al. (2013). Amyloid deposition in early onset versus late onset Alzheimer’s disease. J. Alzheimers Dis. 35, 813–821. doi: 10.3233/jad-121927
Conicella, A. E., and Fawzi, N. L. (2014). The C-terminal threonine of Abeta43 nucleates toxic aggregation via structural and dynamical changes in monomers and protofibrils. Biochemistry 53, 3095–3105. doi: 10.1021/bi500131a
Craig-Schapiro, R., Perrin, R. J., Roe, C. M., Xiong, C., Carter, D., Cairns, N. J., et al. (2010). YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 68, 903–912. doi: 10.1016/j.biopsych.2010.08.025
DeLong, E. R., DeLong, D. M., and Clarke-Pearson, D. L. (1988). Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845. doi: 10.2307/2531595
Fagan, A. M., Mintun, M. A., Mach, R. H., Lee, S. Y., Dence, C. S., Shah, A. R., et al. (2006). Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann. Neurol. 59, 512–519. doi: 10.1002/ana.20730
Folstein, M. F., Folstein, S. E., and McHugh, P. R. (1975). “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198. doi: 10.1016/0022-3956(75)90026-6
Glodzik-Sobanska, L., Pirraglia, E., Brys, M., de Santi, S., Mosconi, L., Rich, K. E., et al. (2009). The effects of normal aging and ApoE genotype on the levels of CSF biomarkers for Alzheimer’s disease. Neurobiol. Aging 30, 672–681. doi: 10.1016/j.neurobiolaging.2007.08.019
Gronning, H., Rahmani, A., Gyllenborg, J., Dessau, R. B., and Hogh, P. (2012). Does Alzheimer’s disease with early onset progress faster than with late onset? A case-control study of clinical progression and cerebrospinal fluid biomarkers. Dement. Geriatr. Cogn. Disord. 33, 111–117. doi: 10.1159/000337386
Hansson, O., Zetterberg, H., Buchhave, P., Londos, E., Blennow, K., and Minthon, L. (2006). Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 5, 228–234. doi: 10.1016/s1474-4422(06)70355-6
Ho, G. J., Hansen, L. A., Alford, M. F., Foster, K., Salmon, D. P., Galasko, D., et al. (2002). Age at onset is associated with disease severity in Lewy body variant and Alzheimer’s disease. Neuroreport 13, 1825–1828. doi: 10.1097/00001756-200210070-00028
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jaworski, J., Psujek, M., and Bartosik-Psujek, H. (2009). Total-tau and phospho-tau(181Thr) in cerebrospinal fluid of neurologically intact population increase with age. Folia Biol. 55, 126–131.
Jesse, S., Steinacker, P., Cepek, L., von Arnim, C. A., Tumani, H., Lehnert, S., et al. (2009). Glial fibrillary acidic protein and protein S-100B: different concentration pattern of glial proteins in cerebrospinal fluid of patients with Alzheimer’s disease and Creutzfeldt-Jakob disease. J. Alzheimers Dis. 17, 541–551. doi: 10.3233/jad-2009-1075
Jing, H., Tan, M. S., Yu, J. T., and Tan, L. (2016). The role of PGRN in Alzheimer’s disease. Mol. Neurobiol. 53, 4189–4196. doi: 10.1007/s12035-015-9358-0
Keller, L., Welander, H., Chiang, H. H., Tjernberg, L. O., Nennesmo, I., Wallin, A. K., et al. (2010). The PSEN1 I143T mutation in a Swedish family with Alzheimer’s disease: clinical report and quantification of Abeta in different brain regions. Eur. J. Hum. Genet. 18, 1202–1208. doi: 10.1038/ejhg.2010.107
Lauridsen, C., Sando, S. B., Shabnam, A., Moller, I., Berge, G., Grontvedt, G. R., et al. (2016). Cerebrospinal fluid levels of amyloid beta 1-43 in patients with amnestic mild cognitive impairment or early Alzheimer’s disease: a 2-year follow-up study. Front. Aging Neurosci. 8:30. doi: 10.3389/fnagi.2016.00030
Le Bastard, N., De Deyn, P. P., and Engelborghs, S. (2015). Importance and impact of preanalytical variables on Alzheimer disease biomarker concentrations in cerebrospinal fluid. Clin. Chem. 61, 734–743. doi: 10.1373/clinchem.2014.236679
Marshall, G. A., Fairbanks, L. A., Tekin, S., Vinters, H. V., and Cummings, J. L. (2007). Early-onset Alzheimer’s disease is associated with greater pathologic burden. J. Geriatr. Psychiatry Neurol. 20, 29–33. doi: 10.1177/0891988706297086
Mattsson, N., Zetterberg, H., Hansson, O., Andreasen, N., Parnetti, L., Jonsson, M., et al. (2009). CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302, 385–393. doi: 10.1001/jama.2009.1064
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. doi: 10.1212/WNL.34.7.939
Morenas-Rodriguez, E., Cervera-Carles, L., Vilaplana, E., Alcolea, D., Carmona-Iragui, M., Dols-Icardo, O., et al. (2016). Progranulin protein levels in cerebrospinal fluid in primary neurodegenerative dementias. J. Alzheimers Dis. 50, 539–546. doi: 10.3233/jad-150746
Olsson, B., Lautner, R., Andreasson, U., Ohrfelt, A., Portelius, E., Bjerke, M., et al. (2016). CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 15, 673–684. doi: 10.1016/s1474-4422(16)00070-3
Ossenkoppele, R., Zwan, M. D., Tolboom, N., van Assema, D. M., Adriaanse, S. F., Kloet, R. W., et al. (2012). Amyloid burden and metabolic function in early-onset Alzheimer’s disease: parietal lobe involvement. Brain 135(Pt 7), 2115–2125. doi: 10.1093/brain/aws113
Petzold, A., Keir, G., Warren, J., Fox, N., and Rossor, M. N. (2007). A systematic review and meta-analysis of CSF neurofilament protein levels as biomarkers in dementia. Neurodegener. Dis. 4, 185–194. doi: 10.1159/000101843
Popp, J., Lewczuk, P., Frommann, I., Kolsch, H., Kornhuber, J., Maier, W., et al. (2010). Cerebrospinal fluid markers for Alzheimer’s disease over the lifespan: effects of age and the APOEepsilon4 genotype. J. Alzheimers Dis. 22, 459–468. doi: 10.3233/jad-2010-100561
Rabinovici, G. D., Furst, A. J., Alkalay, A., Racine, C. A., O’Neil, J. P., Janabi, M., et al. (2010). Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain 133(Pt 2), 512–528. doi: 10.1093/brain/awp326
Saito, T., Suemoto, T., Brouwers, N., Sleegers, K., Funamoto, S., Mihira, N., et al. (2011). Potent amyloidogenicity and pathogenicity of Abeta43. Nat. Neurosci. 14, 1023–1032. doi: 10.1038/nn.2858
Sandebring, A., Welander, H., Winblad, B., Graff, C., and Tjernberg, L. O. (2013). The pathogenic abeta43 is enriched in familial and sporadic Alzheimer disease. PLoS ONE 8:e55847. doi: 10.1371/journal.pone.0055847
Savva, G. M., Wharton, S. B., Ince, P. G., Forster, G., Matthews, F. E., and Brayne, C. (2009). Age, neuropathology, and dementia. N. Engl. J. Med. 360, 2302–2309. doi: 10.1056/NEJMoa0806142
Sutphen, C. L., Jasielec, M. S., Shah, A. R., Macy, E. M., Xiong, C., Vlassenko, A. G., et al. (2015). Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 72, 1029–1042. doi: 10.1001/jamaneurol.2015.1285
Vagberg, M., Norgren, N., Dring, A., Lindqvist, T., Birgander, R., Zetterberg, H., et al. (2015). Levels and age dependency of neurofilament light and glial fibrillary acidic protein in healthy individuals and their relation to the brain parenchymal fraction. PLoS ONE 10:e0135886. doi: 10.1371/journal.pone.0135886
Welander, H., Franberg, J., Graff, C., Sundstrom, E., Winblad, B., and Tjernberg, L. O. (2009). Abeta43 is more frequent than Abeta40 in amyloid plaque cores from Alzheimer disease brains. J. Neurochem. 110, 697–706. doi: 10.1111/j.1471-4159.2009.06170.x
Wennstrom, M., Surova, Y., Hall, S., Nilsson, C., Minthon, L., Hansson, O., et al. (2015). The inflammatory marker YKL-40 is elevated in cerebrospinal fluid from patients with Alzheimer’s but not Parkinson’s disease or dementia with Lewy bodies. PLoS ONE 10:e0135458. doi: 10.1371/journal.pone.0135458
Winblad, B., Palmer, K., Kivipelto, M., Jelic, V., Fratiglioni, L., Wahlund, L. O., et al. (2004). Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 256, 240–246. doi: 10.1111/j.1365-2796.2004.01380.x
Keywords: early-onset Alzheimer’s disease, biomarkers, tau, YKL-40, neurofilament light, glial fibrillary acidic protein, progranulin
Citation: Lauridsen C, Sando SB, Møller I, Berge G, Pomary PK, Grøntvedt GR, Salvesen Ø, Bråthen G and White LR (2017) Cerebrospinal Fluid Aβ43 Is Reduced in Early-Onset Compared to Late-Onset Alzheimer’s Disease, But Has Similar Diagnostic Accuracy to Aβ42. Front. Aging Neurosci. 9:210. doi: 10.3389/fnagi.2017.00210
Received: 03 March 2017; Accepted: 14 June 2017;
Published: 28 June 2017.
Edited by:
Catarina Oliveira, University of Coimbra, PortugalReviewed by:
Gianluigi Zanusso, University of Verona, ItalyCharlotte Elisabeth Teunissen, VU University Amsterdam, Netherlands
Copyright © 2017 Lauridsen, Sando, Møller, Berge, Pomary, Grøntvedt, Salvesen, Bråthen and White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda R. White, bGluZGEud2hpdGVAbnRudS5ubw==